

Abstract
Phospholipase D (PLD) plays important roles in cellular responses to tissue injury that are critical to acute inflammatory diseases, such as the acute respiratory distress syndrome (ARDS). We investigated the expression of PLD isoforms and related phospholipid phosphatases in patients with ARDS, and their roles in a murine model of self-limited acute lung injury (ALI). Gene expression microarray analysis on whole blood obtained from patients that met clinical criteria for ARDS and clinically matched controls (non-ARDS) demonstrated that PLD1 gene expression was increased in patients with ARDS relative to non-ARDS and correlated with survival. In contrast, PLD2 expression was associated with mortality. In a murine model of self-resolving ALI, lung Pld1 expression increased and Pld2 expression decreased 24 h after intrabronchial acid. Total lung PLD activity was increased 24 h after injury. Pld1−/− mice demonstrated impaired alveolar barrier function and increased tissue injury relative to WT and Pld2−/−, whereas Pld2−/− mice demonstrated increased recruitment of neutrophils and macrophages, and decreased tissue injury. Isoform-specific PLD inhibitors mirrored the results with isoform-specific Pld-KO mice. PLD1 gene expression knockdown in human leukocytes was associated with decreased phagocytosis by neutrophils, whereas reactive oxygen species production and phagocytosis decreased in M2-macrophages. PLD2 gene expression knockdown increased neutrophil and M2-macrophage transmigration, and increased M2-macrophage phagocytosis. These results uncovered selective regulation of PLD isoforms after ALI, and opposing effects of selective isoform knockdown on host responses and tissue injury. These findings support therapeutic strategies targeting specific PLD isoforms for the treatment of ARDS.
Keywords: acute lung injury, acute respiratory distress syndrome, microarray, Phospholipase D
1 |. INTRODUCTION
Dysregulated inflammation and altered permeability of alveolar endothelial and epithelial barriers remain central to the pathophysiology of the acute respiratory distress syndrome (ARDS). Despite significant progress in our understanding of ARDS pathogenesis, therapeutic modalities are limited and mortality remains unacceptably high.1
Neutrophils are the first responders to sites of tissue injury to contain and eliminate invading pathogens via intra- and extracellular mechanisms, such as phagocytosis and release of proteases, oxidants, antimicrobial peptides, and neutrophil-extracellular traps (NETs).2 Resident and recruited macrophages participate in these initial inflammatory responses, and orchestrate resolution of inflammation by clearing dead cells and initiating tissue repair.3 In health, regulation of these inflammatory responses ensures a prompt return to homeostasis. As such, excessive neutrophil activation and impaired clearance of the inflammatory milieu can potentiate tissue injury and cause human disease, such as ARDS.4
Phospholipase D (PLD) is necessary for cellular responses to tissue injury and microbial invasion,5 including leukocyte adhesion,6 and phagocytosis.7 Cellular models have shown that PLD1 is highly expressed in leukocytes and regulates several aspects of acute inflammatory responses, including oxidative burst.8 PLD2 plays important roles in regulating phagocytosis and cell replication.9 Both PLD1 and PLD2 are necessary for leukocyte chemotaxis.10 An experimental model of acute pancreatic injury has shown that PLD1 and PLD2 regulate leukocyte recruitment to inflamed tissue and edema formation.11 In a sepsis model with secondary lung injury, PLD2 deficiency afforded lung protection and increased survival.12 Here, we examined PLD involvement in host responses to the important clinical problem of ARDS.
The enzymatically active isoforms of the PLD superfamily, PLD1 and PLD2, hydrolyze phosphatidylcholine to generate choline and phosphatidic acid (PA). The PA generated via PLD activity can play multiple roles in cellular function.13 PA can function in signal transduction events, such as activation of a phosphoinositide kinase, or as a lipid-binding partner to several proteins. In addition, PA production is important in vesicle trafficking, endocytosis, and secretion. PA can also be further converted by PA phosphatases to diacylglycerol, which also signals for cellular processes important to inflammation and tissue repair.14 Apart from its metabolic functions, PLD can also regulate select cellular functions through direct interactions with other proteins.7
Results presented here show that PLD1 expression increased in ARDS patients and was associated with survival. In a murine model of self-resolving ALI, PLD1 gene expression in injured lungs increased, whereas PLD1 deficiency worsened ALI severity by alveolar barrier disruption and decreased macrophage phagocytosis. In contrast, PLD2 expression was associated with mortality in ARDS patients, and PLD2 expression decreased in murine self-resolving ALI. PLD2 deficiency lessened ALI severity, and was associated with increased recruitment of macrophages with enhanced phagocytosis and decreased neutrophil production of reactive oxygen species (ROS). Together, these findings have uncovered counter-regulation by PLD isoforms that influence the nature and severity of tissue injury.
2 |. MATERIALS AND METHODS
2.1 |. Registry of Critical Illness
Details of the Research Registry and Human Sample Repository for the Study of the Biology of Critical Illness, abbreviated as the Registry of Critical Illness (RoCI), have been previously published.15 The RoCI collects demographic, clinical information, and blood specimens from patients with critical illness in the medical intensive care unit of the Brigham and Women‘s Hospital (BWH). RoCI is approved by the Partners Human Research Committee and operates under protocol 2008-P-000495.
Patients are classified by the treating intensive care physician team as sepsis, and sepsis with ARDS (sepsis/ARDS). Sepsis was identified according to the 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference guidelines.16 ARDS was defined according to American-European Consensus Conference on ARDS guidelines.17 All ARDS patients were endotracheally intubated and mechanically ventilated using a low tidal volume ventilation strategy according to ARDSNet protocol.18 The following parameters were collected for each patient for further analyses: age, race, gender, length of hospital stay, APACHE II score, in-hospital mortality, 28-day mortality and overall mortality. APACHE II was calculated on intensive care unit (ICU) admission day for every patient,19 and mortality was assessed using the Partners Health Care Research Patient Data Registry (RPDR) linking deaths to the Social Security Death Index (SSDI).
2.2 |. Human microarray datasets
Peripheral blood with corresponding demographic and clinical information was obtained from adult patients were obtained from subjects enrolled in the RoCI at the Brigham and Women’s Hospital ICU.15 Samples were also obtained from adult patients in the University of Pittsburgh Medical Center ICU,20 and pediatric patients (< 10 years old) admitted to the Cincinnati Children’s Hospital pediatric ICU.21
2.3 |. Microarray preprocessing, normalization, and analysis
All preprocessing and analysis of linear correlation between gene expression levels, ARDS diagnosis, and 28-day survival after adjustment for age, sex, race, and age were performed in R 3.2/Bioconductor.22,23 To evaluate the concordance of gene expression patterns across different microarray platforms, we mapped each microarray probe to its genomic coordinates on the UCSC Human Genome version 18 (hg18) using the R/Bioconductor biomaRt package.24,25 We next matched probes to those on different platforms using the nearest genomic start position and filtering out probes that were not represented across all platforms.25,26 The number of probes in the pooled dataset carried forward for normalization was 18,694. We used the R package, CONOR, to perform distance weighted discrimination cross-platform normalization, to account for differences in probe length, probe type, number of probes, labeling methods, and detection methods.27 PLD genes are expressed as PLD1 and PLD2 in humans, and Pld1, Pld2 in murine datasets.
2.4 |. Mice
C57BL/6 (18–22 week old, male:female ratio 1:1, body weights 20–25 g; Charles River Laboratories, Wilmington, MA), Pld1−/− and Pld2−/− were housed in isolation cages in pathogen-free conditions on a light-dark cycle with light from 7:00 to 20:00 at 25°C. Pld1−/− and Pld2−/− were obtained from Dr. Gilbert Di Paolo (Department of Medicine, College of Physicians and Surgeons, Columbia University, New York, New York)28 and Dr. Yasunori Kanaho (Department of Physiological Chemistry, University of Tsukuba, Tsukuba, Japan).29 The Pld1−/− and Pld2−/− mice were backcrossed with C57bl/6 mice for > 7 generations.29,30 Mice were fed a standard diet (Laboratory Rodent Diet 5001; PMI Nutrition International, St. Louis, MO) containing 4.5% total fat with 0.3% ω-3 fatty acids and <0.02% C20:4 and were provided water ad libitum.
2.5 |. Murine model of acid-induced acute lung injury
All mice were maintained under specific pathogen-free conditions. All studies were reviewed and approved by the Harvard Medical Area standing committee on animals (protocol no. 03618). Mice were anesthetized with i.p. injections of ketamine (100 mg/kg body wt; Phoenix Scientific, St. Joseph, MO) and xylazine (10 mg/kg body wt; Phoenix Scientific). As in Abdulnour et al.,31 hydrochloric acid (0.1 N HCl, pH 1.5, 50 μL, endotoxin free; Sigma–Aldrich, St Louis, MO) was instilled selectively into the left mainstem bronchus via a 24-G angio-catheter inserted intratracheally.
