

Abstract
Yeast surface display (YSD) enables efficient screening and selection of single chain variable fragments (scFvs) of heavy (VH) and light (VL) chains that bind to target antigen with different affinities. Assembly of a scFv library from cDNA usually involves adding different primers and linkers (Gly4/Ser)3 through multiple rounds of PCR amplification and purification. We describe here a simplified scFv assembly method by creating a modified YSD vector with a built-in linker that reduces the time of assembly and decreases accumulated base exchanges due to PCR errors. In addition, we describe a bias screening strategy toward maximizing novel antibodies of interest by a combination of memory B cell selection and depletion by binding to mutant antigens that do not bind to previously identified monoclonal antibodies.
Keywords: Yeast surface display, Human monoclonal antibody isolation, Hepatitis C virus, Virus neutralization, Mutant antigen selection
1. Introduction
Comprehensive reviews and protocols describe yeast surface display (YSD) [1] as a powerful tool for isolating monoclonal antibodies and engineering these antibodies to fine tune their binding properties [2–5]. This protocol is focused on YSD application on the isolation of human monoclonal antibodies (HMAbs) from hepatitis C virus (HCV) infected individuals with modifications from previously described methods, which have been incorporated in three sections of the YSD system.
To facilitate scFv assembly in YSD library construction, we first modified a yeast display vector wherein a flexible linker region (Gly4/Ser)3 is presented in the form of a preassembled scFv. By digesting this vector at various restriction digestion sites located inside and outside of the scFv insertion, the variable regions of immunoglobulin gene pool amplified from cDNA are cloned directly into the vector. This allows all of the gene fragments to be cloned in-frame with its own linker in a single step, without the need for multiple steps of adding separate linkers to VH and VL, and then connecting the two as scFv, which are described in other scFv assembly protocols [3, 6–8]. In this way, the scFv assembly process timeline can be shortened and accumulated base exchanges due to PCR errors and/or PCR biased amplification can be minimized. Second, to increase the probability of isolating neutralizing HMAbs against HCV, we enriched a subset of B cells that is more likely to secrete these antibodies by screening the supernatants of small pools of activated B cells in an infectious cell culture-derived HCV virion (HCVcc) neutralization assay [9, 10]. This step allows for the collection of cells of interest for the initial RNA extraction. As a result, the YSD library size can be smaller and screening by fluorescence-activated cell sorting (FACS) will be more efficient. Third, to have a greater bias for novel neutralizing HMAbs, we developed a series of HCV envelope mutant constructs that are not able to bind nonneutralizing HMAbs or previously identified neutralizing HMAbs [11, 12]. The sequential FACS separation of scFvs with these antigens increases the likelihood of discovering new antibodies. Using this method, we obtained higher affinity neutralizing HCV HMAbs to a new cluster of overlapping epitopes, designated as antigenic domain D, that previously were masked by more immunodominant clusters, designated as antigenic domains A and B [10].
2. Materials
2.1. Reagents
2.1.1. Cells and Plasmids
EBY100 cells (see Note 1).
pYD2.A2 plasmid (see Note 2).
DH5α electroporation competent cells.
2.1.2. Chemicals, Reagents and Buffers
YPD media (Yeast Extract Peptone Dextrose media): Add 10 g yeast extract and 20 g peptone to 600 mL of deionized water (see Note 3). Adjust the final volume to 900 mL with deionized water. Autoclave and cool to 55 °C. Add 100 mL filtered 20% dextrose to bottle. Store at 4 °C.
YPD plates: Add 10 g yeast extract, 20 g peptone, and 17 g agar to 600 mL of deionized water (see Note 3). Adjust the final volume to 900 mL with deionized water. Autoclave and cool to 55 °C. Add 100 mL filtered 20% dextrose to bottle. Pour into 100 mm plates. Store at 4 °C.
SD-CAA media (Synthetic Dextrose minimal media with Casein Amino Acids): Add 7 g yeast nitrogen base (see Note 4), 5.4 g sodium phosphate dibasic, 7.4 g sodium phosphate monobasic anhydrous and 5 g casein amino acids to 600 mL of deionized water. Adjust the final volume to 888 mL with deionized water (see Note 3). Autoclave and cool to 55 °C (see Note 5). Add 100 mL filtered 20% dextrose, 10 mL filtered 0.6% leucine, 1 mL filtered 50 mg/mL kanamycin, and 1 mL filtered 15 mg/mL tetracycline to the contents in the bottle. Store at 4 °C.
SD-CAA plates: Add 7 g yeast nitrogen base (see Note 4), 5.4 g sodium phosphate dibasic, 7.4 g sodium phosphate monobasic anhydrous, 5 g casein amino acids, and 17 g agar to 600 mL of deionized water. Adjust the final volume to 888 mL with deionized water (see Note 3). Autoclave and cool to 55 °C (see Note 5). Add 100 mL filtered 20% dextrose, 10 mL filtered 0.6% leucine, 1 mL filtered 50 mg/mL kanamycin, and 1 mL filtered 15 mg/mL tetracycline to the contents in the bottle. Pour into 150 mm plates. Store at 4 °C.
SG-CAA induction liquid media (Synthetic Galactose Casein Amino Acids): Add 7 g yeast nitrogen base (see Note 4), 5.4 g sodium phosphate dibasic, 7.4 g sodium phosphate monobasic anhydrous and 5 g casein amino acids to 600 mL of deionized water. Adjust the final volume to 888 mL with deionized water (see Note 3). Autoclave and cool to 55 °C (see Note 5). Add 100 mL filtered 20% galactose, 10 mL filtered 0.6% leucine, 1 mL filtered 50 mg/mL kanamycin, and 1 mL filtered 15 mg/ mL tetracycline to the contents in the bottle. Store at 4 °C.
Yeast Freezing Media: Add 30 mL glycerin to 70 mL autoclaved water. Store at room temperature.
LB carbenicillin Media: Add 10 g tryptone, 5 g yeast extract, and 10 g NaCl to 600 mL of deionized water. Adjust the final volume to 1 L with deionized water. Autoclave and cool to 55 °C (see Note 6). Add 1 mL of filtered 100 mg/mL carbenicillin to contents in the bottle. Store at 4 °C.
LB carbenicillin plates: Add 10 g tryptone, 5 g yeast extract, 10 g NaCl, and 15 g agar to 600 mL of deionized water. Adjust the final volume to 1 L with deionized water. Autoclave and cool to 55 °C (see Note 3). Add 1 mL of filtered 100 mg/mL carbenicillin to contents in the bottle. Pour into 100 mm plates. Store at 4 °C.
Yeast electroporation buffer: 1 M sorbitol and 1 mM calcium chloride. Add 91.1 g sorbitol and 50 mL 100 mM calcium chloride to 400 mL of deionized water. Adjust the final volume to 500 mL with deionized water. Filter contents with 0.22 μM filter. Store at room temperature.
Yeast resuspension media: 0.1 M lithium acetate (LiAc) and 10 mM dithiothreitol (DTT). Add 1 mL 1 M LiAc and 100 μL 1 M DTT to 8.9 mL of deionized water. Filter contents with 0.22 μM filter. Store at room temperature.
PBS (Phosphate-buffered saline): Add 8 g sodium chloride, 0.2 g potassium chloride, 1.44 g disodium phosphate, and 0.24 g monopotassium phosphate to 600 mL of deionized water. Adjust the final volume to 1 L with deionized water and adjust pH to 7.4. Filter contents with 0.22 μM filter. Store at room temperature.
