

Abstract
When lung cells experience hypoxia, the functional response, termed hypoxic pulmonary vasoconstriction, activates a multitude of pathways with the goal of optimizing gas exchange. While previously controversial, overwhelming evidence now suggests that increased reactive oxygen species – produced at complex III of the mitochondrial electron transport chain and released into the intermembrane space – is the cellular oxygen signal responsible for triggering hypoxic pulmonary vasoconstriction. The increased reactive oxygen species (ROS) activate many downstream targets that ultimately lead to increased intracellular ionized calcium concentration and contraction of pulmonary arterial smooth muscle cells. While the specific targets of ROS signals are not completely understood, it is clear that this signalling pathway is critical for development and for normal lung function in newborns and adults.
Keywords: hypoxia, mitochondria, redox signaling, pulmonary hypertension, pulmonary circulation
The smooth muscle cells of pulmonary arteries contract during acute hypoxia in a response called hypoxic pulmonary vasoconstriction (HPV). It is likely that this process evolved in mammals from the need to maintain fetal oxygenation during gestation. During fetal development, the lung is filled with hypoxic amniotic fluid. Gas exchange occurs in the placenta, so the blood returning to the fetal heart is relatively well oxygenated. Perfusion of the hypoxic lung with blood that has a high would result in loss of O2 from pulmonary capillary blood to the amniotic fluid. HPV augments pulmonary vascular resistance to a point where it exceeds systemic vascular resistance. This limits blood flow to the lungs and diverts most of the output of the right ventricle through the ductus arteriosus to the aorta. At birth, the initiation of lung ventilation results in a rapid increase in alveolar . The vascular cells of the distal pulmonary arteries detect the change in O2 tension and trigger relaxation of the pulmonary arteries, while the increase in in blood triggers contraction of smooth muscle cells in the ductus arteriosus. These responses enable the shift from placental to lung respiration at birth. O2 sensing by lung and vascular cells is therefore required for that transition.
In the adult lung, smooth muscle cells retain the ability to constrict in response to hypoxia. HPV can help to optimize pulmonary gas exchange by matching pulmonary perfusion to ventilation when hypoxic regions in the lung develop. Pulmonary smooth muscle cells sense a decrease in alveolar O2 tension and decrease local perfusion, thereby redirecting blood flow to other regions with higher alveolar . HPV, however, plays a minimal role in normal healthy lungs where basal pulmonary vascular tone is low. Few regions of hypoxia exist in the normal lung, such that application of pulmonary vasodilators does not alter the efficiency of alveolar ventilation–perfusion matching or arterial . HPV may play a more important role in matching pulmonary perfusion to ventilation in diseases such as asthma and acute respiratory distress syndrome where regional differences in alveolar can develop. However, HPV can also be detrimental in cases of generalized alveolar hypoxia when the HPV response can cause vasoconstriction throughout the lung, resulting in pulmonary hypertension. This can occur in healthy lungs at high altitude, as well as in diseases that produce large areas of alveolar hypoxia such as chronic obstructive pulmonary disease, sleep apnoea and pulmonary fibrosis.
Acute HPV and chronic hypoxia‐induced vascular remodelling
The hypoxic responses of pulmonary vessels have been studied in a wide variety of species and experimental conditions (Sylvester et al. 2012). It has been established that exposure of pulmonary arteries to hypoxia results in a biphasic vasoconstrictor response (Bennie et al. 1991; Leach et al. 1994, 2001; Weissmann et al. 2001, 2004; Sylvester et al. 2012). Phase 1 of the pressor response involves an early vasoconstriction corresponding to acute HPV, while phase 2 represents a subsequent sustained and progressive increase in pulmonary arterial pressure (Weissmann et al. 2001, 2004). Phase 1 of HPV generally begins within seconds of the hypoxic exposure and peaks within 5 min, while phase 2 develops gradually, reaching its sustained maximum at 30–60 min (Weissmann et al. 2001; Sylvester et al. 2012). The degree of constriction during phase 1 can be quite variable, but several studies demonstrated an increase in vascular resistance by 10–300% over basal levels (Rodman et al. 1989; Sylvester et al. 2012). Many studies have demonstrated that the amplitude of the phase 2 contraction is smaller than in phase 1, but other studies suggest that the phase 2 contraction could equal or even surpass that of phase 1 (Sylvester et al. 2012). Investigators simultaneously measuring tension and intracellular Ca2+ concentration ([Ca2+]i) have demonstrated that phase 1 of HPV is associated with an increase in [Ca2+]i, whereas the tension increase in phase 2 occurs without a further increase in [Ca2+]i (Robertson et al. 1995). These data indicate that Ca2+ sensitization is likely involved in phase 2 of HPV. Experimental evidence has shown that prolonged hypoxia can cause an elevation in pulmonary arterial pressure that persists after return to normoxia (Weissmann et al. 2001), which is important from a clinical standpoint because lung diseases associated with chronic hypoxia lead to prolonged elevation of pulmonary arterial pressures that are refractory to increases in the fraction of inspired O2. In addition to changes in Ca2+ sensitivity, chronic hypoxia leads to downregulation of K+ channel expression and upregulation of transient receptor potential canonical (TRPC) ion channel protein expression, which have been interpreted as being associated with store‐operated Ca2+ channel (SOCC) activity (Wang et al. 1997, 2006). These increases in [Ca2+]i lead to pulmonary arterial smooth muscle cell (PASMC) contraction, proliferation and pulmonary vascular remodelling. Chronic hypoxia and sustained HPV can therefore lead to increased pulmonary vascular resistance, pulmonary vascular remodelling and increased right heart afterload.
