

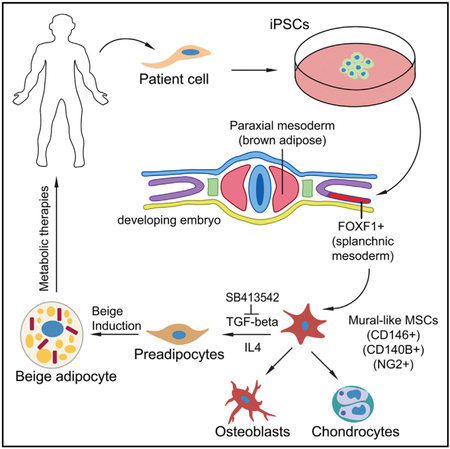
SUMMARY
Molecular- and cellular-based therapies have the potential to reduce obesity-associated disease. In response to cold, beige adipocytes form in subcutaneous white adipose tissue and convert energy stored in metabolic substrates to heat, making them an attractive therapeutic target. We developed a robust method to generate a renewable source of human beige adipocytes from induced pluripotent stem cells (iPSCs). Developmentally, these cells are derived from FOXF1+ mesoderm and progress through an expandable mural-like mesenchymal stem cell (MSC) to form mature beige adipocytes that display a thermogenically active profile. This includes expression of uncoupling protein 1 (UCP1) concomitant with increased uncoupled respiration. With this method, dysfunctional adipogenic precursors can be reprogrammed and differentiated into beige adipocytes with increased thermogenic function and anti-diabetic secretion potential. This resource can be used to (1) elucidate mechanisms that underlie the control of beige adipogenesis and (2) generate material for cellular-based therapies that target metabolic syndrome in humans.
Graphical Abstract
In Brief
Su et al. demonstrate a method for producing beige adipocytes from human induced pluripotent stem cells in a stepwise manner through defined precursor lineages. This renewable resource provides a developmental framework to study human beige adipogenesis and can be used to develop treatments for obesity-related disorders.
INTRODUCTION
The consumption of high-caloric food coupled with a sedentary lifestyle has triggered a global increase in obesity, which correlates with an increased risk for diabetes, stroke, and heart disease (Harms and Seale, 2013; Malik et al., 2013). In chronically obese individuals, diet and exercise alone are often not enough to sustain long-term weight loss due to biological adaptations that undermine beneficial lifestyle modifications (Ochner et al., 2015). During weight gain, periods of prolonged overeating result in lipid storage in white adipose tissue (WAT), leading to inflammation, cellular stress, insulin resistance, and, potentially, diabetes (Lumeng and Saltiel, 2011). New therapeutic strategies to address the public health risk of obesity are focusing on brown and beige adipose tissue. Activation of both tissues correlates positively with a reduced risk for metabolic syndrome, making them appealing therapeutic targets (Harms and Seale, 2013).
Brown and beige adipocytes become metabolically activated in response to cold-stimulated release of norepinephrine by the sympathetic nervous system, where they expend energy stored in glucose and lipids to generate heat. This process, known as non-shivering thermogenesis, likely evolved in mammals to increase neonatal survival and provide warmth in cold temperatures (Cannon and Nedergaard, 2004). Brown adipose tissue (BAT) develops during the fetal period as a permanent tissue, whereas beige adipose tissue is induced in subcutaneous WAT in response to cold and other thermogenic activators (Cousin et al., 1992; Guerra et al.,1998). In humans, BAT was originally thought to be restricted to the fetal and neonatal periods; however, recent studies show that BAT is present in adults, and its activity correlates inversely with BMI (Cypess et al., 2009). Brown and beige adipocytes have multilocular lipid droplet morphology, high mitochondrial content, and express uncoupling protein-1 (UCP1). UCP1 uncouples oxidative phosphorylation and increases proton leak across the inner mitochondrial membrane, resulting in increased thermogenesis and energy expenditure. In addition to its active role in thermogenesis, a number of secreted factors derived from BAT have a positive impact on metabolic dysfunction in mice by targeting adipose tissue, skeletal muscle, and liver in a paracrine or endocrine manner (Wang et al., 2015a). Thus, active brown and beige adipose tissue may play a natural role in the maintenance of metabolic homeostasis and energy balance.
Modifying obesity and diabetes in humans by stimulating energy expenditure in adipose tissue with drugs has largely been unsuccessful. The β3 agonist Myrbetriq, used in the treatment of overactive bladder, stimulates BAT activity in humans, but would likely lead to minimal weight loss at the currently approved dose (Cypess et al., 2015). One potential alternative to drugs is to generate cell-based therapies to supplement obese patients with additional brown or beige adipose tissue, their adipogenic precursors, or secreted factors derived from these cells. Studies in mice have demonstrated that BAT transplantation increases insulin sensitivity, prevents high-fat diet-induced weight gain, and can reverse preexisting obesity (Liu et al., 2013). In humans, BAT becomes more limited or absent with increasing age and weight gain and requires invasive methods to procure (Graja and Schulz, 2015; Wang et al., 2015b). In contrast, beige adipogenic precursors found in subcutaneous WAT are easier to procure than precursors found in BAT. However, they have limited expansion potential, and precursors from obese patients show a decreased capacity for adipocyte differentiation and a compromised ability for beige adipogenesis (Carey et al., 2014; Chung et al., 2017). One approach to overcome these obstacles is to generate patient-matched brown or beige adipocytes from induced pluripotent stem cells (iPSCs). This process requires an understanding of the developmental origins of brown and beige adipose tissue and the creation of robust and efficient methods for their differentiation from iPSCs.
In mice, classical BAT arises developmentally from an Myf5+ precursor population of paraxial mesoderm, whereas the developmental origins of beige adipocytes are more enigmatic and simply described as being derived from an unknown Myf5− precursor population (Hassan et al., 2012; Wang and Seale, 2016). This population of cells ultimately gives rise to mural cells that generate a beige adipogenic precursor population (Wang and Seale, 2016). Compared to the evidence from infants, evidence from adult humans suggests that several of the anatomical locations for classical BAT also consist of adipocytes with a molecular signature that is more similar to beige adipose tissue (Lidell et al., 2014). Because brown and beige adipose depots may be stimulated by different signals and represent potentially distinct therapeutic targets, it is important to develop cellular models that represent both tissue types (Lidell et al., 2014). Previous methods inducing mature beige and brown adipocytes from iPSCs have relied on exogenous gene transfer or short-circuiting mesoderm directly into adipocytes, rather than through the natural progression of a well-defined and expanding mesenchymal stem cell (MSC) population (Ahfeldt et al., 2012; Guénantin et al., 2017; Nishio et al., 2012). Here, we describe a multistage methodology for generating highly expandable mural-like MSCs from iPSCs, their conversion into adipogenic precursors, and their subsequent differentiation into beige adipocytes. These beige adipocytes are formed largely using commercially available defined serum-free or serum-low medium conditions, which should increase reproducibility and provide a more accurate representation of beige cell development as it occurs naturally in mammals.
RESULTS
Generation of FOXF1+ Splanchnic Mesoderm from Human iPSCs
To develop a method for generating human beige adipocytes, we differentiated iPSCs derived from human skin fibroblasts that were reprogrammed using a non-integrating method. The results were additionally replicated using iPSCs derived from cord blood and urine cells (shown in the Supplemental Information, where indicated in text). Commercially available serum-free mesoderm induction medium (MIM) was used to generate mesoderm from human iPSCs during a 5-day monolayer cell culture period. The resulting mesoderm expresses high levels of the canonical mesodermal transcripts Brachyury (T) and MIXL1 and display limited to no expression of the ectoderm marker SOX1 or the endoderm marker SOX17 (Figure 1A). Analysis of differentiating cultures (day 5) for transcripts expressed in specific mesoderm layers indicated high expression of lateral plate mesoderm markers vascular endothelial growth factor receptor 2+ (VEGFR2+) and odd skipped related 1+ (OSR1+) (Mugford et al., 2008) and low to absent expression of somatic (Iroquois related homeobox 3 [IRX3]) and paraxial (platelet-derived growth factor receptor alpha [PDGFRα]) mesoderm markers, respectively (Figure 1B). The highest expression was observed for the transcript forkhead box F1 (FOXF1), which is specific to the splanchnic layer that develops within the lateral plate mesoderm and gives rise to numerous tissues, including perivascular and smooth muscle cells (Mahlapuu et al., 2001a). Consistent with this, FOXF1 protein increased substantially after 2 days of culture in MIM and remained expressed throughout day 5 (Figure 1C). In addition, day 5 mesoderm was homogeneously positive for FOXF1 protein (>95% positive), as determined by immunostaining, indicating that MIM may be specific for generating splanchnic mesoderm (Figure 1E). Other markers necessary for the development of splanchnic mesoderm also showed increased expression by day 5 compared to day 0 iPSCs, including BAPX1 and HOX11 (Roberts et al., 1995; Tribioli et al., 1997) (Figures S1A and S1B). Using a combinatorial screening approach with a number of growth factors and small molecule pathway inhibitors, we were able to generate an in-house version of MIM by supplementing STEMdiff APEL-2 iPSC differentiation medium with the WNT agonist CHIR99021 and recombinant BMP4 and VEGFA proteins (Figures S1C–S1F). This medium was highly specific for the generation of FOXF1+ splanchnic mesoderm. Further addition of the transforming growth factor beta (TGF-β) inhibitor SB431542 (SB) resulted in a switch from the splanchnic to the somatic mesoderm, resulting in a loss of FOXF1 expression and increased expression of IRX3 (Figures S1E–S1G). Overall, these results demonstrate the differentiation of iPSCs toward cells with a molecular profile that resembles the splanchnic mesoderm.
Figure 1. Generation of FOXF1+ Splanchnic Mesoderm from Human iPSCs.

(A) Treatment of iPSCs in mesoderm induction medium (MIM) from day 1 to day 5 increases the expression of mesoderm-specific transcripts, as determined by qPCR. Means ± SDs shown.
(B) iPSCs differentiated in MIM show predominant expression of FOXF1 transcript (day 5, qPCR), indicative of splanchnic mesoderm formation Means ± SDs of three replicates shown.
(C) iPSCs differentiated in MIM induce expression of FOXF1 protein, as determined by western blot. β-Tubulin is the loading control.
(D) Treatment with MIM results in >95% FOXF1+ cells (red), as determined by immunofluorescence staining (n = 3, 20× images, representative image shown). Scale bar, 100 μm. Counterstaining with DAPI (blue).
Generation of Mural-like FOXF1-Derived MSCs
Serum-containing or serum-free MSC medium can aid in the differentiation of iPSC-derived mesoderm into MSCs, with the potential (albeit low) to give rise to adipocytes (Hafner and Dani, 2014). Likewise, we hypothesized that continuous culture and passage of FOXF1+ mesoderm in serum-free MSC medium (MesenCult-ACF [STEMCELL Technologies]) would generate FOXF1-derived MSCs (FD-MSCs) with a mural cell phenotype. After 3–6 passages (20–30 days) in MesenCult-ACF, we obtained highly pure cell populations positive for the markers CD105, CD73, and CD90, a characteristic immunophenotype of human MSCs (Figure 2A). In addition, FD-MSCs display surface markers that are consistent with mural cells, including CD146, PDGFRβ, and NG2 (Figure 2A; Crisan et al., 2008). Transcript analysis during the developmental time course from iPSC to MSC indicates increased expression of α-smooth muscle actin (αSMA) and smooth muscle protein 22-α (SM22α), but not actin from skeletal muscle (ACTA1), which derives from paraxial mesoderm (Figure 2B). This is in agreement with positive immunostaining of FD-MSCs for αSMA and SM22α (Figure 2C). A characteristic feature of MSCs is their ability to differentiate into osteoblasts, chondrocytes, and adipocytes. FD-MSCs were able to differentiate into osteoblasts and chondrocytes using serum-free differentiation medium (Figures 2D and 2E), demonstrating their multipotent ability. However, initial attempts to derive adipocytes were unsuccessful.
