

Introduction
Fabry disease is an X-linked lysosomal storage disease, resulting from a deficiency of the enzyme α-galactosidase A and subsequent cellular storage of the enzyme substrate globotriaosylceramide (Gb3) [1]. Estimates of the incidence of Fabry disease vary markedly, from 1:<5000 male births in a newborn screening study in Italy [2] to 1:117 000 male births in Australia [3] and 1:833 000 male births in northern Portugal [4]. In general, hemizygous males are more severely affected than heterozygous females. In males, life expectancy is reduced by an average of 20 years [5] and in females by 15 years [6]. Although males tend to suffer symptoms earlier than females, both boys and girls can be affected from an early age [7]. Death usually occurs due to renal, cardiovascular or cerebrovascular complications [5,6,8], with renal dysfunction being the main cause of death in men before the development of renal failure requiring dialysis and transplantation [9].
As enzyme replacement therapy (ERT) has recently become available, it is important to recognize the signs and symptoms of Fabry disease so that early treatment can be started before irreversible organ damage occurs. This short review outlines the renal manifestations of Fabry disease and the results of ERT.
Clinical features
Natural history
Urinary concentration defects, despite being rare, may be the earliest manifestation of renal dysfunction, and are responsible for polyuria and nycturia. Otherwise, the first clinical manifestation of Fabry-related renal disease is proteinuria. In their review of the natural history of renal involvement in 105 male patients, Branton et al. found that 50% of patients presented with proteinuria by 35 years of age and 100% by 52 years of age [9]. In patients with Fabry nephropathy in FOS—the Fabry Outcome Survey—the usual level of proteinuria is 0.5–2.0 g per 24 h. True nephrotic syndrome is not frequent, even in patients who have proteinuria in the nephrotic range [9]. Microscopic haematuria is present in 30% of patients.
Chronic renal insufficiency (CRI) develops concomitantly to proteinuria in the majority of male patients. Branton et al. found that 50% of male patients presented with CRI by 42 years of age. After the development of CRI, there was a rapid decline in glomerular filtration rate (GFR) of 12.2 ml/min/year (range, 3–34 ml/min/year), leading to end-stage renal disease (ESRD) after further 4.1 years [9]. It is of note that only 18 of the 105 patients studied had received angiotensin antagonist therapy at any time (6 for hypertension alone, 1 for hypotension and proteinuria, 5 for hypertension with CRI, 3 with proteinuria alone, 1 with congestive heart failure, 2 for reasons unknown) and 11 of the 31 hypertensive patients had medically controlled hypertension. Unpublished data from the Necker Hospital in Paris suggest that the rate of decrease in GFR may be slower in patients with Fabry disease who are receiving these nephroprotective measures as standard. It is interesting that the progressive decline in GFR reported by Branton and colleagues is similar to that seen in untreated diabetic nephropathy and occurs more rapidly than in primary glomerulonephritis [10]. Both these diseases are multisystemic and their nephropathies are caused initially by metabolic deposits—Gb3 in Fabry disease and end-glycosylation products in diabetes. They may both progress in a similar manner to chronic non-diabetic proteinuric nephropathies via a common pathophysiological pathway that is relatively independent of the initial renal insult [11,12]. Contrary to patients with diabetes type I or II, nearly all male patients with Fabry disease will develop CRI during their lifetime. Hypertension is observed in 60% of patients with diabetic nephropathy and, if untreated, contributes to the progression of CRI [13]. Similarly, hypertension has been found in 57% of men and 47% of women with Fabry disease [14]. Finally, as for other monogenic diseases, there is a remarkable variability in the rate of progression of nephropathy [15] that may be partly explained by the role of modifier genes.
A renal biopsy may be used to diagnose Fabry disease in patients with renal symptoms or to follow treatment with ERT; however, diagnosis is often based on non-renal manifestations [9]. Such manifestations include characteristic skin lesions (angiokeratomas), acroparaesthesia, cornea verticillata and left ventricular hypertrophy. A diagnosis may also be made as a result of a family history of the disease. Confirmation of the diagnosis by measurement of the plasma and leukocyte activity of α-galactosidase A in male patients and genetic analysis in female patients is necessary.
Because Fabry disease is a rare, often unrecognized disease, the nephropathy is often already present by the time diagnosis is made. For example, Branton et al. reported that the mean age at diagnosis of Fabry disease in 44 patients was 16 ± 13 years, even though these patients had a known family history. The age at diagnosis of 50 patients without a known family history of Fabry disease was even higher, at 28 ± 12 years, despite signs of the disease being apparent well before diagnosis. Other studies and data from FOS have also highlighted the delay between onset of symptoms in childhood and diagnosis [5,7], pointing to the need for physicians to rediscover the disease because of the availability of ERT and early nephroprotective measures.
Other newly recognized aspects of Fabry nephropathy have been reported recently. In a study of male children with Fabry disease, Ries et al. found that 20 out of 25 patients had microalbuminuria and that 13 out of 25 had an estimated GFR (using the Schwartz formula) above 140 ml/min, suggestive of hyperfiltration, a well-known risk factor for glomerular injury that is responsible for increased glomerular permeability to albumin [16].
Moreover, another recent study has shown that there may already be evidence of CRI (estimated GFR of 60–89 ml/min/1.73 m2) in some untreated children with Fabry disease (n = 2/24) [17]. There is clear evidence that the burden of Fabry nephropathy is already present in boys with Fabry disease and probably starts with a (presumed) silent phase, including hyperfiltration and/or microalbuminuria (stage II Fabry nephropathy; Table 1), which progresses to overt proteinuria, CRI and, finally, ESRD, an evolution closely resembling that seen in diabetic nephropathy (Figure 1) [9]. Renoprotective treatment in patients with Fabry disease should therefore be the same as that for patients with type 1 and type 2 diabetes; i.e. these patients should be treated during the silent phase of the disease to slow, halt or even reverse the pathological changes that lead to renal failure. The literature suggests that the course of kidney disease is generally less severe in females; however, some of them will progress to ESRD before the age of 40 years [18].
