

Abstract
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a rare multisystem autosomal recessive disorder. The disease is clinically heterogeneous with gastrointestinal symptoms of intestinal dysmotility and cachexia as well as neurological symptoms of ophthalmoplegia, neuropathy, sensorineural hearing impairment, and diffuse leukoencephalopathy being most prominent. MNGIE is caused by mutations in TYMP , a gene that encodes thymidine phosphorylase (TP)—a cytosolic enzyme. Mutations in TYMP lead to very low TP catalytic activity, resulting in dramatically increased thymidine and deoxyuridine in plasma. We describe the clinical, biochemical, and neuroimaging findings of three boys with MNGIE from a Pakistani family with a novel homozygous mutation, c.798_801dupCGCG p. (Ala268Argfs*?), in exon 7 of TYMP .
Keywords: mitochondrial neurogastrointestinal encephalomyopathy, Pakistani patients, novel mutation, magnetic resonance imaging of brain, TYMP
Introduction
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) (OMIM no. 603041) is a rare, autosomal recessive disease caused by homozygous/compound heterozygous mutations in TYMP , a gene located on chromosome 22q13.33 that encodes thymidine phosphorylase (TP). This enzyme catalyzes the breakdown of the nucleosides thymidine and deoxyuridine to thymine and uridine, respectively. 1 The accumulation of these nucleosides has been postulated to cause an imbalanced mitochondrial nucleotide pool that, in turn, causes mitochondrial DNA instability. 1
Clinically MNGIE is characterized by progressive external ophthalmoplegia, severe gastrointestinal dysmotility, cachexia, peripheral neuropathy, sensorineural hearing impairment, diffuse leukoencephalopathy, and evidence of mitochondrial dysfunction in the form of histological, biochemical, and genetic abnormalities of the mitochondria. 2 Increased concentrations of thymidine (> 3 µmol/L) and deoxyuridine (> 5 µmol/L) in plasma or a decrease in buffy coat TP activity to < 8% relative to controls is considered sufficient to diagnose MNGIE. 3 About 60% of patients manifest symptoms before 20 years of age, though the initial symptoms can occur between the first and fifth decades. 4 Clinical phenotypes indistinguishable from MNGIE have been reported in patients with mutations in the RRM2B gene. 5 Recently, POLG1 mutations were identified in three patients with a MNGIE-like phenotype without leukoencephalopathy. 6
We report the clinical, biochemical, and neuroimaging findings in three brothers with MNGIE due to a novel homozygous mutation c.798_801dupCGCG p. (Ala268Argfs*?) in exon 7 of TYMP .
Case Presentations
Patient 1
The index patient was a 17-year-old adolescent boy referred for difficulty in walking and impaired vision. He was the second born child of first-cousin parents after an uneventful, full-term pregnancy. No concerns were noted by the parents until 12 years of age at which time he began to have poor attentiveness in school. Bilateral sensorineural hearing loss (SNHL) was detected on brainstem evoked response audiometry (BERA). Two years later, he developed vomiting and diarrhea leading to cachexia. Vision impairment and difficulty in walking was noted 4 and 2 months prior to his presentation. On examination, he was 166 cm tall, weighed 25 kg, and thus had a body mass index (BMI) of 9.07 kg/m 2 . Neurological examination revealed ophthalmoparesis without ptosis, muscle wasting, and weakness more marked in distal muscles. Deep tendon reflexes were depressed. The patient demonstrated stepping gait. Severe axonal peripheral neuropathy was detected on nerve conduction studies (NCS). A T2-weighted magnetic resonance imaging (MRI) of the brain showed diffuse high signal intensity of the cerebral white matter. The corpus callosum, internal capsule, and optic radiation were spared ( Fig. 1 ). Based on the clinical picture, neuroradiological and neurophysiological findings, MNGIE was suspected and plasma pyrimidine analysis was performed using Agilent 1200 high-performance liquid chromatography (HPLC), equipped with a reverse phase column and coupled with a multi-wavelength diode array detector. It showed a markedly elevated plasma thymidine of 14 µmol/L and deoxyuridine of 25 µmol/L, supporting the diagnosis of MNGIE. TYMP sequencing was performed on genomic DNA extracted from peripheral blood using a QIA amp DNA mini kit (Qiagen, Mississauga, Ontario, Canada). All nine coding exons and flanking introns were amplified by specific primers. The polymerase chain reaction (PCR) products were purified using a QIA quick PCR purification kit (Qiagen, Mississauga, Ontario, Canada). Bidirectional sequencing was performed using BigDye Terminator Cycle Sequencing v3.1 (Applied Biosystems, Foster City, California, United States) followed by separation on a fluorescent Genetic Analyzer ABI 3500 (Applied Biosystems, Foster City, California, United States). DNA sequences were analyzed for any changes in comparison to the published sequence of TYMP (GeneBank NM_001113755) using SeqScape software v3.0. The mutation found was not reported in the Human Genome Mutation Database (HGMD). MutationTaster was used to assess pathogenicity ( http://www.mutationtaster.org/ ), and prediction of protein dysfunction was performed by using PredictProtein (PP) ( https://predictprotein.org/ ).