Experimental studies using isolated PASMC and isolated pulmonary vascular rings denuded of endothelium have demonstrated that the phase 1 constriction response is intrinsic to smooth muscle cells (Murray et al. 1990; Demiryurek et al. 1991; Madden et al. 1992; Sham et al. 2000). While these studies indicate a lack of need for circulating mediators, studies using pulmonary vascular rings with intact endothelium or precision‐cut lung slices demonstrate an enhanced response to hypoxia (Murray et al. 1990; Leach et al. 1994; Paddenberg et al. 2006), which suggests that phase 2 hypoxic contraction requires an intact endothelium (Sylvester et al. 2012). Studies in several species have demonstrated that endothelin‐1 (ET‐1) released from endothelial cells enhances the hypoxic reactivity of PASMCs (Oparil et al. 1995; Liu et al. 2001). Indeed, treatment with ET‐1 antagonists inhibits HPV in intact animals suggesting that release of ET‐1 may be required to achieve a full HPV response (Oparil et al. 1995; Liu et al. 2001). Additionally, production of nitric oxide (NO), which promotes vasodilatation, by endothelial cells is decreased in hypoxia (Le Cras & McMurtry, 2001). Together, these data suggest that pulmonary arterial endothelial cells modulate the response of PASMC via the release of vasoactive factors to complement the overall functional response to changes in O2 tension. Indeed, hypoxia also increased endothelial monolayer permeability allowing vasoactive factors to reach PASMC (Partridge, 1995; Yang et al. 2016a). Thus, O2 sensing mechanisms in multiple vascular cell types contribute to the activation of diverse responses that collectively define the responses to acute and chronic hypoxia (Gao et al. 2016).
Characteristics of an O2 sensor
Since its discovery by von Euler and Liljestrand in 1946, HPV has been studied by many investigators and its relevance to health and disease has been defined. The exact mechanism by which cells in the lung detect a decrease in and translate that into a biological response is still not fully established. Identifying how lung cells sense changes in O2 will enhance our understanding of the physiology and pathophysiology of pulmonary circulation and offer insight into how other tissues sense and respond to changes in . In light of this, the use of HPV as a model provides a unique opportunity to study and identify the underlying O2 sensor(s).
Complex organisms cannot rely on diffusion alone to supply cells with molecular O2 and nutrients. Evolution from single celled organism to metazoan species was associated with the development of elegant and elaborate systems to ensure adequate delivery of O2 to meet metabolic demand. Sudden changes in metabolic demand in multicellular organisms require a dynamic system with the ability to respond rapidly (López‐Barneo et al. 2001). Responses to hypoxia can be generalized into two categories. Post‐translational responses limit metabolic energy consuming processes, such as protein synthesis, and enhance glycolysis by translocating glucose transport proteins to the cell membrane. Transcriptional responses involve de novo expression of glycolytic enzymes, glucose transporters and genes that enhance the ability of cells to maintain ATP production in the absence of O2. De novo gene expression requires transcription, translation and often post‐translational modification. Completion of these processes takes a significant amount of time. In general, a response to hypoxia requiring cell proliferation or remodelling is not likely to protect the organism from acute hypoxic stress that can develop in a matter of seconds. Additionally, post‐translational modifications that allow for rapid translocation of glucose transporters to the cell surface are not an optimal response to chronic hypoxia where glucose levels may become depleted (Zhang et al. 2015). Therefore, O2 sensors must be able to respond accordingly to both acute and chronic hypoxic stress in order to coordinate responses with differing time constraints (Schumacker, 2014). Indeed, the ability of these sensors to detect early initial decreases in and to trigger adaptive responses quickly is essential to prevent cellular injury and to lessen the decline in .
Mitochondria as O2 sensors
Mitochondria are responsible for the vast majority of the O2 consumed by the cell. Therefore, they represent an appealing site for an O2 sensor. Given that a major role of mitochondria is the production of ATP, it is conceivable that decreased ATP production due to decreased oxidative phosphorylation may be the signal that initiates the hypoxic response. However, studies in isolated cells as well as intact lungs have demonstrated that hypoxia responses are initiated at values ranging from 25 to 75 mmHg (Murray et al. 1990; Madden et al. 1992). However, mitochondrial respiration can continue normally even at very low O2 levels, and only begins to become O2 supply‐limited at values less than ∼7 mmHg (Chandel et al. 1997). Given that mitochondrial respiration can continue normally at such low O2 levels without a decrease in ATP production, it seems unlikely that ATP concentration could trigger cellular responses to hypoxia (Buescher et al. 1991). If decreases in ATP are not the trigger for hypoxic responses, another possibility is that O2‐dependent production of reactive oxygen species (ROS) by mitochondria is the initiating signal for the hypoxic response.
ROS as signalling molecules
Mammalian cells have developed many signalling systems to relay information including post‐translational modifications such as phosphorylation, acetylation and ubiquitinylation, as well as signalling systems regulated by Ca2+. ROS generated by mitochondria have historically been seen as toxic by‐products of the electron transport chain (ETC) that cause cell damage and injury, such as in ischaemia–reperfusion injury or other disorders. However, significant evidence points to the role of low levels of ROS as signalling molecules that play important physiological roles in a variety of biological processes, such as O2 sensing (McCord, 1985; Cross et al. 1987; Brigelius‐Flohe & Flohe, 2011).
The mitochondrial ETC is composed of four multiprotein complexes in the mitochondrial inner membrane. It generates an electrochemical gradient across the membrane which is used by the ATP synthase to generate ATP during oxidative phosphorylation. Complexes I and II of the ETC each pass a pair of electrons to the electron carrier ubiquinone, which then becomes ubiquinol. The two electrons are sequentially removed from ubiquinol at the Qo ubiquinol binding site in complex III. The Riske iron–sulfur protein (RISP) in complex III receives the first electron from ubiquinol and passes the electron on to cytochrome c 1, to cytochrome c and finally to complex IV where it is transferred to molecular O2 to form H2O. Removal of the first electron from ubiquinol results in the formation of the transient free radical ubisemiquinone at the Qo site. The second electron is then rapidly removed by the b cytochromes and ubiquinone returns to the membrane pool.