Table 1.
Proposed stages of progression of Fabry nephropathy in male patients
| Time of evolution | |||||
|---|---|---|---|---|---|
| (years) | Urinanalysis | Level of GFR | Pathology | Efficacy of ERT | |
| Stage I | 0–5 | Normal | <90 ml/min <140 ml/min | Gb3 deposits mainly in podocytes and capillaries +/++ | Incomplete clearance of deposits after 5 years |
| Stage II | 6–19 | Microalbuminuria (30–300 mg/day) | >140 ml/min | Gb3 deposits ++/+++ Mesangial expansion + | Possible reversibility of hyperfiltration; no data on microalbuminuria Point of no return? |
| Stage III | 20–29 | Overt proteinuria (>300 mg/day) | >60 ml/min <140 ml/min | Gb3 deposits ++/+++ Mesangial expansion ++ Glomerulosclerosis +/++ Tubular atrophy + Arterial remodelling +/++ | No significant effect on proteinuria Stabilization of GFR in the majority of patients |
| Stage IV | >30 | Overt proteinuria Proteinuria of nephrotic range Nephrotic syndrome (rare) | <60 ml/min to ESRD | Gb3 deposits ++/+++ Glomerulosclerosis ++/+++ Interstitial fibrosis ++/+++ Tubular atrophy ++/+++ Arterial remodelling ++/+++ | No stabilization but slows the rate of progression of chronic renal insufficiency |
CRI, chronic renal insufficiency; ESRD, end-stage renal disease; ERT, enzyme replacement therapy; Gb3, globotriaosylceramide; GFR, glomerular filtration rate; +, mild; ++, moderate; +++, severe.
Fig. 1.

The interrelationships between functional and morphological markers in diabetic and Fabry nephropathy. Great similarities are found between these conditions.
Pathophysiology
Progressive accumulation of Gb3 in the kidney results in mesangial expansion and glomerulosclerosis, tubular atrophy and interstitial fibrosis—non-specific anomalies seen in most glomerular diseases and diabetic nephropathy [19]. Vascular injury due to Gb3 deposition in the capillaries and arterial wall probably plays a central role in the loss of nephrons, via ischaemic degenerative alterations. As such, Fabry nephropathy is not only a disease associated with deposition, it should also be considered a vascular disease, like diabetic nephropathy. However, it is likely that other mechanisms, secondary to the progressive accumulation of Gb3, are also involved in the pathogenesis of Fabry nephropathy.
In patients with Fabry nephropathy, all types of renal glomerular cells show lipid inclusions—as do the endothelial and smooth muscle cells of renal vessels, epithelial cells of the proximal tubule, loop of Henle, distal tubule and interstitial cells [20]. Podocytes, in particular, contain high levels of glycosphingolipid deposits early during childhood, although it remains unclear whether the glomerular filtration barrier is physically compromised, as many women with Fabry disease have podocyte deposits without proteinuria throughout their life. Nevertheless, GFR declines over time, mainly due to ischaemic degenerative changes provoked by vascular involvement and progressive expansion of the mesangial matrix, which occurs concomitant to the development of overt proteinuria. Expansion of the mesangial matrix, in turn, leads to thickening of the glomerular basement membrane and ultimately to glomerulosclerosis [21]. In order to evaluate the renal response to ERT, a scoring system for chronic glomerular, vascular, tubular and interstitial alterations is needed.
A series of guidelines for the recognition, evaluation and surveillance of disease-associated morbidities and therapeutic strategies for Fabry disease have been proposed by an international panel of experts [22]. These guidelines recommend that evaluation of kidney function, including estimates of GFR and proteinuria, should be carried out in every patient with Fabry disease. Furthermore, they note that a multidisciplinary approach to treatment of Fabry disease is required, necessitating cooperation and participation between geneticists, nephrologists, cardiologists, neurologists and other clinicians.
Treatment for Fabry nephropathy
Transplantation
Kidney transplantation, with cadaveric or living donor, is considered the optimal therapy for ESRD in suitable patients. A recent analysis of the United States Renal Data System (USRDS) database found 93 incident cases of Fabry-related ESRD, where patients underwent renal transplantation between 1988 and 1998 [23]. One-, five-, and ten-year graft survival in recipients with Fabry disease (91%, 76% and 56%, respectively) was statistically similar to rates of graft survival in patients without Fabry disease (88%, 67% and 49%, respectively). Furthermore, survival rate and the risk of cardiovascular death in untreated (i.e. not receiving ERT) patients with Fabry disease were not statistically different from those of other renal transplant recipients, with cumulative 10-year Kaplan–Meier patient survival estimate being 67% in recipients with untreated Fabry disease and 63% in controls. Despite these surprisingly good results, which may be due to the selection of excellent candidates for transplantation for the study, transplanted patients should also be treated with ERT, as renal transplantation does not correct the underlying metabolic deficit in other organs.
Enzyme replacement therapy
The introduction of ERT with recombinant α-galactosidase A offers the prospect of altering the natural course of Fabry disease. It is still important, however, to systematically consider adjunctive renoprotective treatments (ACE inhibitors and/or ARBs) that are known to be effective in slowing disease progression in other chronic proteinuric kidney diseases [24].
Two forms of α-galactosidase A have been approved in Europe for use in patients with Fabry disease: agalsidase alfa (Replagal®; Shire Human Genetic Therapies, Boston, MA, USA) and agalsidase beta (Fabrazyme®; Genzyme Corp., Cambridge, MA, USA). Agalsidase alfa is produced in a continuous human cell line. It is administered as an intravenous (i.v.) infusion over 40 min at a dose of 0.2 mg/kg body weight every 2 weeks. Agasidase beta is produced in Chinese hamster ovary (CHO) cells and is a chimeric protein. It is given as an i.v. infusion over ∼4.7 h for a 70 kg patient at a dose of 1.0 mg/kg body weight every 2 weeks. These recommended doses for agalsidase alfa and agalsidase beta were determined using dose-finding studies. In the case of agalsidase alfa, a range of doses (0.007–0.1 mg/kg) were administered to male patients with Fabry disease (n = 2 for each dose) [25]. Pharmacokinetic profiles and tolerability were comparable at all doses and levels of urinary sediment Gb3 were decreased by similar amounts [26]. Based on these observations, a dose of 0.2 mg/kg every 2 weeks was tested in longer-term studies and was found to be well tolerated in patients with Fabry disease [27]. In the case of agalsidase beta, the effects of 0.3, 1.0 and 3.0 mg/kg once every 2 weeks and 1.0 and 3.0 mg/kg once every 2 days were evaluated [25]. A reduction of Gb3 was observed in the kidney, heart, skin and plasma at all doses. Plasma Gb3 was cleared in a dose-dependent manner, but was less consistent at the dose of 0.3 mg/kg. Infusion-associated reactions were also dose dependent [25].