2.6 |. Preparation of lung samples prior to quantitative real-time PCR, Western blot, and PLD activity determinations
Left lungs from C57BL/6 mice of 3 genotypes (WT, PLD1−/−, or PLD2−/−) were injured with 0.1 N HCl intrabronchially. Lungs were collected at time zero (no injury) and at 24 h after injury, cut longitudinally and flash frozen. On the day of the analyses, tissue samples were weighed and an appropriate volume (between 70 and 120 μL) of lysis buffer (for Western blot or PLD enzyme assays) or buffer A from RNeasy mini kit (for quantitative real-time PCR[qRT-PCR] assays) was added to reach ~1 mg/mL solutions. Lysis buffer composition for Western blots and enzyme assays was: 50 mM HEPES, pH 7.2,100 μM Na3VO4, 0.1% Triton X-100 and 1 mg/mL each of protease inhibitors (aprotonin and leupeptin). Lung tissue was ground with a micromotorized pestle homogenizer, followed by sonication (10 s) on ice. Samples were spun for 5 min at 250 × g on a refrigerated centrifuge (4°C) to remove debris and supernatants were taken for further analyses
2.7 |. Gene expression measurement by qRT-PCR
Reverse transcription coupled to qPCR was performed following published technical details.32 Total RNA was isolated from cells with the RNeasy minikit per the manufacturer’s protocol (Qiagen, Valencia, CA). RNA concentrations were determined using a NanoDrop 2000c UV-Vis Spectrophotometer (Thermo Scientific, Wilmington, DE) and samples were normalized to a fixed concentration of 50 ng/μL RNA. Reverse transcription was performed with 210 ng RNA, 210 ng random hexamers, 500 μM dNTPs, 84 U RNase OUT and 210 units of Superscriptll reverse transcriptase (Life Technologies, Carlsbad, CA) and incubated at 42°C for 55 min. All qPCR reactions were run with 100 ng total input RNA, 2 μL of PLD1 gene expression assay (FAM-labeled) or 2 μL of the PLD2 gene expression assay (FAM-labeled) multiplexed with the housekeeping gene (β-Actin) (FAM-labeled) with the final concentrations being 200 pmol and 400 pmol for the primers and probe, respectively. All primers and fluorescent probes were synthesized by ThermoFisher Scientific (Life Technologies). The qPCR conditions for the Stratagene Cycler were: 95°C for 3 min and then 50 cycles of the next 3 steps: 30 s 95°C, 1 min 60°C, and then 1 min 72°C. The “cycle threshold” Ct values were chosen from the linear part of the PCR amplification curves where an increase in fluorescence is detected at >10 S.E.M. above the background signal, and referred to housekeeping gene (GAPDH).
2.8 |. Phospholipase activity assay
The measurement of total PLD activity was reported previously.6,33 Briefly, the assay was performed in liposomes of 1,2-dioctanoyl-sn-glycero-3-phosphocholine (PC8) and [3H]-butanol. The following reagents (final concentrations): 3.5 mm PC8 phospholipid, 45 mM HEPES, pH 7.8, and 1.0 μ Ci of n-[3H]-butanol were added in a liposome form as indicated in.34 Samples were incubated for 20 min at 30°C with continuous shaking. The addition of 0.3 mL of ice-cold chloroform/methanol (1:2) stopped the reactions. Lipids were isolated, dried (under N2) and suspended in chloroform:methanol (9:1) and then spotted on thin-layer chromatography plates along with 1,2-dipalmitoyl-sn-glycero-3-phosphobutanol (PBut) (Avanti polar lipids, Inc., AL) authentic standards. The amount of [3H]-phosphatidylbutanol ([3H]-PBut) that co-migrated with PBut standards (Rf~0.45 + 0.36) was measured by scintillation spectrometry and background subtracted. Results were expressed as total PLD enzymatic activity as dpm/mg protein/min.
2.9 |. Bronchoalveolar lavage
At timed intervals, mice were euthanized and the trachea exposed. A 20-gauge angiocatheter was inserted into the trachea and the lungs were lavaged with 2 separate 1 mL volumes of ice-cold PBS with 0.6 mM EDTA. The bronchoalveolar lavage (BAL) fluid was pooled, centrifuged at 500 × g for 5 min at 4°C to pellet the cell fraction. The cell pellet was suspended in cold PBS, and the supernatant was used for albumin, protein, and cytokine analysis. BAL neutrophil count was highest at 24 h after ALI (Supplemental Fig. S1 and,31 therefore that time point was used for determinations after ALI.
2.10 |. Assessment of vascular leakage after pharmacologic isoform-specific PLD inhibition
Evans blue dye (EBD, 40 mL/kg) was injected into the tail vein of mice 30 min before termination of the experiment to assess vascular leak. In brief, lungs were lavaged and perfused free of blood with DPBS before being excised en bloc. The left lungs were dried in an oven (56°C) for 48 h and weighed. The lungs were then homogenized in DPBS (500 μL), incubated with 2 vol formamide (18 h, 56°C), and centrifuged at 5000 × g for 30 min, and the optical density of the supernatant was determined by spectrophotometry at 620 nm. Extravasated EBD in lung homogenates (μg EBD per lung) was calculated with an EBD standard curve. To inhibit PLD isoforms, animals were treated with a specific PLD1 inhibitor (VU0155069),35 PLD2 inhibitor (VU0364739),29 or vehicle (saline in 10% vol/vol DMSO) given by intraperitoneal injections on days –3, –2, –1, and 0 (1 h prior to ALI, 10 mg/kg in 10% DMSO).
2.11 |. FACS analysis and cell sorting
BAL cells were obtained after centrifugation of pooled BAL fluid, and the cell pellet was stained with fluorochrome-labelled antibodies to obtain a leukocyte differential as in Abdulnour et al.36; neutrophils (CD45+ F4/80− Ly6G+ CD11b+), infiltrating macrophages (iMacs; CD45+ SScHigh F4/80+ CD11cLow CD11b+ SiglecF–Ly6C+), exudative macrophages (ExMacs; CD45+ SScHigh F4/80+ CD11cHigh CD11b+ SiglecF+ Ly6C+), and resident alveolar macrophages (rAM; CD45+ F4/80+ SScHigh CD11cHigh CD11b− SiglecF+ Ly6C−). Cell counts were obtained by adding a known amount of fluorescent counting beads (CountBright; Life Technologies) to the BAL cell pellet prior to flow cytometry as per the manufacturer’s recommendations, and normalized to lavage. FACSCanto II (BD Biosciences, San Jose, CA, USA) and FlowJo Ver. 10 software (Tree Star, Ashland, OR) were used for analyses. In some experiments, whole murine lungs were homogenized 24 h after ALI and a cell suspension was prepared as in.31 Neutrophils (CD45+ Ly6G+), macrophages (CD45+ SScHigh F4/80+), lung epithelial cells (CD45− CD31− CD326+), and lung endothelial cells (CD45− CD31+) were sorted with >95% purity, using FACSAria (BD Biosciences, San Jose, CA, USA). The sorted populations were used for RNA extraction and RT-PCR.
2.12 |. Determination of bioactive molecule levels in BALF
Select cytokines were measured in aliquots of pooled BALF using a bead-based immunoassay as per the manufacturer’s instructions (LEG-ENDPlex by Biolegend, San Diego, CA).
2.13 |. Histopathology
Lung tissue was fixed by inflation with Zinc Fixative (BD Pharmingen, San Diego, CA) at a trans-pulmonary pressure of 20 cm H2O and embedded in paraffin. For histologic analysis, lungs were collected 0, 24, and 48 h after Escherichia coli instillation and paraffin-embedded 5-μm sections of lungs were cut and stained with H&E for light microscopy. Leica DFC480 and Leica QWin Standard V3.4.0 (Leica Microsystems Ltd., Buffalo Grove, IL, USA) were used for microscopic analysis.
