

Abstract
The H-atom transfer (HAT) reactivity of a corrolazine cobalt superoxide with weak O–H and N–H substrates has been demonstrated. Kinetic analysis shows relatively fast rates of HAT with diphenylhydrazine (DPH). A kinetic isotope effect (KIE) and Eyring activation parameters are consistent with an HAT mechanism.
Table of Contents
The H-atom transfer reactivity of a porphyrinoid cobalt superoxide with weak O-H and N-H substrates has been demonstrated.
The activation of dioxygen at both heme and nonheme metal sites in biology is the first step in a wide range of substrate oxidation reactions.1 The binding of O2 at a transition metal center (Mn+) typically involves one electron transfer from the metal to the bound O2 molecule, giving a metal-superoxide species (Mn+1(O2•–)). The controlled addition of electrons and protons may eventually result in O–O cleavage, giving a high-valent metal-oxo species that reacts via oxidation with substrates of interest. However, the initial Mn+1(O2•–) intermediate in certain systems may be responsible for direct attack of the substrate, obviating the need for multielectron/proton transfer prior to substrate oxidation. For example, mechanistic proposals for an intriguing class of heme dioxygenases (tryptophan dioxygenase (TDO), 2,3-indoleamine dioxygenase (IDO)), involve substrate dioxygenation beginning with the direct attack of the indole substrate by the superoxo adduct FeIII(O2•–)(porphyrin)(His).2 Similarly, current proposals for the heme enzyme nitric oxide synthase (NOS) suggest a superoxo or peroxo-iron species initiates the attack on an N-hydroxy-L-arginine substrate, ultimately producing L-citrulline and NO products.3 An iron(III)-superoxo intermediate in nonheme iron enzymes, including cysteine dioxygenase,4 myo-inositol oxygenase,5 and isopenicillin-N-synthase,6 are also postulated to be active oxidants that carry out direct attack on substrate.
Synthetic metalloporphyrin complexes were shown to reversibly bind dioxygen via metastable metal-superoxide complexes since the early 1970s.7 More recent elaboration of the porphyrin scaffold has provided access to O2-derived FeIII(O2•–) species which can be converted into ferric-peroxide and ferric-hydroperoxide species under controlled conditions.8 The related cobalt porphyrins and phthalocyanines are also known to reversibly bind O2.9 However, little is known about the reactivity of these Mn+1(O2•–) porphyrinoid species in terms of the direct oxidation of organic substrates.10 Well-defined nonheme metal-superoxide complexes are also known, but their reactivity toward organic substrates is also poorly understood. It was not until recently that a well-defined, nonheme, cobalt(III)-superoxide was shown to mediate H-atom abstraction from an organic substrate.11 To our knowledge, there are as of yet no reports on the analogous cobalt-superoxide reactivity in a porphyrinoid system. Herein we describe a CoIII(O2•–) porphyrinoid complex that abstracts H-atoms from activated O–H and N–H bond substrates.
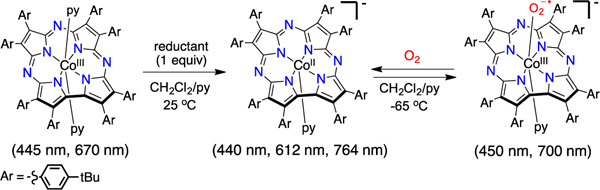
The formation of a cobalt(III)-superoxide species with the octa-(4-tert-butylphenyl)corrolazine) (TBP8Cz) ligand was reported previously.12 The [CoIII(py)(O2)(TBP8Cz)]- species (py = pyridine) was prepared via addition of excess dioxygen to [CoII(py)(TBP8Cz)]- at low temperature. The superoxide complex was characterized by EPR spectroscopy, giving a spectrum similar to other cobalt(III)-superoxides.9b, 9d However, no other spectroscopic characterization was obtained. Herein we provide new low-temperature UV-vis and 1H NMR spectroscopy on the [CoIII(py)(O2)(TBP8Cz)]- complex, as well as on the related CoII and CoIII(py)2 complexes. The spectroscopic data fully support the structural assignments of these complexes. These low-temperature spectroscopic methods also provide a means to monitor the reactivity of the superoxo species with organic substrates. It is shown that [CoIII(py)(O2)(TBP8Cz)]- is capable of abstracting H-atoms from certain N–H donors, and kinetic studies provide insight into the mechanism of H-atom transfer (HAT).
