

Abstract
Development of the pulmonary circulation is a critical component of fetal lung development, and continues throughout infancy and childhood, marking an extended window of susceptibility to vascular maldevelopment and maladaptation. Perinatal vascular insults may result in abnormal vascular structure or function, including decreased angiogenic signaling and vascular endowment, impaired vasoreactivity through increased pulmonary artery endothelial dysfunction and remodeling, or enhanced genetic susceptibility to pulmonary vascular disease through epigenetic modifications or germline mutations. Although some infants develop early onset pulmonary hypertension, due to the unique adaptive capabilities of the immature host many do not have clinically evident early pulmonary vascular dysfunction. These individuals remain at increased risk for development of late‐onset pulmonary hypertension, and may be particularly susceptible to secondary insults. This review will address the role of perinatal vascular insults in the development of late pulmonary vascular dysfunction with an effort to highlight areas of critical research need.
Keywords: Developmental programming, Pulmonary artery, Epigenetics, Vascular dysfunction, Angiogenesis
Introduction
Although the development of pulmonary vascular disease (PVD) is thought to result from multiple converging insults, the role of perinatal pulmonary insults in driving long‐term pulmonary vascular dysfunction remains poorly understood. A number of prenatal and postnatal vascular and alveolar insults may increase the later risk of developing PVD, even in the absence of clinically evident pulmonary vascular dysfunction in the neonate. The Barker Hypothesis, or the Developmental Origins of Health and Disease Hypothesis, was originally proposed to explain developmental priming of the cardiovascular system (Barker, 1995, 2007). However, its tenets are highly relevant to the developing pulmonary vasculature as well (Maron & Abman, 2017). Key concepts include the interplay between adaptive and maladaptive responses to environmental cues in a developmentally plastic host, effects of nutrition and fetal stress, and the role of epigenetic modifications (Wadhwa et al. 2009). This review will address the role of perinatal vascular insults in the development of late pulmonary vascular dysfunction with an effort to highlight areas of critical research need.
Normal vascular development
Development of the pulmonary vascular circulation is a critical component of fetal lung development, and occurs through both vasculogenesis and angiogenesis under highly regulated conditions (Peng & Morrisey, 2013; de Wijs‐Meijler et al. 2017). Vasculogenesis is the process of blood vessel formation by de novo production of endothelial cells, giving rise to the heart and the first primitive vascular plexus within the embryo. Angiogenesis follows, and is responsible for the remodelling and expansion of the pulmonary vascular network through both endothelial sprouting and intussusceptive growth (Peng & Morrisey, 2013; Gao et al. 2016). During the canalicular stage of lung development (human gestation weeks 16–26), there is a dramatic rise in the number of lung capillaries to form the first air–blood interface, which continue to mature during the saccular stage (weeks 24–38). By the alveolar stage (week 36 through infancy), the immature double‐capillary fetal network has fused to form a single capillary layer allowing for efficient gas exchange (Burri, 1999). From birth to adulthood, the pulmonary capillary surface area increases an additional 20‐fold, and capillary volume increases 35‐fold, forming an extensive pulmonary microvascular network. Finally, lung alveolar development is closely intertwined with vascular development, and a multitude of animal studies demonstrate that specific disruption of alveolar development impairs vascular development, and vice versa, confirming that early life alveolar disorders may also impair early pulmonary vascular development (Thebaud & Abman, 2007).
Impaired pulmonary vascularization
The role of prenatal pulmonary vascular insults
Impairments in angiogenic signalling during any of these critical windows of development can reduce pulmonary vascular density and ultimately total adult lung vascular endowment (Fig. 1). A number of prenatal conditions are characterized by impaired pulmonary angiogenesis signalling, including conditions of fetal insufficiency such as pre‐eclampsia, fetal hypoxia and intrauterine growth restriction (IUGR), as well as inflammatory conditions such as chorioamnionitis and fetal infection, or from fetal drug and toxin exposure. Other prenatal conditions may also result in grossly abnormal structural development of the pulmonary vasculature, as seen in congenital diaphragmatic hernia and Down syndrome (Bush et al. 2017; Kardon et al. 2017).
Figure 1. Perinatal pulmonary vascular insults contribute to late‐onset pulmonary vascular dysfunction through multiple mechanisms.