Fig. 1.
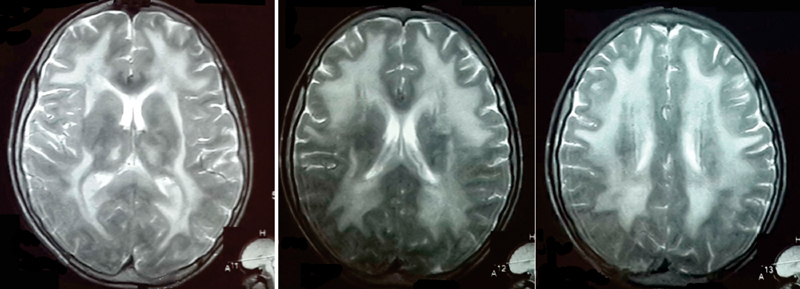
MRI brain T2WI of patient 1 showing diffuse high-intensity signals in white matter.
A novel homozygous frameshift mutation c.798_801dupCGCG p. (Ala268Argfs*?) in exon 7 of TYMP was detected ( Fig. 2 ). Both parents were shown to be heterozygous carriers. In silico analysis by MutationTaster predicted the p.(Ala268Argfs*?) mutation to be disease causing. This mutation causes a change in the amino acid sequence, resulting in an elongated polypeptide chain ( Fig. 3A ). Using HomoloGene ( https://www.ncbi.nlm.nih.gov/homologene ), we identified changes in many amino acid residues in highly conserved regions ( Fig. 3B ). The PredictProtein (PP) predicted a change in a protein-protein binding site and protein structure ( Fig. 4 ).
Fig. 2.
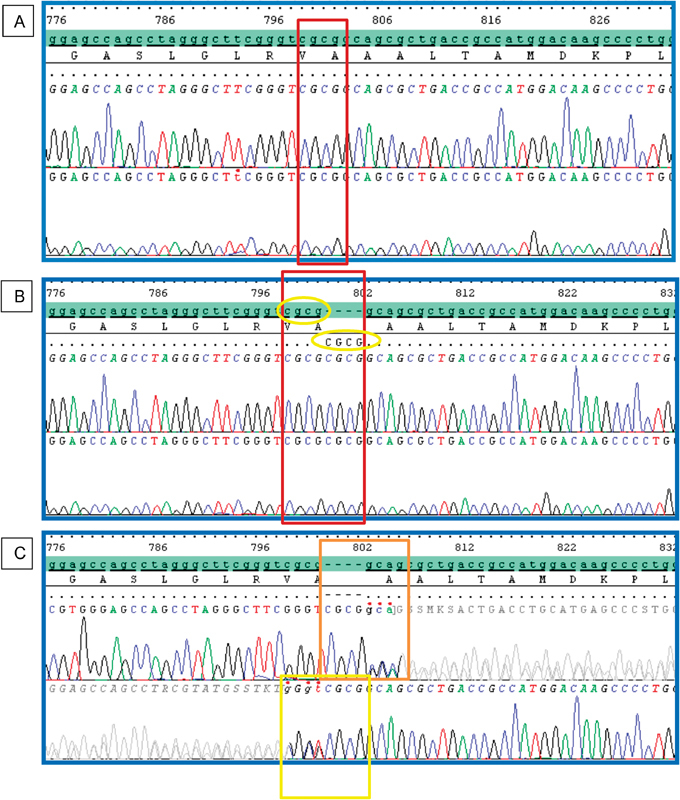
Electropherograms of TYMP exon 7 sequence. ( A ) Normal control sequence, a wild-type CGCG sequence is shown in a red box. ( B ) Patient with homozygous c.798_801dupCGCG. ( C ) Heterozygous duplication of CGCG detected at the same position in both parents as shown in bidirectional sequencing of forward (upper) and reverse (lower).
Fig. 3.