2.14 |. Gene silencing
Human promyelomonocytic HL60 cells were differentiated into neutrophils (CD66b,+CD11b/CD16b+), with 4 days of 1.25% DMSO continuously in culture. Human promonocytic U937 cells were differentiated into macrophages (CD14+,CD68+, CD71+) with 48 h of Vitamin D3 (40 ng/mL) followed by 24 h in control medium (resting period to allow full adhesion). The resulting macrophages were further incubated with 20 ng/mL IL-4to induce an M2 phenotype. Leukocytes were then Amaxa-electroporated 2 × 106 HL-60 cells/100 μL in nucleofector and transfected with either 100 nM (final concentration) of either scrambled siRNA (control), or PLD1 siRNA (PC-PLD1 siRNA, catalog sc-4400), or PLD2 siRNA (PC-PLD1 siRNA, catalog sc-4401), from Santa Cruz Biotechnology, Inc., Dallas, TX. Control siRNA is a 19 bp scrambled sequence with 3′ dT overhangs certified not to have significant homology to any known gene sequences from mouse, rat or human and causes no significant changes in gene expression of transfected cells after 48 h at the same concentration as the siRNA in test. After nucleofection, cells were immediately plated into 6-well plates and incubated at 37°C for 48 h to allow for maximum gene expression silencing.
2.15 |. Neutrophil superoxide release
Neutrophil ROS production, specifically, superoxide anion release was measured by the reduction of cytochrome c with several neutrophil agonists as indicated, with PMA (50 ng/mL) as the positive control and SOD was used to stop the reactions.
2.16 |. Leukocyte chemotaxis
Chemotaxis against IL-8 (10 nM; neutrophils) or MCP-1 (30 nM; macrophages) was measured in Transwell (5 μm diameter pore for neutrophil, 8 μm diameter pore for macrophage) for specified lengths of time; cells migrated at the bottom of the wells were stained and counted. Averages from 7 fields were plotted.
2.17 |. Macrophage phagocytosis
Macrophage phagocytosis was determined using fluorescent-labeled GFP zymosan A (Saccharomyces cerevisiae) (Invitrogen, Carlsbad, CA) as in Ref. 37. Briefly, the particles were opsonized at 37°C for 1h using the zymosan A BioParticles opsonizing reagent (derived from highly purified rabbit polyclonal IgG antibodies) (Invitrogen) and were applied to 1 × 107 cells such that there were approximately 20 zymosan particles per each cell. After application of GFP-Zymosan, the 6-well plates containing the cells were centrifuged at 800×g for 5 min and then were incubated at 37°C for 15 min. Fluorescent-labeled zymosan particles ingested by macrophages were counted manually under the microscope in 5 different fields, and the averages and SEM calculated.
2.18 |. Neutrophil-extracellular traps
To detect NETs in the airway, cell-free supernatant of BAL fluid was used to quantify extracellular DNA citrullinated histone H3, which is a well-established marker of NETs. QuantIt Picogreen DNA quantification assay was used for the quantification of DNA. Rabbit anti-citrullinated histone H3 (citH3) (ab5103; Abcam, Cambridge, MA, USA) was used to perform immunoblots.
2.19 |. Statistical analysis
All data are expressed as means ± SEM. Comparisons between groups were conducted using ANOVA and Student’s t-test as appropriate. A level of P ≤ 0.05 was considered to indicate statistical significance. Statistics were performed using Graphpad Prism 6.0 for Windows (San Diego, CA).
3 |. RESULTS
3.1 |. PLD genes are regulated in patients with ARDS
To determine gene expression profiles in ARDS, microarray analysis was performed on peripheral blood obtained from patients with ARDS (N = 19) and from clinically matched patients without ARDS (Non-ARDS, N = 29) enrolled in a RoCI (Table 1). In addition to PLD1 and PLD2, the expression profile of phospholipid phosphatases (PLPP), a family of genes that includes PA phosphatases, was determined (Supplemental Fig. S2a). Gene expression is represented in log2, where positive values represent increased expression, and negative values represent decreased expression. On presentation, gene expression of PLD1 was increased in patients with ARDS relative to Non-ARDS (log2 relative change = 0.91), and PLD2 expression was unchanged between the 2 groups (log2 relative change = −0.07; Fig. 1A). At day 7, PLD1 expression was similar between the 2 groups (PLD1 log2 relative change = 0.02), and PLD2 expression was slightly increased in ARDS patients (PLD2 log2 relative change = 0.23; Fig. 1B). Of note, the variance in PLD1 and PLD2 gene expression in ARDS patients on presentation showed little change from presentation to day 7; log2 relative change day 7/presentation was 0.07 for PLD1, and 0.13 for PLD2. To confirm these determinations in a replicate cohort, publicly available microarray datasets from patients with ARDS (N = 99) and controls (N = 166) were merged in silico and normalized (see Materials and Methods). Peripheral blood gene expression analysis showed increased PLD1 (8.4 log2 relative change) and PLD2 expression (6.0 log2 relative change) at presentation in patients with ARDS relative to controls (Fig. 1C). In addition, patients with ARDS demonstrated significantly higher PLD1 expression relative to PLD2 expression (1.11 log2 relative change). PLPP gene expression analysis in RoCI patients demonstrated selective regulation as well (Supplemental Figs. S2b and S2c). For example, PLPP2 expression in ARDS patients was increased on presentation (log2 relative change = 2.19; Supplemental Fig. S2b) and decreased on day 7 (log2 relative change = −0.48; Supplemental Fig. S2c).
TABLE 1.
RoCI demographics
| Diagnosis | ARDS | Non-ARDS | P values |
|---|---|---|---|
| Number of patients | 19 | 29 | |
| Age (yrs) | 51 | 61 | 0.03 |
| Sex (%M) | 42.1 | 48.3 | 0.9 |
| APACHEII (avg) | 30 | 25 | 0.002 |
| Hospital stay (days) | 23 | 15 | 0.001 |
| 28-day mortality (%) | 47.4 | 34.5 | 0.03 |
FIGURE 1. PLD genes are differentially regulated in peripheral blood during ARDS.
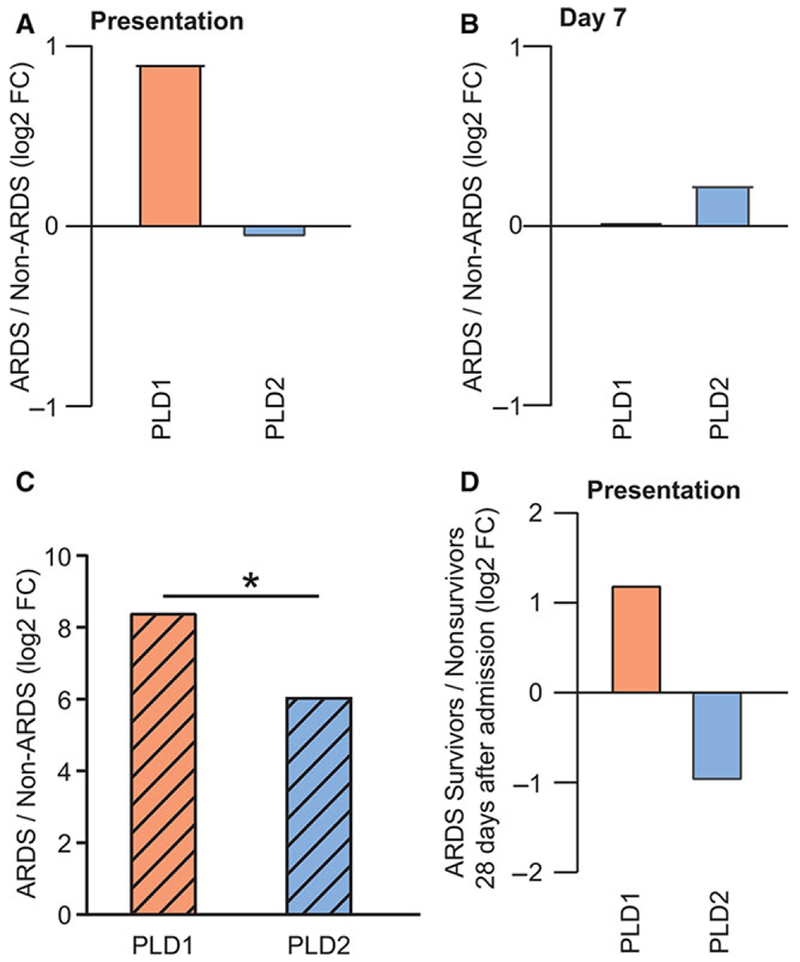
PLD1 and PLD2 gene expression fold change in ARDS/Non-ARDS (A) at presentation to the ICU, and (B) at day 7. (C) PLD1 and PLD2 gene expression fold change in ARDS/Non-ARDS patients from pooled microarray datasets obtained in external cohorts, *P < 0.05. (D) PLD1 and PLD2 gene expression fold change at presentation in ARDS survivors/nonsurvivors. Gene expression is expressed in log2
3.2 |. PLD1 and PLD2 are differentially expressed in human peripheral blood from ARDS survivors
To examine the relationship between gene expression and ARDS outcome, ARDS survivors were identified in the BWH RoCI, and peripheral blood gene expression at presentation was compared with ARDS nonsurvivors. PLD1 and PLD2 expression were changed 1.2 and −1.0 fold (log2) respectively in ARDS survivors (Fig. 1D). Of note, PLPP2 expression was also increased in ARDS survivors (1.84 log2 FC; Supplemental Fig. S2d). Together, these results indicate differential expression of PLD1 and PLD2 genes in peripheral blood leukocytes from ARDS patients. Specifically, PLD1 expression was associated with survival, whereas PLD2 expression was associated with mortality in ARDS patients.