Figure 2. Generation of Mural-like FOXF1-Derived MSCs.

(A) Flow cytometry analysis of MSCs and perivascular markers (red) of iPSCs differentiated into FD-MSCs (pass 6, day 30) with MesenCult-ACF. Isotype controls (blue).
(B) qPCR results demonstrate increased smooth muscle marker (αSMA and SM22α) expression during differentiation of FOXF1+ mesoderm toward MSCs. Means ± SDs of three replicates shown.
(C) Immunostaining of FD-MSCs with anti-smooth muscle marker antibodies. Counterstaining with DAPI (blue). Scale bar, 25 μm.
(D) Alizarin red staining (calcium deposition) of FD-MSCs differentiated into osteoblasts. Scale bar, 200 μm.
(E) Alcian blue staining (proteoglycan deposition) of FD-MSCs differentiated into chondrocytes. Scale bar, 50 μm.
TGF-β Signaling in FD-MSCs Inhibits Their Differentiation into Adipocytes
Recent studies suggest that iPSC-derived MSCs have low adipogenic potential, which can be overcome with the TGF-β inhibitor SB (Hafner et al., 2016). We monitored the expression of TGF-β ligands and receptors during the development of FD-MSCs from iPSCs and found increasing transcript levels for TGF-β1, TGF-β2, and TGF-β3 and the receptors TGF-βR1 and TGF-βR2 with subsequent passages (Figure 3A). Furthermore, the majority of FD-MSCs display cell-surface expression of TGF-βR1 and TGF-βR2 as determined by flow cytometry (Figure 3B). Treatment of FD-MSCs in endothelial growth medium (EGM2) with a modified cocktail of factors for brown adipogenesis (Figure 3C) produced only a limited amount of lipid droplet formation that is minimally discernible by day 12 of differentiation, as determined by boron-dipyrromethene (BODIPY) lipid stain (Figure 3D, upper left). Treatment of FD-MSCs with SB for 2 days before 12 days of differentiation led to the presence of lipid accumulation (Figure 3D, upper right). Treatment of FD-MSCs with SB throughout the 12-day adipogenic differentiation period, but not before, increased lipid content further (Figure 3D, lower left, and quantitated in Figure 3E). Treatment of FD-MSCs with SB for 2 days before and throughout the 12-day adipogenic differentiation period resulted in the greatest amount of lipid accumulation (Figure 3D, lower right, and Figure 3E). Both Perilipin (PLIN) and peroxisome proliferator-activated receptor gamma 2 (PPARγ2) staining of SB-treated cultures indicates that approximately 87%–88% of the cells in culture consisted of mature adipocytes (Figures 3F–3H). When FD-MSCs were pretreated for 2 days in MesenCult-ACF medium with SB before adipocyte differentiation, the constitutive phosphorylation of SMAD2 was inhibited and the cell surface expression of known beige adipogenic precursor markers PDGFRα (Lee et al., 2012) and mesenchymal stem cell antigen 1 (MSCA1) (Estève et al., 2015) increased (Figures 3I–3J). In addition, SB pretreatment for 2 days before the adipogenic differentiation cocktail significantly increased nuclear accumulation of early B cell factor 2 (EBF2), a transcription factor that is present in committed PDGFRα+ adipogenic precursors known to give rise to beige and brown adipocytes (Figures 3K and 3L; Wang and Seale, 2016). These results suggest that the inhibition of TGF-β signaling promotes the transition of FD-MSCs into adipogenic precursors, in addition to its previously described role in promoting mature beige and brown adipocytes (Hafner et al., 2016; Yadav and Rane, 2012).
Figure 3. TGF-β Signaling in FD-MSCs Inhibits Their Differentiation into Adipocytes.

(A) qPCR analysis of TGF-β ligands and receptors during the generation of FD-MSCs and continued subculture. Means ± SDs of three replicates shown.
(B) Flow cytometry of TGF-β receptor expression in FD-MSCs (red) with isotype controls (blue). Representative plots at passage 6 shown.
(C) Schematic diagram illustrating the differentiation protocol used for adipogenic differentiation.
(D) Fluorescence microscopy of BODIPY-stained FD-MSCs differentiated for 12 days with and without 5 μM SB431542 (SB) during pretreatment (2 days) or induction (12 days). Representative images shown. Scale bar, 100 μm.
(E) Quantitation of lipid accumulation in differentiating FD-MSCs with and without SB (as shown in Figure 3D). ImageJ software was used to measure relative integrated density. Means ± SDs shown (n = 4, 20× images).
(F) PLIN (red) and PPARγ2 (green) staining of FD-MSCs induced into mature adipocytes with SB before and during adipogenic differentiation (left). Phase contrast microscopy (right) showing morphology of FD-MSC derived adipocytes treated with SB before and during adipogenic differentiation. Scale bars, 50 μm.
(G) PPARγ2+ staining (green) of FD-MSCs induced into mature adipocytes with SB. DAPI (blue) at left and PPARγ2+/DAPI overlay at right. Scale bar, 50 μm.
(H) Quantitation of PLIN+ and PPARγ2+ cells (as shown in Figures 3F and 3G) expressed as the means ± SDs (n = 4, 20× images each).
(I) Western blot analysis of phosphorylated SMAD2 before and after 3 days of SB treatment. β-Actin serves as a loading control.
(J) Flow cytometry analysis showing increased expression of beige adipogenic precursor markers in FD-MSCs treated with SB (2 days). Isotype controls (blue).
(K) Immunofluorescence staining of EBF2 (top, green) of FD-MSCs untreated (left) or treated (right) with SB (2 days). DAPI (blue, bottom). Scale bar, 50 μm.
(L) Quantitation of (K) expressed as means ± SDs (n = 4, 20× images). Student’s p value shown.
FD-MSCs Differentiate into Beige Adipocytes
PDGFRβ+, αSMA+, and NG2+ mural cells that reside within the vasculature of subcutaneous WAT have been identified as potential precursors of PDGFRα+-EBF2+ preadipocytes that give rise to mature beige adipocytes (Berry et al., 2013; Wang and Seale, 2016). Thus, we hypothesized that FD-MSCs would differentiate into beige adipocytes. A major characteristic of beige adipocytes is the expression of UCP1, a mitochondrial membrane protein that uncouples oxidative phosphorylation and increases proton leak across the inner mitochondrial membrane, resulting in increased thermogenesis and energy expenditure. qPCR analysis of UCP1 transcription during the differentiation time course shows a significant increase in UCP1 in cells differentiated in the presence of SB (Figure 4A). Primary human subcutaneous white adipocytes also exhibit increased UCP1 transcript levels when treated with SB, suggesting that enhanced beige adipogenesis by TGF-β inhibition extends beyond iPSC-derived adipogenic precursors (Figure S2A). Adipocyte differentiation of FD-MSCs results in the increased expression of key proteins that are associated with thermogenically active beige and brown adipocytes, including the mitochondrial uncoupling and respiratory chain proteins UCP1 and cytochrome c oxidase-IV (COX-IV) (Figures 4B and 4C). The lipid droplet-associated protein PLIN, necessary for fatty acid mobilization, was also increased and correlated with increased lipid droplet formation (Figure 4B). FD-MSCs that originated from additional iPSC cell lines (CD34+ cord blood- and urine-derived cells) displayed the same phenotypic and molecular changes as the ones derived from skin-derived iPSCs (Figures S3 and S4). iPSC-derived adipocytes also displayed similar or higher expression of UCP1 compared to differentiated adipocytes derived from human subcutaneous (subQ) and fetal interscapular BAT (iBAT) precursors (Figure S2B). In iPSC-derived adipocytes, expression of transcription factors that regulate beige and brown adipocyte development also increased during differentiation, including CCAAT/enhancer-binding protein alpha/beta (CEBPA/B), EBF2, PGC1A, PRDM16, and PPAR-γ (Figure 4D; Wang and Seale, 2016). Differentiating FD-MSCs display increased transcription of beige-enriched genes such as TMEM26, CITED1, and CD137, but not ZIC1, a classical brown adipocyte marker (Figure 4D; Bartesaghi et al., 2015; Harms and Seale, 2013). Mass spectrometry analysis of human fetal interscapular brown adipogenic precursors differentiated into mature adipocytes display a strong myosin protein expression signature that is consistent with their paraxial mesoderm origin (Figure S2C; Timmons et al., 2007). This signature was absent from human subcutaneous and FD-MSC adipogenic precursors induced to become beige adipocytes. Finally, beige adipogenesis of FD-MSCs results in the expression of genes and proteins that encode secreted factors (fibroblast growth factor 21 [FGF21], NRG4, interleukin 6 [IL-6], and adiponectin, C1Q, and collagen domain containing [ADIPOQ]) shown in mice to have a positive impact on metabolic dysfunction (Figures 4E and 4F; Wang et al., 2015a). These data suggest that FD-MSCs have the potential to differentiate into adipocytes that express a beige molecular profile.
Figure 4. FD-MSCs Differentiate into Beige Adipocytes.

(A) qPCR time course analysis (UCP1) of FD-MSCs during the generation of adipocytes with or without SB. Means ± SDs of three replicates shown.
(B) Protein expression during differentiation of FD-MSCs into adipocytes (12 days), as determined by western blot. β-Tubulin serves as a loading control.
(C) Immunofluorescence staining of FD-MSCs (left, day 0) or FD-MSC-derived adipocytes (right, day 12) with antibodies against the mitochondrial proteins UCP1 (green) and COX-IV (red). Scale bar, 50 μm.
(D) qPCR time course of FD-MSCs differentiating into adipocytes shows a beige enriched expression pattern. Means ± SDs of three replicates shown.
(E) qPCR time course for transcripts encoding secreted anti-diabetic factors. *Values for ADIPOQ were divided by 10,000 for scaling purposes. Means ± SDs of three replicates shown.
(F) ELISA-based measurement of anti-diabetic proteins secreted into culture medium (2 days) by FD-MSCs, iPSC-beige, and primary subcutaneous (subQ) adipocytes. Means ± SDs of three replicates shown.
(G) qPCR of iPSC-beige adipocytes that were initially pretreated with SB (5 μM) and/or IL-4 (10 nM) for 2 days before adipogenic induction. Means ± SDs of three replicates shown.
(H) Western blot analysis time course of FD-MSCs differentiating into iPSC-beige adipocytes pretreated with SB and IL-4 (2 days) before adipogenic induction.