PBSB (Magnetic-activated cell sorting buffer): Add 8 g sodium chloride, 0.2 g potassium chloride, 1.44 g disodium phosphate, 0.24 g monopotassium phosphate, 4 mL 0.5 M EDTA, and 5 g bovine serum albumin to 600 mL of deionized water. Adjust the final volume to 1 L with deionized water and adjust pH to 7.4. Filter contents with 0.22 μM filter. Store at 4 °C.
TAE (Tris–acetate–ethylenediaminetetraacetic acid) buffer: 40 mM Tris base (pH 7.6), 20 mM glacial acetic acid, 1 mM Ethylenediaminetetraacetic acid (EDTA). Add 242 g Tris base, 57.1 mL glacial acetic acid, and 100 mL 0.5 M EDTA (pH 8.0) to 600 mL of deionized water. Adjust the final volume to 1 L with deionized water to prepare a liter of 50x TAE. Store at room temperature. Prepare 1x TAE by adding 20 mL 50x TAE to 980 mL deionized water. Store at room temperature.
DNA Gel: Add 1 g of agarose to 100 mL TAE buffer. To prepare 1% DNA gel. Microwave to dissolve agarose into TAE buffer (see Note 7). Before pouring, add SYBR Safe DNA gel stain to the agarose at a 1:10,000 dilution. Swirl to incorporate the gel stain and then pour.
Restriction enzyme NcoI.
Restriction enzyme SalI.
Restriction enzyme BspEI.
Restriction enzyme NotI.
T4 DNA Ligase.
Phusion polymerase.
10 mM dNTP mix.
Magnetic-activated Cell sorting beads (see Note 8).
Anti-V5.
Fluorescence labeled antibodies (see Note 9).
Biotinylated CBH-4G.
CBH-4G (available upon request).
Biotinylated HCV-E2.
S.O.B. (Super Optimal Broth) media.
S.O.C. (Super Optimal broth with Catabolite repression) media.
Molecular grade water.
Autoclaved water.
Double distilled water.
2.2. Kits
QIAGEN RNeasy Mini kit.
Invitrogen Superscript III First-Strand Synthesis System.
QIAGEN QiAquick Gel Extraction kit.
QIAGEN QiAquick PCR purification kit.
QIAGEN QiAprep Spin Miniprep kit.
QIAGEN QiAfilter Midi kit.
2.3. Material and Equipment
0.22 μM filter.
PCR machine.
PCR tubes
Microcentrifuge tubes.
Cryotubes.
17 × 100 mm polypropylene round-bottom tubes.
50 mL polypropylene conical tubes.
Gel electrophoresis unit.
SYBR safe gel stain.
Microwave.
Autoclave.
Spectrophotometer.
MACS equipment: MidiMACS separation unit, LS Columns and magnetic stand.
5 mL round bottom polystyrene test tube with cell strainer snap cap.
96-deep well plate.
Pre-sterilized breathable sealing film.
96 V-bottom well plate.
3 mm glass beads.
425–600 μm acid-washed glass beads.
Electroporation equipment: Electroporator and cuvettes.
37 °C incubator shaker.
37 °C water bath.
30 °C incubator shaker.
Centrifuge.
18 °C incubator shaker.
Tube shaker and rotator.
Vortex.
Flow cytometry machine.
Vector NTI software.
3. Methods
3.1. Construction of Yeast Display Library
3.1.1. Gene Cloning: RNA Extraction and cDNA Synthesis
A schematic overview describing all stages in this section is shown in Fig. 2.
Fig. 2. Illustration of method 3.1 section: Construction of yeast display library.
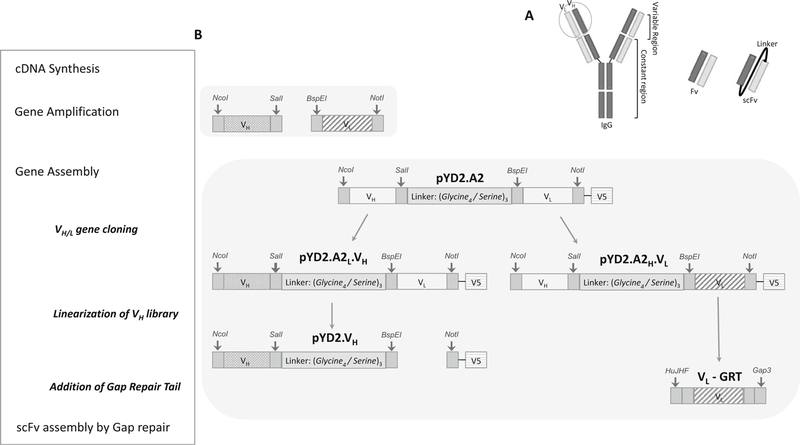
(a) Schematic representation of a scFv fragment. During library construction, VH and VL are randomly paired via a flexible linker to form scFvs. The quality of a library construction is dependent on the degree of scFv pairing diversity. (b) Schematic overview of scFv construction is boxed on the left, with a list of major steps involved in scFv construction. The corresponding diagram on the right illustrates the designated products derived from PCR, restriction digestion and ligation reactions. Vector pYD2.A2 displaying A2-scFv was created by modifying the pYD2 vector, which comprises a (Gly4/Ser)3 linker region carrying SalI and BspEI restriction sites, and NcoI and NotI restriction sites flanking the inserted A2-scFv. An in-frame V5 epitope allows detection of scFv fusion product and normalization of scFv surface expression through immunofluorescence labeling of V5-tag. Patterned filled bars represent amplified VH and VL from all of the heavy and light chain family genes using partially degenerated primers
Extract total RNA from selected B cells isolated from HCV infected individuals (see Note 10) using Qiagen RNeasy Mini kit according to the manufacturer’s instruction. Store RNA sample at −80 °C or use for cDNA synthesis immediately.
Set up five cDNA synthesis reactions for each library using Invitrogen kit according to the manufacturer’s instruction. The following mixes are for a single reaction.
Denature 1–1.5 μg RNA in a 10 μL reaction containing 1 mM dNTP and 50 μM oligo(dT)20 for 5 min at 65 °C and immediately place the reaction on ice for 1 min to relax the secondary structures of RNA, which often inhibit reverse transcriptase and primer annealing.
Add 10 μL of reverse transcriptase mix to each denatured RNA/oligo(dT)20 mixture as described in the table below. Incubate for 50 min at 50 °C.
| Reagent | 1 Rxn (μL) |
|---|---|
| 10× RT buffer | 2 |
| 25 mM MgCl2 | 4 |
| 0.1 M DTT | 2 |
| RNaseOUT (40 U/μL) | 1 |
| SuperScript III RT (200 U/μL) | 1 |
| Final vol | 10 |
Heat-inactivate reverse transcriptase for 5 min at 85 °C to terminate the reaction.
Add 1 μL (2 U) of RNase H to each reaction and incubate the reactions for 20 min at 37 °C to remove the RNA template from the cDNA/RNA products to achieve more efficient gene amplification by PCR.
Combine the five reactions to a total of 100 μL of cDNA and store at−20 °C or use for PCR immediately.