( A ) The reference protein (above) and the mutated protein (below), in which the amino acids that differ are highlighted in red and bold. Normal stop codon is shifted out of frame in mutated protein with additional tail of new amino acids of unknown length, producing an elongated polypeptide chain. ( B ) The amino acid conservation between different organisms. Highly conserved amino acids (highlighted in gray) are replaced by other amino acids in our patients.
Fig. 4.
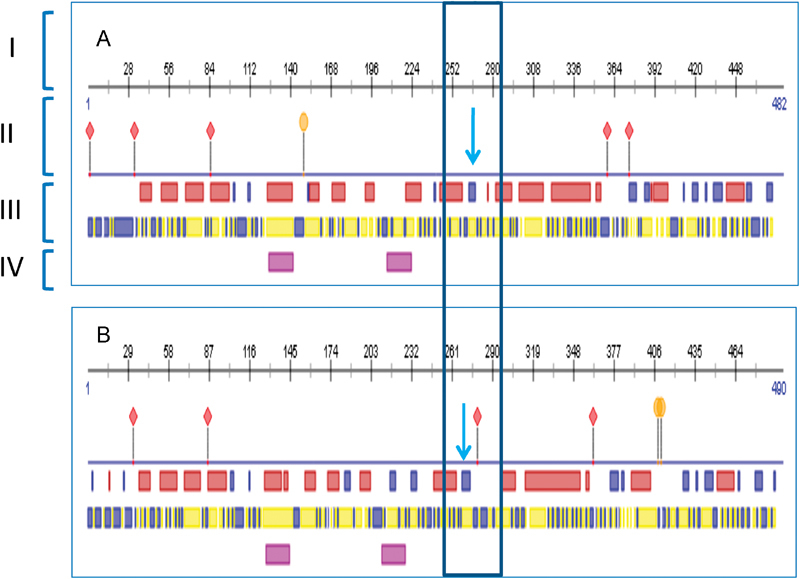
A TYMP protein structure prediction by PredictProtein (PP) showed differences in ( A ) normal protein sequence and ( B ) mutated protein sequence. A mutation of p. (Ala268Argfs*?) is located in the region marked by a blue arrow. The mutation was caused a frameshift that made a protein a bit longer (490 amino acids in B , instead of a normal protein of 482 amino acids in A ). The mutation was also predicted to change the TYMP protein structure (III) and created a new protein-binding region as highlighted in a box. The binding sites have also changed markedly. Note: I: Protein numbering, II: Binding Sites, III: Protein Structure (Helix (red box)/strand [blue box]), IV: Helical Transmembrane Region, Protein binding region, Poly-nucleotide binding region.
Patient 2
The 20-year-old elder brother of patient 1 was born after a full-term, uneventful pregnancy. His parents did not notice any major concerns until adolescence, when he developed frequent diarrhea and progressive weight loss. No visual impairment, hearing impairment, or gait disturbance was noted. On examination, patient 2 was 176 cm tall, weighed 27 kg, and had a BMI of 8.72 kg/m 2 . He was cachectic with normal gait, muscle tone, and reflexes with no ophthalmoparesis or ptosis.
NCS and BERA were normal. Plasma thymidine and deoxyuridine were markedly elevated at 13 µmol/L and 16 µmol/L, respectively. Homozygosity for the familial mutation c.798_801dupCGCG p.(Ala268Argfs*?) in exon 7 of TYMP confirmed the diagnosis of MNGIE.
Patient 3
The 15-year-old younger brother of patient 1 was born after a full-term, uneventful pregnancy. His parents did not notice any major concerns until he was 14 years old, when he reported frequent diarrhea. No visual impairment, hearing impairment, or gait disturbance was noted. On examination, he was 147 cm tall, weighed 25.5 kg, and had a BMI of 11.8 kg/m 2 . Neurological examination was normal except for muscle wasting. No ophthalmoparesis or ptosis was noted. BERA and NCS were normal. Plasma thymidine and deoxyuridine analysis showed mark elevation of 15µmol/L and 27 µmol/L, respectively. Homozygosity for the familial mutation c.798_801dupCGCG p.(Ala268Argfs*?) in exon 7 of TYMP gene confirmed the diagnosis of MNGIE.
Discussion
Mitochondrial neurogastrointestinal encephalomyopathy is an autosomal recessive multisystem disease, which was first described in 1976 by Okamura et al as “congenital oculoskeletal myopathy with abnormal muscle and liver mitochondria.” 7 Based on the age of onset, MNGIE is divided in two categories: early onset, before 40 years of age and late onset, after 40 years of age. 8 The natural course of MNGIE is progressive with death occurring at mean age of 37 years. Allogeneic hematopoietic stem cell transplant (HSCT) has been recommended as the treatment for individuals with MNGIE who have either a human leukocyte antigen (HLA)–identical sibling or a 10/10 allele–matched unrelated donor. 9 In the family presented here, two asymptomatic sisters and both parents were heterozygous for the familial TYMP mutation. However, none of them had an HLA match with any of the three affected males. HSCT from unrelated donors is not currently performed in Pakistan. Transient symptomatic improvement has been reported in some affected individuals with continuous ambulatory peritoneal dialysis 10 and total parenteral nutrition.