Mitochondria can generate ROS at complexes I, II and III (Fig. 1; Misra & Fridovich, 1972; Turrens & Boveris, 1980; Turrens et al. 1985; Garcia‐Ruiz et al. 1995; Genova et al. 2001; Leach et al. 2001). In complexes I and II, escape of electrons from flavins and iron–sulfur groups results in superoxide formation and release into the mitochondrial matrix. In complex III, an increased lifetime of ubisemiquinone due to delayed removal of the last electron represents a possible mechanism for increased ROS generation during hypoxia. Oxidants formed at the Qo site of complex III are released into the intermembrane space, due to the high electrical field strength within the inner membrane (Sabharwal et al. 2013). The principal ROS species produced in cells is superoxide (O2 −), which is dismuted to hydrogen peroxide (H2O2) either enzymatically or spontaneously. Superoxide dismutases (SODs) combine two superoxides to produce H2O2 in the cytosol or intermembrane space (via Cu–Zu SOD) and in the mitochondrial matrix (via MnSOD) (Murphy, 2009; Murphy et al. 2011). H2O2 is an important cellular signalling molecule that interacts with proteins by reversibly oxidizing thiol groups on cysteine or methionine residues resulting in altered protein structure and function (Brigelius‐Flohe & Flohe, 2011). Increases in ROS production are thought to be the triggering mechanism of HPV (Rounds & McMurtry, 1981; Weir et al. 1983; Leach et al. 2001; Waypa et al. 2001, 2006; Liu et al. 2003; Wang et al. 2007).
Figure 1. Signalling in hypoxic pulmonary vasoconstriction.
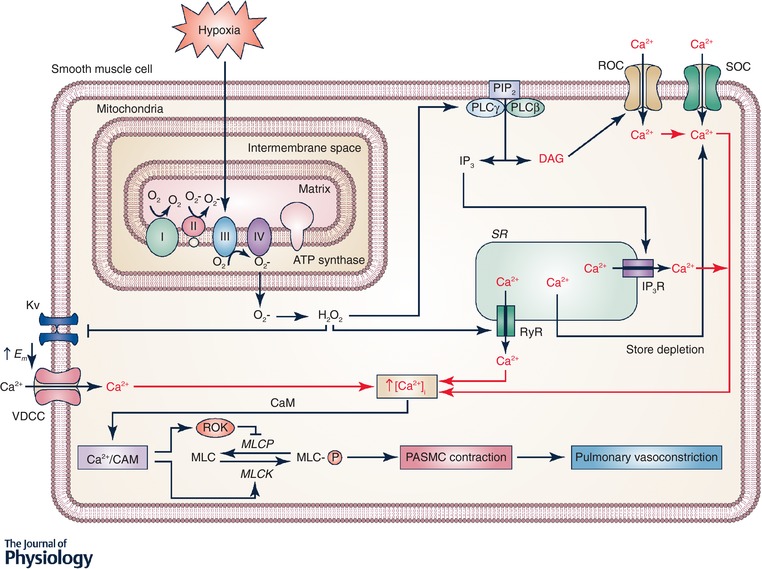
Hypoxia increases production at mitochondrial complex III of reactive oxygen species (ROS), which are released into the mitochondrial intermembrane space. ROS signals (superoxide and H2O2) can move from the intermembrane space to the cytosol where the superoxide is converted to H2O2. H2O2 can then activate many downstream signalling pathways. H2O2 can activate phospholipase C (PLC) leading to production of inositol 1,4,5‐trisphosphate (IP3) and diacylglycerol (DAG) from phosphatidylinositol bisphosphate (PIP2). IP3 activates IP3 receptors resulting in Ca2+ release from the sarcoplasmic reticulum (SR) and DAG activates receptor‐operated Ca2+ channels (ROCC). H2O2 also directly activates ryanodine receptors (RyR) by oxidizing cysteine residues, leading the Ca2+ release from SR. Release of Ca2+ from the SR causes store depletion, which activates Ca2+ entry via activation of calcium release‐activated Ca2+ (CRAC) channels. H2O2 also inhibits KV channels, which increases the membrane potential (E m) and opens voltage‐dependent Ca2+ channels (VDCC). Together, the increased intracellular Ca2+ concentration ([Ca2+]i) causes Ca2+ to bind calmodulin (CaM) and activate myosin light chain kinase (MLCK), which phosphorylates myosin (MLC) and leads to contraction of pulmonary arterial smooth muscle cells (PASMC).
Hypoxia‐induced increases in mitochondrial ROS
The redox theory of HPV originally proposed by Archer and colleagues postulated that ROS generated by the mitochondrial ETC decreases in response to hypoxia in pulmonary arterial smooth muscle cells as a consequence of decreased O2 availability (Archer et al. 1989; Archer et al. 1993). However, since then a significant body of evidence has emerged which indicates that increases in mitochondrial ROS develop in response to hypoxia, which trigger HPV (Leach et al. 2001; Liu et al. 2003; Guzy et al. 2005; Waypa et al. 2006, 2010, 2013; Wang et al. 2007; Rathore et al. 2008; Jung et al. 2013; Yang et al. 2016b; Song et al. 2017). The controversy surrounding these opposing mechanisms stems mainly from the different techniques used to measure ROS generated in response to hypoxia. In the late 1980s and early 1990s, Archer and colleagues employed luminol and lucigenin in isolated perfused lungs (Archer et al. 1989, 1993). As these reagents may be membrane impermeant and also reside in the extracellular space, the decline in chemiluminescence they detected during hypoxia, which was interpreted by that group as reflecting a decrease in mitochondrial ROS generation, instead likely reflects decreases in extracellular superoxide levels, which are less relevant for intracellular signalling.
More recently, several reagents have been developed to measure intracellular ROS in specific subcellular compartments. Using a FRET‐based redox sensor, several studies demonstrated an increase in oxidation following acute hypoxia (Guzy et al. 2005; Mansfield et al. 2005). To further understand the changes in oxidant signalling following hypoxia, Waypa and colleagues targeted redox‐sensitive proteins to specific subcellular compartments of PASMCs (Waypa et al. 2010). These sensors (roGFPs) are mutants of green fluorescent protein that provide ratiometric assessments of thiol oxidation; they reveal that oxidation is significantly decreased in the mitochondrial matrix during hypoxia but is increased in the mitochondrial intermembrane space and cytosol (Waypa et al. 2010). Similar results were seen using these sensors in precision cut murine lung slices (Desireddi et al. 2010). In cultured PASMCs, scavenging of ROS during hypoxia inhibited the hypoxia‐induced increase in Ca2+ in the cytosol ([Ca2+]i) (Waypa et al. 2001, 2002, 2006). Moreover, adenoviral expression of peroxiredoxin‐5, an H2O2 scavenger, in the intermembrane space attenuated ROS signalling in the intermembrane space and cytosol, and also inhibited stabilization of hypoxia‐inducible factor 1α (HIF‐1α) in acute hypoxia in PASMCs (Sabharwal et al. 2013). Together, these studies demonstrate that ROS signalling is required for the activation of pathways linking hypoxia to the contractile response in PASMCs.