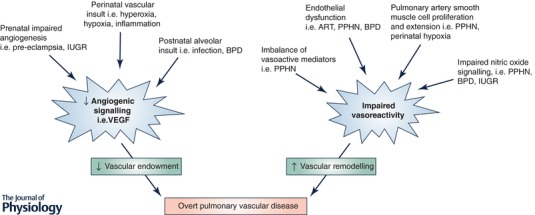
IUGR: intrauterine growth restriction. BPD: bronchopulmonary dysplasia. VEGF: vascular endothelial growth factor. PPHN: persistent pulmonary hypertension of the newborn. ART: assisted reproductive technology.
Vascular endothelial growth factor A (VEGF) is a key regulator of pulmonary vascular development, and its absolute requirement is demonstrated by murine studies showing lethality with targeted VEGF inactivation or knockout (Carmeliet et al. 1996; Ferrara et al. 1996). A number of studies have implicated impaired VEGF signalling and its receptor, excess soluble fms‐like tyrosine kinase‐1 (sFlt‐1), in the pathogenesis of pre‐eclampsia, a progressive multisystem disorder of pregnancy characterized by maternal and placental vascular dysfunction (Levine et al. 2004). The impaired placental vascular perfusion in these pregnancies further increases the risk for development of bronchopulmonary dysplasia and PVD after birth (Mestan et al. 2014).
Several conditions related to placental insufficiency may impair vascular development. For example, studies in IUGR sheep, most commonly defined as fetal weight below the 10th percentile for gestational age, demonstrate decreased fetal pulmonary alveolarization, stunted pulmonary vascular growth, and impaired in vitro pulmonary artery endothelial cell migration, tube formation and nitric oxide production (Rozance et al. 2011). Long term, a human twin study demonstrated that IUGR with a birth weight of less than 2500 g corresponded to a 43% increase in the hazard for developing PVD in adolescence or young adulthood (Class et al. 2014). Beyond placental insufficiency, fetal hypoxia can also drive impaired angiogenesis, and may result from residence at altitude, maternal tobacco use, or anaemia. Antenatal hypoxia results in a cascade of maladaptive consequences, including impaired angiogenesis and vascularization, endothelial barrier disruption and dysfunction, altered pulmonary vasoreactivity and decline in vascular resistance at birth, and pulmonary artery muscularization (Papamatheakis et al. 2013).
Finally, fetal inflammation and infection may affect vascular development, though the type and timing of inflammatory events appear to be important. For example, inflammatory mediators such as interleukin‐6 in amniotic fluid or chronic low‐grade infection with ureaplasma and mycoplasma are identified in a significant number of preterm births, yet these factors paradoxically decrease the risk for moderate to severe respiratory distress syndrome at birth, potentially by accelerating lung maturation (Watterberg et al. 1996; Hannaford et al. 1999; Shimoya et al. 2000). However, for the subset of preterm infants requiring mechanical ventilation, the risk for bronchopulmonary dysplasia is increased (Van Marter et al. 2002), suggesting amplification of the maladaptive response after fetal inflammatory priming (see Kramer et al. 2009 for additional review).
Effects of postnatal pulmonary vascular insults
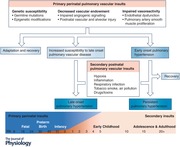
Given that such a large component of pulmonary vascular development occurs after birth, infancy and childhood remain a window of susceptibility for abnormal development and overall endowment. Common direct postnatal vascular insults include premature birth, nutritional deficiency, hypoxia, inflammation, infection and environmental exposures such as cigarette smoke or air pollution. Importantly, postnatal pulmonary vascular insults may serve as the initial primary vascular insult, or alternatively as secondary insults in a vulnerable host, in which case they may further potentiate maldevelopment or maladaptation (Fig. 2).
Figure 2.