3.1.2. Gene Cloning: Amplification of Heavy and Light Chain Variable Genes (VH, Vκ, and Vλ) (Fig. 2b: Gene Amplification)
Prepare separate VH, Vκ, and Vλ PCR master mixes for 8× 50 μL reactions, as described in the table below, to generate a minimum of 10 μg PCR products for each gene. A total of seven to nine primers is included to amplify all of the heavy and light chain family genes.
| Reagent | 1 Rxn (μL) |
|---|---|
| 5× Phusion buffer | 10 |
| 10 mM dNTP mix | 1 |
| Phusion polymerase | 1 |
| Primer forward/primer reversea | 2 |
| cDNA | 2 |
| Molecule grade water | 34 |
| Final vol | 50 |
See Table 1
Aliquot 50 μL for each PCR tube and run the following conditions for VH and Vκ: 95 °C for 30 s, 25 cycles of 95 °C for 10 s, 55 °C for 20 s, 72 °C for 20 s, and 1 cycle of 72 °C for 5 min for final extension. Run the following condition for Vλ amplification: 95 °C for 30 s, 25 cycles of 95 °C for 10 s, 48 °C for 20 s, 72 °C for 20 s, and 1 cycle of 72 °C for 5 min ofor final extension (see Note 11).
Combine the eight PCR reactions and run 2 μL of PCR product on a 1% agarose gel (wt/vol) to ensure the desire bands are present, approximately 350 bp band for VL and 390 bp band for VH various with the length of CDR3 junction.
To purify PCR products, run all the samples on a 1% agarose gel (wt/vol). Excise the correct size bands and elute from agarose using QiAquick Gel Extraction Kit according to the manufacturer’s instruction.
Measure purified DNA concentrations from the A260 reading of a spectrophotometer. For pure double stranded DNA: A260 of 1.0 ¼ 50 mg/mL.
3.1.3. scFv Assembly: Digestion and Purification of Candidate Insert Genes (VH, Vκ, and Vλ) and Vector pYD2.A2 (Fig. 2b: Gene Assembly)
Prepare separate digestion master mixes for three purified PCR products from Subheading 3.1.2, step 4 and vector pYD2.A2, as described in the table below.
| VH digestion | Vκ/Vλ digestion | |||
|---|---|---|---|---|
| Vectora | VH insertb | Vector | Vκ insert | Vλ insert |
| 10 μg | 3 μg | 10 μg | 3 μg | 3 μg |
| NcoI/SalI | NcoI/SalI | BspEI/NotI | BspEI/NotI | BspEI/NotI |
Vector pYD2.A2, see Subheading 2.1.1
Inserts of VH, Vκ, and Vλ are the purified PCR products from Subheading 3.1.2, steps 4 and 5
Set up 10 μg of vector pYD2.A2 and 3 μg of each PCR products of VH, Vκ, and Vλ in microcentrifuge tubes.
For VH and its corresponding vector pYD2.A2 digestion, add 10 μL of 10x NEB buffer #3, 10 μL of 10x BSA, 4 μL of each NcoI and SalI to preset microcentrifuge tubes (step 2), and then add molecular grade water to a final volume of 100 μL.
For VL and its corresponding vector pYD2.A2 digestion, add 10 μL of 10x NEB buffer #3, 10 μL of 10x BSA, 4 μL of each BspEI and NotI to preset microcentrifuge tubes (step 2), and then add molecular grade water to a final volume of 100 μL.
Incubate the vector digestion reaction overnight in a 37 °C water bath and incubate the insert gene digestion reactions of VH, Vκ, and Vλ for 5 h in a 37 °C water bath.
VH digestion reactions generate vector designated as pYD2A2L that is ready for ligation with VH insert at NcoI/SalI restriction sites. VL digestion reactions generate vector designated as pYD2A2H that is ready for ligation with Vκ or Vλ inserts at BspEI/NotI restriction sites.
Purify the digested DNA fragments as the same in Subheading 3.1.2, step 4.
Measure purified linearized DNA concentration of vector by the same calculation as in Subheading 3.1.2, step 5.
3.1.4. scFv Assembly: Ligation
A threefold molar excess of insert to vector ratio are used for ligation. As vector pYD2.A2 is approximately 5670 bp and the average insert of VH, Vκ, or Vλ is 400 bp, for 1 μg of vector ligation reaction add 0.2 μg insert DNA (see Note 12).
Prepare separate ligation master mixes (2 × 50 μL) for each digested VH, Vκ, or Vλ from Subheading 3.1.3, steps 3 and 4 as described in the table below.
| Ligation Rxn | Vector* | Insert* | Final product |
|---|---|---|---|
| VH | pYD2.A2L | Steps 6–8 | pYD2.A2L.VH |
| Vκ | pYD2.A2H | Steps 6–8 | pYD2.A2H.Vκ |
| Vλ | pYD2.A2H | Steps 6–8 | pYD2.A2H.Vλ |
Both digested vectors and inserts are purified products from Subheading 3.1.3 Steps 6–8.
Add 5 μL of T4 DNA ligase buffer, 1 μL of vector (1 μg/μL), 0.2 μg insert DNA (applicable volume), 2 μL of T4 ligase, and molecular grade water to a final volume of 50 μL.
Incubate the ligation reactions overnight at 16 °C (see Note 13).
Purify ligation reactions using QiAquick PCR purification kit according to the manufacturer’s instruction (see Note 14).
Measure purified ligated DNA concentration by the same calculation as in Subheading 3.1.2, step 5. Adjust DNA concentration to 0.1 μg/μL for transformation.
3.1.5. scFv Assembly: Transformation and Diversity Evaluation of Prepared VH, Vκ, and Vλ
Ligation reactions are transformed into DH5α cells via electroporation using ElectroMAX DH5α-E cells according to the manufacturer’s instruction with the following modifications.
Set up four prechilled microcentrifuge tubes for each VH, Vκ, and Vλ transformation reaction.
Mix 1 μL of ligation reaction (0.1 μg/μL) and 20 μL electrocompetent DH5α cells in each tube.
Transfer the cell–DNA mixture into 0.2 cm cuvette and electroporate using BioRad GenePulser II electroporator with the following conditions 2.5 kV, 200 Ω, 25 μF for 0.2 cm cuvette.
To the cells in the cuvette, add 1.0 mL of S.O.B. medium and transfer the solution to a 15 mL snap-cap tube. Flush the cuvette with an additional 1.0 mL of S.O.C.
Shake at 225 rpm (37 °C) for 1 h.
Add 2 mL of transformed cells into prewarmed 50 mL LB containing 100 μg/mL of carbenicillin.
Incubate overnight with shaking at 225 rpm and at 37 °C. This results in plasmid pools designated as pYD2.A2L.VH, pYD2. A2H Vκ, and pYD2.A2H.Vλ.
To titer the transformation, plate 1.0, 10, and 50 μL of transformed culture from step 7 on LB plate containing 100 μg/mL of carbenicillin.
Incubate the plates overnight at 37 °C.
For diversity evaluation, pick ten colonies from VH, Vκ, and Vλ transformed plates and grow them individually in a 15-mL round bottom tube containing 5 mL of LB and carbenicillin (100 μg/mL) overnight with shaking at 225 rpm and at 37 °C.
Isolate plasmid DNA from the overnight 5 mL culture using QiAprep Spin Miniprep kit according to the manufacturer’s instruction.