It has been suggested that the increase in ROS generation during hypoxia is due to increased ROS production at complex III (Waypa et al. 2016). However, several studies suggest that inhibition of complex I or complex III can inhibit HPV in rat lungs (Leach et al. 2001; Waypa et al. 2001, 2006). Genetic studies have identified RISP as essential for ROS production at complex III in hypoxia, because in the absence of this protein complex III cannot oxidize ubiquinol to ubisemiquinone (Guzy et al. 2005). Depletion of RISP from PASMCs has been shown to abolish hypoxia‐induced increases in ROS generation in the mitochondrial intermembrane space, and hypoxia‐induced increases in [Ca2+]i (Guzy et al. 2005). Additionally, deletion of RISP blocks the acute increase in right ventricular systolic pressure in response to hypoxia (Waypa et al. 2013). As decreases in electron flux through complex I can decrease electron entry into complex III, these findings suggest that electron flux through complex III is a critical signalling event in the response of pulmonary vascular cells to hypoxia.
While excessive oxidant stress is known to contribute to cellular dysfunction and death under conditions such as ischaemia–reperfusion (Schriewer et al. 2013), the levels of ROS involved in redox signalling in hypoxia are much lower and their effects on protein thiol redox state are eventually reversed by reductases such as thioredoxin and glutaredoxin (Sabharwal & Schumacker, 2014). Similar low levels of oxidant signalling mediate other physiological effects, such as the mitogenic response to growth factors (Finkel, 2011).
Other sources of ROS
Although an abundance of data support the role of mitochondria as the site of the increase in ROS signalling during hypoxia, other sources of ROS may also be engaged to amplify that response. NADPH oxidases are membrane‐bound multiprotein complexes that transfer electrons from NADH or NADPH to molecular O2 to produce superoxide. These systems have been suggested to function as O2 sensors in a variety of cell types (Wolin et al. 2007). However, studies have demonstrated that inhibition of NOX2 in mice did not alter the pulmonary response to hypoxia (Archer et al. 1999; Weissmann et al. 2006b). These studies suggest that while NADPH oxidases may contribute to the signalling pathways activated in response to hypoxia, they are not essential for triggering the response.
Downstream mechanisms of HPV
In biology, many pathways have redundant and compensatory mechanisms that protect the organism and allow for amplification of the signal. Increased ROS generation is appropriate to consider as a signalling mechanism in HPV because ROS are involved in regulating many processes including Ca2+ release, activation of AMP kinase, activation of Ca2+ channels and other signalling pathways. The following sections will highlight the role of ROS in the downstream mechanisms of HPV.
KV channels
The debate as to whether hypoxia increases or decreases ROS notwithstanding, it has long been known that hypoxia inactivates KV channels (Post et al. 1992; Yuan et al. 1993). Inhibition of KV channels results in depolarization of the plasma membrane which can induce Ca2+‐dependent action potentials, increases in [Ca2+]i and vasoconstriction. Several studies have suggested that increased ROS production leads to inhibition of KV channels (Cogolludo et al. 2006; Mittal et al. 2012; Sahoo et al. 2012). These studies demonstrated that increased production of H2O2 plays a role in KV channel inhibition and the contractile response of pulmonary arteries (Cogolludo et al. 2006; Mittal et al. 2012). Additionally, a recent study demonstrated that hypoxia causes inhibition of KV channels, which can be mimicked by application of H2O2 in PASMCs (Sommer et al. 2017). Together, these data suggest that hypoxia‐induced increases in mitochondrial ROS production can inhibit KV channels resulting in cellular membrane depolarization and HPV (Fig. 1). These data also challenge the idea that decreases in ROS lead to KV channel inhibition through a reductive (as opposed to oxidative) stress pathway (Michelakis et al. 2004).
Increased [Ca2+]i
Hypoxia causes a rapid increase in [Ca2+]i in PASMCs that leads to smooth muscle contraction. As discussed above, increased ROS production following hypoxia inhibits KV channels and causes cellular membrane depolarization. This leads to the opening of voltage‐dependent Ca2+ channels (VDCCs) (Post et al. 1992; Yuan et al. 1993). VDCCs are important for Ca2+ influx following membrane depolarization, but are not the only source of increased [Ca2+]i in response to hypoxia. Studies have shown that blockade of VDCCs can only partially inhibit HPV in rat pulmonary arteries, suggesting that other sources of increased [Ca2+]i play a role in the response to hypoxia (Robertson et al. 2000b). The hypoxia‐induced increase in [Ca2+]i is also mediated by ryanodine receptors, which, when activated, release Ca2+ from the sarcoplasmic reticulum. ROS have been shown to interact with and activate ryanodine receptors (RyRs) in PASMCs leading to increased [Ca2+]i (Lin et al. 2007; Liao et al. 2011). Ca2+ release from the sarcoplasmic reticulum can also come from activation of inositol 1,4,5‐trisphosphate (IP3) receptors. H2O2 has been shown to activate phospholipase C, which generates diacylglycerol (DAG) and IP3 from phosphatidylinositol bisphosphate (PIP2) (Gonzalez‐Pacheco et al. 2002). This suggests a mechanism by which hypoxia‐induced ROS can cause Ca2+ release from IP3‐sensitive intracellular stores. Additionally, DAG activation of receptor‐operated Ca2+ channels (ROCCs), such as transient receptor potential channel 6 (TRPC6), can also increase [Ca2+]i either directly by conducting ionized calcium, or indirectly by conducting sodium ions that contribute to the membrane depolarization. Studies show that application of a DAG analogue caused vasoconstriction under normoxic conditions, but had no effect in TRPC6 knockout mice (Weissmann et al. 2006a; Fuchs et al. 2011; Smith et al. 2015). The release of Ca2+ from the sarcoplasmic reticulum from RyR and IP3 receptors can lead to store depletion and the opening of store‐operated Ca2+ channels (SOCCs). A study by Mungai and colleagues demonstrated that hypoxia induces ROS‐dependent release of calcium from intracellular stores, leading to the activation of SOCCs and increased [Ca2+]i in PASMCs (Mungai et al. 2011). Several studies have reported that inhibition of SOCCs can inhibit HPV, which underscores the importance of this mechanism in mediating the pulmonary vascular response to hypoxia. Collectively, these data demonstrate that several mechanisms involving hypoxia‐induced increases in ROS can lead to increased [Ca2+]i and HPV.