IL-4 Enhances Differentiation of FD-MSCs into Beige Adipocytes
During cold exposure, group 2 innate lymphoid cells secrete IL-4, which directly stimulates PDGFRα+ adipogenic precursors within subcutaneous adipose tissue and enhances their differentiation into beige adipocytes (Lee et al., 2015). To determine whether IL-4 has an effect on FD-MSCs, cells were pretreated with IL-4 for 2 days, with or without SB, before adipogenic differentiation. Pretreatment with IL-4 and SB lead to a synergistic increase in UCP1 transcription and an earlier induction of UCP1 protein expression when compared to SB alone (Figures 4G and 4H). Because IL-4 was only present in cultures before adipogenic differentiation, we reasoned that it may act with SB to enhance the transition of FD-MSCs into FD-beige adipogenic precursors. IL-4 had little effect alone, but when added with SB during the pretreatment, we observed the increased transcription of beige adipogenic precursor markers, including PDGFRα+ and EBF2 (Figures S5A and S5B). Furthermore, PPAR-γ is enriched in adipogenic precursors, and, similarly, we observed an increased expression of all three PPAR-γ isoforms when FD-MSCs were treated with both SB and IL-4 for 2 days (Figure S5C; Tang et al., 2008). These data show that IL-4 and TGF-β inhibition cooperates to enhance differentiation of FD-MSCs into iPSC-derived beige (iPSC-beige) adipocytes.
iPSC-Beige Adipocytes Display Enhanced Respiratory Activity and Uncoupling
To determine whether the molecular profile associated with iPSC-beige adipocytes results in enhanced metabolic activity, we subjected the mature cells to Seahorse XF metabolic analysis. Compared to undifferentiated FD-MSCs, mature iPSC-beige adipocytes display increased basal, ATP-linked, proton leak-linked (>10-fold), maximal, and non-mitochondrial respiration, comparable to or higher than differentiated beige or white adipocytes derived from human subcutaneous adipogenic precursors (Figures 5A and 5B). In agreement with increased proton leak-linked respiration, JC-1 dye-stained iPSC-beige adipocytes show an accumulation of green fluorescent monomers, indicative of a shift in mitochondrial depolarization compared to untreated FD-MSCs, which contain red fluorescent J-aggregates at hyperpolarized membrane potentials (Figures 5C and 5D). These results suggest that iPSC-beige adipocytes display enhanced respiratory activity and uncoupling consistent with UCP1+ human subcutaneous-derived beige adipocytes (Bartesaghi et al., 2015). A hallmark feature of beige adipocytes is their ability to increase thermogenesis and respiration in response to β-adrenergic agonists, although the response in humans is known to be weaker than that observed in rodent adipocytes (Liu et al., 2017). iPSC-beige adipocytes expressed both β1- and β3-adrenergic receptors and increased expression of UCP1 upon stimulation with the β3-adrenergic agonist CL316,243 in a manner comparable to primary subcutaneous adipocytes (Figures 5E and 5F). CL316,243 also increased UCP1 expression during the initial differentiation of iPSC-beige adipocytes, but the effect was masked or inhibitory in the presence of rosiglitazone, possibly due to overstimulation of the cells (Figure 5G). Mature iPSC-beige adipocytes whitened in the absence of rosiglitazone for 4 days (as determined by a loss of UCP1) and then treated with CL316,243 for 4 hr increased overall respiration and proton leak to a greater extent than subcutaneous-derived beige adipocytes (Figures 5H–5J). Finally, iPSC-beige adipocytes that were whitened during a period of 6 days in the absence of rosiglitazone increased fatty acid (FA) uptake after treatment with CL316,243 (Figure 5J), suggesting that exogenous fatty acids may represent a major energy substrate for thermogenesis in these cells. These results demonstrate that iPSC-beige adipocytes possess the necessary cellular machinery required for increased thermogenesis in response to β-adrenergic stimuli.
Figure 5. FOXF1-Derived Beige Adipocytes Display Enhanced Respiratory Activity and Uncoupling.

(A) Seahorse XF analyzer profile of FD-MSCs and iPSC-beige adipocytes (day 14) treated with 1.25 μM oligomycin (Oligo), 1 μM para-trifluoromethoxy carbonyl cyanide phenylhydrazone (FCCP), and 2 μM rotenone/antimycin (Rot/Anti) at the indicated times (arrowheads). Means ± SDs of three replicates per time point shown.
(B) Quantitative summary of the Seahorse XF analysis shown in (A) with the addition of two primary subcutaneous beige and white cell lines. Means ± SDs of three replicates per time point shown.
(C) JC-1 assay indicates increased mitochondrial membrane polarization in iPSC-beige adipocytes (green) compared to FD-MSCs (red). Representative image shown. Scale bar, 100 μm.
(D) Quantitation of JC-1 staining shown in (C). Means of three experiments ± SDs and Student’s p value shown.
(E) Western blot time course of adrenoceptor beta 1 (ADRB1) and ADRB3 during iPSC-beige adipocyte differentiation, with β-tubulin as a loading control. HEPG2 and HL-60 cell lines serve as positive controls.
(F) qPCR of iPSC-beige adipocytes and primary subcutaneous beige cell lines treated with CL316,243 (1 μM) for 4 hr. Means ± SDs of three replicates shown. *p < 0.05 using Student’s t test.
(G) qPCR of differentiating iPSC-beige adipocytes treated with CL316,243 (1 μM) for 24, 48, and 72 hr with and without rosiglitazone (1 μM). Means ± SDs of three replicates shown. *p < 0.03 using Student’s t test.
(H) Western blot analysis time course of iPSC-beige adipocytes with rosiglitazone removed from maintenance medium from days 12 to 20. β-Tubulin serves as a loading control.
(I) Quantitative summary of the Seahorse XF analyzer profile of iPSC-beige adipocytes after 4-hr treatment with CL316,243 (1 μM, day 16). Means ± SDs of three replicates per time point and Student’s p value shown.
(J) Quantitative summary of the Seahorse XF analyzer profile of primary subcutaneous beige cells (line 1) after 4-hr treatment with CL316,243 (1 μM, day 16). Means ± SDs of three replicates per time point and Student’s p value shown.
(K) Quantitative summary of fluorescence microplate kinetic reading of iPSC-beige and -whitened beige adipocytes treated with and without CL316,243 (1 μM) for 2 hr before fatty acid uptake reading for an additional 2 hr. Means ± SDs of four replicates per time point and Student’s p value shown.
Developmental Reprogramming in Patients with Compromised Beige Adipogenesis
To determine whether our method can be used to generate a source of autologous beige adipocytes from patients who lack beige adipogenic potential, we reprogrammed and differentiated subcutaneous adipogenic precursors from a 76-year-old patient with type 2 (T2) diabetes. These primary patient-derived adipogenic precursors (T2 primary-APs) were reprogrammed with a non-integrating mRNA cocktail of pluripotency-related transcription factors to form Tra-1-60+ iPSCs (Figure 6A). These were then differentiated into iPSC-derived adipogenic precursors (T2 iPSC-APs) using our method (Figure 6B). Compared to T2 primary-adipogenic precursors, T2 iPSC-adipogenic precursors displayed a similar expression of CD105, CD73, CD90, PDGFRβ, and PDGFRα. T2 iPSC-adipogenic precursors also displayed increased surface expression of the mural cell markers CD146 and NG2 and the adipogenic precursor marker MSCA1 (Figure 6B). After beige induction of adipogenic precursors, T2 primary and iPSC-beige adipocytes both exhibited similar percentages of total PPARγ2 nuclear staining that were not significantly different from each other (Figures 6C and 6D). However, T2 iPSC-beige adipocytes displayed a >6-fold increase in lipid accumulation as compared to T2 primary adipocytes (Figures 6E and 6F). Upon differentiation, T2 primary-adipogenic precursors failed to display a transcriptional profile consistent with the formation of beige adipocytes, whereas their reprogrammed T2 iPSC-beige adipocyte counterparts displayed the full complement of beige adipogenic biomarkers (Figure S6A). T2 iPSC-beige adipocytes also exhibit higher protein levels of UCP1, PLIN, and COX-IV and increased metabolic activity, including increased basal, ATP-linked, proton leak-linked, maximal, and non-mitochondrial respiration (Figures 6G–6I). T2 iPSC-beige adipocytes also expressed comparable UCP1 protein expression to that of non-diabetic and young primary subcutaneous beige adipocytes (Figure S6B). Similar results were observed with reprogrammed beige adipocytes that were generated from subcutaneous and omental fat depots of 34- and 63-year-old patients with T2 diabetes, respectively (Figure S7, all donor characteristics summarized in Figure S7A), including increased lipid droplet formation, elevated UCP1 expression, and increased metabolic activity (Figures S7B–S7H). To test whether T2 iPSC-beige adipocytes secrete anti-diabetic factors that can be used to increase insulin sensitivity, T2 subcutaneous primary adipocyte cultures from the 76-year-old patient were treated with T2 iPSC-beige adipocyte conditioned medium for 4 days before insulin challenge (Figure 7A). Conditioned medium from T2 iPSC-beige adipocytes resulted in an approximately 3-fold increase in insulin sensitivity, as determined by phosphorylation of AKT, a downstream signaling component of insulin receptor signaling (Figures 7B and 7C). Furthermore, T2 iPSC-beige adipocyte conditioned medium treatment resulted in a significant increase in glucose uptake in the T2 primary adipocytes upon insulin challenge (Figure 7D). Similar results were observed with subcutaneous primary adipocytes treated with conditioned medium from autologous iPSC-beige adipocytes generated from the 34-year-old T2 diabetes patient (Figures S8A–S8C). Overall, these results indicate that adipogenic precursors with an inherently low beige adipogenic potential can be reprogrammed and differentiated with our method to produce iPSC-derived beige adipocytes with increased thermogenic and anti-diabetic potential.
Figure 6. Developmental Reprogramming in a Patient with Compromised Beige Adipogenesis.

(A) Tra-1-60+ live cell staining (upper) and phase contrast (lower) of a representative iPSC colony generated from subcutaneous adipogenic precursors of a 76-year-old patient with type 2 diabetes. Scale bar, 100 μm.
(B) Flow cytometry of MSC and adipogenic precursor (AP) markers expressed on T2 primary-adipogenic precursors and T2-iPSC-adipogenic precursors. FD-MSCs were treated with SB (5 μM) for 2 days to generate T2-iPSC-adipogenic precursors.
(C) PPARγ2+ staining (green) of T2 primary and T2 iPSC-beige adipogenic precursors induced into mature adipocytes with the beige adipogenic cocktail. Scale bar, 50 μm.
(D) Quantitation of PPARγ2+ cells (as shown in C) expressed as means ± SDs (n = 3, 20× images each). Student’s p value shown.
(E) Fluorescence microscopy of BODIPY-stained T2 primary-adipogenic precursors and T2-iPSC-adipogenic precursors after differentiation into adipocytes with the beige induction protocol (12 days). Representative images shown. Scale bar, 100 μm.
(F) Quantitation of lipid accumulation (as shown in E) by ImageJ software, as measured by relative integrated density expressed as means ± SDs (n = 3, 20× images). Student’s p value shown.
(G) Western blot time course of T2 primary-adipogenic precursors and T2-iPSC-adipogenic precursors differentiated into mature adipocytes.
(H) Quantitation of UCP1 protein expression shown in (G); three replicate samples pooled per data point. Data are normalized relative to β-tubulin.