Sequence eluted plasmid DNA with the pYD2.A2 forward primer (GAP5, see Table 1). Compare the CDRs sequences derived from randomly picked ten plasmid insert sequences using IMGT/V-QUEST: http://www.imgt.org/IMGT_vquest/share/textes/imgtvquest.html and Vector NTI software.
Table 1.
Primers for gene cloning and scFv assembly
| VH-forward | |
| HuVH1a | CGGGGCCATGGCCCAGGTGCAGCTGGTGCAGTCTGG |
| HuVH2b | CGGGGCCATGGCCCAGGTCACCTTGAAGGAGTCTGG |
| HuVH3a | CGGGGCCATGGCCGAGGTGCAGCTGGTGGAGTCTGG |
| HuVH4a | CGGGGCCATGGCCCAGGTGCAGCTGCAGGAGTCGGG |
| HuVH5b | CGGGGCCATGGCCAGGTGCAGCTGGTGCAGTCTGG |
| HuVH6a | CGGGGCCATGGCCCAGGTACAGCTGCAGCAGTCAGG |
| HuVH7a | CGGGGCCATGGCCCAGGTGCAGCTGGTGCAATCTGG |
| VH-reverse | |
| HuJH1,2 | CACCTGTCGACCCTGAGGAGACGGTGACCAGGGTGCC |
| HuJH3 | CACCTGTCGACCCTGAAGAGACGGTGACCATTGTCCC |
| HuJH4,5 | CACCTGTCGACCCTGAGGAGACGGTGACCAGGGTTCC |
| HuJH6 | CACCTGTCGACCCTGAGGAGACGGTGACCGTGGTCCC |
| Vκ-forward | |
| HuVk1a | GTGGCTCCGGAGGTGGCGGATCGGACATCCAGATGACCCAGTCTCC |
| HuVk2a | GTGGCTCCGGAGGTGGCGGATCGGATGTTGTGATGACTCAGTCTCC |
| HuVk2b | GTGGCTCCGGAGGTGGCGGATCGGATATTGTGATGACCCAGATCCC |
| HuVk3a | GTGGCTCCGGAGGTGGCGGATCGGAAATTGTGTTGACGCAGTCTCC |
| HuVk4a | GTGGCTCCGGAGGTGGCGGATCGGACATCGTGATGACCCAGTCTCC |
| HuVk5a | GTGGCTCCGGAGGTGGCGGATCGGAAACGACACTCACGCAGTCTCC |
| HuVk6a | GTGGCTCCGGAGGTGGCGGATCGGAAATTGTGCTGACTCAGTCTCC |
| Vκ-reverse | |
| HuJk 1 | CGCCTGCGGCCGCACGTTTGATTTCCACCTTGGTCCC |
| HuJk 2 | CGCCTGCGGCCGCACGTTTGATCTCCAGCTTGGTCCC |
| HuJk 3 | CGCCTGCGGCCGCACGTTTGATATCCACTTTGGTCCC |
| HuJk 4 | CGCCTGCGGCCGCACGTTTGATCTCCACCTTGGTCCC |
| HuJk 5 | CGCCTGCGGCCGCACGTTTAATCTCCAGTCGTGTCCC |
| Vλ-forward | |
| HuVl1 | GTGGCTCCGGAGGTGGCGGATCGCAGTCTGTSBTGACGCAGCCGCC |
| HuVl3 | GTGGCTCCGGAGGTGGCGGATCGTCCTATGWGCTGACWCAGCCAC |
| HuVl38 | GTGGCTCCGGAGGTGGCGGATCGTCCTATGAGCTGAYRCAGCYACC |
| HuVl4 | GTGGCTCCGGAGGTGGCGGATCGCAGCCTGTGCTGACTCARYC |
| HuVl78 | GTGGCTCCGGAGGTGGCGGATCGCAGDCTGTGGTGACYCAGGAGCC |
| HuVl9 | GTGGCTCCGGAGGTGGCGGATCGCAGCCWGKGCTGACTCAGCCMCC |
| HuVl11 | GTGGCTCCGGAGGTGGCGGATCGTCCTCTGAGCTGASTCAGGASCC |
| HuVl13 | GTGGCTCCGGAGGTGGCGGATCGCAGTCTGYYCTGAYTCAGCCT |
| HuVl15 | GTGGCTCCGGAGGTGGCGGATCGAATTTTATGCTGACTCAGCCCC |
| Vλ-Reverse | |
| HuJl12 | CGCCTGCGGCCGCTAGGACGGTSASCTTGGTCC |
| HuJl7 | CGCCTGCGGCCGCGAGGACGGTCAGCTGGGTGC |
| Light chain amplification | |
| HuJH-F | TCAGGGTCGACAGGTGGAG |
| Sequence | |
| PYDF | AGTAACGTTTGTCAGTAATTGC |
| PYDR | GTCGATTTTGTTACATCTACAC |
| GAP5 | TTAAGCTTCTGCAGGCTAGTG |
| GAP3 | GAGACCGAGGAGAGGGTTAGG |
For library size (diversity) determination, count total colonies from the plates (steps 9–11) and calculate library size using the general formula: cfu (colonies on the plate, step 9) x 101, 102 or 103 (dilution factor) x total vol (total volume in the LB flask, step 7) = library size (diversity) (see Note 15).
3.1.6. scFv Assembly: Generation of Gap Repair Ready Vκ and Vλ
Purify plasmid DNA from Subheading 3.1.5, steps 7 and 8 (4× 50 mL overnight bacterial cells cultures) using QiAfilter Midi kit according to the manufacturer’s instruction.
Measure purified plasmid DNA concentration by the same calculation as in Subheading 3.1.2, step 5.
Amplify Vκ and Vλ genes from prepared plasmid pYD2H.A2.Vl to add nucleotides (GRT primers, Table 1) that are homologs to pYD2.A2 for gap-repair mediated integration of the light chain genes into the vector plasmid carrying VH genes.
Set up PCR master mixes (4× 50 μL) for amplification of Vκ and Vλ from steps 1 and 2, as described in the table below.
| Reagent | 1 Rxn (μL) |
|---|---|
| 5x Phusion buffer | 10 |
| 10 mM dNTP mix | 1 |
| Phusion polymerase | 1 |
| HuJH-F | 2 |
| Gap3 | |
| Plasmidsa (350 ng) | 2 |
| Molecule grade water | 34 |
| Final vol | 50 |
Plasmids pYD2.A2H.Vκ and pYD2.A2H.Vλ from steps 1 and 2
Aliquot 50 μL for each PCR tube and run the following conditions for Vκ: 98 °C for 30 s, 25 cycles of 98 oC for 10 s, 55 °C for 20 s, 72 oC for 30 s, and 1 cycle of 72 oC for 5 min for final extension. Run the following condition for Vλ amplification: 98 oC for 30 s, 25 cycles of 98 °C for 10 s, 48 °C for 20 s, 72 °C for 20 s, and 1 cycle of 72 °C for 5 min for final extension.
Isolate the 400 bp PCR product as described in Subheading 3.1.2, steps 3–5.
3.1.7. scFv Assembly: Digestion of Vector pYD2. A2L.VH for Gap Repair
Set up 20 μg of pYD2.A2L.VH plasmid prepared from Subheading 3.1.6, steps 1 and 2 in a microcentrifuge tube.
Add 10 μL of 10× NEB buffer #3, 10 μL of 10× BSA, 7 μL of each BspEI and NotI to the tube and add molecular grade water to a final volume of 100 μL.
