

Abstract
Background
Metronidazole is one of the first-line drugs of choice in the standard triple therapy used to eradicate Helicobacter pylori infection. Hence, the global emergence of metronidazole resistance in Hp poses a major challenge to health professionals. Inactivation of RdxA is known to be a major mechanism of conferring metronidazole resistance in H. pylori. However, metronidazole resistance can also arise in H. pylori strains expressing functional RdxA protein, suggesting that there are other mechanisms that may confer resistance to this drug.
Methods
We performed whole-genome sequencing on 121 H. pylori clinical strains, among which 73 were metronidazole-resistant. Sequence-alignment analysis of core protein clusters derived from clinical strains containing full-length RdxA was performed. Variable sites in each alignment were statistically compared between the resistant and susceptible groups to determine candidate genes along with their respective amino-acid changes that may account for the development of metronidazole resistance in H. pylori.
Results
Resistance due to RdxA truncation was identified in 34% of metronidazole-resistant strains. Analysis of core protein clusters derived from the remaining 48 metronidazole-resistant strains and 48 metronidazole-susceptible identified four variable sites significantly associated with metronidazole resistance. These sites included R16H/C in RdxA, D85N in the inner-membrane protein RclC (HP0565), V265I in a biotin carboxylase protein (HP0370) and A51V/T in a putative threonylcarbamoyl–AMP synthase (HP0918).
Conclusions
Our approach identified new potential mechanisms for metronidazole resistance in H. pylori that merit further investigation.
Keywords: Helicobacter pylori, metronidazole, antibiotic resistance, whole-genome sequencing, core protein clusters, sequence alignment
Introduction
Helicobacter pylori is a Gram-negative microaerophilic bacterium that persistently colonizes the human gastric mucosa. Common clinical manifestations of H. pylori chronic infection include dyspepsia, chronic gastritis, gastric atrophy and peptic ulceration. In more severe but less common cases, H. pylori infection may cause gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue (MALT) [1, 2]. Consequently, H. pylori has been classified as a type 1 carcinogen by the World Health Organization [3].
In the recent Maastricht V consensus report, administration of standard triple therapy remains the first-line treatment for eradication of H. pylori infection in areas of low clarithromycin resistance [4]. Standard triple therapy consists of a proton pump inhibitor and two antibiotics (amoxicillin and either clarithromycin or metronidazole). The later antibiotic, metronidazole, is a prodrug commonly used for the treatment of parasitic infections, including trichomoniasis and giardiasis. It is effective against anaerobic and certain microaerophilic microorganisms, including H. pylori [5–7]. However, the widespread use of metronidazole has resulted in the emergence of resistant H. pylori strains. In a recent meta-analysis of antibiotic resistance in H. pylori, the overall occurrence of metronidazole resistance was found to be as high as 47.22%, substantially undermining the efficacy of H. pylori eradication therapy [8].
The current literature indicates that resistance to metronidazole in H. pylori primarily involves inactivation of the RdxA gene. This gene encodes for an oxygen-insensitive NADPH nitroreductase that catalyses the reduction of metronidazole by the transfer of four electrons to form hydroxylamine—a potent mutagen that is toxic to H. pylori [9]. Apart from nonsense and frameshift mutations, amino-acid substitutions in the RdxA protein including R16H, Y46H, P51L and A67V have been reported as potentially contributing to metronidazole resistance [10–12]. Furthermore, C19Y, T49K and C159A/S mutations have been experimentally demonstrated to confer metronidazole resistance in H. pylori [13, 14].
FrxA, the second nitroreductase protein in H. pylori, also plays a role in metronidazole resistance. It has been shown that the level of resistance in Hp with RdxA mutation could be enhanced further by FrxA inactivation and that the high expression level of FrxA could deter the development of metronidazole resistance attributed to RdxA inactivation [15]. In addition, mutations in several genes including HP0630 (MdaB), HP1087 (RibF), HP1027 (Fur), HP1382 and HP0922 and the overexpression of SodB superoxide dismutase via down-regulation of the Fur gene were shown to enhance resistance to metronidazole [16, 17].
In our collection of metronidazole-resistant H. pylori clinical strains from Barry Marshall’s H. pylori Research Laboratory and University of Malaya Helicobacter Research Laboratory, some were found not to harbour any of the above-mentioned mutational changes. This suggested that there may be additional, as-yet uncharacterized, mechanisms of metronidazole resistance. To uncover these mechanisms, we sequenced and analysed the draft genomes of 73 metronidazole-resistant and 48 metronidazole-susceptible H. pylori clinical strains.