(I) Quantitative summary of the Seahorse XF analyzer profile of T2 primary and T2 iPSC-beige adipocytes (day 14). Means ± SDs of three replicates per time point shown. *p < 0.05 and **p < 0.01 using Student’s t test.
Figure 7. iPSC-Derived Beige Adipocytes Secrete Factors that Improve Insulin Sensitivity and Glucose Uptake.

(A) Schematic representation showing experimental design to test for anti-diabetic secretion potential of iPSC-beige adipocytes.
(B) Western blot analysis of phospho-AKT in T2 primary adipocytes treated with insulin after culture in T2 primary adipocyte conditioned medium (lanes 4–8) or T2 iPSC-beige adipocyte conditioned medium (lanes 9–12).
(C) Quantitation of phospho-AKT protein expression (shown in B). Data are normalized to β-actin protein. Means ± SDs of four replicates and Student’s p value shown.
(D) Glucose uptake analysis of T2 primary adipocytes treated with insulin in the presence of T2 primary adipocyte conditioned medium or T2 iPSC-beige adipocyte conditioned medium. Means ± SDs of six replicates and *p < 0.05 using Student’s t test shown.
DISCUSSION
An increased understanding of the development and regulation of beige adipose has the potential to enhance metabolic health through the generation of new treatments for metabolic syndrome. Considering that procurement of primary beige adipogenic precursors from humans requires invasive methods and that these cells have limited renewability, direct reprogramming of adult cells into iPSCs has raised the possibility of producing an unlimited number of patient-matched beige adipocytes for their study and use in cellular therapies. Here, we demonstrate that human beige adipocytes can be generated from iPSCs using a defined, multistage culture system (iPSC ≤ mesoderm ≥ MSC ≥ adipogenic precursor ≥ beige adipocyte). In addition, we have identified FOXF1+ mesodermal precursor cells as a developmental source of beige adipocytes. Culture of FOXF1+ precursors in MSC medium generates murallike MSCs that can be expanded through multiple passages to produce adipocytes with molecular and metabolic profiles that are consistent with beige adipocytes, including expression of UCP1 and proton leak-linked respiration. Moreover, iPSC reprogramming in conjunction with our method can greatly increase the browning potential of primary adipogenic precursors that possess an inherently low adipogenic capacity to form a more robust, renewable, and metabolically active source of beige adipocytes.
The developmental origins of adipocytes are not well defined, yet this information may shed light on the distribution of distinct adipose tissue depots, help determine insights into their metabolic differences, and be used to develop strategies to engineer different types of adipose tissue for cellular therapies (Sanchez-Gurmaches et al., 2016). Along these lines, methods that generate adipocytes from iPSCs without the exogenous transfer of adipogenic genes can be exploited to determine the developmental origins of specific adipose tissue depots that arise in humans. A few studies have focused on the development of beige and brown adipocytes. In one such study, iPSC-derived classical brown adipocytes were generated using a two-stage hematopoietic growth factor cocktail that includes bone morphogenetic protein 4 (BMP4) and BMP7 (Nishio et al., 2012). These growth factor-treated iPSCs differentiate through the paraxial mesoderm and subsequently myoblast lineages (Nishio et al., 2012). The generation of iPSC-derived beige adipocytes has also been recently reported (Guénantin et al., 2017). With this method, hematopoietic cell medium supplemented with BMP4 and Activin A was used to generate mesoderm that was directly induced into adipocytes with a beige molecular signature. However, the mesodermal lineage was not characterized in detail. Mature adipocytes normally arise through the commitment of MSCs to adipogenic precursors and their terminal differentiation (Cristancho and Lazar, 2011). While the two above-mentioned methods produce mature brown or beige adipocytes from iPSCs rapidly (12 and 20 days, respectively), natural development through a multipotent and expandable MSC precursor is absent and renewability is only achieved at the level of the iPSCs. In another study, embryoid bodies from iPSCs were formed on non-adherent plates in the presence of transient retinoic acid treatment with subsequent outgrowth on adherent plates to generate an expanding population of MSCs (Hafner et al., 2016; Mohsen-Kanson et al., 2014). These MSCs differentiate into adipocytes with a molecular profile that is indicative of both brown and beige characteristics. While their mesodermal origin is unknown, these cells may derive from either the PAX3+ neural crest or myogenic lineages.
Fate-mapping studies in mice have demonstrated that classical brown adipocytes derive from Myf5+ -Pax7+ precursors that arise from the dermomyotome (Harms and Seale, 2013). Other lineage-tracing studies in mice have demonstrated that a significant portion of visceral white adipocytes are derived from a Wt1+ precursor population that arises from the lateral plate mesoderm, which is distinct from the origin of subcutaneous adipocytes (Chau et al., 2014). New evidence based upon specific transcriptional and epigenomic patterns in mice suggests that beige adipocytes represent a distinct lineage, which upon whitening, retain an epigenomic memory of prior cold exposure that aids in inducing a rapid thermogenic response upon additional cold challenge (Roh et al., 2018). However, a precursor population that can be used to lineage trace the origin of beige adipocytes during embryonic development has remained elusive. White adipocytes arise from precursors present in the mural cell compartment of the adipose vasculature, and it has been speculated that they originate from an Myf5− precursor originating from the lateral plate mesoderm (Hassan et al., 2012; Tang et al., 2008). Our results demonstrate that beige adipocytes can be generated from a homogeneous population of FOXF1+ cells that normally originate from within the splanchnic mesoderm. During embryonic development, Foxf1 is expressed initially throughout the lateral plate mesoderm and becomes restricted to and required for separation of the splanchnic mesoderm through the repression of somatically expressed Irx3 (Mahlapuu et al., 2001b). Consistent with this, our differentiating cultures show low to absent expression of IRX3. Generation of an Foxf1 reporter mouse line and lineage tracing may shed more light onto the developmental origins of beige adipocytes that arise within distinct subcutaneous WAT depots.
Studies in mice have shown that the inhibition of TGF-β signaling results in increased browning of WAT and improved metabolic function (Yadav and Rane, 2012). In addition, iPSC-derived MSCs show vastly increased adipogenic potential in the presence of the TGF-β inhibitor SB (Hafner et al., 2016). We have confirmed these results and demonstrated that MSCs generated with our method express TGF-β ligands and receptors that activate TGF-β signaling via phosphorylated SMAD2. In addition to increasing beige adipogenesis, SB alone directed the differentiation of FD-MSCs toward an adipogenic precursor phenotype, including increased expression of PDGFRα, MSCA1, PPAR-γ, and EBF2. Moreover, when induced with an adipogenic cocktail of factors, these precursors display an enhanced ability to form UCP1-expressing beige adipocytes.
Cold exposure in mice leads to the activation of type 2 immune cells, including eosinophils, which secrete IL-4 and IL-13 to alternatively activate macrophages toward a polarized M2 fate (Qiu et al., 2014). This anti-inflammatory subset of macrophages may in turn contribute to thermogenesis in WAT through the secretion of catecholamines (van den Berg et al., 2017). In addition to activating macrophages, IL-4 can act directly on PDGFRα+ adipogenic precursors isolated from subcutaneous WAT and commit these precursors to a beige rather than a WAT molecular phenotype (Lee et al., 2015; Lizcano et al., 2017). Treatment of FD-MSCs with IL-4 in our study also increased beige adipogenesis, as assessed by the expression of UCP1. A more recent study calls into question whether M2 macrophages secrete significant amounts of catecholamines and suggests that IL-4 plays no role in the adaptive thermogenesis of adipose tissue (Fischer et al., 2017). Whether IL-4 promotes beige adipogenesis at the level of the adipogenic precursor or through catecholamine secretion by macrophages remains unclear. It could depend on past environmental exposures of the animal or length of cold induction, which may influence whether beige adipogenesis results from the browning of preexisting mature adipocytes or from de novo differentiation of adipogenic precursors (Wang and Seale, 2016). In mice, long-term cold exposure appears to favor de novo beige formation from PDGFRβ+ mural cell adipogenic precursors (Vishvanath et al., 2016). Along these lines, our study indicates that IL-4 can significantly enhance the differentiation of mural-like FD-MSCs (PDGFRβ+) into committed beige adipogenic precursors. Differentiation of these precursors displays enhanced beige adipogenesis, including earlier induction and increased expression of UCP1. Thus, IL-4 may exert its effects by promoting the commitment of the PDGFRβ+ mural cell lineage to beige adipocytes during chronic cold exposure.
Our method produces large numbers of thermogenically active adipocytes and has several advantages over other existing approaches. While our method takes longer than others to generate beige adipocytes from iPSCs, once obtained, the FD-MSCs are highly expandable (>15 passages) and require only 9–12 days of adipogenic differentiation to form fully mature beige adipocytes. Thus, our method is renewable at both the iPSC and MSC stages. Because the three mediums used to generate iPSC-derived beige adipocytes are produced commercially under high-quality-controlled conditions, this should facilitate more reproducibility across different laboratories. In addition, FD-MSCs are multipotent and can be used to study the formation of other terminally differentiated cell types that arise from the FOXF1+ mesoderm compartment, including osteoblasts and chondrocytes. Hence, this method can be widely applied to iPSCs that are derived from patients with diseases that affect bone, cartilage, and adipocytes. In this study, we also demonstrated that reprogramming can increase the beige adipogenic potential of adipogenic precursors isolated from an elderly patient with type 2 diabetes. Whether the inability of the primary adipogenic precursors from this patient to form beige adipocytes is due to the age of the individual or type 2 diabetes is unknown. To further characterize this, we analyzed primary adipogenic precursors isolated from the subcutaneous adipose tissue of a 34-year-old patient with type 2 diabetes, which displayed a higher level of beige adipogenesis than the precursors isolated from the 76-year-old patient. However, reprogramming these younger cells into iPSC-derived beige adipocytes further increased browning and the metabolic rate, suggesting that both increasing age and diabetes may play negative roles in beige adipogenic potential. Other primary cell types from patients with metabolic abnormalities may provide a better source of iPSCs for the generation of beige adipocytes, but reprogramming primary adipogenic precursors may be used to find important epigenetic changes that result in dysfunctional beige adipogenesis by comparing epigenetic signatures before and after reprogramming.
In summary, we have identified a method to generate metabolically active beige adipocytes from iPSCs that serves as a model to study beige cell development and regulation. The scalability of this method provides a platform to generate copious numbers of beige adipocytes for high-throughput drug screening, transplantation-directed therapies, and the discovery and isolation of secreted factors that may aid in reducing pathophysiologies that are associated with metabolic syndrome.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Aaron Brown (BROWNA7@MMC.ORG).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Primary cells
All primary cell lines isolated from WAT were generated from outgrowth of the stromal vascular fraction and purchased from commercial vendors. All cell lines were validated to be at least 95% positive for adipogenic precursors as determined by flow cytometry for PDGFRα. Human white preadipocytes (HWP-c) cells, purchased from PromoCell (C-12730), were isolated from subcutaneous adipose tissue of a 27-year old Caucasian male with a BMI of 28. Normal human preadipocytes, purchased from Lonza (PT-5020), were isolated from subcutaneous adipose tissue of a 37 year old black female with a BMI of 21. Type 2 diabetic human preadipocytes, purchased from Lonza (PT-5022), were isolated from subcutaneous adipose tissue of a 76 year old black female with a BMI of 42.1. Type 2 diabetic human preadipocytes, purchased from Zenbio (OPD-F-3), were isolated from omental adipose tissue of a 63 year old white female with a BMI of 38.6. Type 2 diabetic human preadipocytes, purchased from Zenbio (SPD-F-3), were isolated from subcutaneous adipose tissue of a 34 year old black female with a BMI of 53.5. Cryopreserved cells were thawed and seeded in a 100×20 mm TC dish and treated as passage 0. Primary adipogenic precursors were expanded in DMEM containing 10% fetal bovine serum. Medium was changed daily and cells were cultured at 37°C in a 5% CO2 incubator. Upon 80% confluence, cells are split and passaged at a ratio of 1:6 (Passage 1). At 100% confluence, cells were induced to differentiate in beige adipogenic medium as was performed for generating FD-beige adipocytes.