Impact of early pulmonary vascular insults on adult pulmonary vascular endowment and ageing
Because they are born during a window of critical lung maturation (saccular stage), premature infants are among the highest risk group for persistent pulmonary vascular injury. They have dramatically fewer vessels and higher pulmonary vascular resistance than those born at term, and are uniquely susceptible to further insults from mechanical ventilation, oxygen exposure, inflammation and infection (Bland et al. 2000). Those who demonstrate echocardiographic evidence of PVD at 7 days of age are at increased risk to develop bronchopulmonary dysplasia (Mourani et al. 2015), which is frequently considered a PVD in and of itself as it is characterized by significant vascular rarefication (Thebaud & Abman, 2007; Mourani et al. 2015).
Long term, preterm birth is associated with an 8.5‐fold increased risk for developing pulmonary hypertension (defined as a mean pulmonary artery pressure ≥25 mmHg) in childhood and adolescence, and a 3.1‐fold increased risk in adulthood, even after adjusting for confounding factors such as acute pulmonary disorders, congenital heart defects, congenital diaphragmatic hernia and chromosomal disorders (Naumburg et al. 2015, 2017). At rest, otherwise healthy young adults born preterm exhibit increased vascular stiffness with elevations in resting mean pulmonary arterial pressure (Goss et al. 2018). With graded exercise, adults born preterm demonstrate an exaggerated increase in pulmonary artery pressure (Laurie et al. 2018), though it is currently unclear if this is due to endothelial dysfunction such as impaired nitric oxide‐mediated vasodilatation or due to a decreased capillary surface area available for recruitment.
Even infants born at or near term (alveolar stage) remain at risk for postnatal pulmonary vascular injury. Common insults in early life include respiratory infections and pulmonary over‐circulation due to left‐to‐right shunts, which may be further exacerbated by exposure to hypoxia, hyperoxia or mechanical ventilation and worsening inflammation or oxidative stress (de Wijs‐Meijler et al. 2017). Prolonged mechanical ventilation during the alveolar stage, even in the absence of hyperoxia or infection, can inhibit alveolar septation and impair angiogenesis signalling (Mokres et al. 2010). Left‐to‐right shunts, as seen in septal defects or patent ductus arteriosus, may accentuate pulmonary vascular sheer stress and promote vascular remodelling (Gao et al. 2016).
Potential for catch‐up vascularization and lung growth
Each of these early insults results in a decreased pulmonary vascular density with potential to reduce the adult pulmonary vascular endowment (Fig. 2). The degree to which perinatal insults can be overcome by lung ‘catch‐up’ growth is unclear. Individuals born premature or IUGR frequently demonstrate reduced spirometry throughout childhood and into adulthood. Although the data are predominantly negative, one study suggests lung catch‐up growth in those who also demonstrated weight catch‐up growth by age 5 (Friedrich et al. 2007; Kotecha et al. 2010; Vollsaeter et al. 2013; Suresh et al. 2015). Long term, distinct lung function trajectories develop during early childhood that are predictive of adult lung function and risk for adult lung disease such as chronic obstructive lung disease (Belgrave et al. 2018). However, most of these studies did not assess diffusion capacity or vascular density, and the effect on lung vascular growth is unclear.
Intriguingly, rodent studies of postnatal hyperoxia exposure, a commonly employed model of bronchopulmonary dysplasia, demonstrate a robust pulmonary vascular catch‐up by 8 weeks of age, followed by severe vascular pruning when mice are aged beyond 1 year (Yee et al. 2011), which would suggest both catch‐up potential as well as an accelerated ageing phenotype. Studies of pneumonectomy in adult rats demonstrate regenerative alveolarization in the remaining lung through angiocrine growth factors secreted by pulmonary capillary endothelial cells (Ding et al. 2011). The effectiveness of regenerative alveolarization in the setting of perinatal vascular insults and pulmonary endothelial dysfunction remains unclear. Endothelial progenitor cells are essential for vascular repair, and neonatal hyperoxia exposure also results in decreased pulmonary endothelial progenitor cells in the developing lung, increasing risk for development of bronchopulmonary dysplasia and pulmonary hypertension (Balasubramaniam et al. 2007). Whether decreased vascular density from a perinatal insult would impair such long‐term regenerative capacity is unknown, but would add to the concern for an accelerated lung function decline with ageing.