Materials and methods
Bacterial cultures
This study was approved by the Sir Charles Gairdner and Osborne Park Health Care Group Human Research Ethics Committee (HREC No: 2013–007) and the University of Malaya Medical Centre (UMMC) Medical Ethics Committee. Biopsy samples for culturing were obtained with informed and written consent from patients who presented for endoscopy at Sir Charles Gairdner Hospital and UMMC.
H. pylori strains were isolated from human gastric biopsy samples on selective and non-selective agar plates. The non-selective plates used were Columbia blood agar plates (CBA) containing 5% horse blood (PathWest Laboratory Medicine WA Media, Australia). The selective plates were CBA plates with Dent supplement (Oxoid, UK). The plates were incubated for 3–4 days at 37°C in a 10% CO2 environment as previously described [18].
Metronidazole-sensitivity test
A metronidazole-sensitivity test was performed using Etest® strips (BioMérieux, France). Metronidazole resistance was defined with a minimum inhibition concentration (MIC) value of ≥8 mg/L [18].
Illumina library preparation and whole-genome sequencing
Genomic DNA was extracted using a DNeasy® blood & tissue kit (Qiagen, Germany) according to the manufacturer’s instructions. Preparation of the MiSeq library was performed using an Illumina Nextera XT DNA sample preparation kit (Illumina, USA) with minor modifications. In brief, 1 ng of genomic DNA was fragmented in 5 µL of Amplicon Tagment Mix and 10 µL of Tagment DNA buffer. Tagmentation reaction was performed by incubation at 55°C for 5 minutes followed by neutralization with 5 µL of Neutralise Tagment Buffer for 5 minutes. Tagmented DNA (25 µL) was indexed in a 50-µL limited-cycle PCR (12 cycles) as outlined in the Nextera XT protocol and subsequently purified using 25 µL of AMPure XP beads (Beckman Coulter, Australia). The fragment-size distribution of the purified DNA was analysed by the Australian Genome Research Facility utilizing the PerkinElmer LabChip GXII instrument. DNA libraries were adjusted to 2 nmol/L, pooled in equal volumes and then denatured with 0.2 N NaOH. The libraries were sequenced using the 2 × 250 paired-end protocol (MiSeq Reagent Kit v2 for 500 cycles) on an Illumina MiSeq instrument.
Identification and alignment of core protein clusters
The generated MiSeq reads of each H. pylori clinical strain included in this study were assembled using a SPAdes genome assembler (version 3.10.1) with the careful option [19]. Draft genomes were annotated using Prokka (version 1.12) [20]. Clustering of the orthologues was performed using all predicted coding sequences with ProteinOrtho (version 5.15) with the following parameters: -identity = 50 -cov = 95 [21]. In this study, a protein cluster was considered to be a core cluster when an orthologue was identified in at least 98% of input strains. Alignment of each core protein cluster was performed with MAFFT using the following parameters: –localpair –maxiterate 1000 [22]. Consensus sequences were generated. Variable sites including gaps in each alignment were extracted and subjected to further statistical analysis. Both the Prokka-annotated draft genomes and the alignments are available at the public data repository Figshare (https://figshare.com/), with doi: 10.6084/m9.figshare.5271046. All draft genomes have been deposited at DDBJ/ENA/GenBank and all sequencing data generated in this study have been submitted to Sequence Read Archives (SRA) database. Accession numbers are listed in Supplementary Table 1.
Protein-structure search and structural alignment
To better understand the function of hypothetical protein HP0918, a protein-structure search was undertaken using the Phyre2 server [23]. The model for the hit with the highest percentage-identity was then displayed and structurally aligned using the UCSF Chimera package, version 1.11.2 [24].
Statistical analysis
In the first stage, the distributions of RdxA inactivation and FrxA inactivation in both metronidazole-resistant and metronidazole-susceptible strains were analysed using the Fisher’s exact test. A P-value of less than 0.05 was considered significant. In the second stage, involving strains with full-length RdxA, for each gene in the core genomes, the association of each variable site in a protein sequence alignment with a metronidazole-resistant phenotype was statistically examined using Fisher’s exact test with a Bonferroni correction. This involved multiplying the acquired P-value by the number of variable sites in the gene being tested. An adjusted P-value of less than 0.05 was regarded as statistically significant.