The efficacy and, possibly, the safety of ERT critically depend on the infused therapeutic enzyme being recognized as an endogenous compound and transported to its site of activity—the lysosome. Enzymes produced for ERT should therefore resemble as closely as possible their natural counterparts, including the pattern of glycosylation. As glycosylation patterns are species- and cell-type specific, it has been suggested that this may have important implications in the production of recombinant therapeutic proteins [28]. While separate clinical studies have demonstrated the efficacy of both agalsidase alfa and agalsidase beta in slowing the progression of Fabry disease [27,29], there are limited data on the comparative efficacy and tolerability of these preparations when tested in parallel. In the only clinical head-to-head comparison of agalsidase alfa and agalsidase beta (both at 0.2 mg/kg EOW), the clinical potency of these preparations was found to be broadly similar [30]. The one difference reported between the two preparations being that among patients with elevated urinary Gb3 levels at baseline, mean levels were significantly reduced in the agalsidase alfa group (P = 0.04) but not the agalsidase beta group (P = 0.66). However, due to the small number of patients enrolled in this study, inconsistencies in the collection of baseline and follow-up measurements and the observation by the authors that damage in this severely affected older population group will probably not respond to therapy, the relative efficacies of these drugs remains incompletely defined.
As already mentioned, variation in glycosylation patterns between agalsidase alfa and agalsidase beta may have implications for the long-term safety of ERT in patients with Fabry disease. Differences in antibody formation have been reported between these CHO- and human fibroblast-derived enzymes. Data from long-term clinical trials using the approved doses suggest that IgG antibodies develop in a higher proportion of patients receiving agalsidase beta (89.7%) than agalsidase alfa (56%) [31,32]; however, differences in methods of antibody detection make comparison difficult. Antibody formation has also been assessed in two head-to-head trials; however, these studies were on small patient groups and have included a lower dose of agalsidase beta than typically employed clinically, making conclusions limited [30,33]. In the first study, IgG antibodies were reported in 11 out of 18 patients (in 4 out of 7 patients on 0.2 mg/kg agalsidase alfa EOW, 4 out of 6 patients on 0.2 mg/kg agalsidase beta EOW, 3 out of 5 patients on 1.0 mg/kg agalsidase beta EOW) during the first 6–12 months of treatment and all antibodies exhibited neutralizing capacities in vitro and there was complete cross-reactivity [33]. In a second 24-month study comparing both treatments at the same dose (0.2 mg/kg EOW), antibodies developed in four out of eight patients on agalsidase alfa and six out of eight patients on agalsidase beta [30]. While these data should be interpreted with caution, even when receiving a lower dose than typically given clinically, a greater proportion of patients on agalsidase beta appear to develop antibodies. Furthermore, IgE antibodies have been reported after infusion of agalsidase beta [32,34], but not so far in patients given agalsidase alfa.
Several studies have reported that IgG antibody formation interferes with urinary and plasma Gb3 clearance [30,31,33,35–37]; however, the relationship between antibodies, Gb3 levels and clinical response is far from clear. The elevated levels of urinary and plasma Gb3 reported in patients with IgG antibodies suggest that these antibodies may have a neutralizing effect. Consistent with these reports, studies of both enzymes in small patient groups have reported in vitro inhibition of serum enzyme activity in patients with IgG antibodies [33,35]. It has been suggested that higher doses may be needed in patients with antibodies to ensure delivery to the target tissues [36]; however, this has yet to be thoroughly evaluated and it must be remembered that in vitro enzyme inhibition does not necessarily imply a loss of efficacy at the level of the lysosome.
It is also important to note that clinical improvements have been reported in patients with IgG antibodies and elevated Gb3 levels. In the recent head-to-head study, despite the failure of treatment with either product to clear urinary and plasma Gb3 in antibody positive patients, the induction of antibodies did not correlate with the occurrence of treatment failure (defined as progression of renal disease, cardiac disease or occurrence of a new cerebrovascular accident/lacunar infarction) [30]. In the clinical trial by Schiffmann et al., the presence or absence of persistently positive IgG antibodies against agalsidase alfa did not appear to correlate with the magnitude or direction of changes in estimated GFR in individual patients [31]. In fact, the authors report that there were patients with persistent antibodies who demonstrated improvement in estimated GFR. Such data show that treatment may be clinically effective despite a failure to effectively clear Gb3, calling into question the usefulness of Gb3 for monitoring the response to treatment. It is clear that well-controlled studies using comparable methodology are needed to address these issues.
Clinical efficacy
The original studies of the efficacy of agalsidase alfa and agalsidase beta were carried out by Schiffmann et al. [26,27] and Eng et al. [25,29], respectively. Schiffmann et al. undertook a double-blind, randomized, placebo-controlled study of agalsidase alfa, administered for 6 months to 26 patients with Fabry disease. This was followed by a 12-month open-label maintenance study of all patients completing the placebo-controlled study [26,27]. Statistically significant reductions in neuropathic pain (P = 0.02), the primary endpoint of the study, were accompanied by a statistically significant improvement in creatinine clearance compared with placebo (P = 0.02). This improvement in renal function was maintained over a further 12-month follow-up in patients treated with agalsidase alfa, and renal function was stabilized in the patients who were initially given placebo and then switched to ERT.