Reduction of CoIII(py)2(TBP8Cz) to the 5-coordinate anionic [CoII(py)(TBP8Cz)]- was carried out previously by addition of excess NaBH4 in EtOH/py.12 Other reductant/solvent combinations were tested to determine an optimal protocol for the reduction. Addition of Cr(η6-C6H6)2, Co(η5-(C5(CH3)5)2 or Bu4N+BH4- led to the stoichiometric reduction of the CoIII complex to give [CoII(py)(TBP8Cz)]-. The UV-visible spectrum for CoIII(py)2(TBP8Cz) in CH2Cl2/pyridine 99/1 (v/v) (445, 543, 622, 670 nm) is converted to a new spectrum with loss of the 670 nm Q-band and growth of new peaks at 612 and 764 nm (Figure S1). The reductant Bu4N+BH4- was found to be the most robust and economical, and therefore was employed for all other studies.
Cooling the CoII complex to −65 °C leads to no significant changes in the UV-vis spectrum. However, addition of excess O2 at this temperature causes the immediate disappearance of the spectral signature for the CoII complex, and the appearance of a new spectrum with λmax = 450, 600, and 700 nm, as shown in Figure 1. The new spectrum is assigned to the Co-dioxygen adduct [CoIII(py)(O2)(TBP8Cz)]-. This species is stable for > 5 h at temperatures equal to or lower than −65 °C. However, the Co(III)-superoxo complex slowly converts to the CoIII(py)2 species upon warming above −65 °C, as evidenced by a decrease of the 700 nm peak and a concomitant growth of a peak at 670 nm characteristic of CoIII(py)2(TBP8Cz). Reversible binding of O2 is noted if the solution is sparged with Ar(g) for 20 min at −65 °C, which causes a return of the CoII absorbance spectrum. Further addition of excess O2 regenerates the spectrum for the superoxo species (Figure S2). The generation of the superoxo complex is summarized in Scheme 1.
Fig. 1.
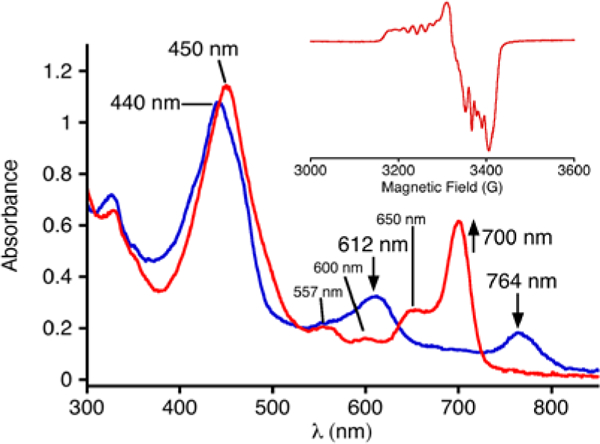
UV-vis spectra for [CoII(py)(TBP8Cz)]- (blue line) and [CoIII(py)(O2)(TBP8Cz)]- (red line), 18 μM in CH2Cl2/pyridine (99/1, v/v) at −65 °C. Inset: X-band EPR spectra at 15 K for [CoIII(py)(O2)(TBP8Cz)]−.
Scheme 1.