Results
Nonsense and frameshift mutations in RdxA and FrxA from metronidazole-resistant and metronidazole-susceptible strains
Of the 121 whole-genome sequenced clinical strains, 73 were resistant to metronidazole, with MIC values ranging from 8–256 mg/L (Supplementary Table 1). RdxA truncation is known to play a predominant role in metronidazole resistance. Hence, a BLASTN alignment of HP0954, which encodes for the oxygen-insensitive NAD(P)H nitroreductase RdxA protein in H. pylori 26695, was performed against all strains. All RdxA sequences were then examined for any mutations that would result in translational defects. All 48 metronidazole-susceptible strains harboured intact RdxA genes that encoded for full-length functional RdxA. However, 25 of the 73 metronidazole-resistant strains had nonsense mutations or frame alterations attributed to nucleotide insertions or deletions in RdxA, resulting in protein truncation or mistranslation and consequently a complete loss of RdxA function (Table 1). The distribution of RdxA inactivation in metronidazole-resistant strains was statistically significant (P < 0.001).
Table 1.
MIC of metronidazole-resistant strains with RdxA nonsense and frameshift mutations
| Strain | MIC (mg/L) | Mutation | Change | Codon position |
|---|---|---|---|---|
| HP11054 | 48 | Nonsense | CAG → TAG | 50 |
| HP12064 | 256 | Nonsense | CAG → TAG | 50 |
| HP13024 | 256 | Nonsense | GGA → TGA | 155 |
| HP14016 | 256 | Nonsense | GAA → TAA | 75 |
| HP14052 | 256 | Nonsense | GAG → TAG | 175 |
| HP14056 | 256 | Nonsense | GAG → TAG | 175 |
| HP15012 | 64 | Nonsense | GAG → TAG | 107 |
| HP15015 | 256 | Nonsense | CAA → TAA | 102 |
| HP15026 | 256 | Nonsense | CAA → TAA | 130 |
| HP11043 | 256 | Frameshift | 7A → 8A | 65 |
| HP13012 | 256 | Frameshift | 7A → 8A | 65 |
| HP13013 | 24 | Frameshift | 3A → 5A | 14 |
| HP13028 | 64 | Frameshift | AAG → AATG | 20 |
| HP13061 | 256 | Frameshift | −CAGCGTTAAT | 81 |
| HP13072 | 256 | Frameshift | AAG → AG | 190 |
| HP15002 | 256 | Frameshift | AAA → AATA | 8 |
| HP15011 | 256 | Frameshift | GAT → GAAGAAATGAT | 77 |
| HP15022 | 256 | Frameshift | AGG → TAGG | 41 |
| HP15031 | 256 | Frameshift | 7A → 8A | 65 |
| HP15032 | 256 | Frameshift | −TCAAAAGTTGATGCGATTAC | 202 |
| HP15034 | 256 | Frameshift | 7A → 6A | 64 |
| HP15059 | 256 | Frameshift | 7A → 6A | 64 |
| HP15067 | 64 | Frameshift | GGT → ATTGGGT | 189 |
| HP16004 | 256 | Frameshift | 7A → 6A | 64 |
| HP16056 | 256 | Frameshift | AAG → TAAG | 60 |
In a transposon mutagenesis study conducted by Moore and Salama [25], one of the metronidazole-resistant mutants was shown to harbour an insertion positioned 25 bp upstream of the RdxA gene, prompting us to investigate the occurrence of this mutation in our strains that carry a full-length RdxA gene. No such mutation was observed. However, there was only one metronidazole-resistant strain in which its Shine-Dalgarno sequence had altered from AGGA to ATGA. Between the AGGA sequence and ATG start codon, nucleotide variations were found in a few metronidazole-resistant and metronidazole-susceptible strains. Nevertheless, we were unable to conclude whether these changes would affect the transcription or translation of RdxA.
To investigate whether FrxA inactivation could be associated with H. pylori metronidazole resistance, a BLASTN search of HP0642 was performed. Of the 48 metronidazole-resistant and 48 metronidazole-susceptible strains carrying complete RdxA genes, 31 of the metronidazole-resistant strains and 19 of the metronidazole-susceptible strains harboured a frameshift or a nonsense mutation in FrxA (Table 2). The distribution of FrxA inactivation was significantly greater in the metronidazole-resistant strains (P = 0.008), indicating that FrxA truncation might play a minor role in conferring metronidazole resistance.