3.3 |. Pld1 and Pld2 gene expression are differentially regulated in lungs after self-limited acute tissue injury
To further characterize the roles of these phospholipases in response to acute lung injury (ALI), lung Pld1 and Pld2 gene expression was determined after sterile lung injury using a model of ARDS from gastric acid aspiration, an important clinical risk factor for ARDS (see Materials and Methods; Fig. 2A). Pld1 and Pld2 were expressed in neutrophils, macrophages, epithelial cells, and endothelial cells, with relatively more Pld2 expression in macrophages and endothelial cells (Supplemental Fig. S3). Lung Pld1 expression significantly increased 2.91 fold (±0.72) relative to control (Fig. 2B), whereas lung Pld2 expression significantly decreased 0.12 fold (±0.09; Fig. 2C). Expression of PLPP genes was also regulated; Plpp1 and Plpp2 expression increased 12.4 fold (±1.83; Fig. 2D) and 3.3 fold (±0.46; Fig. 2E) in injured lungs relative to control. Plpp6 expression was also increased in injured lungs (Fig. 2F).
FIGURE 2. PLD isoforms and phospholipid phosphatase genes are differentially regulated in lungs after acute lung injury.
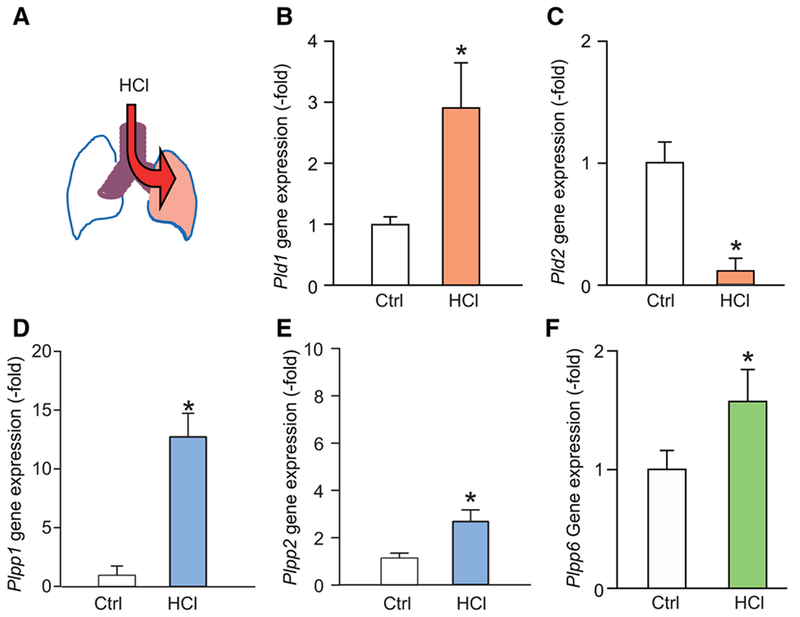
(A) Model of selective intratracheal HCl instillation to the left lung, and tissue harvesting at 24 h. Gene expression of (B) Pld1, (C) Pld2, (D) Plpp1, (E) Plpp2, and (F) Plpp6 in control and lungs 24 h after intratracheal HCl. Gene expression is expressed in fold change relative to housekeeping gene (GAPDH, see Materials and Methods). Results are expressed as mean ± SEM, N > 10, *P < 0.05
3.4 |. PLD activity increases after ALI
We next examined total lung PLD activity and the relative contributions of Pld isoforms (see Materials and Methods). To this end, intra-bronchial acid was administered to wild-type mice (WT), Pld1 deficient mice (Pld1−/−), and Pld2-deficient mice (Pld2−/−). At baseline, lungs from WT gave significantly higher PLD activity (1.39 ± 0.15) relative to lungs from Pld1−/− (0.36 ± 0.05) and Pld2−/− (0.62 ± 0.10; Fig. 3A). PLD activity was significantly increased 24 h after intrabronchial HCl in WT mice (2.34 ± 0.37; Fig. 3A). After injury, PLD activity in lungs from Pld1−/− (1.13 ± 0.18) and Pld2−/− (0.99 ± 0.16) also significantly increased but remained lower than lungs from WT (Fig. 3A).
FIGURE 3. PLD deficiency impacts alveolar barrier disruption.
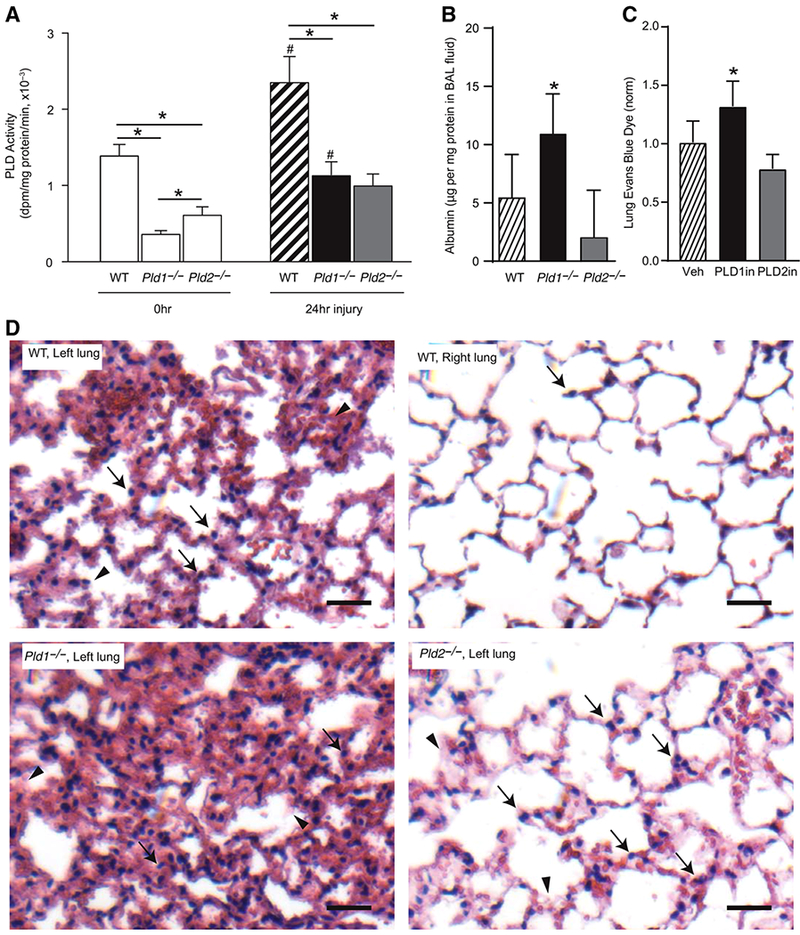
(A) Total lung PLD activity in wild-type (WT) mice, and mice deficient in Pld1 and Pld2 gene, at t = 0 and t = 24 h after intrabronchial HCl. Results are expressed as mean ± SEM, N ≥ 10, *P < 0.05. (B) Albumin levels in BAL fluid from WT, Pld1−/−, and Pld2−/− animals 24 h after intrabronchial HCl. (C) Evans blue dye levels in left lung homogenate 24 h after intrabronchial HCl and treatment with a selective inhibitor of PLD1 (PLD1in, black) or PLD2 (PLD2in, gray), or vehicle (Veh, hatched). Results are expressed as mean ± SEM, N ≥ 10, *P < 0.05. (D) Representative lung sections from WT (injured left lung and uninjured right lung from same animal), Pld1−/−, and Pld2−/− animals
3.5 |. Pld1 deficiency is associated with impaired barrier function after ALI
To determine the impact of Pld isoforms on alveolar barrier function after acid-induced lung injury, levels of extravasated albumin were quantified in the alveolar compartment. Albumin concentration in BAL fluid obtained 24 h after intrabronchial acid was significantly higher in Pld1−/− (10.9 μg/mg protein ± 1.3 SEM) relative to WT (5.5 μg/mg protein ± 1.4 SEM) and Pld2−/− (2.0 μg/mg protein ± 1.4 SEM; Fig. 3B). Because the WT control animals were not the result of heterozygote mating (see Materials and Methods), another line of evidence for PLD isoform specific actions was determined with selective inhibitors. A selective pharmacologic inhibitor of PLD1 increased extravasation of intravascular EBD into lung interstitium (Fig. 3C). Selective inhibition of PLD2 did not significantly impact barrier integrity after ALI (Fig. 3C). Histology obtained 24 h after acid administration showed demonstrable tissue leukocyte infiltration and alveolar and interstitial edema in the left lung of WT that were in sharp contrast to the uninjured right lung (Fig. 3D). Relative to WT mice, alveolar edema was worse in lungs from Pld1−/− mice, and diminished in lungs from Pld2−/− mice (Fig. 3C). Of interest, determination of BAL fluid cytokine levels 24 h after injury gave significantly higher levels of IL-1α and GM-CSF in Pld2−/− mice relative to Pld1−/− (Supplemental Fig. S4). IL-6 was increased in Pld2−/− mice relative to WT and Pld1−/− mice but statistical significance was not reached. Levels of TNF α and IL-10 were not significantly different (Supplemental Fig. S4).