Induced pluripotent stem cell (iPSC) lines
Integration-free iPSCs, purchased from Applied StemCell (ASE-9202), were derived from human skin fibroblasts. Integration free iPSCs, purchased from GIBCO (A18945), were derived from cord blood-derived CD34+ progenitors. Cryopreserved cells were thawed and seeded as a monolayer on hESC-qualified Matrigel-coated plates and treated as passage 0. iPSCs were expanded in NutriStem hPSC XF Medium and, upon reaching 80% confluence, were passaged with ReLeSR passaging reagent. Medium was changed daily and cells were cultured at 37°C in a 5% CO2 incubator. Cell lines were authenticated by confirming expression of Oct4, Sox2, SSEA4 and TRA-1-60.
Reprogrammed iPS cell lines
Integration- and xeno- free iPSCs were generated from urine-derived cells obtained from a de-identified 60-year old female patient (with patient informed consent) with no history of cancer through the Maine Medical Center BioBank via the MMC BioBank’s IRB approved protocol (#2526). These cells were expanded and reprogrammed using the integration-free StemRNA-NM_Reprogramming kit and protocol for urine-derived cells following the manufacturers’ instructions. iPSCs were also generated from passage 1 subcutaneous white adipose adipogenic precursors (Lonza PT-5022, Zenbio OPD-F-3, Zenbio SPD-F-3) using the StemRNA NM reprogramming kit following the manufacturers’ instructions for the fibroblast protocol to generate integration- and xeno- free iPSCs. iPSCs generated in house were live cell-stained to evaluate pluripotency with anti-TRA-1-60-Vio488 Live Cell Stain following the manufacturers’ instructions and imaged with an epifluorescence microscope (Leica).
Human Tissue
Human fetal interscapular brown adipose tissue (huFe iBAT) from 18-week gestation was obtained from the University of Pittsburgh Health Sciences Tissue Bank through an honest broker system after approval from the University of Pittsburgh Institutional Review Board (IRB number: 0702050) and in accordance with the University of Pittsburgh anatomical tissue procurement guidelines. Adipose tissue was dissociated using the Adipose Tissue Dissociation Kit enzyme digest cocktail. Isolated cells were seeded in a 100×20 mm dish in DMEM containing L-glutamine and 10% FBS for expansion. Medium was changed daily and cells were maintained at 37°C in a 5% CO2 incubator.
METHOD DETAILS
For culture of iPSCs, mesoderm induction, generation of iPSC-derived MSCs and differentiation of MSCs into beige adipocytes, please refer to the Methods S1 section.
Generation of chondrocytes
Expand MSCs in a 6 well plate to 90% confluence. Conduct cell chondrocyte differentiation in a laminar flow cabinet.
Warm up cell wash buffer, ACF enzymatic dissociation solution, and Chondrogenesis Differentiation Medium to room temperature.
Aspirate MesenCult-ACF Plus medium from cell culture wells and wash once with PBS without calcium or magnesium. Remove PBS and replace with 500 μl/well ACF enzymatic dissociation solution.
Place cell culture plate in a 37°C incubator for 5 minutes and tap side of plate firmly on hard surface several times to sheer cells from plate surface (Figure M2). Check that cells have dislodged from the plate under a light microscope. If not, extend incubation time in 1-minute increments until noticeable detachment.
Add 500 μL of ACF enzymatic inhibition solution to each well of a 6 well plate. Further suspend cells in 2 mL of cell wash buffer and gently remove cells from the plate by washing with a pipette. Transfer cell suspension to a 15 mL centrifuge tube and centrifuge at 300 g for 5 minutes. Check for the appearance of a cell pellet at the bottom of the tube.
Remove the supernatant and resuspend pelleted cells in 2ml of Chondrogenesis Differentiation Medium. Centrifuge tube at 300 g for 5 minutes to pellet cells.
Once pelleted, place tube in a 5% CO2 incubator at 37°C for 3 days with a loose cap to facilitate gas exchange. On day 3, and every 3 days up to day 24, change 100% of medium and gently flick tube to ensure that the cell pellet is not attached to the tube.
Generation of osteoblasts
Prior to osteoblast differentiation, MSCs grown in well culture plates must be at 100% confluence. Conduct osteoblast differentiation in a laminar flow cabinet.
Aspirate MesenCult-ACF Plus medium and wash once with PBS without calcium or magnesium.
-
Remove PBS and replace with OsteoLife Complete Osteogenesis Medium in the following amounts based upon well size.
96 well plate – 100 μl/well
24 well plate – 500 μl/well
6 well plate – 2000 μl/well
Change 100% of the medium every 3 days for 21 days.
Isolation and culture of primary preadipocytes
Recipe for differentiation mediums
Beige adipocyte induction medium:
| Factor | stock concentration | working concentration | for 10 ml of medium |
|---|---|---|---|
| DMEM | 1X | 1X | 9 ml |
| FBS | 10X | 1X | 1 ml |
| Insulin | (10 μg/ml = 1.7 mM) | 170 nM | 1 μl |
| T3 | 1 μM | 2 nM | 20 μl |
| Rosiglitazone | 5 mM | 1 μM | 2 μl |
| SB | 10 mM | 5 μM | 5 μl |
| IBMX | 500 mM | 0.5 mM | 10 μl |
| Dexamethasone | 5 mM | 5 μM | 10 μl |
| Indomethacin | 125 mM | 125 μM | 10 μl |
| AA2P | 50 mg/ml | 50 μg/ml | 10 μl |
Beige adipocyte maintenance medium:
| Factor | stock concentration | working concentration | for 10 ml of medium |
|---|---|---|---|
| DMEM | 1X | 1X | 9 ml |
| FBS | 10X | 1X | 1 ml |
| Insulin | 1.7 mM | 170 nM | 1 μl |
| T3 | 1 μM | 2 nM | 20 μl |
| Rosiglitazone | 5 mM | 1 μM | 2 μl |
| SB | 10 mM | 5 μM | 5 μl |
| AA2P | 50 mg/ml | 50 μg/ml | 10 μl |
Cell isolation, expansion and differentiation of interscapular brown adipose tissue preadipocytes
Prior to adipose cell isolation, warm DMEM, FBS and cell wash buffer to room temperature.
In a laminar flow cabinet for cell culture, place 5 g of adipose tissue on a 100×20 mm dish. Suspend tissue in 1 mL of Adipose Tissue Dissociation Kit enzyme digest cocktail and finely mince with a razorblade.
Transfer cell suspension slurry to a 5 mL polystyrene round bottom tube and add an additional 1.5 mL of enzyme digest cocktail to tube.
Incubate tube at 37°C on a rotating nutator for 30 minutes. During incubation period, briefly vortex sample for 5 s in 10-minute intervals.
Transfer cells to a 15 mL tube and add 5 mL of DMEM. Filter cell suspension through a 100 μm strainer into a new 15 mL tube and centrifuge at 300 g for 5 minutes to form a pellet.
Remove supernatant and leave behind <100 μL of DMEM. Resuspend cell pellet in 10 mL of DMEM containing 10% FBS and plate in a 100×20 mm dish for expansion. Agitate plate to spread cells evenly on well surface and incubate plate at 37°C in a 5% CO2 incubator undisturbed. Change 100% of medium every day.
Upon 80% confluence, split and passage cells at a ratio of 1:6.
Prepare for passage by warming cell wash buffer and cell culture medium (DMEM containing 10% FBS) to room temperature. Warm TrypLE dissociation reagent to 37°C.
Aspirate cell culture medium from wells and wash once with PBS. Remove PBS and add 500 μL of pre-warmed TrypLE reagent to each well of a 6 well plate. Scale TrypLE amount for larger tissue culture plates accordingly.
Incubate culture plate at 37°C for 5 minutes. Check that most cells have been dislodged from the plate surface with a light microscope. If not, extend incubation time in 1-minute increments until noticeable cell detachment.
Add cell wash buffer to each well and gently resuspend cells from the plate by washing with a pipette. Check that most cells have been dislodged from the plate with a light microscope. If not, pipette up and down as needed.
Transfer cells to an appropriate tube and centrifuge at 300 g for 5 minutes. Check for the appearance of a cell pellet at the bottom of the tube.
Remove the supernatant and resuspend cells in an appropriate amount of DMEM containing 10% FBS. Plate cells at a ratio of 1:6 by adding 2 mL of cell suspension to each well of a 6 well plate. Agitate to spread cells evenly on well surface. Incubate plate at 37°C in a 5% CO2 incubator undisturbed. Change 100% of medium every 2 days.
Differentiation can be performed when cells reach 100% confluence.
-
Aspirate DMEM containing 10% FBS and wash once with PBS. Remove PBS and replace with beige adipocyte induction medium in the following amounts based upon well size.
96 well plates – 150 μl/well
24 well plates – 1000 μl/well
6 well plates – 3000 μl/well
-
After 72 hours of induction treatment, aspirate beige adipocyte induction medium from wells and replace with beige adipocyte maintenance medium in the following amounts based upon well size.
96 well plates – 150 μl/well
24 well plates – 1000 μl/well
6 well plates – 3000 μl/well
Change 100% of beige adipocyte maintenance medium every 48 hours for 4 days. Note: huFe iBAT-derived beige adipocytes often times reach full maturity following only 7 days of differentiation.
Cell expansion and differentiation of commercial preadipocytes isolated from subcutaneous or omental adipose tissue
Thaw and plate preadipocytes isolated from subcutaneous or omental adipose tissue following the manufacturers’ recommen-dations in a laminar flow cabinet for cell culture.
Upon 80% confluence, split and passage cells at a ratio of 1:6 (see Cell isolation, expansion and differentiation of interscapular BAT preadipocytes above).
Differentiation can be performed when cells reach 100% confluence (see Cell isolation, expansion and differentiation of interscapular BAT preadipocytes above).
Change 100% of beige adipocyte maintenance medium every 72 hours for 9 days. Note: commercial preadipocytes-derived beige adipocytes often reach full maturity between 9 and 12 days of differentiation.
Medium conditioning and glucose uptake
Conditioning medium
Prior to conditioned medium experiments, iPSC-derived beige adipocytes and type 2 diabetic subcutaneous adipocytes must be differentiated to full maturity (day 12) (see Generation of beige adipocytes and Culture differentiation of commercial preadipocytes isolated from subcutaneous or omental adipose tissue above). Differentiate iPSC-derived beige adipocytes in 24 well plates (experimental medium conditioning) and type 2 diabetic subcutaneous adipocytes in both 24 well plates (control medium conditioning) and 96 well plates (for glucose uptake).