Contraction mechanisms
An increase in [Ca2+]i is a major trigger for PASMC contraction and acute pulmonary vasoconstriction (Fig. 1). When [Ca2+]i is increased, Ca2+ binds to calmodulin (CaM) and forms a Ca2+–CaM complex. The Ca2+–CaM complex activates myosin light chain kinase (MLCK), which phosphorylates myosin and leads to contraction of PASMCs. The Ca2+–CaM complex can also coordinate with RhoA to activate Rho kinase (ROK), which phosphorylates myosin light chain phosphatase (MLCP). Phosphorylation inactivates MLCP also causing smooth muscle contraction. Hypoxia has been shown to activate RhoA in PASMC and endothelial cells leading to activation of ROK (Robertson et al. 2000a; Wang et al. 2001; Gosal et al. 2015). ROK induces Ca2+ sensitization of contractile proteins by activating the MLCP‐inhibitor protein, CPI‐17 (Koyama et al. 2000). This allows for a small increase in [Ca2+]i to cause a significant increase in contraction of PASMCs.
Chronic hypoxia‐induced signalling
Hypoxia activates hypoxia‐inducible factors (HIFs) which are oxygen‐sensitive transcription factors involved in regulating a variety of physiological and pathological mechanisms (Shimoda & Laurie, 2014). HIF transcriptional activity requires formation of a heterodimer composed of an oxygen‐regulated component (HIF‐1α) and an O2‐independent component (HIF‐1β, ARNT). Under normoxic conditions, HIF‐1α degradation is initiated by HIF prolyl hydroxylase enzymes. Multiple studies have shown that mitochondrially derived ROS signals regulate HIF‐1α stabilization by controlling the activity of HIF prolyl hydroxylase (Chandel et al. 1998; Guzy et al. 2005). However, given that pulmonary arteries contract almost immediately after sensing a decrease in O2 tension, HIF‐mediated changes in transcription are unlikely to regulate the acute phase of HPV (Shimoda & Laurie, 2014). Some support for a role for HIF in phase 1 of HPV comes from patients with Chuvash polycythemia, a rare genetic condition which results in stabilization of HIF under normoxic conditions, who exhibit enhanced HPV (Smith et al. 2006; Shimoda & Laurie, 2014). The exact role of HIF in the acute phase of HPV remains to be determined, but it seems likely that basal HIF activity contributes to the permissive transcriptional control of multiple components that contribute to the HPV response. Indeed, HIF‐1α heterozygous mice show blunted hypoxia responsiveness in multiple tissues (Shimoda et al. 2001). HIF also plays a role in the development of pulmonary hypertension, which arises from various aetiologies including chronic hypoxia. HIF mediates the hypoxia‐induced increase in TRPC channel expression and the decrease in K+ channel expression which leads to pulmonary vasoconstriction and pulmonary vascular remodelling (Wang et al. 1997, 2006; Shimoda & Laurie, 2014). Additionally, ET‐1 is a HIF target (Bodi et al. 1995) that contributes to sustained activation of ROK. Prolonged hypoxia can thereby increase ROS signalling, leading to HIF stabilization, ET‐1 release, ROK activation, Ca2+ sensitization and pulmonary vasoconstriction (Chi et al. 2010). Together, these data demonstrate a role of HIF in the pulmonary vascular remodelling and the development of pulmonary hypertension in response to chronic hypoxia.
Conclusion
Hypoxia triggers a diverse set of cellular responses involving transcriptional and post‐translational mechanisms in the lung, which are triggered through the activation of O2 sensors. Abundant evidence supports the concept that increased ROS production at complex III is a required event in the activation of these mechanisms that comprise the functional responses to hypoxia in the pulmonary circulation. While the molecular details linking hypoxia to the increase in ROS following hypoxia are not yet fully elucidated, it is clear that release of ROS into the intermembrane space is involved in the hypoxia response. Further understanding of these pathways and how they are activated may lead to the identification of targets that could be exploited for treatment of lung disorders that involve cellular hypoxia, including lung cancer, pulmonary hypertension and high‐altitude pulmonary oedema.
Additional information
Competing interests
The authors have no competing interests to report.
Funding
This work was supported by NIH grants HL35440, HL122062 and HL109478 (P.T.S.) and a Parker B. Francis Fellowship (K.A.S.).
Author contributions
K.A.S. wrote the initial draft of the manuscript. P.T.S. reviewed and revised the manuscript. All authors approved the final version of the manuscript. Both authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Biographies
Kimberly A. Smith studied biology at Illinois Wesleyan University and earned her Master's degree from Southern Illinois University, and her PhD from Northwestern University. During her postdoctoral fellowship in P.T.S.’s lab, she studied the molecular mechanisms of hypoxia and pulmonary hypertension.

Paul T. Schumacker is the Patrick M. Magoon Professor of Neonatology Research in the Department of Pediatrics at Northwestern University Feinberg School of Medicine and the Ann and Robert H. Lurie Children's Hospital, with joint appointments in the Departments of Medicine (Pulmonary and Critical Care), Cellular and Molecular Biology, and the Northwestern University Comprehensive Cancer Center. His laboratory studies oxygen sensing, mitochondrial redox signalling, and cellular responses to hypoxia in health and disease. He has published more than 200 articles, mostly in the field of respiratory biology.