The existence of long‐term impairments in pulmonary vascular signalling merits further study. VEGF inhibition in adult rat models leads to air space enlargement, pulmonary vascular pruning and emphysema, suggesting ongoing need for VEGF signalling to maintain both alveolar and vascular structures (Kasahara et al. 2000). In a study of systemic microvascular density in human infants, 3‐month‐old offspring of hypertensive pregnancies had increased circulating anti‐angiogenic factors and decreased vascularization (Yu et al. 2016). However, the degree to which VEGF and other angiogenic signalling is impaired later in life after perinatal insults remains unknown. While lung function studies in adults born premature offer conflicting evidence of an accelerated lung function decline, human studies evaluating adult pulmonary vascular density after early pulmonary vascular insults are lacking (Doyle et al. 2006; Vollsaeter et al. 2013). This is in part due to a still relatively small number of adult survivors of severe perinatal pulmonary vascular insults who are beyond their mid‐twenties, when normal declines in age‐related lung function begin. Therefore, there is a critical need to better understand the pulmonary vascular ageing process in these individuals.
Altered pulmonary vasoreactivity and function
Even in the absence of an interruption of normal pulmonary angiogenesis and development, a number of perinatal conditions may impair normal pulmonary vasoreactivity (Fig. 1). Normally at birth, there is a progressive fall in pulmonary vascular resistance, rise in systemic vascular resistance, and closure of fetal shunts such as the ductus arteriosus, resulting in a successful transition from fetal to postnatal circulation (Hillman et al. 2012). In the setting of persistent pulmonary hypertension of the newborn, the pulmonary vascular resistance remains abnormally elevated at birth, resulting in right‐to‐left shunting through fetal pathways and severe hypoxaemia. Mechanisms are multifactorial and include decreased vascularity, but also abnormal muscularization of the small pulmonary arteries and an imbalance of vasoconstrictive mediators (Murphy et al. 1981; Christou et al. 1997; Pearson et al. 2001). Although initially considered primarily a failure to transition from a fetal to a postnatal pulmonary circulation, the multifactorial nature suggests a more long‐lasting imprint on the pulmonary vasculature. Indeed, a history of persistent pulmonary hypertension of the newborn is likely over‐represented in childhood pulmonary hypertension registries, and is associated with significantly increased pulmonary vasoreactivity in adulthood (Sartori et al. 1999).
An emerging perinatal cause of long‐term abnormal pulmonary vasoreactivity is assisted reproductive technology. Compared to control children, healthy children conceived with assisted reproductive technology demonstrated 30% higher pulmonary artery pressures and increased right ventricular dysfunction when acutely exposed to altitude (Scherrer et al. 2012; von Arx et al. 2015). Intriguingly, a 4‐week pretreatment with the antioxidants Vitamin C and E improved endothelial function, evidenced by enhanced nitric oxide bioavailability, improved flow‐mediated vasodilatation, and attenuated hypoxic pulmonary hypertension in these children, suggesting a component of reversible redox dysregulation (Rimoldi et al. 2015).
Finally, perinatal hypoxia exposure may also drive abnormal long‐term vasoreactivity. In humans, a history of perinatal hypoxia increases susceptibility to pulmonary vascular dysfunction more than 6‐fold in young adults living at high altitude (Julian et al. 2015). Intriguingly, a mouse study of perinatal hypoxia demonstrated persistent alteration in the nitric oxide–cyclic GMP pathway persisting into adulthood, which was ameliorated by treatment with inhaled nitric oxide during perinatal hypoxic exposure (Peyter et al. 2014). Given that more hypoxic neonates are being treated with inhaled nitric oxide, additional studies of its long‐term effects on vascular function in humans are warranted.
Genetic predisposition and epigenetic modifications
Insults to the pulmonary vasculature may occur on a background of increased genetic susceptibility to PVD (Austin & Loyd, 2014). For example, children with Down syndrome who die from cardiopulmonary disease exhibit abnormal pulmonary vascular development, including persistence of a double capillary network in the distal lung, increased pulmonary arterial smooth muscle thickness, and prominent intrapulmonary bronchopulmonary anastomoses (Bush et al. 2017). Other mutations, such as mutations in the gene encoding for bone morphogenetic protein receptor 2 (BMPR2), exhibit reduced penetrance, variable expressivity, and are insufficient for the development of PVD alone but may contribute greatly to the overall susceptibility to secondary insults.