In line with the stabilization in renal function, histopathological assessments showed a 21% increase in the proportion of normal glomeruli and a 33% decrease in mesangial widening following agalsidase alfa treatment. In contrast, the placebo-treated group saw a 27% decrease in the proportion of normal glomeruli and a 69% increase in mesangial widening. Furthermore, a significant decrease in glycolipid deposits in vascular endothelial cells and a significant fall in Gb3 concentrations in plasma and urinary sediment were seen in patients treated with agalsidase alfa compared with those given placebo at the end of the 12-month follow-up study. These metabolic improvements were maintained throughout 48 months of treatment with agalsidase alfa [31]. In addition, this study showed that long-term treatment appeared to stabilize kidney function in patients with mild-to-moderate chronic kidney disease at baseline (GFR ≥ 60 ml/min/1.73 m2), and to slow the decline in renal function in the subgroup of adult male patients with an estimated GFR of 30–59 ml/min/1.73 m2 at baseline compared with historical controls [9].
Eng et al. conducted a double-blind, randomized, placebo-controlled study in 58 male patients with Fabry disease who were treated with agalsidase beta every 2 weeks for 20 weeks. The primary endpoint of the study was clearance of Gb3 deposits in interstitial capillary endothelial cells [29]. Clearance was achieved in 20 of the 29 patients receiving active therapy compared with 0 out of 29 patients given placebo. Further histopathological analysis of kidney biopsy samples from 48 of the 58 patients showed nevertheless that Gb3 clearance in podocytes and the distal tubular epithelium was limited, compared with other renal cell types [38]. In the open-label extension trial that followed, Wilcox et al. also demonstrated that renal function, as measured by mean serum creatinine and estimated GFR which was normal at baseline, remained normal over 30–36 months of treatment with agalsidase beta [32]. Of note, only 10 patients in this trial had estimated GFR values below 90 ml/min/1.73 m2 before treatment and renal function was either stabilized or improved in seven of this group by the end of the study. Furthermore, as in the study by Schiffmann et al., there is no information in this study on whether patients were receiving standard nephroprotective measures (ACE inhibitors and/or ARBs) during this study.
Following publication of the baseline characteristics of 366 patients registered in FOS, an outcomes database for patients with Fabry disease who are receiving, or may receive, ERT with agalsidase alfa [8], Beck et al. assessed the overall effects of 12 and 24 months of agalsidase alfa treatment [39]. Renal disease (GFR < 90 ml/min/1.73/m2 and/or proteinuria) was present in 84% (n = 274) of the patients, for whom data were available. No data are presented on the use of ACE inhibitors or ARBs in these patients. Renal function was stabilized in those patients who had a mild or moderate deterioration in renal function at baseline (GFR between 60 and 90 ml/min/1.73/m2 or 30 and 60 ml/min/1.73/m2, respectively) following 1 and 2 years of ERT (no data were reported for proteinuria). This should be interpreted in the context of the rapid decline in GFR (by 12 ml/min/year) that occurs in untreated patients with CRI [9]. In the study by Branton et al. (2002), 18 out of 105 received an angiotensin agonist at any time. ERT with agalsidase alfa may thus help to arrest the progression to irreversible kidney failure in patients with mild or moderate Fabry nephropathy according to data from 2 years of treatment, but a longer follow-up is needed to evaluate the long-term benefit on GFR. Interestingly, pancreatic transplantation can reverse the glomerulosclerosis seen in diabetic nephropathy in patients with type I diabetes, but this requires more than 5 years of normoglycaemia [40].
Recently, Schwarting et al. reported a longitudinal analysis of data from FOS, on the progression of renal dysfunction in 201 patients with Fabry disease treated with agalsidase alfa [41]. Among this population, data were available from 1 year before treatment, at the start of treatment and 1 year after starting treatment in 20 patients (12 with a baseline GFR of 60–89 ml/min/1.73 m2; 8 with a baseline GFR of 30–59 ml/min/1.73 m2), and 2 years after starting treatment in 13 patients of this group (8 with a baseline GFR of 60–89 ml/min/1.73 m2; 5 with a baseline GFR of 30–59 ml/min/1.73 m2).
The results of the analysis revealed that the statistically significant (P < 0.05) decline in renal function, observed over the year prior to commencing ERT in patients with a GFR of 60–89 ml/min/1.73 m2 at baseline, was halted after 1 year of treatment with agalsidase alfa. Furthermore, renal function remained stable in patients with both mild and moderate renal disease at baseline treated with agalsidase alfa for 2 years (Figure 2). Analysis of proteinuria before and after ERT showed inconsistent findings with some patients showing an increase and others a decrease. Persistence of significant proteinuria (≥1 g/day) is a well-known factor in the progression of CRI, whatever the aetiology of the nephropathy [11,12]. Of the 20 patients followed for 2 years, 5 used ACE inhibitors or ARBs at baseline, another 5 used these drugs at any time and the information was missing for 10 patients. A multivariate analysis on data collected from 201 patients with serum creatinine levels below 2 mg/dl (<180 mmol/l) revealed a negative correlation between serum creatinine and time on agalsidase alfa (P < 0.05), further supporting the conclusion that agalsidase alfa may prevent deterioration of renal function in patients with Fabry disease who have mild or moderate renal impairment [41].
Fig. 2.