Edited by: Larissa Shimoda & Harold Schultz
References
- Archer SL, Huang J, Henry T, Peterson D & Weir EK (1993). A redox‐based O2 sensor in rat pulmonary vasculature. Circ Res 73, 1100–1112. [DOI] [PubMed] [Google Scholar]
- Archer SL, Nelson DP & Weir EK (1989). Simultaneous measurement of O2 radicals and pulmonary vascular reactivity in rat lung. J Appl Physiol (1985) 67, 1903–1911. [DOI] [PubMed] [Google Scholar]
- Archer SL, Reeve HL, Michelakis E, Puttagunta L, Waite R, Nelson DP, Dinauer MC & Weir EK (1999). O2 sensing is preserved in mice lacking the gp91 phox subunit of NADPH oxidase. Proc Natl Acad Sci U S A 96, 7944–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennie RE, Packer CS, Powell DR, Jin N & Rhoades RA (1991). Biphasic contractile response of pulmonary artery to hypoxia. Am J Physiol 261, L156–L163. [DOI] [PubMed] [Google Scholar]
- Bodi I, Bishopric NH, Discher DJ, Wu X & Webster KA (1995). Cell‐specificity and signaling pathway of endothelin‐1 gene regulation by hypoxia. Cardiovasc Res 30, 975–984. [DOI] [PubMed] [Google Scholar]
- Brigelius‐Flohe R & Flohe L (2011). Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 15, 2335–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher PC, Pearse DB, Pillai RP, Litt MC, Mitchell MC & Sylvester JT (1991). Energy state and vasomotor tone in hypoxic pig lungs. J Appl Physiol (1985) 70, 1874–1881. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Budinger GR, Choe SH & Schumacker PT (1997). Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J Biol Chem 272, 18808–18816. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC & Schumacker PT (1998). Mitochondrial reactive oxygen species trigger hypoxia‐induced transcription. Proc Natl Acad Sci U S A 95, 11715–11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi AY, Waypa GB, Mungai PT & Schumacker PT (2010). Prolonged hypoxia increases ROS signaling and RhoA activation in pulmonary artery smooth muscle and endothelial cells. Antioxid Redox Signal 12, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogolludo A, Frazziano G, Cobeno L, Moreno L, Lodi F, Villamor E, Tamargo J & Perez‐Vizcaino F (2006). Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann N Y Acad Sci 1091, 41–51. [DOI] [PubMed] [Google Scholar]
- Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM & Harman D (1987). Oxygen radicals and human disease. Ann Intern Med 107, 526–545. [DOI] [PubMed] [Google Scholar]
- Demiryurek AT, Wadsworth RM & Kane KA (1991). Effects of hypoxia on isolated intrapulmonary arteries from the sheep. Pulm Pharmacol 4, 158–164. [DOI] [PubMed] [Google Scholar]
- Desireddi JR, Farrow KN, Marks JD, Waypa GB & Schumacker PT (2010). Hypoxia increases ROS signaling and cytosolic Ca2+ in pulmonary artery smooth muscle cells of mouse lungs slices. Antioxid Redox Signal 12, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T ( 2011). Signal transduction by reactive oxygen species. J Cell Biol 194, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs B, Rupp M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Gudermann T, Dietrich A & Weissmann N (2011). Diacylglycerol regulates acute hypoxic pulmonary vasoconstriction via TRPC6. Respir Res 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Chen T & Raj JU (2016). Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am J Respir Cell Mol Biol 54, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Ruiz C, Colell A, Morales A, Kaplowitz N & Fernandez‐Checa JC (1995). Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor‐kappa B: studies with isolated mitochondria and rat hepatocytes. Mol Pharmacol 48, 825–834. [PubMed] [Google Scholar]
- Genova ML, Ventura B, Giuliano G, Bovina C, Formiggini G, Parenti Castelli G & Lenaz G (2001). The site of production of superoxide radical in mitochondrial Complex I is not a bound ubisemiquinone but presumably iron‐sulfur cluster N2. FEBS Lett 505, 364–368. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Pacheco FR, Caramelo C, Castilla MA, Deudero JJ, Arias J, Yague S, Jimenez S, Bragado R & Alvarez‐Arroyo MV (2002). Mechanism of vascular smooth muscle cells activation by hydrogen peroxide: role of phospholipase C gamma. Nephrol Dial Transplant 17, 392–398. [DOI] [PubMed] [Google Scholar]
- Gosal K, Dunlop K, Dhaliwal R, Ivanovska J, Kantores C, Desjardins JF, Connelly KA, McNamara PJ, Jain A & Jankov RP (2015). Rho kinase mediates right ventricular systolic dysfunction in rats with chronic neonatal pulmonary hypertension. Am J Respir Cell Mol Biol 52, 717–727. [DOI] [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U & Schumacker PT (2005). Mitochondrial complex III is required for hypoxia‐induced ROS production and cellular oxygen sensing. Cell Metab 1, 401–408. [DOI] [PubMed] [Google Scholar]
- López‐Barneo J, Pardal R & Ortega‐Sáenz P (2001). Cellular mechanism of oxygen sensing. Annu Rev Physiol 63, 259–287. [DOI] [PubMed] [Google Scholar]
- Jung HJ, Kim Y, Chang J, Kang SW, Kim JH & Kwon HJ (2013). Mitochondrial UQCRB regulates VEGFR2 signaling in endothelial cells. J Mol Med (Berl) 91, 1117–1128. [DOI] [PubMed] [Google Scholar]
- Koyama M, Ito M, Feng J, Seko T, Shiraki K, Takase K, Hartshorne DJ & Nakano T (2000). Phosphorylation of CPI‐17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho‐kinase. FEBS Lett 475, 197–200. [DOI] [PubMed] [Google Scholar]
- Le Cras TD & McMurtry IF (2001). Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol 280, L575–L582. [DOI] [PubMed] [Google Scholar]
- Leach RM, Hill HM, Snetkov VA, Robertson TP & Ward JPT (2001). Divergent roles of glycolysis and the mitochondrial electron transport chain in hypoxic pulmonary vasoconstriction of the rat: identity of the hypoxic sensor. J Physiol 536, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach RM, Robertson TP, Twort CH & Ward JP (1994). Hypoxic vasoconstriction in rat pulmonary and mesenteric arteries. Am J Physiol 266, L223–L231. [DOI] [PubMed] [Google Scholar]