Beyond germline mutations, epigenetic modifications such as DNA methylation and histone modifications likely play a critical role in imprinting the developing pulmonary vasculature. Specifically, differential hypermethylation of pulmonary artery smooth muscle cells leads to a deficiency in superoxide dismutase‐2, creating a hyperproliferative, apoptosis‐resistant vasculature, and is one potential mechanism for long‐term perinatal priming of the pulmonary vasculature (Archer et al. 2010). In rats, offspring of mothers fed restrictive diets during pregnancy develop exaggerated right ventricular hypertrophy and hypoxic pulmonary hypertension, associated with altered lung DNA methylation. These effects are attenuated by administration of histone deacetylase inhibitors to offspring, or alternatively by administration of the nitroxide tempol to calorie‐restricted mothers during gestation (Rexhaj et al. 2011). Whether these are targetable modifications of the genetic code to decrease susceptibility to secondary insults and late PVD in humans merits further investigation. However, early success in modulating alveolar development through maternal vitamin C supplementation in infants exposed to in utero tobacco smoke was recently demonstrated (McEvoy et al. 2017), and suggests at least the future potential to favourably modulate vascular development.
Effect of early pulmonary vascular insults on right ventricular development
The cardiac inflow tract, pulmonary vascular smooth muscle, and proximal vascular endothelium all arise from a common multipotent cardiopulmonary mesoderm progenitor in utero, and thus direct insults to the developing pulmonary vasculature could also serve as direct insults to the right ventricle (Peng et al. 2013). However, this hypothesis remains poorly studied. Overall, the best evidence for a shared insult is that of premature birth. Infants born preterm, even in the absence of clinically evident PVD, have a distinct cardiac structure characterized by increased biventricular hypertrophy, with the right ventricle more affected than the left (Aye et al. 2017). This persists into early adulthood when early impairments in right ventricular function are clinically detectable (Lewandowski et al. 2013a,b). In a rodent model of bronchopulmonary dysplasia using postnatal hyperoxia exposure, rats aged to 12 weeks develop an adaptive type of right ventricular hypertrophy and are more tolerant to secondary insults such as hypoxia exposure (Goss et al. 2015a,b). However, when rats are aged to 1 year, they develop a similar degree of right ventricular dysfunction as seen in humans, associated with impaired mitochondrial function and biogenesis. Since these changes occur in the absence of substantial deterioration in pulmonary vascular pressures, they suggest late transition to a maladaptive right ventricle (Goss et al. 2017). Whether the right ventricular dysfunction observed in humans and animal models is truly due to a direct and independent right ventricular injury, or is secondary to a lifetime of increased cardiac work from mild increases in pulmonary vascular compliance, merits further study.
Increased susceptibility to late pulmonary vascular insults
Clearly, perinatal pulmonary insults increase the risk to develop late PVD through a number of mechanisms. Given that the development of pulmonary hypertension is frequently thought to result from multiple insults to the pulmonary vasculature, these perinatal insults also increase the susceptibility to established later pulmonary vascular insults such as hypoxia, cigarette smoke, infections, drugs or toxins. Beginning in infancy, postnatal alveolar insults also drive the risk for pulmonary vascular dysfunction. For example, in rodent models of bronchopulmonary dysplasia, postnatal hyperoxia exposure results in an increased susceptibility to viral illnesses and cigarette smoke (McGrath‐Morrow et al. 2011; Buczynski et al. 2013). In humans, lower respiratory tract infection in childhood is associated with increased airflow obstruction and chronic obstructive pulmonary disease in adulthood (Chan et al. 2015; Hayden et al. 2015). Early life lower respiratory tract infection therefore may be an unrecognized risk factor for later PVD as well. Additional childhood alveolar disorders are also associated with an increased risk for PVD with age. For example, severe asthmatics may have evidence of pulmonary vascular pruning (Ash et al. 2018), and adults with cystic fibrosis who develop pulmonary hypertension have significantly higher mortality (Hayes et al. 2014).