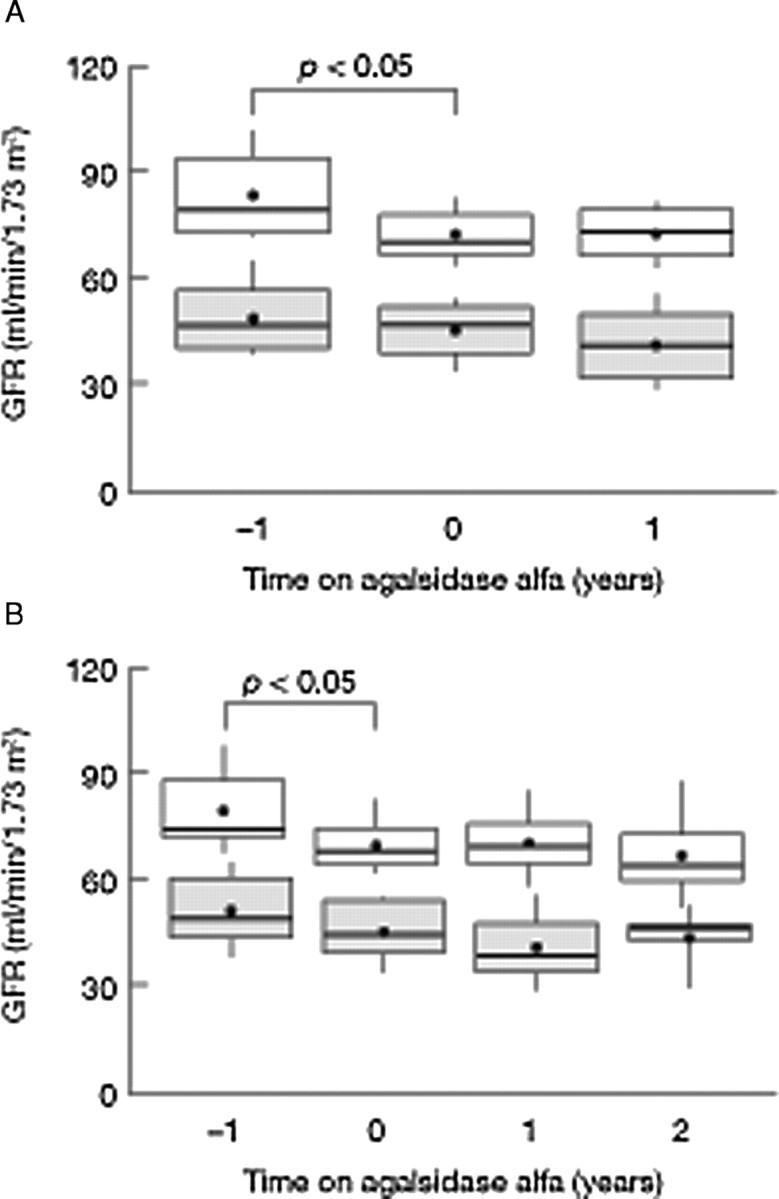
(A) Change in estimated glomerular filtration rate (eGFR) in 20 patients before and 1 year after the initiation of enzyme replacement therapy (ERT) with agalsidase alfa. eGFR 1 year before treatment, at baseline and 1 year after the start of ERT in 12 patients with a mean baseline GFR of 60–89 ml/min/1.73 m2 (top) and 8 patients with a mean eGFR of 30–59 ml/min/1.73 m2 (bottom). (B) Change in eGFR in 13 patients before and during 2 years after the initiation of ERT with agalsidase alfa. eGFR 1 year before treatment, at baseline and 1 and 2 years after the start of ERT in 8 patients with a mean baseline GFR of 60–89 ml/min/1.73 m2 (top) and 5 patients with a mean eGFR of 30–59 ml/min/1.73 m2 (bottom). Data are shown as mean (dot), median (rule) and 10th, 25th, 75th and 90th percentiles (box and whiskers). Adapted with permission from Schwarting et al. [41], courtesy of Dustri–Verlag.
In a more recent open-label extension trial, Germain et al. reported longitudinal renal data in 58 patients (56 male) with Fabry disease with normal baseline GFR, receiving agalsidase beta for up to 54 months [42]. They showed that median serum creatinine and estimated GFR remained stable (normal) at month 54 in the 41 patients for whom data were available. Renal disease progressed in only six patients (decline in estimated GFR ranging from −3.5 to −12.1 ml/min per 1.73 m2/year), four of whom were over 40 years of age and had significant proteinuria and evidence of sclerotic glomeruli at baseline. Five of this group were receiving ACE inhibitors/ARBs during the trial (a further 18 patients also received ACE inhibitors/ARSs over the study). Subgroup analyses revealed that high proteinuria or glomerulosclerosis at baseline contributed to the rate of decline in estimated GFR. So the effect of ERT combined with ACE inhibitors/ARBs on proteinuria in Fabry disease is not clear. In addition, the study by Germain et al. showed that Gb3 was cleared (as indicated by a score of zero) from renal interstitial capillary endothelial cells in 47 out of 49 patients after 6 months of treatment, and in 8 out of 8 patients after 54 months of treatment. In the available small samples (range, n = 5–8), total clearance of Gb3 was also reported in the distal convoluted tubules/collecting ducts, glomerular endothelial cells, mesangial cells and non-capillary cells of the kidney; however, clearance was not complete in the interstitial cells and non-capillary smooth muscle. Taken together, these data suggest that early and continued intervention with agalsidase beta can preserve renal function in patients with normal renal function (and without significant proteinuria or severe glomerulosclerosis) at the start of treatment.
A recent study investigating the agressive use of ACE inhibitors/ARBs in patients with Fabry disease showed that proteinuria was reduced by this therapy and that this effect was maintained during ERT [43]. Although there was an initial decline in estimated GFR after the introduction of ACE inhibitors/ARBs (which was significant for those with stage 3 or 4 kidney disease at baseline and probably due to lowering of blood pressure), following the start of ERT, renal function as measured by estimated GFR remained stable. The average progression rate of estimated GFR during treatment with agalsidase beta (mean, 30.3 months; range, 16–43 months) was 1.18 ± 2.78 ml/min per 1.73 m2 per year in patients with stage 1 or 2 kidney disease and −0.23 ± 1.12 ml/min per 1.73 m2 per year in patients with stage 3 or 4 kidney disease at baseline. Although this study did not include a control group receiving ERT alone, these data support the combined use of ACE inhibitors/ARBs and ERT in the treatment of renal disease in patients with Fabry disease.