- Liao B, Zheng YM, Yadav VR, Korde AS & Wang YX (2011). Hypoxia induces intracellular Ca2+ release by causing reactive oxygen species‐mediated dissociation of FK506‐binding protein 12.6 from ryanodine receptor 2 in pulmonary artery myocytes. Antioxid Redox Signal 14, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Yang XR, Cao YN & Sham JS (2007). Hydrogen peroxide‐induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 292, L1598–L1608. [DOI] [PubMed] [Google Scholar]
- Liu JQ, Sham JS, Shimoda LA, Kuppusamy P & Sylvester JT (2003). Hypoxic constriction and reactive oxygen species in porcine distal pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 285, L322–L333. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sham JS, Shimoda LA & Sylvester JT (2001). Hypoxic constriction of porcine distal pulmonary arteries: endothelium and endothelin dependence. Am J Physiol Lung Cell Mol Physiol 280, L856–L865. [DOI] [PubMed] [Google Scholar]
- Madden JA, Vadula MS & Kurup VP (1992). Effects of hypoxia and other vasoactive agents on pulmonary and cerebral artery smooth muscle cells. Am J Physiol 263, L384–L393. [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT & Simon MC (2005). Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF‐α activation. Cell Metab 1, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord JM ( 1985). Oxygen‐derived free radicals in postischemic tissue injury. N Engl J Med 312, 159–163. [DOI] [PubMed] [Google Scholar]
- Michelakis ED, Thebaud B, Weir EK & Archer SL (2004). Hypoxic pulmonary vasoconstriction: redox regulation of O2‐sensitive K+ channels by a mitochondrial O2‐sensor in resistance artery smooth muscle cells. J Mol Cell Cardiol 37, 1119–1136. [DOI] [PubMed] [Google Scholar]
- Misra HP & Fridovich I (1972). The univalent reduction of oxygen by reduced flavins and quinones. J Biol Chem 247, 188–192. [PubMed] [Google Scholar]
- Mittal M, Gu XQ, Pak O, Pamenter ME, Haag D, Fuchs DB, Schermuly RT, Ghofrani HA, Brandes RP, Seeger W, Grimminger F, Haddad GG & Weissmann N (2012). Hypoxia induces Kv channel current inhibition by increased NADPH oxidase‐derived reactive oxygen species. Free Radic Biol Med 52, 1033–1042. [DOI] [PubMed] [Google Scholar]
- Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK & Schumacker PT (2011). Hypoxia triggers AMPK activation through reactive oxygen species‐mediated activation of calcium release‐activated calcium channels. Mol Cell Biol 31, 3531–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP ( 2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, Rhee SG, Thornalley PJ, Partridge L, Gems D, Nystrom T, Belousov V, Schumacker PT & Winterbourn CC (2011). Unraveling the biological roles of reactive oxygen species. Cell Metab 13, 361–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray TR, Chen L, Marshall BE & Macarak EJ (1990). Hypoxic contraction of cultured pulmonary vascular smooth muscle cells. Am J Respir Cell Mol Biol 3, 457–465. [DOI] [PubMed] [Google Scholar]
- Oparil S, Chen SJ, Meng QC, Elton TS, Yano M & Chen YF (1995). Endothelin‐A receptor antagonist prevents acute hypoxia‐induced pulmonary hypertension in the rat. Am J Physiol 268, L95–L100. [DOI] [PubMed] [Google Scholar]
- Paddenberg R, Konig P, Faulhammer P, Goldenberg A, Pfeil U & Kummer W (2006). Hypoxic vasoconstriction of partial muscular intra‐acinar pulmonary arteries in murine precision cut lung slices. Respir Res 7, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge CA ( 1995). Hypoxia and reoxygenation stimulate biphasic changes in endothelial monolayer permeability. Am J Physiol 269, L52–L58. [DOI] [PubMed] [Google Scholar]
- Post JM, Hume JR, Archer SL & Weir EK (1992). Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol 262, C882–C890. [DOI] [PubMed] [Google Scholar]
- Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS & Wang YX (2008). Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS‐PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 45, 1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson TP, Aaronson PI & Ward JP (1995). Hypoxic vasoconstriction and intracellular Ca2+ in pulmonary arteries: evidence for PKC‐independent Ca2+ sensitization. Am J Physiol 268, H301–H307. [DOI] [PubMed] [Google Scholar]
- Robertson TP, Dipp M, Ward JP, Aaronson PI & Evans AM (2000a). Inhibition of sustained hypoxic vasoconstriction by Y‐27632 in isolated intrapulmonary arteries and perfused lung of the rat. Br J Pharmacol 131, 5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson TP, Hague D, Aaronson PI & Ward JP (2000b). Voltage‐independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol 525, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodman DM, Yamaguchi T, O'Brien RF & McMurtry IF (1989). Hypoxic contraction of isolated rat pulmonary artery. J Pharmacol Exp Ther 248, 952–959. [PubMed] [Google Scholar]
- Rounds S & McMurtry IF (1981). Inhibitors of oxidative ATP production cause transient vasoconstriction and block subsequent pressor responses in rat lungs. Circ Res 48, 393–400. [DOI] [PubMed] [Google Scholar]
- Sabharwal SS & Schumacker PT (2014). Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer 14, 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharwal SS, Waypa GB, Marks JD & Schumacker PT (2013). Peroxiredoxin‐5 targeted to the mitochondrial intermembrane space attenuates hypoxia‐induced reactive oxygen species signalling. Biochem J 456, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo N, Schönherr R, Hoshi T & Heinemann SH (2012). Cysteines control the N‐ and C‐linker‐dependent gating of KCNH1 potassium channels. Biochim Biophys Acta 1818, 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriewer JM, Peek CB, Bass J & Schumacker PT (2013). ROS‐mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia‐reperfusion. J Am Heart Assoc 2, e000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacker PT ( 2014). Cellular and molecular mechanisms of O2 sensing In High Altitude: Human Adaptation to Hypoxia, ed. Swenson ER & Bärtsch P, pp. 1–22. Springer, New York. [Google Scholar]
- Sham JS, Crenshaw BR Jr, Deng LH, Shimoda LA & Sylvester JT (2000). Effects of hypoxia in porcine pulmonary arterial myocytes: roles of KV channel and endothelin‐1. Am J Physiol Lung Cell Mol Physiol 279, L262–L272. [DOI] [PubMed] [Google Scholar]