Secondary insults such as hypoxia are also important. Acutely, hypoxia exposure results in inhibition of pulmonary artery smooth muscle cell potassium channel activity, resulting in depolarization, calcium entry and ultimately hypoxic pulmonary vasoconstriction. Over time, the elevated calcium level promotes smooth muscle cell proliferation and hypertrophy, leading to increased muscularization and chronic vasoconstriction (Weir et al. 2005). Pathological conditions such as sleep disordered breathing or high altitude exposure may activate these pathways. For example, children and adults born premature have a blunted hypoxic ventilatory drive making them at increased risk for intermittent nocturnal hypoxia due to sleep disordered breathing (Hibbs et al. 2008; Montgomery‐Downs et al. 2010; Bates et al. 2014). Hypoxia is a well‐established cause of pulmonary hypertension, and in rat models, adult exposure to hypobaric hypoxia following postnatal hyperoxia results in an exaggerated hypoxic pulmonary hypertension (Goss et al. 2015a).
Finally, there is concern for the effect of later exposure to pulmonary vascular toxins in individuals with a history of perinatal pulmonary vascular insults. For example, tobacco exposure results in pulmonary vascular remodelling in smokers, even prior to the development of clinical lung disease (Santos et al. 2002; Weissmann et al. 2012). Another significant unanswered question is whether stimulants may serve as secondary insults in at‐risk children and young adults with attention deficit disorder, especially given the increased use and growing recognition of amphetamines as a cause of pulmonary hypertension (Zamanian et al. 2018), though this remains largely untested. The decreased vascular density, vascular remodelling and epigenetic modifications that follow perinatal pulmonary vascular insults likely make these individuals uniquely susceptible to stressors later in life, and future studies should address this potential synergy.
Knowledge gaps
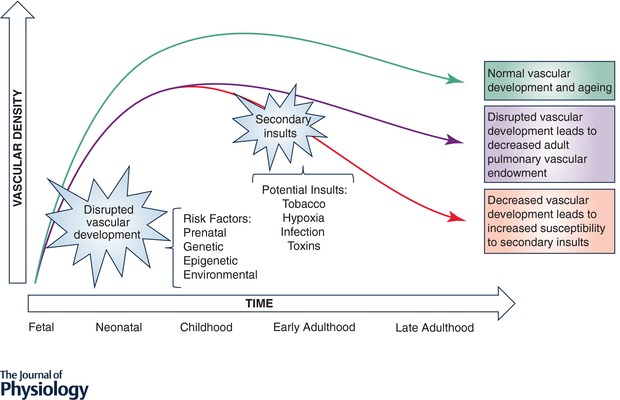
A number of questions and research gaps regarding the long‐term pulmonary vascular outcomes after perinatal insults remain (Fig. 3). First, there is a critical need to better understand the adaptive and maladaptive responses to perinatal pulmonary vascular insults. Whether the adaptive and regenerative capacity of the developing lung can be harnessed in childhood to improve vascularization and ultimately adult pulmonary vascular endowment remains to be seen. Modulating vascular angiogenesis will require careful study of safe therapeutic windows, given that mouse studies of VEGF overexpression have resulted in development of leaky capillaries and pulmonary oedema (Kaner et al. 2000). Second, longitudinal biomarkers are needed to identify individuals at high risk for late PVD. Given the prominent role that epigenetics likely play in modifying pulmonary vascular risk after early pulmonary insults, future studies are warranted to identify high‐risk individuals for early intervention, limiting the morbidity and mortality of late PVD and right ventricular dysfunction. Third, a better characterization of the susceptibility to secondary insults for specific perinatal insults would allow for personalized risk avoidance. Fourth, there is a critical need to understand the role of perinatal pulmonary vascular insults on the pulmonary vascular ageing process, and slowing vascular ageing may also improve alveolar ageing. Finally, given the significant impact of right ventricular dysfunction on all‐cause mortality, additional study of the effects of perinatal pulmonary vascular insults on the transitioning right ventricle are warranted.
Figure 3.

Current knowledge gaps
Conclusions
Based on the number of frequently concomitant perinatal pulmonary vascular insults, it is not surprising that one third of pediatric PVDs are considered multifactorial (del Cerro Marin et al. 2014). However, perinatal factors have not been assessed in adult pulmonary hypertension registry studies. In an age of personalized medicine, these factors truly deserve inclusion. Future studies should address both neonatal interventions to prevent early injury, as well as early diagnosis and management of high‐risk individuals to prevent late morbidity and mortality. Given the remarkable improvements in neonatal care over the past several decades, resulting in growing numbers of infants with moderate to severe perinatal pulmonary insults now reaching early adulthood, the time is now.