Table 2.
FrxA frameshift and nonsense mutations
| Mutation | Change | Affected codon position | No. of strains |
|
|---|---|---|---|---|
| MR (n = 48) | MS (n = 48) | |||
| Frameshift | −GATTTGCTGCAAAAAAATACGATCC | 13 | 0 | 1 |
| Frameshift | 7A → 6A | 18 | 19 | 11 |
| Frameshift | −G | 20 | 1 | 0 |
| Frameshift | 4G → 3G | 38 | 1 | 0 |
| Frameshift | −TT | 52 | 0 | 1 |
| Frameshift | +TG | 60 | 1 | 0 |
| Frameshift | −C | 70 | 2 | 2 |
| Frameshift | 6G → 7G | 70 | 1 | 1 |
| Frameshift | 6G → 5G | 70 | 2 | 0 |
| Frameshift | GAC → TAAT | 92 | 1 | 0 |
| Frameshift | −G | 106 | 1 | 0 |
| Frameshift | +TATC | 145 | 1 | 0 |
| Frameshift | −G | 168 | 1 | 0 |
| Frameshift | +A | 200 | 0 | 1 |
| Nonsense | CGA → TGA | 13 | 0 | 1 |
| Nonsense | CGA → TGA | 86 | 0 | 1 |
| Total | 31 | 19 | ||
MR, metronidazole-resistant; MS, metronidazole-susceptible.
RdxA amino-acid substitutions
We next examined the 96 full-length RdxA sequences for amino-acid substitutions, following which statistical analysis (Fisher’s exact test) was performed on each site. Among the 72 variable sites identified, 19 were present only in metronidazole-resistant strains, including four occurrences each at Cys-19 and Gly-163, three occurrences each at Ser-43 and Ala-80, two occurrences each at Pro-44, Ala-67, Ser-81, Met-84, Gly-145 and Gly-189, and one occurrence each involving Ser-29, Ala-40, Asn-48, Cys-87, Cys-148, 157D, Lys-190, Glu-194 and Ser-202 (Supplementary Table 2). However, no statistical significance could be established. Conversion of R16H was identified in 13 metronidazole-resistant strains and R16C conversion was identified in five metronidazole-resistant strains. Although these R16 conversions were also identified in four metronidazole-susceptible strains, amino-acid substitution of Arg-16 was shown to be statistically significant (adjusted P = 0.038). In addition to the findings above, 19 substitutions were found exclusively in the metronidazole-susceptible strains (R10K, S30R/N, A37V/S, V57A, D61G, H69R/Y, S70R, E74K, E75D/W, K78E, S92I, K110R, I114L, V123L/M, M154I, P166A, L167V, K168R and A193T/V), suggesting that these amino-acid residue changes do not significantly perturb RdxA function.
Distribution of variable sites in core protein-cluster alignments
To identify additional mutations that might play a role in enhancing H. pylori resistance against metronidazole in the presence of a full-length functional RdxA protein, we conducted further analysis of the draft genomes of 96 H. pylori strains that contained an intact rdxA gene. Clustering of orthologous genes was initially performed on all predicted coding sequences using ProteinOrtho. We specified that a core protein cluster must have an orthologue existing in at least 98% of input strains. Thus, we acquired a total of 1035 core protein clusters for further protein multiple sequence alignment using MAFFT. In each alignment, the distribution of every variable site in both metronidazole-resistant and metronidazole-susceptible strains was statistically examined.
Using this approach, four protein clusters were found to harbour a variable site in which the distribution of amino-acid variants was significantly greater among the metronidazole-resistant strains than the metronidazole-susceptible strains (Table 3). These substitutions included the R16H/C in RdxA (adjusted P = 0.038) described above, D85N in the inner-membrane protein RclC (adjusted P = 0.021), V265I in a biotin carboxylase protein (adjusted P = 0.047) and A51V/T in HP0918 (adjusted P = 0.006). HP0918 is a hypothetical protein that is likely to be involved in N6-L-threonylcarbamoyladenosine37-modified tRNA biosynthesis, specifically catalysing the formation of the L-threonylcarbamoyladenylate intermediate compound. The putative function of HP0918 is further discussed below.
Table 3.