Banikazemi et al. studied the effects of ERT in 51 patients with advanced Fabry disease (defined as having serum creatinine measurements between 106 μmol/l and 265 μmol/l or an estimated creatinine clearance <80 ml/min) receiving agalsidase beta every 2 weeks for up to 35 months [44]. The results of the study showed that ERT was able to slow progression to the composite clinical outcome (defined as any clinical event) of renal, cardiac and cerebrovascular complications and death compared with placebo (n = 31), even in patients with overt kidney dysfunction. This indicates that progression to severe manifestations was slowed. Nevertheless, the authors stress that they feel that therapeutic intervention before the onset of irreversible organ damage is important.
An open-label study of treatment with agalsidase beta in 25 patients (mean age, 41 years) with severe symptoms and organ manifestations (19 men, 6 women; mean age, 30 years) presented data on renal function after a mean treatment time of 23 ± 8 months in 20 patients (three with functioning kidney grafts) [45]. These authors reported clear differences in the response to ERT depending on the baseline characteristics of the patients. While GFR appeared to be stabilized by ERT in patients with normal renal function at baseline (GFR > 90 ml/min/1.73 m2), GFR was not stabilized (but the rate of progression was reduced compared with historical cases) in those patients with impaired renal function at baseline (mean GFR, 71 ± 17 ml/min/1.73 m2), suggesting that there is a point of no return where kidney damage is irreversible and CRI progresses, despite ERT.
A recent study by Schiffman et al. provides evidence that it may be possible to reverse renal impairment by increasing the dose and frequency of infusions [46]. In this study, weekly infusions of 0.2 mg/kg agalsidase alfa were given to a subgroup of 11 adult male patients, in whom there was a continuing decline in renal function (estimated GFR decline > 5 ml/min per 1.73 m2/year) despite 2–4 years of conventional agalsidase alfa therapy (0.2 mg/kg EOW). After switching to weekly infusions, there was a slowing in the rate of decline of estimated GFR in six patients and an improvement in three patients. Analysis confirmed that the main factor responsible for this was weekly infusions of agalsidase alfa, with a weaker contribution from the concomitant use of ACE inhibitors/ARBs. Of note, although the three patients for whom estimated GFR improved had low proteinuria upon switching to the weekly dose, five of the seven patients with baseline proteinuria in excess of 1000 mg/24 h showed a substantial slowing of the loss of GFR after switching. These data suggest that for a certain subgroup of patients, an increased dose or frequency of infusion could be beneficial. However, caution is needed before considering a switch from agalsidase alfa to agalsidase beta in such patients, as data from the study by Germain, discussed earlier, suggest a significant loss of GFR despite a dose of 1.0 mg/kg of agalsidase beta in some patients with normal baseline GFR who had proteinuria or glomerulosclerosis [42]. There is clearly a need for further prospective studies in which the risk factors for progressive CRI in patients with Fabry disease are better defined.
It is interesting to note that an autopsy study of a patient who had been treated with agalsidase beta for 2.5 years, showed continued tissue Gb3 storage despite long-term ERT [47,48]. Gb3 immunostaining was detected in the cell membrane and cytoplasm of endothelial cells, even in the absence of lysosomal inclusions. This illustrates that Gb3 immunoreactivity remains in cells and tissues even after years of ERT and highlights how much we still have to learn about the mechanisms at work in Fabry disease.
Currently, there is only one published study of the effects of ERT on renal function in children [17]. A recent 6-month open-label study of treatment with agalsidase alfa examined renal function in a group of 24 children with Fabry disease (19 boys, 5 girls) with, on average, normal renal function at baseline (mean estimated GFR, 121 ± 5.0 ml/min/ 1.73 m2) and found that renal function remained normal after 6 months of treatment (mean estimated GFR, 116 ± 3.9 ml/min/1.73 m2). Interestingly, in a subset of seven patients who had hyperfiltration at baseline, estimated GFR came back to within the normal reference range following ERT. Only one patient, who had gross proteinuria at baseline, was receiving an ACE inhibitor. Further studies of longer-term administration of ERT in children are needed to evaluate whether the progressive decline in renal function that is characteristic of Fabry disease can be prevented by treatment.
A recent study has investigated the normal distribution of α-galactosidase A in renal cells and the manner in which recombinant enzyme is taken up into cells. In the normal human kidney, α-galactosidase A is found in virtually all tubular cells and interstitial cells but not renal endothelial cells and podocytes. In mice with Fabry disease, uptake of recombinant α-galactosidase A occurs in podocytes, proximal tubules and interstitial cells [49], but not in vascular endothelial cells. A potential explanation for the clearance is a rapid turnover of renal endothelial cells, which has been demonstrated, at least for the capillary endothelial cells [24], possibly combined with lower circulating amounts of Gb3 as a consequence of ERT [38].
Effects of ERT in patients receiving renal replacement therapy
Data from the USA and Europe have shown that patients with Fabry-related ESRD who are receiving dialysis have a lower survival rate than non-diabetic controls [18,50], highlighting the requirement for ERT in these patients. In an open-label prospective study of the effects of agalsidase beta in nine patients receiving dialysis (n = 6 haemodialysis, n = 3 peritoneal dialysis), treatment was well tolerated and associated with an improvement in extrarenal symptoms and slower progression of left ventricular hypertrophy [51]. That the beneficial effects of ERT are apparent in other organ systems in patients receiving dialysis suggests that these patients should be treated with ERT.
Home infusion
In the long-term study by Schiffmann et al., ERT with agalsidase alfa was well tolerated [31]. The relatively low level of infusion reactions allowed patients to make the transition to home infusion regimens. As ERT is a life-long treatment, being able to use it at home for 40 min one evening every 2 weeks offers considerably less disruption to a patient's life than having to travel to receive treatment in hospital; this may in itself enhance compliance. Milligan et al., in a questionnaire survey of 20 patients receiving ERT for Fabry disease, discovered that 95% preferred home-based therapy because it was more comfortable, less stressful, more effective and had less impact on family life than hospital-based treatment [52]. Linthorst et al. also reported that home treatment with agalsidase alfa and agalsidase beta, 0.2 mg/kg was feasible, safe and reduced both the burden related to chronic i.v. therapy and healthcare costs [53]. However, home use of agalsidase beta at its recommended dose of 1.0 mg/kg was not reported.