Additional information
Competing interests
None declared.
Author contributions
Sole author.
Funding
K.G. is supported by the University of Wisconsin Clinical and Translational Science Award (CTSA) program, through the NIH National Center for Advancing Translational Sciences (NCATS), grant NIH UL1TR000427 (PI Drezner; 4KL2TR000428‐10), and by a Parker B. Francis Fellowship Award.
Acknowledgements
The author would like to thank Dr Tim Lahm for constructive feedback during the initial drafting of the manuscript.
Biography
Kara Goss is an Assistant Professor of Medicine and Pediatrics at the University of Wisconsin in Madison, WI, USA. Trained in Internal Medicine, Pediatrics and Adult Pulmonary and Critical Care, she specializes in pulmonary hypertension and right ventricular failure. Her research is focused on the long‐term effects of prematurity and postnatal oxygen exposure on the cardiopulmonary unit. Current studies are investigating the acute and chronic adaptive and maladaptive responses of the right ventricle and pulmonary vasculature in both adults born premature and a rodent model utilizing postnatal hyperoxia exposure.

Edited by: Larissa Shimoda & Harold Schultz
References
- Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JRB, Gomberg‐Maitland M, Thébaud B, Husain AN, Cipriani N & Rehman J (2010). Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121, 2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash SY, Rahaghi FN, Come CE, Ross JC, Colon AG, Cardet‐Guisasola JC, Dunican EM, Bleecker ER, Castro M, Fahy JV, Fain SB, Gaston BM, Hoffman EA, Jarjour NN, Mauger DT, Wenzel SE, Levy BD, Estepar RSJ, Israel E & Washko GR (2018). Pruning of the pulmonary vasculature in asthma. The Severe Asthma Research Program (SARP) Cohort. Am J Respir Crit Care Med 198, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED & Loyd JE (2014). The genetics of pulmonary arterial hypertension. Circ Res 115, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aye CYL, Lewandowski AJ, Lamata P, Upton R, Davis E, Ohuma EO, Kenworthy Y, Boardman H, Wopperer S, Packham A, Adwani S, McCormick K, Papageorghiou AT & Leeson P (2017). Disproportionate cardiac hypertrophy during early postnatal development in infants born preterm. Pediatr Res 82, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniam V, Mervis CF, Maxey AM, Markham NE & Abman SH (2007). Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292, L1073–L1084. [DOI] [PubMed] [Google Scholar]
- Barker DJ (1995). Fetal origins of coronary heart disease. BMJ 311, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ (2007). The origins of the developmental origins theory. J Intern Med 261, 412–417. [DOI] [PubMed] [Google Scholar]
- Bates ML, Farrell ET & Eldridge MW (2014). Abnormal ventilatory responses in adults born prematurely. N Engl J Med 370, 584–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgrave DCM, Granell R, Turner SW, Curtin JA, Buchan IE, Le Souef PN, Simpson A, Henderson AJ & Custovic A (2018). Lung function trajectories from pre‐school age to adulthood and their associations with early life factors: a retrospective analysis of three population‐based birth cohort studies. Lancet Respir Med 6, 526–534. [DOI] [PubMed] [Google Scholar]
- Bland RD, Albertine KH, Carlton DP, Kullama L, Davis P, Cho SC, Kim BI, Dahl M & Tabatabaei N (2000). Chronic lung injury in preterm lambs: abnormalities of the pulmonary circulation and lung fluid balance. Pediatr Res 48, 64–74. [DOI] [PubMed] [Google Scholar]
- Buczynski BW, Yee M, Martin KC, Lawrence BP & O'Reilly MA (2013). Neonatal hyperoxia alters the host response to influenza A virus infection in adult mice through multiple pathways. Am J Physiol Lung Cell Mol Physiol 305, L282–L290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burri P (1999). Lung development and pulmonary angiogenesis In Lung Development, ed. Gaultier C, Bourbon J. & Post M, pp. 122–151. Oxford University Press, New York. [Google Scholar]