Incubate the digestion reaction overnight in a 37 °C water bath.
Purify the digested DNA fragments by the same protocol as in Subheading 3.1.2, step 4.
Measure purified linearized DNA concentration of vector by the same calculation as in Subheading 3.1.2, step 5.
3.1.8. Yeast Transformation, Tittering and Freezing
EBY100 Saccharomyces cerevisiae is the host strain for transformation. Streak from frozen EBY100 stock onto an YPD plate and grow at 30 °C for 48 h.
Inoculate single or a few colonies from the newly streaked YPD plate into 10 mL of YPD medium. The starting concentration should range between 0.05 and 0.1 OD600, which can be performed by using a spectrophotometer (see Note 16). Grow overnight the yeast culture in the shaker incubator at 250 rpm and 30 °C.
After 12–16 h of growth, determine the concentration (OD600) of the yeast culture by spectrophotometer (see Note 17).
Inoculate an aliquot of the overnight culture in 100 mL YPD medium to achieve an OD600 of 0.3.
Incubate the flask in the shaking incubator at 250 rpm and 30 °C until OD600 is at 1.6 (see Note 18).
Pellet the cells at 3000 x g for 5 min at 20 °C and aspirate the supernatant. Wash the cell pellet twice by resuspending cells first in 25 mL autoclaved water, centrifuge and aspirate the supernatant, and once in 50 mL of ice-cold electroporation buffer (1 M sorbitol–1 mM CaCl2).
The cell pellet is resuspended in 20 mL 0.1 M LiAc/10 mM DTT, transferred to a 100 mL culture flask and then incubated in a shaker incubator at 250 rpm and 30 °C for 30 min.
Collect the cells by centrifugation again, wash once in 50 mL ice-cold electroporation buffer and then resuspend the cell pellet in 100–200 μL electroporation buffer, then adjust to a final volume of 1 mL (see Note 19).
Combine 4 μg of digested vector and 6 μg of DNA insert for each 400 μL electroporation reaction. The DNA mixture in water should be less than 50 μL. Reduce the volume by precipitation and resuspend in a smaller volume if necessary. See table below.
| Reagent | pYD2.VH.Vκa | pYD2.VH.Vλ | Control |
|---|---|---|---|
| pYD2.A2L.VH/BspEI/NotIb | 4 μg | 4 μg | 4 μg |
| Vκ with gap tailc | 6 μg | ||
| Vλ with gap tailc | 6 μg | ||
| EBY100 | 400 μL | 400 μL | 400 μL |
| Final vol | <450 μL | <450 μL | <450 μL |
The volume of DNA mixture should be less than 50 μL
pYD2.A2L.VH/BspEI/NotI is the product from Subheading 3.1.7, step 5
Vκ and Vλ with gap tails are the purified PCR products from Subheading 3.1.6, steps 3–6
Gently mix 400 μL electrocompetent cells and DNA and transfer to a prechilled BioRad GenePulser cuvette (0.2 cm electrode gap). Tap the suspension to the bottom and keep on ice for 5 min until electroporation.
Set the Gene Pulser Xcell conditions as the following: C = 25 μF, R = 200 Ω, and V = 2.5 kV. Before pulsing, prepare 8 mL of 1:1 mixture of 1 M sorbitol and YPD medium in a sterile 17 × 100 mm tube per sample. Place the cuvette in the ShockPod. Push the chamber lid down to close and pulse once.
Remove the cuvette from the chamber and immediately add 1.0 mL of the 1 M sorbitol–YPD medium to the cuvette. Gently transfer the diluted cells into the tube with the remaining 7 mL of sorbitol–YPD medium.
Check and record the pulse parameters. The voltage should be approximately 2.5 kV. Typical time constant ranges 3.5–4.5 ms.
Incubate the transformant tube on a platform shaker at 225 rpm and 30 °C for 1 h.
Collect cells by centrifugation and discard the supernatant. For the negative control sample, the transformants can be resuspended in 220 μL autoclaved water and then plate 20 and 200 μL onto SD-CAA plates. For the library construction samples, resuspend the pellet in 300 mL SD-CAA medium in a 1.5 L flask, which is the total library of transformants. To titer the transformation, dilute the transformants at 1:104, 1:105, and 1:106 by plating 30 μL, 3.0 μL, or 0.3 μL from the 300 mL transformation to SD-CAA agar plates. The liquid cultures are incubated with shaking at 250 rpm and 30 °C for 16–24 h or longer until the OD600 reaches 2.5–3. The titration plates are incubated at 30 °C for 2–3 days.
For library titer determination, count the colonies on the titer plates and multiply by the dilution factor (104, 105, or 106) to obtain the yeast library size (see Note 20). The negative control plate may show some colonies but should be less than 0.1% of the library size.
To preserve the library, pool 100 OD600 yeast cells into 50 mL conical tubes and collect the pellet by centrifugation at 3000 x g for 5 min at 20 °C and aspirate the supernatant. Prepare the freezing buffer by mixing sterile 50% glycerol with SD-CAA medium at a ratio of 2:3 to get 20% glycerol/SDCAA media. Resuspend the pellet to a concentration of 100 OD600 yeast cell/mL, aliquot into cryotubes, and store at −80 °C.
3.2. Library Sorting
3.2.1. Library Growth and Induction
Quickly thaw aliquots of frozen scFv yeast library (see Note 21) at 30 °C and resuspend in SD-CAA media to obtain an OD600 of 0.2–0.5. Grow the culture overnight with shaking at 250 rpm and 30 °C.
Determine the growth of yeast by measuring the OD600. Pellet at least a 10x representation of library diversity from freshly passaged culture at 3000 x g for 5 min in 50 mL conical tubes. For a library size of 1 × 108, a total of 1 × 109 yeast cells are needed for induction, which equals to 100 OD600 yeast cells.
For induction, resuspend the cells in SG-CAA media to an absorbance of 0.5 OD600/mL (see Note 22) and grow the cells for 48 h at 18 °C and 250 rpm (see Note 23).
3.2.2. Magnetic-Activated Cell Sorting (MACS)
Pellet freshly induced yeast cells of at least 10x representation of library diversity by centrifugation at 3000 x g for 5 min at 20 °C and aspirate the supernatant. Wash cells once by resuspending in 25 mL PBSB buffer, centrifuge as described and aspirate the supernatant.
Wild-type (wt) HCV E2 glycoprotein or E2 mutant proteins are used for selection. The preparation of secreted E2 glycoprotein or mutants is described in our publication [10] (see Note 24). Prepare dilution of E2 in 10 mL of PBSB buffer. Resuspend the yeast cells in the E2 solution by gentle mixing.
Incubate cell suspension at room temperature with gentle rotation for 60 min and then followed by 10 min incubation on ice. All subsequent steps should be done with ice-cold buffer or at 4 °C (see Note 25).
Pellet and wash the cells with 25 mL PBSB buffer. Repeat wash one more time. Resuspend the cell pellet in 5 mL PBSB buffer with biotinylated E2-specific detection antibodies (see Note 26). Incubate on ice for 30 min. If the biotinylated wt E2 or mutant E2 is used to label the library in step 2, then skip step 4 and directly go to step 5.
Pellet and wash cells with 5 mL PBSB buffer. Repeat wash one more time.
Resuspend the cell pellet in 5 mL PBSB buffer with 25 μL of MACS streptavidin magnetic microbeads from Miltenyi (see Note 27).