The importance of follow-up studies
The relatively low incidence of Fabry disease means that recruitment of patients into randomized controlled clinical trials is problematic for ethical reasons, particularly given the heterogeneity and progressive nature of this disorder. However, it is important to carry out long-term follow-up studies to assess the long-term efficacy and safety of ERT and to gather information on the natural history of this disorder. The systematic collection of clinical data from treatment centres worldwide—as in observational studies such as FOS and the Fabry Registry—allows large amounts of information to be collated over a long period. The relatively broad inclusion criteria, including coexisting illnesses and variations in disease severity, often mean that patients enrolled in these studies are representative of the patient population as a whole. Furthermore, the centralization of data from participating centres worldwide allows comparison of clinical practices and efficacy data in different regions as well as extending our experience of treating patients from a range of backgrounds. As such, the evidence contained within these outcome databases may contribute to clinical decision making and promote standardization of clinical practices to ensure that all patients with Fabry disease receive the required standard of care.
Finally, in view of the multisystemic nature of Fabry disease, it is important that patients with Fabry disease receive integrated follow-up from the appropriate specialists, to ensure that they receive treatment in a timely and appropriate manner and to ensure that the standard of their care is high.
Conclusions
Fabry nephropathy is characterized by different levels of disease and a rate of progression of CRI very similar to diabetic nephropathy. According to the experience gained with diabetic nephropathy, renoprotective measures should be introduced as soon as hyperfiltration and/or microalbuminuria are detected. Both enzymes have been shown to be effective in halting the progression of renal manifestations of Fabry disease in patients with mild or moderate Fabry nephropathy and in slowing the progression of CRI in patients with advanced CRI. Significant proteinuria and marked glomerulosclerosis at baseline in patients with normal GFR are the best predictors of progression of Fabry nephropathy, despite ERT. From the 5-year follow-up of ERT, we have learned that ERT and nephroprotective measures should be introduced as soon as possible in male patients with Fabry disease, i.e. during infancy/adolescence before organ damage becomes irreversible. As there is no accepted biomarker for evaluating the response to ERT in Fabry disease, ongoing surveillance studies, with large numbers of patients and comprehensive data collection and analysis, should help to determine the long-term effects of these enzymes on morbidity and mortality in patients with Fabry disease.
Conflict of interest statement. Frédéric Barbey has received support from TKT Europe-5S (Shire Human Genetic Therapies) and travel grants from Genzyme Corporation. Olivier Lidove has received support from Shire HGT, Actelion Pharmaceuticals and travel grants from Genzyme Corporation. Andreas Schwarting has received support from TKT Europe-5S (Shire Human Genetic Therapies).
References
- 1.Brady RO, Gal AE, Bradley RM, et al. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276:1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- 2.Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meikle PJ, Hopwood JJ, Clague AE, et al. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 4.Pinto R, Caseiro C, Lemos M, et al. Prevalence of lysosomal storage diseases in portugal. Eur J Hum Genet. 2004;12:87–92. doi: 10.1038/sj.ejhg.5201044. [DOI] [PubMed] [Google Scholar]
- 5.MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38:750–760. doi: 10.1136/jmg.38.11.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry outcome survey. Acta Paediatr. 2006;95:86–92. doi: 10.1080/08035250500275022. [DOI] [PubMed] [Google Scholar]
- 8.Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest. 2004;34:236–242. doi: 10.1111/j.1365-2362.2004.01309.x. [DOI] [PubMed] [Google Scholar]
- 9.Branton MH, Schiffmann R, Sabnis SG, et al. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 2002;81:122–138. doi: 10.1097/00005792-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 10.Ruggenenti P, Perna A, Gherardi G, et al. Chronic proteinuric nephropathies: outcomes and response to treatment in a prospective cohort of 352 patients with different patterns of renal injury. Am J Kidney Dis. 2000;35:1155–1165. doi: 10.1016/s0272-6386(00)70054-0. [DOI] [PubMed] [Google Scholar]
- 11.Rennke H, Anderson S, Brenner BM. Structural and functional correlations in the progression of renal disease. In: Tisher CC, Brenner BM, et al., editors. Renal Pathology. Philadelphia, PA: Lippincott; 1989. pp. 43–66. [Google Scholar]
- 12.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339:1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- 13.Contreras F, Rivera M, Vasquez J, et al. Diabetes and hypertension physiopathology and therapeutics. J Hum Hypertens. 2000;14(Suppl 1):S26–S31. doi: 10.1038/sj.jhh.1000983. [DOI] [PubMed] [Google Scholar]
- 14.Kleinert J, Dehout F, Schwarting A, et al. Prevalence of uncontrolled hypertension in patients with Fabry disease. Am J Hypertens. 2006;19:782–787. doi: 10.1016/j.amjhyper.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Verovnik F, Benko D, Vujkovac B, et al. Remarkable variability in renal disease in a large Slovenian family with Fabry disease. Eur J Hum Genet. 2004;12:678–681. doi: 10.1038/sj.ejhg.5201184. [DOI] [PubMed] [Google Scholar]
- 16.Ries M, Gupta S, Moore DF, et al. Pediatric Fabry disease. Pediatrics. 2005;115:e344–e355. doi: 10.1542/peds.2004-1678. [DOI] [PubMed] [Google Scholar]
- 17.Ries M, Clarke JT, Whybra C, et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics. 2006;118:924–932. doi: 10.1542/peds.2005-2895. [DOI] [PubMed] [Google Scholar]
- 18.Thadhani R, Wolf M, West ML, et al. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002;61:249–255. doi: 10.1046/j.1523-1755.2002.00097.x. [DOI] [PubMed] [Google Scholar]