3.6 |. Pld2 deficiency is associated with increased leukocyte tissue infiltration after ALI
To determine the role of Pld isoforms in acute lung inflammation, cellular host responses were examined by flow cytometric analysis of total and differential leukocyte counts in BALF after intrabronchial HCl in Pld1−/−, Pld2−/−, and WT mice (Fig. 4A, as in Ref. 36). At baseline, total and differential leukocyte counts were similar between the 3 genotypes (Figs. 4B–4G). After injury, total leukocyte counts increased in Pld2−/− (220.5 ± 24.4 × 103 cells per BALF), whereas they decreased in WT (97.2 ± 12.7 × 103 cells per BALF) and were not significantly different in Pld1−/− (132.4 ± 17.3 × 103 cells per BALF; Fig. 4B). Neutrophil (Fig. 4C) and recruited macrophage (exMacs in Fig. 4F, and iMacs in Fig. 4G) counts increased, whereas resident alveolar macrophage counts decreased 24 h after injury (rAMs; Fig. 4E).
FIGURE 4. PLD1 and PLD2 regulate leukocyte recruitment after ALI.
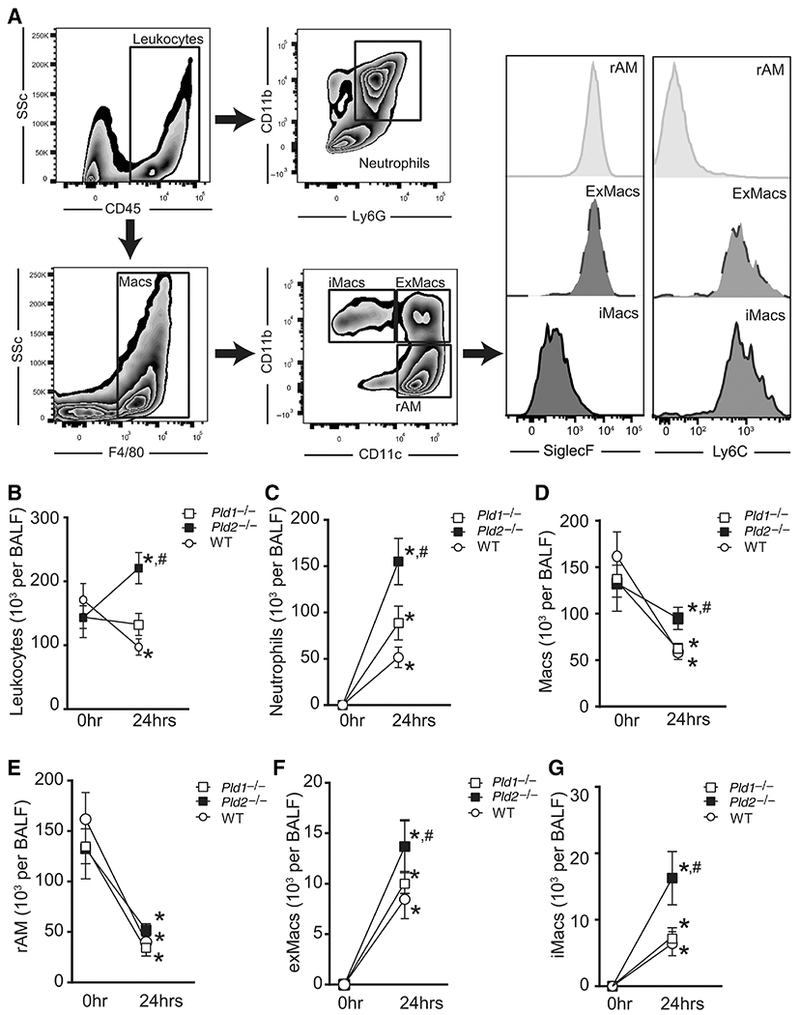
(A) Representative flow cytometry dot plots and histograms from BAL gated on total cell population. Neutrophils, CD45+ CD11b+ Ly6G+; Macs, total macrophages, CD45+ F4/80+; iMacs; inflammatory macrophages, CD45+ F4/80+ CD11c− CD11b+ SiglecF− Ly6C+; exMacs; exudative macrophages, CD45+ F4/80+ CD11c+ CD11b+ SiglecF+ Ly6C+; rAM, resident alveolar macrophages, CD45+ F4/80+ CD11c+ CD11b− SiglecF+ Ly6C−. (B–G) Enumeration of (B) total cell count and differential cell count including (C) neutrophils, (D) total macrophages, (E) rAM, (F) ExMacs, and (G) iMacs, by flow cytometry analysis of BAL fluid after intrabronchial acid. Results are expressed as mean ± SEM. n = > 10 per group. *P < 0.05
The difference in direction of total leukocyte responses to injury at this time point suggests Pld isoform-specific influence on neutrophil and macrophage responses (Fig. 4B). To this end, neutrophil and alveolar macrophage subset counts were compared after lung injury between genotypes. Total leukocyte counts were significantly higher in Pld2−/− relative to Pld1−/− (P = 0.009) and WT (P = 0.0004; Fig. 4B). Specifically, deficiency in Pld2 was associated with increased BAL neutrophils relative to WT (155 ± 25.0 × 103 vs. 51.6 ± 11.1 × 103 cells per BALF, P = 0.003; Fig. 4C) and Pld1−/− (88.6 ± 18.2 × 103 cells per BALF, P = 0.04; Fig. 4D), and increased total macrophage counts relative to WT (94.9 ± 12.0 × 103 vs. 59.0 ± 8.2 × 103 cells per BALF, P = 0.04; Fig. 4D) and Pld1−/− (61.3 ± 6.8 × 103 cells per BALF, P = 0.04; Fig. 4D). Macrophage subset determination demonstrated that iMacs were significantly increased in BAL fluid from Pld2−/− relative to WT (16.3 ± 4.0 × 103 vs. 6.4 ± 1.8 × 103 cells per BALF, P = 0.05; Fig. 4G), and trended higher relative to Pld1−/− (7.4 ± 1.4 × 103 cells per BALF, P = 0.07; Fig. 4D). ExMacs trended higher in Pld2−/− relative to WT (13.7 ± 2.3 × 103 vs. 7.1 ± 1.5 × 103 cells per BALF, P = 0.07; Fig. 4F). Together, these results indicate that deficiency in Pld2 is associated with increased neutrophil and iMac recruitment to injured lung.
3.7 |. Pld2 deficiency is associated with increased iMac-neutrophil interactions during experimental ARDS
Recruited alveolar macrophages, iMacs and ExMacs engage with neutrophils for efferocytosis.36 To determine if Pld2 regulates cell-cell interactions between macrophages and neutrophils, macrophage-bound neutrophils in BALF were quantified by flow cytometry (Fig. 5A, as in Ref. 36). Deficiency in Pld2 was associated with significantly increased absolute Ly6G+ iMac counts relative to WT (14.3 ± 3.5 × 103 vs. 5.7 ± 1.6 × 103 cells per BALF, P = 0.04; Fig. 5B), and higher percentage of iMacs that were Ly6G+ (87.4 ± 3.2 vs. 74.0 ± 5.5% of iMacs, P = 0.05; Fig. 5C). The absolute Ly6G+ exMacs counts appeared higher in Pld2−/− relative to WT (16.3 ± 3.9 × 103 vs. 8.4 ± 1.7 × 103 cells per BALF, P = 0.08; Fig. 5D), and the percentage of exMAcs that were Ly6G+ was very high in both genotype groups (95.1 ± 1.3 vs. 91.6 ± 2.6% of exMacs, P = 0.12; Fig. 5E).