Incubate on ice for 10 min with gentle mixing by inverting the tube several times.
Pellet cells at 3000 x g for 5 min at 4 °C, aspirate supernatant and resuspend the pellet in 50 mL PBSB buffer.
Place a LS column onto the magnetic stand. Equilibrate the column by flowing through the column by gravity 3 mL of ice-cold PBSB buffer.
Vortex and pass the cells through a cell strainer cap tube to achieve a single-cell suspension and immediately load onto the Miltenyi LS column. Add 7 mL of cell suspension to the column. After the cells have flowed through the column, briefly remove the column from the magnet and immediately place back on the magnet (see Note 28). With the column back in the magnet, add 1 mL of PBSB wash buffer and let it flow through. Repeat until all of the cells have been loaded.
Once the cells are loaded on the column, wash the column with 3 mL of PBSB wash buffer. Remove column from magnet and immediately place it back, as noted in step 10. Repeat this wash two additional times.
To elute the cells, remove the column and place it over a collection tube. Add 7 mL SD-CAA medium to the column and use the plunger supplied with the column to push the remaining cells through.
Propagate eluted yeast for subsequent rounds of sorting. Add SD-CAA medium to the eluted cell suspension to a final volume of 50 mL in a 250 mL flask and grow overnight at 30 °C. An optional step is another round of MACS by repeating steps 1–13.
3.2.3. FACS Library Sorting
After expansion, the MACS output cells are further enriched by cell sorting.
MACS usually enriches at least tenfold of the original library. Figure 3 illustrates an example [10]. For FACS sorting, 108 cells can be used as the input, which should cover at least tenfold of the diversity of the MACS output. Pellet and wash the cells once with PBSB buffer. Resuspend the pellet in 1 mL PBSB including both HCV wt E2 glycoprotein (or E2 mutant) at 5 μg/mL and anti-V5 antibody (1:5000). Besides the E2 staining, other controls can be employed, as described in the table below. This will help to monitor specific binding against the E2 antigen (see Note 29).
Fig. 3. An example of cell sorting on the sample after two rounds of MACS enrichment (R1 and R2).
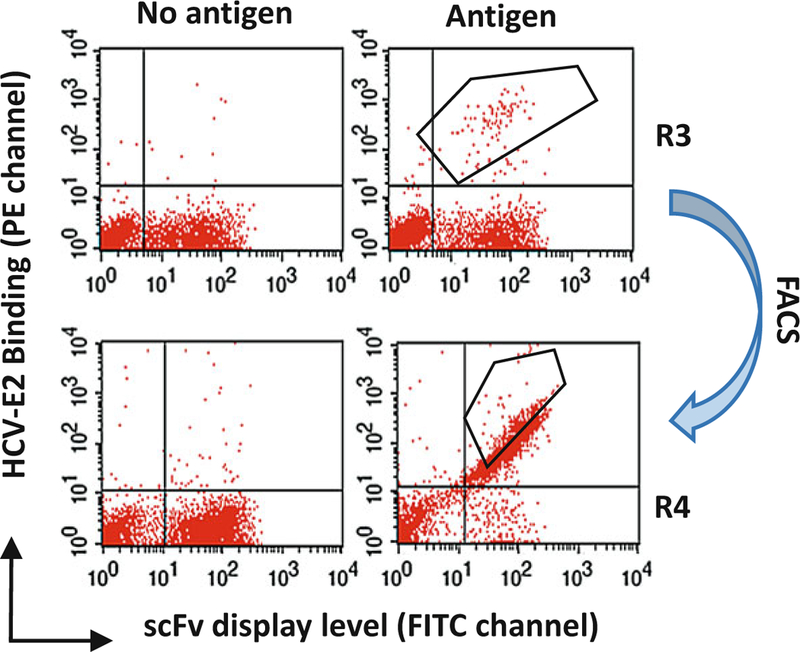
To perform 3rd round sorting (R3): 107 MACS isolated cells (from R2) were incubated with E2 mutant (E2Y632A), and then incubated with anti-V5 and HC-33.1 (an anti-E2 HMAb to a defined epitope) at 10 μg/ml. The cells were then labeled with FITC-anti-mouse and PE-anti-human (Fc-specific). The labeled cells, 107 cells/ml, were used for sorting by flow cytometry. The sorting gates were set to collect the desired double positive cells (the upper right graph). The ‘‘no antigen’’ control (the upper left graph) provided guidance on setting the sorting gates. After R3 selection, the double positive cells are highly enriched as showing in the lower right graph
| Yeast | Sample | Ctrl 1 | Ctrl 2 | Ctrl 3 | Ctrl 4 | Unstained |
|---|---|---|---|---|---|---|
| Biotin-E2 | + | − | − | − | − | − |
| Anti-V5 | + | + | + | − | − | − |
| SA-APC | + | + | − | + | − | − |
| Anti-mouse-FITC | + | + | + | − | + | − |
Incubate at room temperature for 1 h and then followed by 10 min on ice.
Pellet and wash the cells with 1 mL PBSB. Incubate the cells with anti-mouse-FITC and streptavidin-APC, both at 1:200 dilution, for 30 min at 4 °C.
Pellet the labeled cells, wash and sort. In a typical quadrant gated setting, the RU region corresponds to specific antigen binding and fully scFv expressed cells. Draw an appropriate region to gate in the double-positive quadrant for cell sorting. Apply “Purity mode” for the sorting. An example of enrichment of positive binders after going through MACS and FACS is shown in Fig. 3.
Grow the collected cells in 5 mL SD-CAA medium that is used for analysis or the next round of sorting after induction in SG-CAA, as described above.
The enrichment of double stained cells can be analyzed on the collected samples (presorted and postsorted samples) by staining as outlined in step 2. Repeat steps 2–7 until the population of double positive binding cells is above 20% of total cells. Typically, this will require 2–3 rounds of FACS sorting. At this point, the sorted cells are ready for monoclonal yeast screening in 96-well plates.
3.2.4. Clonal Analysis
The cell sorter will calculate the sorted yeast cell number during the process of sorting. After the final round of sorting, directly plate a few hundreds to one thousand cells on a SD-CAA plate. At least several plates can be prepared.
Incubate the plates at 30 °C for 48 h or until the yeast colonies can be seen and easily picked.
Prepare 96-deep well (2 mL vol/well) plates with 0.6 mL/well of SD-CAA medium. Inoculate freshly grown single yeast cells into each well. For each plate, inoculate a negative control well of yeast transformed with pYD2.A2 (see reagent). Seal the plate with breathable sealing film and shake at 250 rpm at 30 °C for 16 h until the OD600 is >5.
Similar to step 3, prepare another 96-deep well plate with 0.54 mL/well of SG-CAA medium. Subculture 60 μL SD-CAA culture into 540 μL SG-CAA medium, which will make a 1:10 dilution of each individual yeast culture. The yeast density will be 0.5–1 OD600, and under good condition for induction. Grow at 30 °C for 20 h to induce expression of scFv.
We routinely use 96 V-bottom well plates for monoclonal screening by flow cytometry. Prepare 1 × 106 yeast (~50 μL) in each well. Pellet the cell by 3000 x g for 5 min. Resuspend the induced yeast with 200 μL of PBSB buffer, wash once and pellet again as described.
Resuspend the yeast cells in 100 μL PBSB buffer containing anti-V5 mAb (1:5000) and biotinylated HCV-E2 (5 μg/mL).