- 19.Schieppati A, Remuzzi G. Proteinuria and its consequences in renal disease. Acta Paediatr Suppl. 2003;92:9–13. doi: 10.1111/j.1651-2227.2003.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 20.Sessa A, Tosoni A, Nebuloni M, et al. Renal ultrastructural findings in Anderson-Fabry disease. J Nephrol. 2002;15:109–112. [PubMed] [Google Scholar]
- 21.Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002;13(Suppl 2):S134–S138. [PubMed] [Google Scholar]
- 22.Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8:539–548. doi: 10.1097/01.gim.0000237866.70357.c6. [DOI] [PubMed] [Google Scholar]
- 23.Ojo A, Meier-Kriesche HU, Friedman G, et al. Excellent outcome of renal transplantation in patients with Fabry's disease. Transplantation. 2000;69:2337–2339. doi: 10.1097/00007890-200006150-00020. [DOI] [PubMed] [Google Scholar]
- 24.Warnock DG, West ML. Diagnosis and management of kidney involvement in Fabry disease. Adv Chronic Kidney Dis. 2006;13:138–147. doi: 10.1053/j.ackd.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Eng CM, Banikazemi M, Gordon RE, et al. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001;68:711–722. doi: 10.1086/318809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiffmann R, Murray GJ, Treco D, et al. Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci USA. 2000;97:365–370. doi: 10.1073/pnas.97.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiffmann R, Kopp JB, Austin HA, 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- 28.Grabenhorst E, Schlenke P, Pohl S, et al. Genetic engineering of recombinant glycoproteins and the glycosylation pathway in mammalian host cells. Glycoconj J. 1999;16:81–97. doi: 10.1023/a:1026466408042. [DOI] [PubMed] [Google Scholar]
- 29.Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 30.Vedder AC, Linthorst GE, Houge G, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS ONE. 2007;2:e598. doi: 10.1371/journal.pone.0000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiffmann R, Ries M, Timmons M, et al. Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant. 2006;21:345–354. doi: 10.1093/ndt/gfi152. [DOI] [PubMed] [Google Scholar]
- 32.Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linthorst GE, Hollak CE, Donker-Koopman WE, et al. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- 34.Germain DP. Fabry disease: recent advances in enzyme replacement therapy. Expert Opin Investig Drugs. 2002;11:1467–1476. doi: 10.1517/13543784.11.10.1467. [DOI] [PubMed] [Google Scholar]
- 35.Whitfield PD, Calvin J, Hogg S, et al. Monitoring enzyme replacement therapy in Fabry disease - role of urine globotriaosylceramide. J Inherit Metab Dis. 2005;28:21–33. doi: 10.1007/s10545-005-4415-x. [DOI] [PubMed] [Google Scholar]
- 36.Ohashi T, Sakuma M, Kitagawa T, et al. Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. Mol Genet Metab. 2007;92:271–273. doi: 10.1016/j.ymgme.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Young E, Mills K, Morris P, et al. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr Suppl. 2005;94:51–54. doi: 10.1111/j.1651-2227.2005.tb02112.x. [DOI] [PubMed] [Google Scholar]
- 38.Thurberg BL, Rennke H, Colvin RB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002;62:1933–1946. doi: 10.1046/j.1523-1755.2002.00675.x. [DOI] [PubMed] [Google Scholar]
- 39.Beck M, Ricci R, Widmer U, et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur J Clin Invest. 2004;34:838–844. doi: 10.1111/j.1365-2362.2004.01424.x. [DOI] [PubMed] [Google Scholar]
- 40.Fioretto P, Steffes MW, Sutherland DE, et al. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–75. doi: 10.1056/NEJM199807093390202. [DOI] [PubMed] [Google Scholar]
- 41.Schwarting A, Dehout F, Feriozzi S, et al. Enzyme replacement therapy and renal function in 201 patients with Fabry disease. Clin Nephrol. 2006;66:77–84. [PubMed] [Google Scholar]
- 42.Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18:1547–1557. doi: 10.1681/ASN.2006080816. [DOI] [PubMed] [Google Scholar]
- 43.Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and Fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol. 2007;18:2609–2617. doi: 10.1681/ASN.2006121400. [DOI] [PubMed] [Google Scholar]
- 44.Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- 45.Breunig F, Weidemann F, Strotmann J, et al. Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int. 2006;69:1216–1221. doi: 10.1038/sj.ki.5000208. [DOI] [PubMed] [Google Scholar]
- 46.Schiffmann R, Askari H, Timmons M, et al. Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol. 2007;18:1576–1583. doi: 10.1681/ASN.2006111263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schiffmann R, Rapkiewicz A, Abu-Asab M, et al. Pathological findings in a patient with Fabry disease who died after 2.5 years of enzyme replacement. Virchows Arch. 2006;448:337–343. doi: 10.1007/s00428-005-0089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Askari H, Kaneski CR, Semino-Mora C, et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007;451:823–834. doi: 10.1007/s00428-007-0468-6. [DOI] [PubMed] [Google Scholar]
- 49.Christensen EI, Zhou Q, Sorensen SS, et al. Distribution of alpha-galactosidase A in normal human kidney and renal accumulation and distribution of recombinant alpha-galactosidase A in Fabry mice. J Am Soc Nephrol. 2007;18:698–706. doi: 10.1681/ASN.2006080822. [DOI] [PubMed] [Google Scholar]
- 50.Obrador GT, Ojo A, Thadhani R. End-stage renal disease in patients with Fabry disease. J Am Soc Nephrol. 2002;13(Suppl 2):S144–S146. [PubMed] [Google Scholar]
- 51.Pisani A, Spinelli L, Sabbatini M, et al. Enzyme replacement therapy in Fabry disease patients undergoing dialysis: effects on quality of life and organ involvement. Am J Kidney Dis. 2005;46:120–127. doi: 10.1053/j.ajkd.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 52.Milligan A, Hughes D, Goodwin S, et al. Intravenous enzyme replacement therapy: better in home or hospital? Br J Nurs. 2006;15:330–333. doi: 10.12968/bjon.2006.15.6.20681. [DOI] [PubMed] [Google Scholar]
- 53.Linthorst GE, Vedder AC, Ormel EE, et al. Home treatment for Fabry disease: practice guidelines based on 3 years experience in The Netherlands. Nephrol Dial Transplant. 2006;21:355–360. doi: 10.1093/ndt/gfi221. [DOI] [PubMed] [Google Scholar]