Generation of [CoIII(py)(O2)(TBP8Cz)]− (1)
The EPR spectrum of the CoII complex (Figure S3) matches that previously reported for [CoII(py)(TBP8Cz)]- generated in a different solvent system with a different reductant.12 The EPR spectra of CoIII(O2-) complexes are well-documented and follow a characteristic pattern.9b, 9d The spectrum of [CoIII(py)(O2)(TBP8Cz)]- (Figure 1 inset, red line) follows this pattern and matches what was reported previously.12 The NMR spectra for the three cobalt species are shown in Figure 2. The bis(pyridine) complex CoIII(py)2(TBP8Cz) exhibits a diamagnetic 1H NMR spectrum, as expected for a low-spin d6 ion. Upon reduction to the CoII species, a paramagnetic spectrum appears, consistent with a low-spin d7 (S = ½) complex. Addition of O2 causes the 1H NMR spectrum of the CoII complex to disappear and be replaced by a spectrum with broadened, poorly defined peaks. This spectrum is consistent with the presence of a paramagnetic superoxo species with spin delocalization over the Co and O2 moieties. The spectrum for CoII is regenerated upon sparging with Ar(g), as also seen in the UV-vis data. Taken together, the EPR and NMR data confirm that the reduced CoII complex is quantitatively converted into the cobalt(III)-superoxo complex, [CoIII(py)(O2)(TBP8Cz)]-.
Fig. 2.
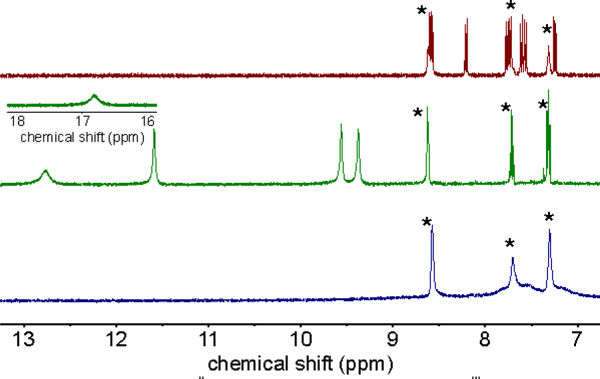
1H NMR spectra of CoIII(py)2(TBP8Cz) (red line) at 25 °C, [CoII(py)(TBP8Cz)]− (green line) at 25 °C, and [CoIII(py)(O2)(TBP8Cz)]− (blue line) in CD2Cl2/pyridine-d5 (20/1 v/v) at −65 °C, showing the aromatic region. * = pyridine peaks
The reactivity of [CoIII(py)(O2)(TBP8Cz)]- with H-atom donors was examined. Initial experiments were focused on determining if the superoxo complex could oxidize C–H substrates. We began with potential H-atom donors with relatively weak C–H bonds, such as dihydroanthracene (BDE(C–H) = 80.6 kcal/mol)13 or xanthene (BDE(C–H) = 77.9 kcal/mol).13 Addition of an excess of C-H substrate (25 – 100 equiv) to the superoxo complex in CH2Cl2/py at −65 °C resulted in no spectroscopic change by UV-vis over 2 h, indicating that the CoIII(O2•-) species was unreactive toward these C-H substrates. However, reaction of O–H and N–H substrates led to different results. Addition of excess TEMPOH under the same conditions led to the isosbestic conversion of the superoxo complex to CoIII(py)2(TBP8Cz) (Figure S5). The production of the bis(pyridine)-ligated complex was consistent with an initial H-atom transfer (HAT) from TEMPOH to give a cobalt(III)-hydroperoxo complex, that rapidly loses hydroperoxide via displacement by the excess pyridine. The stability of the low-spin, d6 CoIII(py)2 unit, as evidenced by the high formation constant for pyridine (β2 = 9.0 × 107 M−2) measured in CH2Cl2,14 likely provides much of the driving force for the second step.