FIGURE 5. PLD genetic deficiency is associated with increased macrophage–neutrophil interactions.
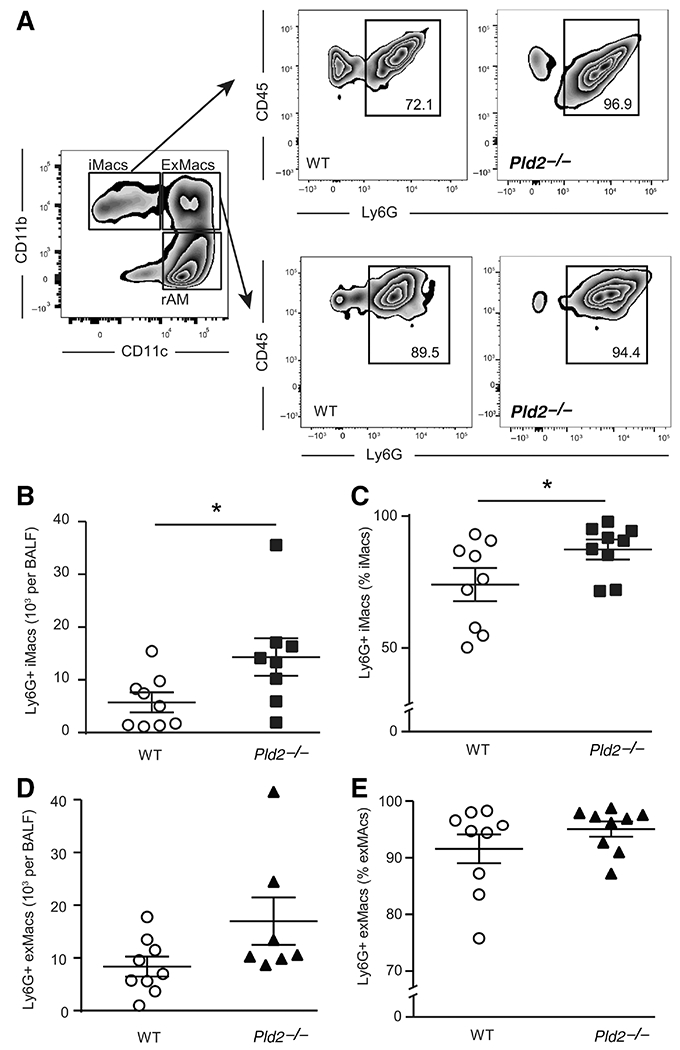
(A) Representative dot plots of macrophage–neutrophil interactions as determined by Ly6G staining gated on macrophage subsets in BAL obtained 24 h after intrabronchial acid. (B) Absolute count and (C) percentage of iMacs that interact with neutrophils. (D) Absolute count and (E) percentage of exMacs that interact with neutrophils. Results are expressed as mean ± SEM. n ≥ 6 per group. *P < 0.05
3.8 |. PLD2 silencing enhances proresolving macrophage functions
Pld2 was associated with changes in murine macrophage recruitment and function in vivo (vide supra). To investigate the impact of PLD on macrophage responses to injury, phagocytosis and chemotaxis were determined in human M2 macrophages (see Materials and Methods). Decreased PLD1 gene expression was associated with decreased macrophage chemotaxis toward MCP-1 relative to control (1.45 ± 0 26 × 103 vs. 2.77 ± 0.42 × 103 cells per HPF, P = 0.01), whereas decreased PLD2 gene expression again gave contrasting results with increased macrophage transmigration (3.92 ± 0.35 × 103 vs. 2.77 ± 0.42 × 103 cells per HPF, P = 0.03) at 360 min after exposure to the chemoattractant (Fig. 6A). In addition, decreased PLD1 gene expression was associated with decreased phagocytosis of latex beads relative to control (2.94 ± 0.33 vs. 4.82 ± 0.37 particle per cell, P = 0.043), whereas decreased PLD2 gene expression gave the opposite response with increased phagocytosis (6.15 ± 0.64 vs. 4.82 ± 0.37 particle per cell, P = 0.006) (Fig. 6B).
FIGURE 6. PLD isoform knockdown is associated with selective regulation of leukocyte function.
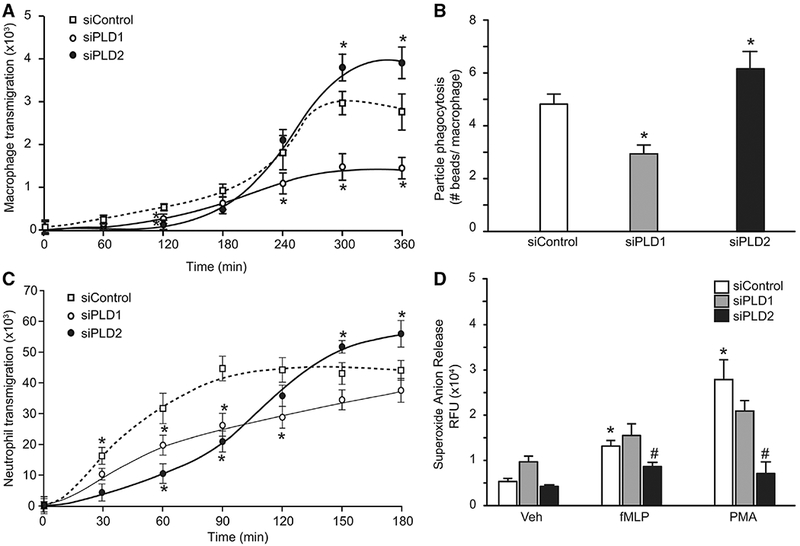
(A and B) Differentiated human M2 macrophages and neutrophils were transfected with either scrambled siRNA (controls), or a pool of PLD1 siRNAs, or a pool of PLD2 siRNAs (see Materials and Methods). (A) Chemotaxis across transwell membrane toward MCP-1(10 nM) was determined for the indicated lengths of time; cells migrated at the bottom of the wells were stained and counted. Averages from 7 fields were plotted. (B) Phagocytosis of green-fluorescent latex beads (0.1 mg/mL) was determined and expressed as the average of beads inside macrophages by fluorescence. (C and D) Differentiated human neutrophils were transfected with either scrambled siRNA (controls), or a pool of PLD1 siRNAs, or a pool of PLD2 siRNAs (see Materials and Methods). (C) Neutrophil transmigration across transwell membrane toward IL-8(10nM) was determined for the indicated lengths of time; cells migrated at the bottom of the wells were stained and counted. Averages from 7 fields were plotted. *P < 0.05 versus control. (D) Neutrophil superoxide anion release in response to vehicle (veh), f-MLP (150 nM), and PMA (50 ng/mL). Data presented are averages + SEM from 4 separate biologic experiments each in duplicate. *P < 0.05 versus veh, #P < 0.05 versus siControl
3.9 |. PLD2 silencing decreased ROS production
Pld2 deficiency was associated with increased neutrophil recruitment and decreased histologic injury after experimental ALI (Figs. 3 and 4). The impact of PLD on neutrophil chemotaxis and ROS production was determined (see Materials and Methods). Decreased PLD2 gene expression gave increased chemotaxis toward IL-8 relative to control (55.8 ± 4.5 × 103 vs. 44.0 ± 2.3 × 103 cells per HPF, P = 0.04), at 180 min after addition of chemoattractant, whereas decreased PLD1 gene expression was associated with decreased chemotaxis relative to control (28.4 ± 2.6 × 103 vs. 44.2 ± 3.1 × 103 cells per HPF, P = 0.035) at 120 min after addition of chemoattractant (Fig. 6C). Although DNA levels were increased in BAL fluid after intrabronchial acid (Supplemental Fig.S5a), NETs were not detected (Supplemental Fig. S5b). Neutrophil ROS production in response to the agonists FMLP and PMA was determined. F-MLP (50 ng/mL) and PMA (50 ng/mL) increased ROS production in control neutrophils (Fig. 6D). PLD1 gene expression did not significantly alter ROS production to agonists. In contrast, decreased PLD2 gene expression decreased ROS production to both agonists.
4 |. DISCUSSION
Results presented herein indicate that PLD isoforms are differentially regulated after ARDS. PLD1 gene expression is upregulated in human peripheral blood from ARDS survivors, and in murine lungs after self-limited ALI. In addition, PLD1 deficiency was associated with increased alveolar barrier disruption and tissue injury, suggesting a protective role for PLD1 in ARDS. Moreover, PLD2 gene expression was associated with ARDS nonsurvivors and was down-regulated in murine lungs after self-limited ALI. Pld2 deficiency was associated with decreased tissue injury, increased recruitment of proresolving macrophages with enhanced phagocytosis, and decreased ROS production by neutrophils. Together, these findings support a pathogenic role for PLD2 that operates in opposition to PLD1 in ARDS.