Use iPSC-derived beige adipocytes and type 2 diabetic adipocytes in 24 well plates to generate conditioned medium. Aspirate beige maintenance medium from wells of 24-well plates and wash once with PBS without calcium or magnesium. Remove PBS and replace with 500 μL of high glucose DMEM (4500mg/l) supplemented with 2% FBS and 10 mM HEPES per well. Allow medium to be conditioned for 24 hours prior to use.
Time 0: After 24 hours collect conditioned medium from 24 well plates and replace with 500ul fresh medium. Prior to use, centrifuge conditioned medium at 300 g for 5 minutes and then transfer supernatant to new tubes. To condition type 2 diabetic subcutaneous adipocytes with conditioned medium, remove medium from wells of 96 well plate and wash once with PBS. Remove PBS and replace with 100 μL of a 50/50 mixture of fresh medium and conditioned medium per well (iPSC-beige or type diabetic for controls). Fresh medium consists of high glucose DMEM (4500mg/l) supplemented with 2% FBS and 10mM HEPES.
Time 24 hours: Repeat step 3 above.
Time 48 hours: Repeat step 3 above, but replace fresh medium on 24 well plates with 500ul DMEM (4500 mg/l) supplemented with 0.5% BSA and 10 mM HEPES per well for conditioning.
Time 72 hours. Remove 100% of medium from 96 well plates and replace with 100 μL of 50% conditioned medium and 50% DMEM (4500 mg/l) supplemented with 0.5% BSA and 10 mM HEPES per well. In this way, medium conditioning for the last 24 hours will be FBS free.
Glucose uptake
Time 96 hours: Remove 100% of medium and wash once with PBS. Remove PBS and replace with no glucose and serum-free DMEM with insulin (0.02 to 20 nM with four replicates each) for one hour prior to glucose uptake tests.
Glucose uptake analysis was performed using the Glucose Uptake-Glo Assay Kit and analyzed on a GloMax luminometer following the manufacturer’s instructions.
Medium conditioning and phospho-AKT assay
Repeat exactly as for glucose uptake assay, but with type 2 diabetic cells grown in 24 well-plate for protein harvest with a 50/50 mixture of fresh medium (250 μl) and conditioned medium (250 μl) per well. At 96 hours, treat with 20 nM insulin for 10 minutes and harvest cells 80 μL RIPA buffer with protease and phosphatase inhibitor cocktail to each well. Dislodge the cells by gently scrapping and washing the surface of the wells with a 200 μL pipet tip. Transfer protein lysate to 1.7 mL eppendorf tubes and centrifuge at 14,000 rpm for 5 minutes to pellet cell debris.
Run samples through a western blot analysis using an anti-phospho-AKT antibody (see protein extraction and immunoblotting for western blot)
Oxygen consumption rate assay
Assay preparation
Expand and culture FD-MSCs to confluence and plate in a 24-well Seahorse V7 culture plate (see Generation of mesenchymal stem cells above).
Once FD-MSCs are 90% confluence, differentiate with 100 μl of beige adipogenic medium for 14 days (see Generation of beige adipocytes above).
Prior to OCR assay, aspirate beige maintenance medium and replace with 180 μl of Seahorse XF assay medium (containing 2 mM glutamine) supplemented with 10 mM pyruvate and 25 mM glucose.
Conducting assay and analyzing results
With a Seahorse XF24 analyzer, measure OCR with small molecule inhibitors added through the injection ports. The following concentrations of the inhibitors were used: 1.25 μM oligomycin, 1 μM FCCP and 2 μM each of antimycin A and rotenone.
Basal, uncoupled and maximal respiration rates can be calculated upon subtraction of the non-mitochondrial oxygen consumption obtained at the end of each assay by the addition of antimycin A and rotenone. The values obtained can be normalized to total μg protein per well as measured by BioRad Protein assay reagent.
Note: For β3-adrenergic response assay, remove rosiglitazone from maintenance medium at day 12 and culture adipocytes for an additional 4 days to whiten. Treat cells with CL316,243 (1 μM) for 4 hours prior to Seahorse XF analysis.
Flow cytometry
Cell preparation
Prior to antibody staining, cells must be dissociated from cell culture plates and pelleted in cell wash buffer (see 1.4: Cell passaging and 2.5: Generation of mesenchymal stem cells above).
In a laminar flow cabinet, remove supernatant and resuspend cells in an appropriate amount of cell wash buffer. For example, when staining with 3 PE/APC pairs, resuspend cell pellet from one well of a 24 well plate in 100 μL of cell wash buffer and split suspension into 4 microcentrifuge tubes, 25 μL each. Label tubes with their respective PE/APC antibody pairs. Separate one tube as an unstained control.
Dilute primary conjugated antibodies 1:10 for staining. Mix cell suspension by gently pipetting. Allow cells to stain undisturbed at 4°C for 15 minutes in the dark.
Following 15 minutes, add 900 μL of cell wash buffer to each sample tube and centrifuge at 300 g for 5 minutes to pellet cells.
Gently remove supernatant and leave behind cell pellet. Resuspend cell in 250 μL of cell wash buffer and transfer to a 5 mL Polystyrene round-bottom tube for flow cytometry analysis and quantification with MACSQuantify.
For TGFBR1 and TGFBR2, fix cells with paraformaldehyde for 10 minutes and permeabilize with methanol for 30 minutes on ice prior to staining. To stain, dilute antibodies 1:10 in 25 μL cell wash buffer for 30 minutes at 4°C.
Immuno- and BODIPY staining
BODIPY Staining
Prior to staining, expand and grow adipocytes to full maturity (day 12) (see Generation of beige adipocytes and Cell expansion and differentiation of commercial preadipocytes isolated from subcutaneous or omental adipose tissue above).
Remove culture medium and stain cells with BODIPY diluted 1:2000 in fresh culture medium for 30 minutes at 37°C.
Following 30 minutes, remove culture medium with BODIPY and gently wash once with PBS. Remove PBS and replace with fresh cell culture medium prior to imaging on an EVOS Fluorescence Microscope.
Quantitate lipid accumulation via the integrated density function with ImageJ software.
Immunofluorescence staining
Prior to staining, expand and grow beige adipocytes to full maturity (day 12) (see Generation of beige adipocytes and Cell expansion and differentiation of commercial preadipocytes isolated from subcutaneous or omental adipose tissue above).
Wash cells with PBS and fix cells for 10 minutes by gently pipetting 4% paraformaldehyde down the side of the wells.
After 10 minutes, remove paraformaldehyde and wash once with PBS. Remove PBS and permeabilize cells with 0.3% Triton X-100 in PBS for 15 minutes at room temperature.
After 15 minutes, remove 0.3% Triton X-100 in PBS and add blocking buffer (10% donkey serum diluted in PBS) for 1 hour at room temperature.
Remove blocking buffer and stain with the following primary antibodies diluted 1:100 in blocking buffer for 24 hours at 4°C: anti-FOXF1, anti-SMA, anti-SM22 alpha, anti-COX-IV, anti-UCP1, anti-PLIN, and anti-EBF2. To avoid evaporation, wrap plate in parafilm during the 24 hour period.
After 24 hours, remove antibodies in blocking buffer and gently wash wells 3 times with PBS.
Prepare appropriate secondary antibodies diluted 1:200 in blocking buffer and add to wells. Allow plate to sit undisturbed for 1 hour at room temperature in the dark.
Remove secondary antibodies in blocking buffer and wash wells 3 times with PBS. Remove and replace with fresh PBS prior to imaging with an epifluorescence microscope (Leica).
Quantitative PCR
Sample preparation
Aspirate medium from wells and add RLT lysis buffer as provided by RNeasy Micro Kit. Gently scrape and wash wells with the RLT lysis buffer using an appropriately sized pipette.
Transfer cell lysate to a microcentrifuge tube. If not immediately used, cell lysate can be stored at −70°C.
Complete manual or automated RNA extraction procedure according to manufacturers’ instructions.
Generate cDNA from 250 ng of RNA with qScript cDNA SuperMix.
For qPCR, amplify gene-specific cDNA transcripts in triplicate with iQ SYBR Green Supermix in a CFX384 Touch Real-Time PCR Detection System.
Normalize data to β-actin expression using the ΔΔCT method. Fold changes will be relative to the vehicle control.
JC-1 assay
Assay preparation
Prepare a 2 μM working solution of JC-1 dye by diluting stock solution in HBSS with 20 mM HEPES buffer. Heat solution on a rotating nutator in a 37°C incubator to facilitate dissolution. Conduct JC-1 assay preparation in a laminar flow cabinet for cell culture.
Aspirate medium from cell cultures and add JC-1 dye solution. Incubate in a 37°C, 5% CO2 incubator for 30 minutes.
Remove and replace working solution with HBSS prior to imaging. Image for fluorescence change at Ex/Em = 490/525 nm and 540/595 nm with an epifluorescence microscope.
Calculate subsequent ratio analysis from images using ImageJ software.
Mass spectrometry
Preparation
Prior to preparation, expand and grow beige or brown adipocytes to full maturity in a monolayer culture in 6 well plates (day 12) (see Generation of beige adipocytes and Cell expansion and differentiation of commercial preadipocytes isolated from subcutaneous or omental adipose tissue above).
Dissociate cells from plate with 0.5 mM EDTA solution, pellet, and extract cells using the Qproteome Mammalian Protein Prep Kit according to manufacturers’ instructions. Additionally, add two magnetic beads and homogenize sample with an Autodisruptor at maximum speed for 60 s during the cell lysis step.
Digest protein samples by adding trypsin as provided by the ProteoExtract digestion kit.
Separate peptides with an Ultimate RSLC system 3000 nanoscale liquid chromatograph and then infuse onto a 5600 TripleTOF mass spectrometer.
Profile protein samples using the SWATH data-independent acquisition method.
Spectra processing and database searching of an ion library consisting of 4091 proteins was performed with ProteinPilot software (Sciex). SWATH runs were extracted and analyzed using PeakView software and MarkerView software was utilized to find statistically relevant relationships through t test comparisons.
Protein Extraction and Immunoblotting
Protein extraction
Aspirate cell culture medium and add RIPA buffer supplemented with Halt Protease & Phosphatase Inhibitor Cocktail according to manufacturers’ instructions.
Gently scrape well surface while washing with lysis buffer with an appropriately sized pipette. Transfer cell suspension to 1.7 μL Eppendorf tubes and centrifuge at 15,000 rpm for 5 minutes to pellet cells. Collect the supernatant to new Eppendorf tubes.
Determine protein concentration using the Pierce BCA Protein Assay Kit following manufacturers’ instructions.
Immunoblotting
Denature protein lysates by heating at 90°C for 5 min
Separate protein with a CriterionTM TGX gels and then transfer onto PVDF membranes.
Incubate membrane in 1X Detector Block Solution for 1 hour at room temperature or overnight at 4°C. Following 1 hour, add primary antibodies diluted at a ratio of 1:1000~2000 in TBS with Tween 20 and 0.2X Detector Block Solution and incubate overnight at 4°C.
Remove solution and wash 3 times with TBS with Tween 20 (5 minutes per wash). Following washes, add secondary antibody diluted at a ratio of 1:1200 and incubate 2 hours at room temperature.