Given that TEMPOH has a BDE(O-H) of 72.1 kcal/mol,13 we examined N-H substrates of similar strength. Both phenylhydrazine (BDE(N–H) = 75.0 kcal/mol) and diphenylhydrazine (DPH) (BDE(N–H) = 71.7 kcal/mol)13 react with the superoxide complex at −65 °C, as seen by the same spectral changes in the UV-vis that were seen for the reaction with TEMPOH. The isosbestic conversion of [CoIII(py)(O2)(TBP8Cz)]- to CoIII(py)2(TBP8Cz) for the reaction with DPH is shown in Figure 3. The absorbance at 670 nm (ε = 5.3 × 104 M−1 cm−1) indicates an 80% yield of the final bis(pyridine) product. A complete loss of the EPR signal from the superoxide complex was also noted. The final EPR-silent species can be assigned to the low-spin CoIII(py)2(TBP8Cz) complex.
Fig. 3.
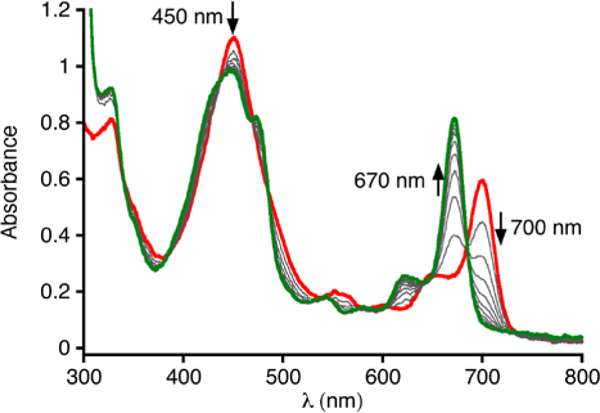
Time-resolved UV-vis spectra (0 – 40 min) for the reaction between [CoIII(py)(O2)(TBP8Cz)]- (18 μM) and DPH (0.33 mM) in CH2Cl2/py (99/1, v/v) at −65 °C.
If the DPH reaction involves HAT to the superoxide complex, it should lead to formation of azobenzene, which is the result of a formal dehydrogenation (i.e. two HAT events) of the substrate. Carrying out the reaction of [CoIII(py)(O2)(TBP8Cz)]- with excess DPH in CD2Cl2/py-d5 (20/1) at −65 °C led to identification of both azobenzene and the diamagnetic CoIII(py)2(TBP8Cz) by 1H NMR spectroscopy (Figure S7). The reaction stoichiometry is [CoIII(py)(O2)(TBP8Cz)]-:DPH 2:1, assuming a single superoxide complex accepts only one hydrogen atom (Scheme 2). A comparison of the integrated areas for the 1H NMR peaks corresponding to azobenzene versus the CoIII product confirmed the expected 2:1 stoichiometry. Similarly, comparison of integrations with the peak for an internal standard (3,5-dimethylanisole) gave a yield of 93 ± 12% for azobenzene, and 87 ± 12% for CoIII(py)2(TBP8Cz).
Scheme 2.
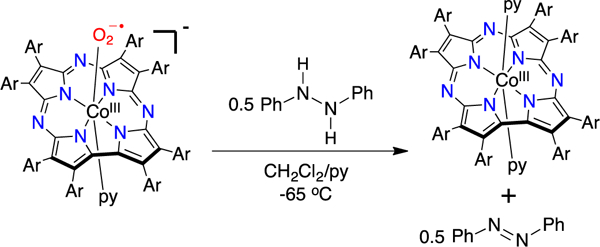
Reaction of [CoIII(py)(O2)(TBP8Cz)]− with the H-atom donor DPH at −65 °C
A proposed mechanism for the observed reactions between H-atom donors and the cobalt-superoxide complex is shown in Scheme 3. The first step involves HAT from the donor to the OOH- ligand is then displaced in a rapid second step by excess pyridine to give the stable CoIII(py)2 species. The isosbestic conversion between the superoxo and bis(pyridine) complexes, and the lack of any spectroscopic (UV-Vis, NMR) evidence for the CoIII(OOH) species, support this mechanism. Attempts were made to independently generate the CoIII(OOH) species by addition of hydrogen peroxide and triethylamine to CoIII(py)2(TBP8Cz), but no reaction was observed, and addition of a large excess of H2O2/Et3N led to bleaching of the solution.