List of amino-acid substitutions that are significantly associated with metronidazole-resistant H. pylori clinical strains
| Clustera | Consensus AA residue and position | No. of strains with consensus AA |
No. of strains with variant AA |
Total variable sites including gaps | Adjusted P-value | Protein description | ||
|---|---|---|---|---|---|---|---|---|
| MR | MS | MR | MS | |||||
| 522 | R16 | 30 | 44 | 13 (H), 5 (C) | 4 (H) | 72 | 0.038 | Oxygen-insensitive NAD(P)H nitroreductase RdxA (HP0954) |
| 902 | D85 | 38 | 47 | 10 (N) | 0 | 33 | 0.021 | Inner-membrane protein RclC (HP0565) |
| 978 | V265 | 35 | 47 | 12 (I) | 1 (I) | 66 | 0.047 | Biotin carboxylase (HP0370) |
| 993 | A51 | 33 | 47 | 9 (V), 5 (T) | 1 (V) | 41 | 0.006 | Putative threonylcarbamoyl–AMP synthase (HP0918) |
AA, amino acid; MR, metronidazole-resistant; MS, metronidazole-susceptible.
aAlignment of each protein cluster is available at the public data repository Figshare (https://figshare.com/), with doi 10.6084/m9.figshare.5271046.
HP0918
A structure-based search using Phyre2 yielded a number of hits to structures related to the protein YrdC, with the highest percentage-identity hit being 1HRU, threonylcarbamoyl–AMP synthase (TsaC) from Escherichia coli. A structural alignment of the model produced by Phyre2 for HP0918 versus the structure used to create that model, 1HRU, revealed that all of the amino acids identified by Teplova et al. [26] being conserved in this class of proteins are also conserved in HP0918 (shown as red stars in Figure 1). On the other hand, HP0918 lacks the Pfam domain that is associated with YrdC (PF01300). However, a final observation is that, when HP0918 was viewed in the BioCyc database, in all H. pylori strains, HP0918 is found within a predicted operon next to carbamoyl-phosphate synthase [27].
Figure 1.

Structural alignment comparing the model of HP0918 computed by Phyre2 based on 1HRU and 1HRU itself. Highlighted with red stars are the conserved amino acids of the TsaC protein family, identified by Teplova et al. [26], which are identical in HP0918. Highlighted in blue are amino acids that are found in the YrdC subgroup, which are also found in HP0918 or for which there are conservative substitutions.
Discussion
Many studies have demonstrated that the development of metronidazole resistance in H. pylori is essentially due to loss-of-function mutations in the RdxA gene, which encodes an oxygen-insensitive nitroreductase exhibiting metronidazole reduction activity under micro-aerobic conditions [9, 11, 28]. Consistently with previous reports, sequence analysis of our collection of clinical strains identified a strong correlation between RdxA-inactivating mutations and metronidazole resistance. Numerous missense mutations were also identified in RdxA sequences from both our metronidazole-resistant and metronidazole-susceptible strains. The crystal structure of RdxA from H. pylori strain 26695 has recently been solved [14]. Based on that study, 5 (C19Y/F, S43L, G145E, G163D and S202L) of the 19 substitutions found only in the metronidazole-resistant strains were proposed to impair RdxA function through destabilization of the RdxA dimer formation or by decreasing the binding affinity of RdxA for the flavin mononucleotide (FMN) cofactor [14]. Conversely, none of the amino-acid residue changes identified only in the metronidazole-susceptible strains was predicted to cause any functional effects on the RdxA protein.
Notably, a significant number of our metronidazole-resistant strains contained a mutation of the Arg-16 residue of RdxA. This is one of several amino-acid residues responsible for binding between the FMN phosphoryl group and RdxA, and thus mutation of Arg-16 may dampen RdxA–FMN interaction and consequently impair the reduction–activation activity involving metronidazole [14]. However, this substitution was also identified in several metronidazole-susceptible strains, suggesting that Arg-16 mutation alone may not be sufficient to confer a metronidazole-resistant phenotype in H. pylori and could therefore involve additional mutations apart from the rdxA gene. Alternatively, the metronidazole-resistant phenotype attributed to Arg-16 substitution could be counteracted by high-level expression of a second nitroreductase, FrxA, as it has been previously demonstrated that high expression of FrxA renders H. pylori susceptible to metronidazole, regardless of RdxA status [29].