Remove secondary antibody solution and wash 3 times with TBS with Tween 20 (5 minutes per wash).
Develop membrane with ClarityTM Western ECL Substrate and detect using a Bio-Rad ChemiDoc Touch Imaging System.
Quantitation of protein was performed using Image Lab software (Bio-Rad).
Fatty acid uptake assay
Preparation
Expand and grow iPSC-beige and whitened iPSC-beige adipocytes to full maturity in a 96 well plate (day 12) (see Generation of beige adipocytes above). To whiten iPSC-beige adipocytes for this assay, induce iPSC-MSCs for three days in the induction cocktail as described in the differentiation protocol (see Generation of beige adipocytes above). Following three days of induction, replace beige induction medium with DMEM containing 10% FBS and insulin (1.7 μM). Change 100% of medium every 3 days until full maturity (day 12).
On day 12, replace culture medium with DMEM containing 10% FBS. Change 100% of medium every 2 days for 6 days.
Following 6 days of rest, remove culture medium and replace with 90 μl of serum free DMEM containing 0.2% BSA. Incubate the plate at 37°C in a 5% CO2 incubator for 1 hour.
After 1 hour, add CL316,243 (1 μM) to designated wells. Allow cells to incubate for 2 hours.
Prepare loading buffer according to manufacturers’ instructions. Following 2 hours of incubation, add 100 μl of 1X loading buffer and immediately analyze with Flexstation 3 (Molecular devices).
Analyze data with SoftMax Pro 7 software continuously for two hours. Set software to record points in 30 s intervals.
QUANTIFICATION AND STATISTICAL ANALYSIS
P values were calculated in Microsoft Excel and derived using a two-tailed homo- or heteroscedastic Student’s t test, which can be found in the individual figure panels and figure legends. In the case of multiple comparisons, Bonferoni correction was used to adjust P values after one-way ANOVA analysis. Significance was defined as p < 0.05. Cell culture experiments were performed with an n ≥ 3 and are indicated in the figure legends in the case of immunofluorescence quantitations. Error bars in graphs are defined in the figure legends and represent the mean ± SD (standard deviation). The mean ± SEM (standard error of mean), which reflects the variability of the mean values, was used in cases where experimental readings were repeated a large number of times (ex. Fatty Acid Uptake assay).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human anti-TRA-1-60-Vio488 live cell stain | Miltenyi | Cat#130-106-872; RRID:AB_2654228 |
| Human anti-CD105-PE | Miltenyi | Cat#130-112-163; RRID:AB_2654424 |
| Human anti-CD73-APC | Miltenyi | Cat#130-097-945; RRID:AB_2661037 |
| Human anti-CD90-APC | Miltenyi | Cat#130-097-935; RRID:AB_2660949 |
| Human anti-CD146-PE | Miltenyi | Cat#130-097-939; RRID:AB_2660768 |
| Human anti-PDGFRβ-APC | Miltenyi | Cat#130-105-322; RRID:AB_2655084 |
| Phospho-Akt (Ser473) (D9E) Rabbit mAb | Cell Signaling Technology | Cat#4060S; RRID:AB_2315049 |
| Human and mouse anti-AN2-PE | Miltenyi | Cat#130-100-468; RRID:AB_2651231 |
| Human anti-MSCA-1-APC | Miltenyi | Cat#130-099-650; RRID:AB_2660803 |
| Human anti-CD140a-APC | Miltenyi | Cat#130-115-338; RRID:AB_2727009 |
| Mouse TGF-beta RI/ALK-5 APC-conjugated Antibody | R&D Systems | Cat#FAB5871A; RRID:AB_10890557 |
| Human TGF-beta RII APC-conjugated Antibody | R&D Systems | Cat#FAB2411A; RRID:AB_10990409 |
| Anti-FOXF1 antibody | Abcam | Cat#ab168383 |
| Anti-alpha smooth muscle Actin antibody | Abcam | Cat#ab5694; RRID:AB_2223021 |
| Anti-SM22 alpha antibody | Abcam | Cat#ab14106; RRID:AB_443021 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7074; RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat#7076; RRID:AB_330924 |
| COX IV (3E11) Rabbit mAb | Cell Signaling Technology | Cat#4850; RRID:AB_2085424 |
| Human/Mouse UCP1 Antibody | R&D Systems | Cat#MAB6158; RRID:AB_10572490 |
| PLIN-1 (D418) Antibody | Cell Signaling Technology | Cat#3470; RRID:AB_2167268 |
| Human/Mouse EBF-2 Antibody | R&D Systems | Cat#AF7006; RRID:AB_10972102 |
| β-Tubulin Antibody | Cell Signaling Technology | Cat#2146; RRID:AB_2210545 |
| β-Actin (8H10D10) Mouse mAb | Cell Signaling Technology | Cat#3700; RRID:AB_2242334 |
| Phospho-Smad2 (Ser465/467) (138D4) Rabbit mAb | Cell Signaling Technology | Cat#3108; RRID:AB_490941 |
| Chemicals, Peptides and Recombinant Proteins | ||
| RPMI 1640 with L-glutamine | Corning | Cat#10-040-CV |
| Matrigel Matrix hESC-Qualified | Corning | Cat#354277 |
| Phosphate Buffered Saline (1X) without calcium and magnesium | Lonza | Cat#BE17-516F |
| NutriStem hPSC XF Medium for Human iPS and ES cells | Stemgent | Cat#01-0005 |
| ReLeSR passaging reagent | StemCell Technologies, Inc. | Cat#05872 |
| Cell wash buffer – PBS containing 2mM EDTA and 0.5% BSA | Miltenyi | Cat#130-091-221 |
| TrypLE Select Enzyme (1X) | Thermo Fisher | Cat#12563-029 |
| InSolution Y-27632 in DMSO | Calbiochem | Cat#688002 |
| STEMdiff Mesoderm Induction Medium | StemCell Technologies, Inc. | Cat#05221 |
| MesenCult-ACF Plus Medium Kit | StemCell Technologies, Inc. | Cat#05445 |
| ACF enzymatic dissociation solution | StemCell Technologies, Inc. | Cat#05427 |
| ACF enzymatic inhibition solution | StemCell Technologies, Inc. | Cat#05428 |
| MesenCult-ACF attachment substrate | StemCell Technologies, Inc. | Cat#07130 |
| Dulbecco’s Modified Eagle’s Medium | ATCC | Cat#30-2002 |
| Fetal Bovine Serum | ATCC | Cat#30-2020 |
| SB | Sigma Aldrich | Cat#616461 |
| Recombinant Human IL-4 | Peprotech | Cat#200-04 |
| 3,3′,5-Triiodo-L-thyronine | Sigma Aldrich | Cat#T2877 |
| Rosiglitazone | Sigma Aldrich | Cat#557366-10MG-M |
| 3-Isobutyl-1-methylxanthine | Sigma Aldrich | Cat#I5879-250MG |
| Dexamethasone | Sigma Aldrich | Cat#D4902-100MG |
| Indomethacin | Sigma Aldrich | Cat#17378-10G |
| EGM-2 Endothelial Cell Growth Medium-2 BulletKit | Lonza | Cat#CC-3162 |
| OsteoLife Complete Osteogenesis Medium | Lifeline Cell Technology | Cat#LM-0023 |
| ChondroLife Complete Chondrogenesis | Lifeline Cell Technology | Cat#LM-0022 |
| Insulin solution human | Sigma Aldrich | Cat#19278-5ML |
| Adipose Tissue Dissociation Kit | Miltenyi | Cat#130-105-808 |
| L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate | Sigma Aldrich | Cat#A8960-5G |
| HEPES solution, 1M | Sigma Aldrich | Cat#SRE0065 |
| DMEM, no glucose | GIBCO | Cat#11966025 |
| DMEM low glucose | GIBCO | Cat#11885092 |
| RIPA buffer | Sigma Aldrich | Cat#R0278 |
| Halt Protease and Phosphatase Inhibitor Cocktail (100X) | Thermo Fisher | Cat#78440 |
| Paraformaldehyde | Sigma Aldrich | Cat#P6148 |
| Triton X-100 | Sigma Aldrich | Cat#X100 |
| Normal Donkey Serum | Jackson Immunoresearch lab | Cat#017-000-121 |
| HBSS without Calcium, Magnesium or Phenol Red | GIBCO | Cat#14175-095 |
| Detector Block Powder | KPL | Cat#72-01-03 |
| Tris Buffer Saline with Tween 20 | Cell Signaling Technology | Cat#9997S |
| CL 316243 disodium salt | Tocris Bioscience | Cat#1499 |
| StemRNA-NM Reprogramming Kit | Stemgent | Cat# 00-0076 |
| Critical Commercial Assays | ||
| ClarityTM Western ECL Substrate | Bio-Rad | Cat#170-5060 |
| QBT Fatty Acid Uptake Assay Kit | Molecular Device | Cat#R8132 |
| Glucose Uptake-Glo Assay Kit | Promega | Cat#J1342 |
| Seahorse XF assay medium, contains 2 mM GlutaMAX, 1 L | Agilent | Cat#102365-100 |
| Seahorse XF 1.0 M glucose solution | Agilent | Cat#103577-100 |
| Seahorse XF 100 mM pyruvate solution | Agilent | Cat#103578-100 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat#103015-100 |
| BioRad Protein assay reagent | Bio-Rad | Cat#500-0114 |
| BODIPY 493/503 | Thermo Fisher | Cat#D3922 |
| qScript cDNA SuperMix | QuantaBio | Cat#95048 |
| iQ SYBR Green Supermix | Bio-Rad | Cat#1708880 |
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| JC-1 Dye | Thermo Fisher | Cat#T3168 |
| Qproteome Mammalian Protein Prep Kit | QIAGEN | Cat#37901 |
| ProteoExtract digestion kit | Calbiochem | Cat#650212 |
| Pierce BCA protein assay kit | Thermo Fisher | Cat#23227 |
| Experimental Models: Cell Lines | ||
| Human: HWPc (subcutaneous) | PromoCell | Cat#C-12730 |
| Human: HprAD (subcutaneous) | Lonza | Cat#PT-5020 |
| Human: D-HprAD (subcutaneous) | Lonza | Cat#PT-5022 |
| Human: DOMM050211 (omental) | Zenbio | Cat#OPD-F-3 |
| Human: DSQM041007 (subcutaneous) | Zenbio | Cat#SPD-F-3 |
| Human: Fetal brown adipose tissue (interscapular) | In house | N/A |
| Human: Episomal iPSC Line (skin fibroblast) | Applied StemCell | Cat#ASE-9202 |
| Human: Episomal iPSC Line (CD34+ cord blood) | Thermo Fisher | Cat#A18945 |
| Human: Urine-derived cells | MMC BioBank | Cat#R16-0809 |
| Oligonucleotides | ||
| See Table S1 for sequences | Integrated DNA Technologies | Custom designed |
| Software and Algorithms | ||
| Excel 2016 | Microsoft | N/A |
| MACSQuantify Software | Miltenyi | Cat#130-094-556 |
| Bio-Rad CFX Manager Software | Bio-Rad | Cat#1845000 |
| Leica LAS X Software | Leica | https://www.leica-microsystems.com/products/microscope-software/details/product/leica-las-x-ls/ |
| Seahorse Wave Desktop | Agilent | https://www.agilent.com/en/products/cell-analysis/software-download-for-wave-desktop |
| ProteinPulot software | Sciex | https://sciex.com/products/software/proteinpilot-software |
| PeakView software | Sciex | https://sciex.com/products/software/peakview-software |
| MarkerView software | Sciex | https://sciex.com/products/software/markerview-software |
| ImageJ software | NIH | https://imagej.nih.gov/ij/download.html |
| Image Lab software | Bio-Rad | http://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| SoftMax Pro 7 Software | Molecular Devices | Cat#SMP7 PROF |
| Other | ||
| 6 well culture plate | Corning | Cat#3516 |
| 24 well culture plate | Corning | Cat#3524 |
| 96 well culture plate | Corning | Cat#3595 |
| CO2 Incubator | Nuaire | Cat#NU-5510 |
| 15 ml PP centrifuge tubes | Corning | Cat#430052 |
| 100×20 mm TC dish | CytoOne | Cat#CC7682-3394 |
| 5 ml Polystyrene round-bottom tube | Corning | Cat#352054 |
| MACS SmartStrainers (100 μm) | Miltenyi | Cat#130-098-463 |
| Microcentrifuge Tubes (1.7 ml) | National Scientific Supply Co | Cat#CA1700-BP |
| Seahorse XF24 V7 PS Cell Culture Microplates | Agilent | Cat#100777-004 |
| PVDF membrane | Bio-Rad | Cat#1704157 |
| CriterionTM TGX gels | Bio-Rad | Cat#5678123 |
Highlights.