Incubate for 60 min at room temperature followed by 10 min on ice. Pellet by 3000 x g for 5 min and wash once with 200 μL of ice-cold PBSB buffer.
Resuspend pellet in 100 μL of secondary reagents: anti-mouseFITC and streptavidin-APC, each at 1:200 dilution. Incubate for 30 min on ice, pellet the cells and wash two times in cold buffer.
Resuspend the cells in 100 μL of ice-cold PBSB buffer and keep on ice in the dark until analysis by flow cytometry. E2-specific binders will show both APC and FITC reactivity.
3.2.5. Plasmid Recovery and Conversion to Full Length IgG (See Note 30)
The method here is based on the Qiagen spin miniprep kit and is described in the literature [2]. The buffers used (P1, P2, N3, EB) are provided in the kit. Pellet approximately 250 μL of yeast cell culture centrifuge at 3000 x g for 5 min, aspirate the supernatant and wash once with double distilled water.
Resuspend the pellet in 250 μL Qiagen buffer P1 and add approximately 100 μL of glass beads. Vortex at high speed for 5 min.
Add 250 μL buffer P2, gently mix by inverting, and incubate at room temperature for 5 min.
Add 350 μL buffer N3 (see Note 31) and spin at 16,000 x g in a microcentrifuge for 10 min.
Apply supernatant to a Qiagen spin miniprep column and spin at 16,000 x g for 1 min. Discard the flow-through.
Add 750 μL buffer PE, and spin at 16,000 x g for 1 min. Discard flow-through and spin at 16,000 x g for 2 min to remove residual buffer PE. Replace collection tube with a clean eppendorf tube and add 50 μL of elution buffer EB and spin at 16,000 x g for 1 min to elute.
The miniprep can be used as template for PCR amplification as in Subheading 3.1.2, step 1. The primer set is PYDF/PYDR, and 5 μL of miniprep DNA is used as template. The PCR product is suitable for DNA sequencing. Use the Gap5 or Gap3 primer to sequence.
After sequencing, the individual antigen-specific yeast clones can be cloned into full IgG mammalian expression vector for IgG production and further characterization [13].
Fig. 1. Vector map and sequence of pYD2.A2 plasmid.
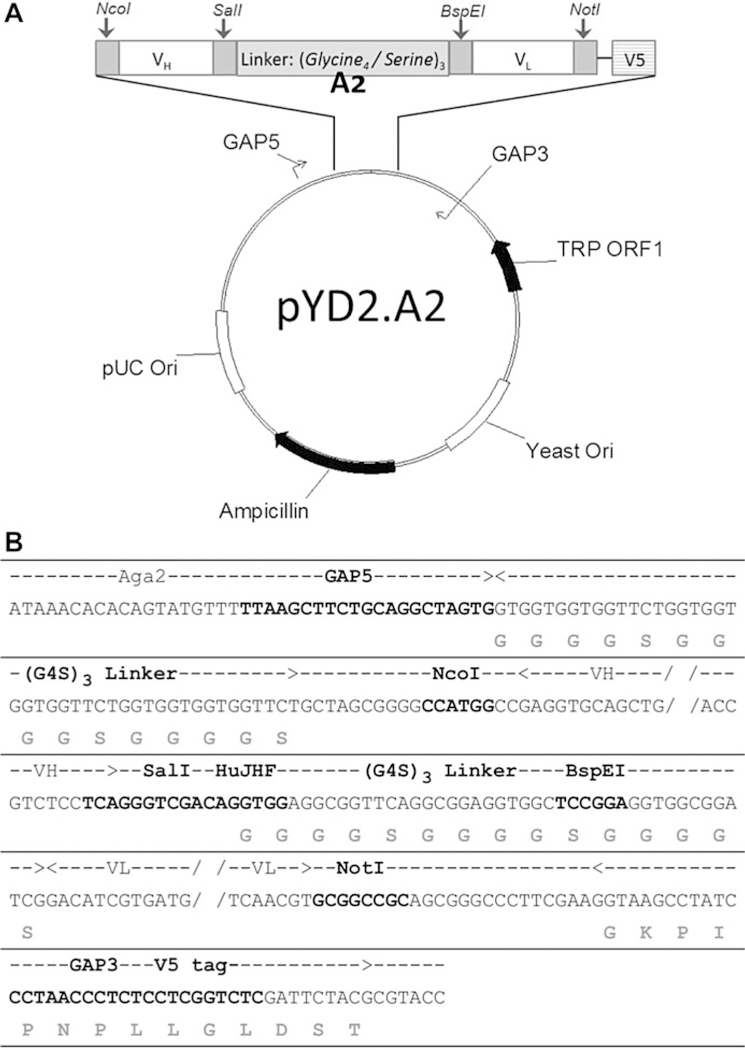
(a) Vector map of pYD2.A2 plasmid showing the relative position of genes, linkers, primer sites and restriction digestion sites. (b) Sequence showing the insert of A2 region in the vector. The bold nucleotides in the middle row indicate 1) GAP5, GAP3 and HuJHF primer binding sites and 2) the restriction digestion sites for NcoI, SalI, BspEI and NotI. The grey amino acids on the bottom row indicate the (G4S)3 linkers and V5 tag. The dashed line with arrows on the top row indicate the span of the (G4S)3 linkers, VH insert, VL insert and V5 tag
Acknowledgments
This work was support in part by National Institute of Allergy and Infectious Diseases/NIH grants R41-AI108024, U19-AI123862, R01-AI132213 and R21-AI126582 to SKHF.
Footnotes
Notes
EBY100 can be purchased from ATCC with ATCC number: MYA-4941.
Vector pYD2 was kindly provided by J.D. Marks (UCSF) [14] and we made further modifications to vector pYD2.A2 (Fig. 1).
Use a stir bar to help incorporate the different components. If pouring plates, it is recommended to use a conical flask because the narrow top makes it easier to hold and pour.
Use yeast nitrogen base without amino acids.
Salts will precipitate at higher temperatures and dissolve back into solution as the temperature drops.
A 0.22 μM filter can be used in lieu of autoclaving.
Use proper PPE gear (i.e., eye protection and hot gloves). Microwave for short durations (i.e., 15–30 s) and swirl the bottle in between heating to see if the agarose is dissolved. Allow the bottle to cool down to 55 °C or cool enough to hold without hot gloves before pouring in gel tray. If the agarose is too hot, it will warp the walls of the gel tray.
Various beads have been used depending on the experimental setup (such as anti-biotin magnetic microbeads and streptavidin magnetic microbeads).
Some detection fluorescence labelled antibodies that we used are FITC-anti-mouse IgG, PE-anti-human IgG (Fcγ specific), PE-streptavidin, and APC-streptavidin.
Peripheral blood memory B cells are plated at different densities (100–1000 cells/well) and then activated by EBV as described [9]. Supernatants are screened by HCVcc neutralization assay [10] and cells from neutralization positive wells are pooled and used for total RNA isolation.
Setting up multiple PCR reactions with minimal cycles of amplification help to obtain 10 μg of final PCR products and reduce biased selection due to over amplification.