Scheme 3.
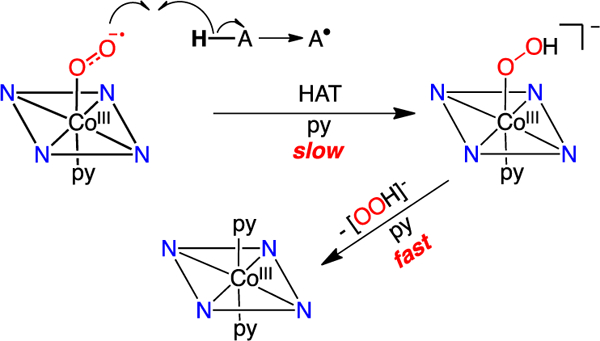
Proposed mechanism for the reaction between the cobalt(III)-superoxide complex and H-atom donors.
To gain further mechanistic insights into the reaction of [CoIII(O2)(py)(TBP8Cz)]- with DPH, kinetic analyses were performed. The rate of the reaction with excess DPH (pseudo-first-order conditions) was followed by the disappearance of the 700 nm peak corresponding to the starting material, as well as the growth of the 670 nm peak corresponding to the CoIII(py)2 product. First-order behaviour was observed (>5 half-lives) and fitting of the data (FIGURE S8) led to first-order rate constants (KOBS) that were shown to vary linearly with substrate concentration. A second-order rate law was implicated and was consistent with HAT being the rate-determining step, with a K2 = 3.6(8) M−1 s-1.
Additional support for an HAT mechanism comes from the observation of a kinetic isotope effect (KIE). Substitution of deuterium for the N–H protons led to a significant decrease in second-order rate constant (DPH, 3.6(8) M−1 s−1; DPH-d2, 0.39(8) M−1 s−1), giving a KIE (kH/kD) = 9.2 (Figure 4a). This relatively large KIE provides compelling evidence that the rate determining step involves N-H bond cleavage, as expected for an H-atom transfer.
Fig. 4.
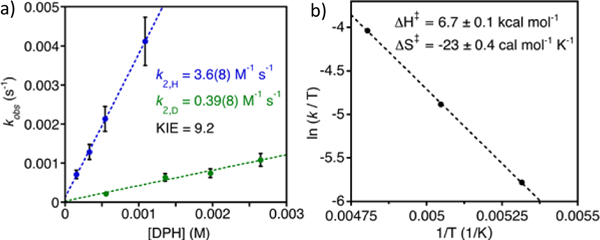
(a) Plot of pseudo-first-order rate constants (kobs) versus [DPH] (blue circles) or [DPH-d2] (green circles) together with best-fit lines. (b) Plot of ln (k/T) versus inverse temperature (Eyring Plot).
An Eyring plot for the reaction with DPH constructed from data at −85 °C, −75 °C and −65 °C yielded the activation parameters ΔH⧧ = 6.7 ± 0.1 kcal mol−1 and ΔS⧧ = −23 ± 0.4 cal mol−1 K−1 (Figure 4b). The relatively large, negative activation entropy is consistent with the bimolecular nature of an HAT reaction and implicates a well-ordered transition state.