Although our statistical analysis showed that FrxA truncation is associated with metronidazole resistance, 19 of the 48 metronidazole-susceptible strains were also found to contain a truncated FrxA. This observation indicates that FrxA inactivation does not play a dominant role in imparting metronidazole resistance. Rather, FrxA inactivation may work in tandem with other mutations to enhance resistance, consistently with a previous study showing that the inactivation of FrxA gene had resulted in a higher level of metronidazole resistance in RdxA-deficient H. pylori cells, but no significant changes in the metronidazole susceptibility of cells containing an intact rdxA gene [29]. On the other hand, of the 48 H. pylori metronidazole-resistant clinical strains carrying full-length functional RdxA, 14 did not have any inactivating mutations in their FrxA gene (Supplementary Table 3). This finding suggests that there could be other genetic determinants involved in conferring metronidazole resistance.
To explore further for genes that are possibly responsible for metronidazole resistance, multiple sequence alignment followed by statistical comparison of the distribution of amino-acid variants at each variable site in both metronidazole-resistant and metronidazole-susceptible groups was performed on orthologous protein clusters. Four variable sites were found to be significantly associated with metronidazole resistance, including D85N in the RclC inner-membrane protein, V265I in a biotin carboxylase protein, A51V/T in a putative threonylcarbamoyl–AMP synthase and R16H/C in RdxA. The successful identification of the frequently reported Arg-16 mutation in RdxA is an important validation of our analytical method, providing further support that the novel mutations associated with metronidazole-resistant phenotype identified in this study are highly reliable.
The RclC inner-membrane protein is referred to as HP0565 in H. pylori 26695. It shares 53.3% amino-acid sequence similarity with the RclC protein in E. coli K-12 substrain MG1655, which plays an essential role in reactive chlorine resistance [30]. In H. pylori, besides the formation of hydroxylamine via the transfer of four electrons to metronidazole by RdxA, metronidazole can also be reduced by single electron transfer [9]. Under such a situation, the molecular oxygen present in the micro-aerobic intracellular compartment would compete with metronidazole radicals for electrons. This allows re-oxidation and restoration of metronidazole to its inactive state, and yet produces DNA-damaging superoxide anion radicals [31]. This process is termed futile recycling. It would therefore be of interest to determine whether HP0565 could play a role in H. pylori resistance against these free radicals besides providing protection against reactive chlorine species. And, if so, does the D85N substitution further enhance such a capacity to facilitate development of metronidazole resistance?
The biotin carboxylase subunit of acetyl coenzyme A (acetyl-CoA), which is designated as HP0370 in 26695, catalyses the ATP-dependent carboxylation of biotin and generates hydrogen ions as one of the end products [32, 33]. We propose that the hydrogen ions could be utilized by the H. pylori superoxide dismutase enzyme (HP0389) to convert superoxide radicals into hydrogen peroxide, which can be further inactivated by AhpC alkyl hydrogen peroxidase into water molecules (Figure 2) [34, 35]. Finally, as discussed above, HP0918 is predicted to be part of a multi-protein complex that facilitates the synthesis of N6-threonylcarbamoyladenosine in tRNAs.37 In particular, we hypothesize that HP0918, like E. coli TsaC, catalyses the formation of L-threonylcarbamoyladenylate, but uses a different mechanism involving carbamoyl phosphate. However, the role of mutations to HP0918 in metronidazole resistance is unclear. Future work should involve gene knockout and overexpression analysis of these identified genes to confirm their role and elucidate the mechanism by which they may mediate H. pylori metronidazole resistance.
Figure 2.
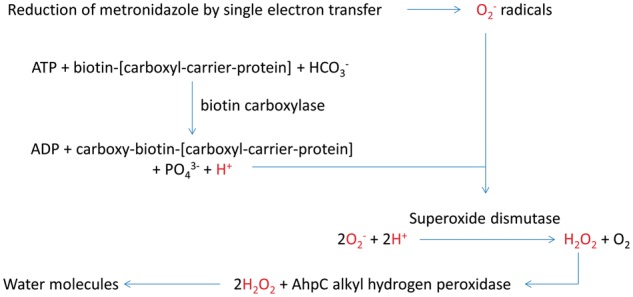
Proposed mechanism of superoxide anion radical neutralization mediated by HP0370, superoxide dismutase and catalase enzymes in H. pylori. In this diagram, we propose that the hydrogen ions generated by biotin carboxylation can be utilized by the superoxide dismutase enzyme to convert superoxide radicals into hydrogen peroxide. This can be further inactivated by AhpC alkyl hydrogen peroxidase into non-deleterious water molecules.