PLD1 and PLD2 are the enzymatically active members of the mammalian PLD superfamily that catalyze the hydrolysis of phosphatidylcholine, the most abundant membrane phospholipid, to generate choline and PA, a second messenger signaling lipid (PA).10 Although animal models and in vitro studies have revealed pathogenic roles for PLD1 and PLD2 in thrombotic disease, cancer, and inflammatory diseases such as sepsis,38 potential roles in human ARDS had yet to be investigated.
Examination of a published human gene expression microarray dataset from our institution15 has enabled here a targeted analysis of PLD and PLD-related gene expression in patients with ARDS. As the number of popular platforms to measure gene expression increases, the ability to compare and integrate gene expression measures across diverse platforms has become correspondingly important. Several studies have shown an overall concordance of gene expression measures across platforms.39,40 A recent study led to development of a new approach to map probes across platforms, allowing investigators to map probes from different microarray platforms.26 The application of this method to the BWH RoCI and publicly available ARDS microarray datasets has increased our power to detect regulation of PLD gene expression, and allowed for external confirmation of findings from the BWH RoCI.
Regulation of PLD gene expression in peripheral blood prompted the investigation of gene expression in diseased lung. As gene expression changes in ALI are comparable between animal models and human disease,41 associations between gene expression and recovery from disease were determined in a self-limited mouse model of acid induced ALI an important cause of ARDS.1 In this model, inflammation peaks and resolution processes are engaged 24 h after intrabronchial acid instillation. Lung PLD gene expression correlated with results from human peripheral blood microarray datasets, indicating that PLD responses to lung injury are systemic, and could serve as potential biomarkers. Pld1 expression was increased during self-resolving lung injury, as was expression of Plpp1 and Plpp2, correlating with the association between gene expression and survival in patients with ARDS. Similarly, decreased lung Pld2 expression in ALI correlated with decreased PLD2 expression in peripheral blood from ARDS patients. Results of the survivor analyses suggested that loss of PLD1 was detrimental, whereas loss of PLD2 was protective.
Indeed, Pld1 gene deletion was associated with worse tissue injury and barrier dysfunction relative to WT mice after intrabronchial acid-induced lung injury. Total PLD activity was decreased in both Pld-deficient animals, indicating that compensatory increases in activity from the other PLD isoform was an unlikely confounding factor. PLD1 was expressed by multiple cell types in the lung, and played important roles in processes central to the development of ALI, such as endothelial barrier function, clearance of alveolar edema, and neutrophil activation. PLD signaling results in ROS-mediated endothelial cytoskeletal dysfunction.42 PLD1 activation by TGF-beta results in internalization of the epithelial sodium channel ENaC, which prevents clearance of alveolar edema and potentiates lung injury.43 In addition, PLD1 activation is linked to superoxide anion generation,5 and neutrophil adhesion.6 Therefore, the detrimental effect of Pld1 deletion is likely related to a separate process. Endothelial cells from Pld1−/− mice demonstrate decreased integrin-dependent cell adhesion, and migration on extracellular matrices with subsequent impairments in angiogenesis.29 In addition, PLD1 regulates beta-catenin signaling,9 a signaling pathway important for lung epithelial repair.44 Intrabronchial acid targets the lung epithelium,45 and loss of PLD1 could disrupt epithelial repair and augment barrier dysfunction. In addition, the lack of impact of Pld1−/− on in vivo cytokine release suggests a direct action on cellular responses rather than an indirect action downstream of cytokines.
As suggested by PLD2 gene expression profiles in ARDS patients and in murine self-limited ALI, Pld2 gene deletion was protective, and associated with less tissue injury. The Pld2−/− mice had significantly increased neutrophil recruitment relative to WT, and PLD2 gene knockdown of human neutrophils increased chemotaxis to IL-8. Increased tissue infiltration by PLD2-deficient neutrophils was also observed in models of pancreatitis,11 colitis,46 and ALI secondary to sepsis.12 PLD2 deficiency is associated with increased NETosis and survival from sepsis-induced ALI.12 DNA levels were increased here with self-limited ALI; however, NETosis was not identified and DNA levels did not differ significantly by Pld genotype.
Deficiency in Pld2 increased macrophage recruitment as well. Further characterization of the alveolar macrophage population demonstrated increased recruitment of iMacs and exMacs. Interactions between these macrophage subtypes and neutrophils, indicators of ongoing efferocytosis,36 were also increased in Pld2-deficient mice. These macrophage subtypes participate in efferocytosis of apoptotic neutrophils for resolution of ALI,36 and release protective cytokines such as IL-1ra,47 and IL-10.48 In addition, PLD2 gene knockdown was associated with increased human macrophage transmigration and phagocytosis by cells of proresolving M2 phenotype.3 These changes in macrophage functions related to PLD2 expression were associated with increased levels of IL-1α and GM-CSF, suggesting PLD2 signaling was upstream of macrophage cytokine expression. Indeed, GM-CSF regulates efferocytosis by alveolar macrophages after lung injury,49,50 and IL-1α promotes CD11bLow alveolar macrophage proliferation and differentiation into CD11bHigh macrophages.51 Together, these findings suggest a regulated interplay between PLD2 and select cytokines that influence macrophage functional responses.
In health, specialized proresolving lipid mediators restrain inflammatory responses and promote resolution of inflammation.52 These enzymatically derived endogenous lipid mediators counter-regulate proinflammatory signals to prevent excessive cellular responses and bystander tissue injury. Of interest, lipoxin A4 (LXA4) inhibits PLD activity and NADPH oxidase assembly by targeting PLPP6.8,53 Of note, activation of PLPP6 can decrease levels of presqualene diphosphate, an inhibitor of PLD, to facilitate neutrophil responses in inflammation8 and ALI.54 Results presented here demonstrated regulation of PLPP6 gene expression in both human and experimental ARDS, supporting a role for this signaling pathway in the control of lung inflammation. Investigation into potential roles for PLD isoform-specific interactions with PLPP6 in ALI remains to be defined.
In summary, our findings have uncovered pathogenic and protective signaling circuits centered on PLD that are isoform specific. Data from ARDS patients and a mammalian model of self-resolving ALI demonstrated that PLD1 expression was associated with protection from tissue injury and increased survival from ARDS, whereas PLD2 expression adversely impacted recruitment of proresolving macrophages and recovery from ALI. Characterization of these distinct pathways offers a potential new host-directed therapeutic approach to ARDS that emphasizes PLD signaling and actions that are isoform specific. The opposing actions of PLD1 and PLD2 provide a mechanism for host tissues to regulate the intensity of inflammation and its resolution.
Supplementary Material
Summary statement:
PLD isoforms are differentially associated with survival in patients with ARDS, and regulate host responses to acute lung injury.
ACKNOWLEDGMENTS
We thank Troy Carlo, Ho Pan Sham, and Guangli Zhu for technical advice and assistance. This research was supported in part by the US National Institutes of Health K08-HL130540 (R.E.A.), R01-HL056653-14 (J.G.C.), and P01-GM095467 (B.D.L.). The content is solely the responsibility of the authors and does not necessarily reflect the official views of NHLBI, NIGMS, or the National Institutes of Health.