- Shimoda LA & Laurie SS (2014). HIF and pulmonary vascular responses to hypoxia. J Appl Physiol (1985) 116, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda LA, Manalo DJ, Sham JSK, Semenza GL & Sylvester JT (2001). Partial HIF‐1α deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 281, L202–L208. [DOI] [PubMed] [Google Scholar]
- Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D & Yuan JX (2015). Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TG, Brooks JT, Balanos GM, Lappin TR, Layton DM, Leedham DL, Liu C, Maxwell PH, McMullin MF, McNamara CJ, Percy MJ, Pugh CW, Ratcliffe PJ, Talbot NP, Treacy M & Robbins PA (2006). Mutation of von Hippel‐Lindau tumour suppressor and human cardiopulmonary physiology. PLoS Med 3, e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer N, Huttemann M, Pak O, Scheibe S, Knoepp F, Sinkler C, Malczyk M, Gierhardt M, Esfandiary A, Kraut S, Jonas F, Veith C, Aras S, Sydykov A, Alebrahimdehkordi N, Giehl K, Hecker M, Brandes RP, Seeger W, Grimminger F, Ghofrani HA, Schermuly RT, Grossman LI & Weissmann N (2017). Mitochondrial complex IV subunit 4 Isoform 2 is essential for acute pulmonary oxygen sensing. Circ Res 121, 424–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T, Zheng YM & Wang YX (2017). Cross talk between mitochondrial reactive oxygen species and sarcoplasmic reticulum calcium in pulmonary arterial smooth muscle cells. Adv Exp Med Biol 967, 289–298. [DOI] [PubMed] [Google Scholar]
- Sylvester JT, Shimoda LA, Aaronson PI & Ward JP (2012). Hypoxic pulmonary vasoconstriction. Physiol Rev 92, 367–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, Alexandre A & Lehninger AL (1985). Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237, 408–414. [DOI] [PubMed] [Google Scholar]
- Turrens JF & Boveris A (1980). Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Juhaszova M, Rubin LJ & Yuan XJ (1997). Hypoxia inhibits gene expression of voltage‐gated K+ channel alpha subunits in pulmonary artery smooth muscle cells. J Clin Invest 100, 2347–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL & Shimoda LA (2006). Hypoxia inducible factor 1 mediates hypoxia‐induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98, 1528–1537. [DOI] [PubMed] [Google Scholar]
- Wang QS, Zheng YM, Dong L, Ho YS, Guo Z & Wang YX (2007). Role of mitochondrial reactive oxygen species in hypoxia‐dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic Biol Med 42, 642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jin N, Ganguli S, Swartz DR, Li L & Rhoades RA (2001). Rho‐kinase activation is involved in hypoxia‐induced pulmonary vasoconstriction. Am J Respir Cell Mol Biol 25, 628–635. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Chandel NS & Schumacker PT (2001). Model for hypoxic pulmonary vasoconstriction involving mitochondrial oxygen sensing. Circ Res 88, 1259–1266. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW & Schumacker PT (2006). Increases in mitochondrial reactive oxygen species trigger hypoxia‐induced calcium responses in pulmonary artery smooth muscle cells. Circ Res 99, 970–978. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, Dokic D, Ball MK & Schumacker PT (2013). Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med 187, 424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D & Schumacker PT (2010). Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res 106, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT & Schumacker PT (2002). Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ Res 91, 719–726. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Smith KA & Schumacker PT (2016). O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol Aspects Med 47–48, 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir EK, Will JA, Lundquist LJ, Eaton JW & Chesler E (1983). Diamide inhibits pulmonary vasoconstriction induced by hypoxia or prostaglandin F2 alpha. Proc Soc Exp Biol Med 173, 96–103. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Akkayagil E, Quanz K, Schermuly RT, Ghofrani HA, Fink L, Hänze J, Rose F, Seeger W & Grimminger F (2004). Basic features of hypoxic pulmonary vasoconstriction in mice. Resp Physiol Neurobiol 139, 191–202. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Mederos Y Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F & Gudermann T (2006a). Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A 103, 19093–19098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann N, Winterhalder S, Nollen M, Voswinckel R, Quanz K, Ghofrani HA, Schermuly RT, Seeger W & Grimminger F (2001). NO and reactive oxygen species are involved in biphasic hypoxic vasoconstriction of isolated rabbit lungs. Am J Physiol Lung Cell Mol Physiol 280, L638–L645. [DOI] [PubMed] [Google Scholar]
- Weissmann N, Zeller S, Schafer RU, Turowski C, Ay M, Quanz K, Ghofrani HA, Schermuly RT, Fink L, Seeger W & Grimminger F (2006b). Impact of mitochondria and NADPH oxidases on acute and sustained hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol 34, 505–513. [DOI] [PubMed] [Google Scholar]
- Wolin MS, Ahmad M, Gao Q & Gupte SA (2007). Cytosolic NAD(P)H regulation of redox signaling and vascular oxygen sensing. Antioxid Redox Signal 9, 671–678. [DOI] [PubMed] [Google Scholar]
- Yang L, Chen X, Simet SM, Hu G, Cai Y, Niu F, Kook Y & Buch SJ (2016a). Reactive oxygen species/hypoxia‐inducible factor‐1alpha/platelet‐derived growth factor‐BB autocrine loop contributes to cocaine‐mediated alveolar epithelial barrier damage. Am J Respir Cell Mol Biol 55, 736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Zhuan B, Yan Y, Jiang S & Wang T (2016b). Roles of different mitochondrial electron transport chain complexes in hypoxia‐induced pulmonary vasoconstriction. Cell Biol Int 40, 188–195. [DOI] [PubMed] [Google Scholar]
- Yuan XJ, Goldman WF, Tod ML, Rubin LJ & Blaustein MP (1993). Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol 264, L116–L123. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Bhattacharya S, Pi J, Clewell RA, Carmichael PL & Andersen ME (2015). Adaptive posttranslational control in cellular stress response pathways and its relationship to toxicity testing and safety assessment. Toxicol Sci 147, 302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]