Conclusion
In this study, metronidazole resistance associated with RdxA inactivation was identified only in approximately 34% of H. pylori clinical strains. Our results also provide additional evidence that FrxA inactivation alone does not result in metronidazole resistance. By conducting whole-genome sequencing followed by core proteome analysis of the metronidazole-resistant and the metronidazole-susceptible H. pylori clinical strains, further genetic elements that are likely to be involved in mediating metronidazole resistance were identified. The results help to explain the varying levels of metronidazole resistance observed in different H. pylori strains. They may also help in the design of PCR-based assay tests for metronidazole resistance. Such tests would remove the need for time-consuming culture and sensitivity testing by clinical microbiologists and allow clinicians to make accurate decisions in tailoring H. pylori eradication treatments for individual patients.
Acknowledgements
Data analysis and drafting the manuscript: E.G.C. and M.J.W. Revision of draft manuscript: K.M.W. and A.W.D. Whole-genome sequencing: F.P. and B.L. Bacterial culturing and metronidazole-sensitivity test: F.P., B.L., E.G.C. and M.F.L. Ethics application: C.Y.T. and M.F.L. Endoscopic examination and biopsy: B.J.M. Funding and resources: B.J.M. and J.V. Read and approved the manuscript: All authors.
Funding
This project was supported by ShenZhen’s Sanming Project (Grant No: SZSM201510050), University of Malaya-Ministry of Education (UM-MoE) High Impact Research (HIR) grant (reference UM.C/625/1/HIR/MoE/CHAN13/3; Account No. H-50001-A000030), a National Health and Medical Research Council (NHMRC) Sir McFarlane Burnett Fellowship grant (572723) to B.J.M., the Vice Chancellor of the University of Western Australia, and the Western Australian Department of Commerce and Department of Health. A.W.D. was supported by an Early Career Research Fellowship from the NHMRC (APP1073250).
Conflict of interest statement: none declared.
Supplementary Material
References
- 1. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984;1:1311–5. [DOI] [PubMed] [Google Scholar]
- 2. Marshall BJ, Windsor HM. The relation of Helicobacter pylori to gastric adenocarcinoma and lymphoma: pathophysiology, epidemiology, screening, clinical presentation, treatment, and prevention. Med Clin North Am 2005;89:313–44. viii. [DOI] [PubMed] [Google Scholar]
- 3.Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum/World Health Organ, Intl Agency Res Cancer 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 4. Malfertheiner P, Megraud F, O’Morain CA et al. . Management of Helicobacter pylori infection-the Maastricht V/Florence Consensus Report. Gut 2017;66:6–30. [DOI] [PubMed] [Google Scholar]
- 5. Edwards DI. Mechanisms of selective toxicity of metronidazole and other nitroimidazole drugs. Br J Vener Dis 1980;56:285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chow AW, Patten V, Guze LB. Susceptibility of anaerobic bacteria to metronidazole: relative resistance of non-spore-forming Gram-positive baccilli. J Infect Dis 1975;131:182–5. [DOI] [PubMed] [Google Scholar]
- 7. Glupczynski Y, Delmee M, Bruck C et al. . Susceptibility of clinical isolates of Campylobacter pylori to 24 antimicrobial and anti-ulcer agents. Eur J Epidemiol 1988;4:154–7. [DOI] [PubMed] [Google Scholar]
- 8. Ghotaslou R, Leylabadlo HE, Asl YM. Prevalence of antibiotic resistance in Helicobacter pylori: a recent literature review. WJM 2015;5:164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olekhnovich IN, Goodwin A, Hoffman PS. Characterization of the NAD(P)H oxidase and metronidazole reductase activities of the RdxA nitroreductase of Helicobacter pylori. Febs J 2009;276:3354–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jenks PJ, Ferrero RL, Labigne A. The role of the rdxA gene in the evolution of metronidazole resistance in Helicobacter pylori. J Antimicrob Chemother 1999;43:753–8. [DOI] [PubMed] [Google Scholar]