The homozygous frameshift mutation c.798_801dupCGCG p.(Ala268Argfs*?) in TYMP detected in this family causes a shift in reading frame leading to addition of amino acids to the protein and alters several highly conserved regions toward C-terminal end of the protein. As a result, an elongated polypeptide chain of unknown length is predicted because the shifted frame does not contain a new stop codon. The resulting altered COOH terminus is likely to disrupt the three-dimensional structure of the enzyme, thus reducing its stability and activity. 11
It has been reported that homozygous mutations in the TYMP gene usually result in almost complete abolition of TP activity 1 ; therefore, the c.798_801dupCGCG frameshift mutation identified here is likely to cause a loss-of-function that subsequently leads to accumulation of the excess amount of the nucleoside substrates of thymidine and deoxyuridine in plasma, as demonstrated in this family by biochemical studies.
The three individuals in this family with the same TYMP genotype manifested variable clinical severity, supporting the clinical heterogeneity of MNGIE with intra- and interfamilial variability reported in the literature. 2
Garone et al reported a cohort of 102 patients with MNGIE and found the disease most prevalent in Europeans (53%) followed by Americans and Asians with 13.7% in each group. 2 It is apparent that despite the diagnostic criteria for MNGIE being established in 1994, the diagnosis of MNGIE is often missed.
Funding Statement
Funding None.
Footnotes
Conflict of Interest None declared.
References
- 1.Hirano M, Lagier-Tourenne C, Valentino M L, Martí R, Nishigaki Y. Thymidine phosphorylase mutations cause instability of mitochondrial DNA. Gene. 2005;354:152–156. doi: 10.1016/j.gene.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 2.Garone C, Tadesse S, Hirano M.Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy Brain 2011134(Pt 11):3326–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martí R, Spinazzola A, Tadesse S, Nishino I, Nishigaki Y, Hirano M. Definitive diagnosis of mitochondrial neurogastrointestinal encephalomyopathy by biochemical assays. Clin Chem. 2004;50(01):120–124. doi: 10.1373/clinchem.2003.026179. [DOI] [PubMed] [Google Scholar]
- 4.Cardaioli E, Da Pozzo P, Malfatti E et al. A second MNGIE patient without typical mitochondrial skeletal muscle involvement. Neurol Sci. 2010;31(04):491–494. doi: 10.1007/s10072-010-0225-5. [DOI] [PubMed] [Google Scholar]
- 5.Shaibani A, Shchelochkov O A, Zhang S et al. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol. 2009;66(08):1028–1032. doi: 10.1001/archneurol.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang S, Dimberg E L, Milone M, Wong L J. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol. 2012;259(05):862–868. doi: 10.1007/s00415-011-6268-6. [DOI] [PubMed] [Google Scholar]
- 7.Okamura K, Santa T, Nagae K, Omae T. Congenital oculoskeletal myopathy with abnormal muscle and liver mitochondria. J Neurol Sci. 1976;27(01):79–91. doi: 10.1016/0022-510x(76)90236-7. [DOI] [PubMed] [Google Scholar]
- 8.Nakhro K, Chung K W, Kim S M et al. Compound mutations of PEO1 and TYMP in a progressive external ophthalmoplegia patient with incomplete mitochondrial neurogastrointestinal encephalomyopathy phenotype. Genes Genomics. 2011;33:431–437. [Google Scholar]
- 9.Halter J, Schüpbach W, Casali C et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46(03):330–337. doi: 10.1038/bmt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yavuz H, Ozel A, Christensen M et al. Treatment of mitochondrial neurogastrointestinal encephalomyopathy with dialysis. Arch Neurol. 2007;64(03):435–438. doi: 10.1001/archneur.64.3.435. [DOI] [PubMed] [Google Scholar]
- 11.Cardaioli E, Sicurelli F, Carluccio M A et al. A new thymidine phosphorylase mutation causing elongation of the protein underlies mitochondrial neurogastrointestinal encephalomyopathy. J Neurol. 2012;259(01):172–174. doi: 10.1007/s00415-011-6113-y. [DOI] [PubMed] [Google Scholar]