Human beige adipocytes are generated from iPSC-derived FOXF1+ mesoderm
iPSC-derived beige adipocytes express UCP1 and exhibit uncoupled respiration
Reprogramming adipocyte precursors from diabetic patients improves beige adipogenesis
iPSC-derived beige adipocytes secrete factors that improve insulin sensitivity
ACKNOWLEDGMENTS
This work was supported by NIH Centers of Biomedical Research Excellence (COBRE) award P20GM121301 (to A.C.B., L.L., and C.J.R.). The project used the services of the Molecular Phenotyping and Progenitor Cell Analysis Core Facility funded by the NIH COBRE award P20GM106391 (R. Friesel, principal investigator [PI]), and the Physiology, Proteomics and Lipidomics, and Histopathology and Histomorphometry core facilities, funded by NIH COBRE awards P20GM106391 and P30GM103392 (D. St. Germain, PI) and P20GM121301 (L.L., PI). Additional support for our core facilities was provided by NIH award U54GM115516 (C.J.R., PI). A Pilot Project awarded under P30GM106391 to Dr. Robert Friesel provided additional support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Methods, eight figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.037.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahfeldt T, Schinzel RT, Lee YK, Hendrickson D, Kaplan A, Lum DH, Camahort R, Xia F, Shay J, Rhee EP, et al. (2012). Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol 14, 209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi S, Hallen S, Huang L, Svensson PA, Momo RA, Wallin S, Carlsson EK, Forslöw A, Seale P, and Peng XR (2015). Thermogenic activity of UCP1 in human white fat-derived beige adipocytes. Mol. Endocrinol 29, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, Stenesen D, Zeve D, and Graff JM (2013). The developmental origins of adipose tissue. Development 140, 3939–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Carey AL, Vorlander C, Reddy-Luthmoodoo M, Natoli AK, Formosa MF, Bertovic DA, Anderson MJ, Duffy SJ, and Kingwell BA (2014). Reduced UCP-1 content in in vitro differentiated beige/brite adipocytes derived from preadipocytes of human subcutaneous white adipose tissues in obesity. PLoS One 9, e91997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau YY, Bandiera R, Serrels A, Martínez-Estrada OM, Qing W, Lee M, Slight J, Thornburn A, Berry R, McHaffie S, et al. (2014). Visceral and subcutaneous fat have different origins and evidence supports a mesothelial source. Nat. Cell Biol 16, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KJ, Chatzigeorgiou A, Economopoulou M, Garcia-Martin R, Alexaki VI, Mitroulis I, Nati M, Gebler J, Ziemssen T, Goelz SE, et al. (2017). A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat. Immunol 18, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin B, Cinti S, Morroni M, Raimbault S, Ricquier D, Pénicaud L, and Casteilla L (1992). Occurrence of brown adipocytes in rat white adipose tissue: molecular and morphological characterization. J. Cell Sci 103, 931–942. [DOI] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. (2008). A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 3, 301–313 [DOI] [PubMed] [Google Scholar]
- Cristancho AG, and Lazar MA (2011). Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol 12, 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. (2009). Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med 360, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elía E, Kessler SH, Kahn PA, English J, Chatman K, Trauger SA, Doria A, and Kolodny GM (2015). Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 21, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estève D, Boulet N, Volat F, Zakaroff-Girard A, Ledoux S, Coupaye M, Decaunes P, Belles C, Gaits-Iacovoni F, Iacovoni JS, et al. (2015). Human white and brite adipogenesis is supported by MSCA1 and is impaired by immune cells. Stem Cells 33, 1277–1291. [DOI] [PubMed] [Google Scholar]
- Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, Kalinovich AV, Petrovic N, Wolf Y, Clemmensen C, et al. (2017). Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med 23, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graja A, and Schulz TJ (2015). Mechanisms of aging-related impairment of brown adipocyte development and function. Gerontology 61, 211–217. [DOI] [PubMed] [Google Scholar]
- Guénantin AC, Briand N, Capel E, Dumont F, Morichon R, Provost C, Stillitano F, Jeziorowska D, Siffroi JP, Hajjar RJ, et al. (2017). Functional human beige adipocytes from induced pluripotent stem cells. Diabetes 66, 1470–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra C, Koza RA, Yamashita H, Walsh K, and Kozak LP (1998). Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J. Clin. Invest 102, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AL, and Dani C (2014). Human induced pluripotent stem cells: a new source for brown and white adipocytes. World J. Stem Cells 6, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner AL, Contet J, Ravaud C, Yao X, Villageois P, Suknuntha K, Annab K, Peraldi P, Binetruy B, Slukvin II, et al. (2016). Brown-like adipose progenitors derived from human induced pluripotent stem cells: identification of critical pathways governing their adipogenic capacity. Sci. Rep 6, 32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, and Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat. Med 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- Hassan M, Latif N, and Yacoub M (2012). Adipose tissue: friend or foe? Nat. Rev. Cardiol 9, 689–702. [DOI] [PubMed] [Google Scholar]
- Lee YH, Petkova AP, Mottillo EP, and Granneman JG (2012). In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 15, 480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, Yun K, Locksley RM, and Chawla A (2015). Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidell ME, Betz MJ, and Enerbäck S (2014). Two types of brown adipose tissue in humans. Adipocyte 3, 63–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng Z, Zhu X, Meng M, Li L, Shen Y, Chi Q, Wang D, Zhang Z, Li C, et al. (2013). Brown adipose tissue transplantation improves whole-body energy metabolism. Cell Res. 23, 851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cervantes C, and Liu F (2017). Common and distinct regulation of human and mouse brown and beige adipose tissues: a promising therapeutic target for obesity. Protein Cell 8, 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano F, Vargas D, Gómez Á, and Torrado A (2017). Human ADMC-derived adipocyte thermogenic capacity is regulated by IL-4 receptor. Stem Cells Int. 2017, 2767916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, and Saltiel AR (2011). Inflammatory links between obesity and metabolic disease. J. Clin. Invest 121, 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlapuu M, Enerbäck S, and Carlsson P (2001a). Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128, 2397–2406. [DOI] [PubMed] [Google Scholar]
- Mahlapuu M, Ormestad M, Enerbäck S, and Carlsson P (2001b).The forkhead transcription factor Foxf1 is required for differentiation of extra-embryonic and lateral plate mesoderm. Development 128, 155–166. [DOI] [PubMed] [Google Scholar]
- Malik VS, Willett WC, and Hu FB (2013). Global obesity: trends, risk factors and policy implications. Nat. Rev. Endocrinol 9, 13–27. [DOI] [PubMed] [Google Scholar]
- Mohsen-Kanson T, Hafner AL, Wdziekonski B, Takashima Y, Villageois P, Carrière A, Svensson M, Bagnis C, Chignon-Sicard B, Svensson PA, et al. (2014). Differentiation of human induced pluripotent stem cells into brown and white adipocytes: role of Pax3. Stem Cells 32, 1459–1467. [DOI] [PubMed] [Google Scholar]
- Mugford JW, Sipilä P, McMahon JA, and McMahon AP (2008). Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev. Biol 324, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Yoneshiro T, Nakahara M, Suzuki S, Saeki K, Hasegawa M, Kawai Y, Akutsu H, Umezawa A, Yasuda K, et al. (2012). Production of functional classical brown adipocytes from human pluripotent stem cells using specific hemopoietin cocktail without gene transfer. Cell Metab. 16, 394–406. [DOI] [PubMed] [Google Scholar]
- Ochner CN, Tsai AG, Kushner RF, and Wadden TA (2015). Treating obesity seriously: when recommendations for lifestyle change confront biological adaptations. Lancet Diabetes Endocrinol. 3, 232–234. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, Palmiter RD, and Chawla A (2014). Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157, 1292–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Sonder AM, Lumsden A, and Korsmeyer SJ (1995). Development expression of Hox11 and specification of splenic cell fate. Am. J. Pathol 146, 1089–1101. [PMC free article] [PubMed] [Google Scholar]
- Roh HC, Tsai LTY, Shao M, Tenen D, Shen Y, Kumari M, Lyubetskaya A, Jacobs C, Dawes B, Gupta RK, et al. (2018). Warming induces significant reprogramming of beige, but not brown, adipocyte cellular identity. Cell Metab. 27, 1121–1137.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Gurmaches J, Hung CM, and Guertin DA (2016). Emerging complexities in adipocyte origins and identity. Trends Cell Biol. 26, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, and Graff JM (2008). White fat progenitor cells reside in the adipose vasculature. Science 322, 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Wennmalm K, Larsson O, Walden TB, Lassmann T, Petrovic N, Hamilton DL, Gimeno RE, Wahlestedt C, Baar K, et al. (2007). Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 104, 4401–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribioli C, Frasch M, and Lufkin T (1997). Bapx1: an evolutionary conserved homologue of the Drosophila bagpipe homeobox gene is expressed in splanchnic mesoderm and the embryonic skeleton. Mech. Dev 65, 145–162. [DOI] [PubMed] [Google Scholar]
- van den Berg SM, van Dam AD, Rensen PC, de Winther MP, and Lutgens E (2017). Immune modulation of brown(ing) adipose tissue in obesity. Endocr. Rev 38, 46–68. [DOI] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, and Gupta RK (2016). Pdgfrβ+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab. 23, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, and Seale P (2016). Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol 17, 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Zhao XY, and Lin JD (2015a). The brown fat secretome: metabolic functions beyond thermogenesis. Trends Endocrinol. Metab 26, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang M, Xu M, Gu W, Xi Y, Qi L, Li B, and Wang W (2015b). Brown adipose tissue activation is inversely related to central obesity and metabolic parameters in adult human. PLoS One 10, e0123795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav H, and Rane SG (2012). TGF-β/Smad3 signaling regulates brown adipocyte induction in white adipose tissue. Front. Endocrinol. (Lausanne) 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.