- 11. Secka O, Berg DE, Antonio M et al. . Antimicrobial susceptibility and resistance patterns among Helicobacter pylori strains from The Gambia, West Africa. Antimicrob Agents Chemother 2013;57:1231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miftahussurur M, Shrestha PK, Subsomwong P et al. . Emerging Helicobacter pylori levofloxacin resistance and novel genetic mutation in Nepal. BMC Microbiol 2016;16:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paul R, Postius S, Melchers K et al. . Mutations of the Helicobacter pylori genes rdxA and pbp1 cause resistance against metronidazole and amoxicillin. Antimicrob Agents Chemother 2001;45:962–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martinez-Julvez M, Rojas AL, Olekhnovich I et al. . Structure of RdxA–an oxygen-insensitive nitroreductase essential for metronidazole activation in Helicobacter pylori. Febs J 2012;279:4306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeong JY, Mukhopadhyay AK, Akada JK et al. . Roles of FrxA and RdxA nitroreductases of Helicobacter pylori in susceptibility and resistance to metronidazole. J Bacteriol 2001;183:5155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsugawa H, Suzuki H, Satoh K et al. . Two amino acids mutation of ferric uptake regulator determines Helicobacter pylori resistance to metronidazole. Antioxid Redox Signal 2011;14:15–23. [DOI] [PubMed] [Google Scholar]
- 17. Albert TJ, Dailidiene D, Dailide G et al. . Mutation discovery in bacterial genomes: metronidazole resistance in Helicobacter pylori. Nat Meth 2005;2:951–3. [DOI] [PubMed] [Google Scholar]
- 18. Tay CY, Windsor HM, Thirriot F et al. . Helicobacter pylori eradication in Western Australia using novel quadruple therapy combinations. Aliment Pharmacol Ther 2012;36:1076–83. [DOI] [PubMed] [Google Scholar]
- 19. Bankevich A, Nurk S, Antipov D et al. . SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014;30:2068–9. [DOI] [PubMed] [Google Scholar]
- 21. Lechner M, Findeiss S, Steiner L et al. . Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 2011;12:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katoh K, Misawa K, Kuma K et al. . MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res 2002;30:3059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelley LA, Mezulis S, Yates CM et al. . The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pettersen EF, Goddard TD, Huang CC et al. . UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- 25. Moore JM, Salama NR. Mutational analysis of metronidazole resistance in Helicobacter pylori. Antimicrob Agents Chemother 2005;49:1236–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Teplova M, Tereshko V, Sanishvili R et al. . The structure of the yrdC gene product from Escherichia coli reveals a new fold and suggests a role in RNA binding. Protein Sci 2000;9:2557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caspi R, Billington R, Ferrer L et al. . The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 2016;44:D471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goodwin A, Kersulyte D, Sisson G et al. . Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol Microbiol 1998;28:383–93. [DOI] [PubMed] [Google Scholar]
- 29. Jeong JY, Mukhopadhyay AK, Dailidiene D et al. . Sequential inactivation of rdxA (HP0954) and frxA (HP0642) nitroreductase genes causes moderate and high-level metronidazole resistance in Helicobacter pylori. J Bacteriol 2000;182:5082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parker BW, Schwessinger EA, Jakob U et al. . The RclR protein is a reactive chlorine-specific transcription factor in Escherichia coli. J Biol Chem 2013;288:32574–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Wouden EJ, Thijs JC, Kusters JG et al. . Mechanism and clinical significance of metronidazole resistance in Helicobacter pylori. Scand J Gastroenterol Suppl 2001;36:10–4. [DOI] [PubMed] [Google Scholar]
- 32. Polakis SE, Guchhait RB, Zwergel EE et al. . Acetyl coenzyme A carboxylase system of Escherichia coli: studies on the mechanisms of the biotin carboxylase- and carboxyltransferase-catalyzed reactions. J Biol Chem 1974;249:6657–67. [PubMed] [Google Scholar]
- 33. Kuhns LG, Benoit SL, Bayyareddy K et al. . Carbon fixation driven by molecular hydrogen results in chemolithoautotrophically enhanced growth of Helicobacter pylori. J Bacteriol 2016;198:1423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seyler RW Jr, Olson JW, Maier RJ. Superoxide dismutase-deficient mutants of Helicobacter pylori are hypersensitive to oxidative stress and defective in host colonization. Infect Immun 2001;69:4034–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker LM, Raudonikiene A, Hoffman PS et al. . Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J Bacteriol 2001;183:1961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.