0.2 μg of insert in this reaction was calculated using the formula [3/1 × 1 μg x 400 bp/5670 bp ¼ 0.2 μg] used: required mass insert (μg) ¼ desired insert–vector molar ratio x mass of vector (μg) x ratio of insert to vector lengths. Please note, in comparison to the gap-repair method in Subheading 3.1.8, step 9, in which 6 μg/reaction of PCR product is required for the library construction, here 0.2 μg/reaction of PCR product is much easier to obtain. It is one of the reasons that we first clone the VH/Vκ/Vλ into an E. coli plasmid rather than directly gap-repair into yeast.
Ligation reaction can be also carried out for 4 h at room temperature.
Gel purification of ligation reaction is very important because salts and buffers in ligation reaction can severely inhibit electroporation.
Example for calculation of the library size: (a) 1 mL transformed cells are added into 100 mL of medium for bacterial growth, (b) plate 100 μL from 100 mL of diluted transformed cells on to LB plate, (c) 74 colonies appear on the LB plate after overnight incubation, (d) calculation of library size is as following: [74/100 μL x 103 × 100,000 μL ¼ 7.4 × 107]. At this stage, three libraries are constructed: pYD2.A2L.VH, pYD2.A2H.Vκ, and pYD2.A2H.Vλ. pYD2.A2L.VH is used as a backbone to be opened for the Vκ/Vλ fragment to be cloned into to form a full scFv. pYD2.A2H.Vκ and pYD2.A2H.Vλ serve as template for the Vκ/Vλ amplification in Subheading 3.1.6, step 3. Another application for VH/VL libraries is that it can be directly used for light chain shuffling for affinity maturation of lower affinity scFvs.
Cell numbers are generally determined by an absorbance reading at 600 nm on a spectrophotometer (OD600). Blank the spectrophotometer with the same buffer without cells. An absorbance of 1 OD600 contains about 1 × 107 cells per mL, hereafter, the yeast OD means the absorbance at 600 nm.
Due to the high density of overnight culture, a 1:10 or 1:20 dilution needs to be prepared for the OD measurement; thus, the OD readout will in a range of 0.5–1. Pipet 50 μL of cells into 0.95 mL of water in a spectrophotometer cuvette, mix thoroughly and measure the OD. Remember to multiply by the dilution factor to determine the titer in the cell culture.
It usually takes around 5 h, which corresponds to approximately 1.6 × 109 yeast cells.
It is not easy to estimate the volume of yeast pellet and residual buffer after centrifugation and aspiration. To prepare competent cells, first add 100–200 μL electroporation buffer to resuspend the pellet and then measure the volume with the pipettor. More buffer can be added to reach 1 mL of yeast suspension. For library construction, 50 OD is required for each transformation. Take into account cell loss during each wash step. The yeast competent cells are sufficient for two electroporation reactions of 400 μL. The cell culture and preparation can be proportionally scaled up if more electrocompetent cells are needed to make larger or more libraries.
Example for calculation of the library size: (a) transformed cells is resuspended and added into 300 mL of medium for yeast growth, (b) plate 3 μL from 300 mL of diluted transformed cells on to SD-CAA plate, (c) 200 colonies appear on the SD-CAA plate after 2–3 day incubation, (d) calculation of library size is as following: [200/3 μL x 300,000 μL ¼2.0 × 107].
To maximize diversity, the starting SD-CAA culture should contain a minimum of a 10x representation of the diversity of the immunized yeast library. For a 108 diverse library, 109 yeast cells are needed to start the culture.
An easier way for yeast induction is to directly dilute the SD-CAA cultured yeast in SG-CAA medium. Please note that the dilution factor should be above 1:10 since 2 g/L dextrose will improve scFv display on yeast surface (higher dextrose concentrations may inhibit scFv display).
The induction temperature can be raised to 30 °C after the first MACS selection.
Selection antigen is based on the experimental design. Besides wt E2 protein, we have employed E2 mutants to avoid the selection of previously identified antibodies [10]. Purified E2 antigens are preferred reagents in this application. Before MACS enrichment, the antigen concentration suitable for MACS needs to be determined by flow cytometry analysis with a positive control of E2 proteins binding scFv displayed yeast cells. The concentration showing a high signal by flow cytometry can be used for MACS selection. In our lab, 5 μg/ mL of purified E2 proteins works well.
Completion of secondary labeling at 4 °C to minimize antigen dissociation from yeast-displayed scFvs.
To isolate neutralizing antibodies, we have employed E2 mutants to avoid the selection of nonneutralizing scFvs. For details, please refer to related publications [10].
As an option, anti-biotin magnetic microbeads can be used. Alternating between different microbeads during subsequent rounds of MACS selection will decrease the probability of obtaining unwanted secondary reagent-specific clones.
This rearranges the iron beads in the column and allows the unbound cells that are physically trapped between the beads to pass through.
It is important to use different secondary antigen-labeling reagents (streptavidin-APC or streptavidin-PE) between FACS sorts to eliminate or reduce unwanted secondary reagent-specific scFv. If PE and FITC are used for double staining the cells, the flow cytometer should be properly compensated to eliminate the overlap between these two fluorophores.
While the E2 binding scFvs are identified by flow cytometry analysis, the unique sequences among the binders will need to be examined. There are different PCR methods to directly amplify the scFv fragment from the yeast colony. We found the most reliable way is to extract yeast plasmid first and then perform PCR as described in the following.
A cloudy precipitate will form.
References
- 1.Boder ET, Wittrup KD (1997) Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol 15:553–557 [DOI] [PubMed] [Google Scholar]
- 2.Bidlingmaier S, Su Y, Liu B (2015) Combining phage and yeast cell surface antibody display to identify novel cell type-selective internalizing human monoclonal antibodies. Methods Mol Biol 1319:51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller KD, Pefaur NB, Baird CL (2008) Construction and screening of antigen targeted immune yeast surface display antibody libraries. Curr Protoc Cytom Chapter 4:Unit 4 7. [DOI] [PubMed] [Google Scholar]
- 4.Sheehan J, Marasco WA (2015) Phage and yeast display. Microbiol Spectr 3:AID-0028 2014 [DOI] [PubMed] [Google Scholar]
- 5.Van Deventer JA, Wittrup KD (2014) Yeast surface display for antibody isolation: library construction, library screening, and affinity maturation. Methods Mol Biol 1131:151–181 [DOI] [PubMed] [Google Scholar]
- 6.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD (2006) Isolating and engineering human antibodies using yeast surface display. Nat Protoc 1:755–768 [DOI] [PubMed] [Google Scholar]
- 7.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JM, Yeung YA et al. (2003) Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol 21:163–170 [DOI] [PubMed] [Google Scholar]
- 8.Feldhaus MJ, Siegel RW (2004) Yeast display of antibody fragments: a discovery and characterization platform. J Immunol Methods 290:69–80 [DOI] [PubMed] [Google Scholar]
- 9.Perkins S, Foung SK (1995) Stabilizing antibody secretion of human Epstein Barr virusactivated B-lymphocytes with hybridoma formation by electrofusion. Methods Mol Biol 48:295–307 [DOI] [PubMed] [Google Scholar]
- 10.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J et al. (2012) Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8:e1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keck ZY, Saha A, Xia J, Wang Y, Lau P, Krey T et al. (2011) Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol 85:10451–10463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S et al. (2000) Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol 74:10407–10416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R et al. (2009) Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc 4:372–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Razai A, Garcia-Rodriguez C, Lou J, Geren IN, Forsyth CM, Robles Y et al. (2005) Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J Mol Biol 351:158–169 [DOI] [PubMed] [Google Scholar]