Abbreviations:
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- BAL
bronchoalveolar lavage
- EBD
Evans blue dye
- HCl
hydrochloric acid
- ICU
intensive care unit
- NETs
neutrophil-extracellular traps
- PA
phosphatidic acid
- PLPP
phospholipid phosphatases
- PLD
phospholipase D
- ROS
reactive oxygen species
- RoCI
Registry of Critical Illness
- qRT-PCR
quantitative real-time PCR
- WT
wild-type
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
- 1.Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. [DOI] [PubMed] [Google Scholar]
- 2.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013;13(3):159–175. [DOI] [PubMed] [Google Scholar]
- 3.Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 2014;306(8):L709–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthay MA,Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012;122(8):2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy BD, Hickey L, Morris AJ, et al. Novel polyisoprenyl phosphates block phospholipase D and human neutrophil activation in vitro and murine peritoneal inflammation in vivo. Br J Pharmacol 2005;146(3):344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speranza F, Mahankali M, Henkels KM, Gomez-Cambronero J. The molecular basis of leukocyte adhesion involving phosphatidic acid and phospholipase D. J Biol Chem 2014;289(42):28885–28897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomez-Cambronero J, Kantonen S. A river runs through it: how autophagy, senescence, and phagocytosis could be linked to phospholipase D by Wnt signaling. J Leuk Biol 2014;96(5):779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy BD, Fokin VV, Clark JM, Wakelam MJ, Petasis NA, Serhan CN. Polyisoprenyl phosphate (PIPP) signaling regulates phospholipase D activity: a ‘stop’ signaling switch for aspirin-triggered lipoxin A4. FASEB J 1999;13(8):903–911. [DOI] [PubMed] [Google Scholar]
- 9.Gomez-Cambronero J Phospholipase D in cell signaling: from a myriad of cell functions to cancer growth and metastasis. J Biol Chem 2014;289(33):22557–22566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez-Cambronero J, Di Fulvio M, Knapek K. Understanding phospholipase D (PLD) using leukocytes: pLD involvement in cell adhesion and chemotaxis. J Leuk Biol 2007;82(2):272–281. [DOI] [PubMed] [Google Scholar]
- 11.Ali WH, Chen Q, Delgiorno KE, et al. Deficiencies of the lipid-signaling enzymes phospholipase D1 and D2 alter cytoskeletal organization, macrophage phagocytosis, and cytokine-stimulated neutrophil recruitment. PloS One. 2013;8(1):e55325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SK, Kim SD, Kook M, et al. Phospholipase D2 drives mortality in sepsis by inhibiting neutrophil extracellular trap formation and down-regulating CXCR2. J Exp Med 2015;212(9):1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenkins GM, Frohman MA. PhospholipaseD: a lipid centric review. Cell Mol Life Sci 2005;62(19-20):2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nanjundan M,Possmayer F. Pulmonary phosphatidic acid phosphatase and lipid phosphate phosphohydrolase. Am J Physiol Lung Cell Mol Physiol 2003;284(1):L1–23. [DOI] [PubMed] [Google Scholar]
- 15.Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 2012;185(11):1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy MM, Fink MP, Marshall JC, et al. SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 2001;31(4):1250–1256. [DOI] [PubMed] [Google Scholar]
- 17.Bernard GR, Artigas A, Brigham KL, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med 1994;149(3 Pt 1):818–824. [DOI] [PubMed] [Google Scholar]
- 18.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The acute respiratory distress syndrome network. N Engl J Med 2000;342(18):1301–1308. [DOI] [PubMed] [Google Scholar]
- 19.Kollef KE, Reichley RM,Micek ST, Kollef MH. Themodified APACHE II score outperforms Curb65 pneumonia severity score as a predictor of 30-day mortality in patients with methicillin-resistant Staphylococcus aureus pneumonia. Chest. 2008;133(2):363–369. [DOI] [PubMed] [Google Scholar]
- 20.Howrylak JA, Dolinay T, Lucht L, et al. Discovery of the gene signature for acute lung injury in patients with sepsis. Physiol Genom 2009;37(2):133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong HR, Shanley TP, Sakthivel B, et al. Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genom 2007;30(2):146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5(10):R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durinck S, Moreau Y, Kasprzyk A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21(16):3439–3440. [DOI] [PubMed] [Google Scholar]
- 25.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 2009;4(8):1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin SH, Beane L, Chasse D, Zhu KW, Mathey-Prevot B, Chang JT. Cross-platform prediction of gene expression signatures. PloS One. 2013;8(11):e79228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudy J, Valafar F. Empirical comparison of cross-platform normalization methods for gene expression data. BMC Bioinformatics. 2011;12:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oliveira TG, Chan RB, Tian H, et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci 2010;30(49):16419–16428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Q, Hongu T, Sato T, et al. Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci Signal. 2012;5(249):ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thielmann I, Stegner D, Kraft P, et al. Redundant functions of phospholipases D1 and D2 in platelet alpha-granule release. J Thromb Haemostasis. 2012;10(11):2361–2372. [DOI] [PubMed] [Google Scholar]
- 31.Abdulnour RE, Dalli J, Colby JK, et al. Maresin 1 biosynthesis during platelet-neutrophil interactions is organ-protective. Proc Natl Acad Sci USA. 2014;111(46):16526–16531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahankali M, Peng HJ, Henkels KM, Dinauer MC, Gomez-Cambronero J. Phospholipase D2 (PLD2) is a guanine nucleotide exchange factor (GEF) for the GTPase Rac2. Proc Natl Acad Sci USA. 2011;108(49):19617–19622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene. 2013;32(49):5551–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liscovitch M, Czarny M, Fiucci G, Tang X. Phospholipase D: molecular and cell biology of a novel gene family. Biochem J 2000;345(Pt 3):401–415. [PMC free article] [PubMed] [Google Scholar]
- 35.Henkels KM, Muppani NR, Gomez-Cambronero J. PLD-Specific small-molecule inhibitors decrease tumor-associated macrophages and neutrophils infiltration in breast tumors and lung and liver metastases. PloS One. 2016;11(11):e0166553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdulnour RE, Sham HP, Douda DN, et al. Aspirin-triggered resolvin D1 is produced during self-resolving gram-negative bacterial pneumonia and regulates host immune responses for the resolution of lung inflammation. Mucosal Immunol 2016;9(5):1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kantonen S, Hatton N,Mahankali M, et al. A novel phospholipase D2-Grb2-WASp heterotrimer regulates leukocyte phagocytosis in a two-step mechanism. Mol Cell Biol 2011;31(22):4524–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelson RK, Frohman MA. Physiological and pathophysiological roles for phospholipase D. J Lipid Res 2015;56(12):2229–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo Y, Affourtit J, Daigle S, et al. A comparison of cDNA, oligonucleotide, and Affymetrix GeneChip gene expression microarray platforms. J Biomol Tech 2004;15(4):276–284. [PMC free article] [PubMed] [Google Scholar]
- 40.Barnes M, Freudenberg J, Thompson S, Aronow B, Pavlidis P. Experimental comparison and cross-validation of the Affymetrix and Illumina gene expression analysis platforms. Nucleic Acids Res 2005;33(18):5914–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweeney TE, Lofgren S, Khatri P, Rogers AJ. Gene expression analysis to assess the relevance of rodent models to human lung injury. Am J Respir Cell Mol Biol 2017;57(2):184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Usatyuk PV, Kotha SR, Parinandi NL, Natarajan V. Phospholipase D signaling mediates reactive oxygen species-induced lung endothelial barrier dysfunction. Pulm Circ 2013;3(1):108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peters DM, Vadasz I, Wujak L, et al. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA. 2014;111(3):E374–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zemans RL, Briones N, Campbell M, et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci USA. 2011;108(38):15990–15995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008;295(3):L379–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou G, Yu L, Yang W, Wu W, Fang L, Liu Z. Blockade of PLD2 ameliorates intestinal mucosal inflammation of inflammatory bowel disease. Mediat Inflamm 2016;2016:2543070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herold S, Tabar TS, Janssen H, et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am J Respir Crit Care Med 2011;183(10):1380–1390. [DOI] [PubMed] [Google Scholar]
- 48.Aggarwal NR, Tsushima K, Eto Y, et al. Immunological priming requires regulatory T cells and IL-10-producing macrophages to accelerate resolution from severe lung inflammation. J Immunol 2014;192(9): 4453–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shyamsundar M, McAuley DF, Ingram RJ, et al. Keratinocyte growth factor promotes epithelial survival and resolution in a human model of lung injury. Am J Respir Crit Care Med 2014;189(12):1520–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berclaz PY, Shibata Y, Whitsett JA, Trapnell BC. GM-CSF, via PU.1, regulates alveolar macrophage Fcgamma R-mediated phagocytosis and the IL-18/IFN-gamma -mediated molecular connection between innate and adaptive immunity in the lung. Blood. 2002;100(12):4193–4200. [DOI] [PubMed] [Google Scholar]
- 51.Huaux F, Lo Re S, Giordano G, Uwambayinema F, Devosse R, Yakoub Y, et al. IL-1alpha induces CD11b(low) alveolar macrophage proliferation and maturation during granuloma formation. J Pathol 2015;235(5):698–709. [DOI] [PubMed] [Google Scholar]
- 52.Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carlo T,Kalwa H, Levy BD. 15-Epi-lipoxin A4 inhibits human neutrophil superoxide anion generation by regulating polyisoprenyl diphosphate phosphatase 1. FASEB J 2013;27(7):2733–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonnans C, Fukunaga K, Keledjian R, Petasis NA, Levy BD. Regulation of phosphatidylinositol 3-kinase by polyisoprenyl phosphates in neutrophil-mediated tissue injury. J Exp Med 2006;203(4):857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.