The H-atom transfer reactivity of a few other discrete, well-defined M(O2•–) complexes with M = Cu,15 Cr,16 Ni,17 and Fe18 has been described. The oxidation of weak C-H bonds is quite rare,18b but the abstraction of hydrogen from either O–H or N–H bonds with relatively low BDEs (66 – 83 kcal/mol) is more common.15, 16c, 18a Interestingly, only two other cobalt-superoxide complexes besides [CoIII(py)(O2)(TBP8Cz)]- have been shown to abstract H-atoms from organic substrates, and both involved weak O–H bonds.11, 19 The latter complexes are derived from nonheme ligand sets. Thus there is limited kinetic information to compare the reactivity of [CoIII(py)(O2)(TBP8Cz)]- with other M(O2•–) complexes involved in HAT reactions. In terms of activation parameters, Meyer has shown that a dicopper μ−1,2-superoxide complex reacts with TEMPOH with ΔH⧧ = 9.03 ± 0.41 kcal mol−1 and ΔS⧧ = −26.8 ± 2 cal mol−1 K−1, which are similar to those reported here for [CoIII(O2)(py)(TBP8Cz)]- and are consistent with a similar HAT mechanism.15b The latter study also describes the reactivity of the Cu2(μ−1,2-O2•–) complex with the N–H substrate phenylhydrazine (BDE(N-H) = 75.0 kcal mol−1), a close analog of DPH. A second-order rate constant of 0.81 M−1 s−1 at −20 °C was reported for the oxidation of phenylhydrazine. A rate constant of k2 = 89.4 M−1 s−1 at the same temperature (−20 °C) is predicted for [CoIII(py)(O2)(TBP8Cz)]- via extrapolation from the measured activation parameters, allowing for a direct comparison of the kinetics of N–H cleavage. The second-order rate constants indicate that the cobalt complex is one hundred-fold more reactive than the dicopper superoxo-bridged species in N–H cleavage. The same CuII2 complex reacts with the O–H donor TEMPOH, which has a BDE(O–H) (72.1 kcal/mol) close to that of DPH (71.7 kcal/mol), with a rate constant for HAT of k2 = 0.13 M−1 s−1, which is seven hundred-fold slower than that seen for [CoIII(py)(O2)(TBP8Cz)]- in reaction with DPH. The origins of this differential reactivity (e.g. steric or electronic factors) remain to be determined.
In summary, we have shown that a porphyrinoid cobalt(III)-superoxide complex is competent to abstract hydrogen atoms from relatively weak O–H and N–H bonds. The CoIII(O2•–) complex is significantly more reactive than a non-heme Cu2(μ−1,2-O2•–) complex in reaction with N–H bonds. These results add to the growing body of evidence that metal-superoxide species in biologically relevant environments may function as oxidants for certain organic substrates, although the oxidation of C–H bonds remains quite challenging for such species. The factors that control the relative reactivity of M(O2•–) species remain poorly understood, providing motivation for further research in this area. The authors acknowledge research support from the NIH (GM101153 to D. P. G.).
Supplementary Material
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Sahu S and Goldberg DP, J Am Chem Soc, 2016, 138, 11410–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Makino R, Obayashi E, Hori H, Iizuka T, Mashima K, Shiro Y and Ishimura Y, Biochemistry, 2015, 54, 3604–3616; [DOI] [PubMed] [Google Scholar]; (b) Booth ES, Basran J, Lee M, Handa S and Raven EL, J. Biol. Chem, 2015, 290, 30924–30930; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lewis-Ballester A, Batabyal D, Egawa T, Lu C, Lin Y, Marti MA, Capece L, Estrin DA and Yeh S-R, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 17371–17376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Stoll S, NejatyJahromy Y, Woodward JJ, Ozarowski A, Marletta MA and Britt RD, J. Am. Chem. Soc, 2010, 132, 11812–11823; [DOI] [PubMed] [Google Scholar]; (b) Huang H, Hah J-M and Silverman RB, J. Am. Chem. Soc, 2001, 123, 2674–2676. [DOI] [PubMed] [Google Scholar]
- 4.Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP and Jameson GNL, Chem. Commun, 2016, 52, 8814–8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C and Bollinger JM Jr., Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 6130–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tamanaha E, Zhang B, Guo Y, Chang W-c., Barr EW, Xing G, St. Clair J, Ye S, Neese F, Bollinger JM Jr., Krebs C, J. Am. Chem. Soc, 2016, 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baglia RA, Zaragoza JP and Goldberg DP, Chem. Rev, 2017, 117, 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Liu J-G, Ohta T, Yamaguchi S, Ogura T, Sakamoto S, Maeda Y and Naruta Y, Angew. Chem. Intl. Ed, 2009, 48, 9262–9267; [DOI] [PubMed] [Google Scholar]; (b) Liu J-G, Shimizu Y, Ohta T and Naruta Y, J. Am. Chem. Soc, 2010, 132, 3672–3673; [DOI] [PubMed] [Google Scholar]; (c) Nagaraju P, Ohta T, Liu JG, Ogura T and Naruta Y, Chem Commun, 2016, 52, 7213–7216. [DOI] [PubMed] [Google Scholar]
- 9.(a) Niederhoffer EC, Timmons JH and Martell AE, Chem. Rev, 1984, 84, 137–203; [Google Scholar]; (b) Jones RD, Summerville DA and Fred B, Chem. Rev, 1979, 79, 139–179; [Google Scholar]; (c) Busch DH and Alcock NW, Chem. Rev, 1994, 94, 585–623; [Google Scholar]; (d) Smith TD and Pilbrow JR, Coord. Chem. Rev, 1981, 39, 295–383. [Google Scholar]
- 10.Early work on Co-porphyrins suggested their Co(O2-) species could abstract H–atoms from phenols. See Wang X-Y, Motekaitis RJ and Martell AE, Inorg. Chem, 1984, 23, 271–275. [Google Scholar]
- 11.Wang CC, Chang HC, Lai YC, Fang H, Li CC, Hsu HK, Li ZY, Lin TS, Kuo TS, Neese F, Ye S, Chiang YW, Tsai ML, Liaw WF and Lee WZ, J. Am. Chem. Soc, 2016, 138, 14186–14189. [DOI] [PubMed] [Google Scholar]
- 12.Ramdhanie B, Telser J, Caneschi A, Zakharov LN, Rheingold AL and Goldberg DP, J. Am. Chem. Soc, 2004, 126, 2515–2525. [DOI] [PubMed] [Google Scholar]
- 13.Warren JJ, Tronic TA and Mayer JM, Chem. Rev, 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramdhanie B, Zakharov LN, Rheingold AL and Goldberg DP, Inorg. Chem, 2002, 41, 4105–4107. [DOI] [PubMed] [Google Scholar]
- 15.(a) Lee JY, Peterson RL, Ohkubo K, Garcia-Bosch I, Himes RA, Woertink J, Moore CD, Solomon EI, Fukuzumi S and Karlin KD, J. Am. Chem. Soc, 2014, 136, 9925–9937; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kindermann N, Gunes CJ, Dechert S and Meyer F, J. Am. Chem. Soc, 2017, 139, 9831–9834; [DOI] [PubMed] [Google Scholar]; (c) Tano T, Okubo Y, Kunishita A, Kubo M, Sugimoto H, Fujieda N, Ogura T and Itoh S, Inorg. Chem, 2013, 52, 10431–10437. [DOI] [PubMed] [Google Scholar]
- 16.(a) Cho J, Woo J and Nam W, J. Am. Chem. Soc, 2010, 132, 5958–5959; [DOI] [PubMed] [Google Scholar]; (b) Goo YR, Maity AC, Cho KB, Lee YM, Seo MS, Park YJ, Cho J and Nam W, Inorg. Chem, 2015, 54, 10513–10520; [DOI] [PubMed] [Google Scholar]; (c) Nemes A and Bakac A, Inorg. Chem, 2001, 40, 746–749. [DOI] [PubMed] [Google Scholar]
- 17.Company A, Yao S, Ray K and Driess M, Chemistry, 2010, 16, 9669–9675. [DOI] [PubMed] [Google Scholar]
- 18.(a) Hong S, Sutherlin KD, Park J, Kwon E, Siegler MA, Solomon EI and Nam W, Nat. Commun, 2014, 5, 5440; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chiang CW, Kleespies ST, Stout HD, Meier KK, Li PY, Bominaar EL, Que L Jr., Munck E and Lee WZ, J. Am. Chem. Soc, 2014, 136, 10846–10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corona T, Padamati SK, Acuna-Pares F, Duboc C, Browne WR and Company A, Chem. Commun, 2017, 53, 11782–11785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.