- Bush D, Abman SH & Galambos C (2017). Prominent intrapulmonary bronchopulmonary anastomoses and abnormal lung development in infants and children with Down syndrome. J Pediatr 180, 156–162.e1. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W & Nagy A (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380, 435–439. [DOI] [PubMed] [Google Scholar]
- Chan JYC, Stern DA, Guerra S, Wright AL, Morgan WJ & Martinez FD (2015). Pneumonia in childhood and impaired lung function in adults: a longitudinal study. Pediatrics 135, 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou H, Adatia I, Van Marter LJ, Kane JW, Thompson JE, Stark AR, Wessel DL & Kourembanas S (1997). Effect of inhaled nitric oxide on endothelin‐1 and cyclic guanosine 5'‐monophosphate plasma concentrations in newborn infants with persistent pulmonary hypertension. J Pediatr 130, 603–611. [DOI] [PubMed] [Google Scholar]
- Class QA, Rickert ME, Lichtenstein P & D'Onofrio BM (2014). Birth weight, physical morbidity, and mortality: a population‐based sibling‐comparison study. Am J Epidemiol 179, 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Cerro Marin MJ, Sabate Rotes A, Rodriguez Ogando A, Mendoza Soto A, Quero Jimenez M, Gavilan Camacho JL, Raposo Sonnenfeld I, Moya Bonora A, Albert Brotons DC & Moreno Galdo A (2014). Assessing pulmonary hypertensive vascular disease in childhood. Data from the Spanish registry. Am J Respir Crit Care Med 190, 1421–1429. [DOI] [PubMed] [Google Scholar]
- de Wijs‐Meijler DP, Duncker DJ, Tibboel D, Schermuly RT, Weissmann N, Merkus D & Reiss IKM (2017). Oxidative injury of the pulmonary circulation in the perinatal period: Short‐ and long‐term consequences for the human cardiopulmonary system. Pulm Circ 7, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, Shido K, Rabbany SY & Rafii S (2011). Endothelial‐derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 147, 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LW, Faber B, Callanan C, Freezer N, Ford GW & Davis NM (2006). Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics 118, 108–113. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver‐Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell‐Braxton L, Hillan KJ & Moore MW (1996). Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380, 439–442. [DOI] [PubMed] [Google Scholar]
- Friedrich L, Pitrez PMC, Stein RT, Goldani M, Tepper R & Jones MH (2007). Growth rate of lung function in healthy preterm infants. Am J Respir Crit Care Med 176, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Cornfield DN, Stenmark KR, Thébaud B, Abman SH & Raj JU (2016). Unique aspects of the developing lung circulation: structural development and regulation of vasomotor tone. Pulm Circ 6, 407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KN, Beshish AG, Barton GP, Haraldsdottir K, Levin TS, Tetri LH, Battiola TJ, Mulchrone AM, Pegelow DF, Palta M, Lamers LJ, Watson AM, Chesler NC & Eldridge MW (2018). Early pulmonary vascular disease in young adults born preterm. Am J Respir Crit Care Med (in press; 10.1164/rccm.201710-2016OC). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KN, Cucci AR, Fisher AJ, Albrecht M, Frump A, Tursunova R, Gao Y, Brown MB, Petrache I, Tepper RS, Ahlfeld SK & Lahm T (2015a). Neonatal hyperoxic lung injury favorably alters adult right ventricular remodeling response to chronic hypoxia exposure. Am J Physiol Lung Cell Mol Physiol 308, L797–L806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KN, Kumari S, Tetri LH, Barton G, Braun RK, Hacker TA & Eldridge MW (2017). Postnatal hyperoxia exposure durably impairs right ventricular function and mitochondrial biogenesis. Am J Respir Cell Mol Biol 56, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KN, Tepper RS, Lahm T & Ahlfeld SK (2015b). Increased cardiac output and preserved gas exchange despite decreased alveolar surface area in rats exposed to neonatal hyperoxia and adult hypoxia. Am J Respir Cell Mol Biol 53, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannaford K, Todd DA, Jeffery H, John E, Blyth K & Gilbert GL (1999). Role of Ureaplasma urealyticum in lung disease of prematurity. Arch Dis Child Fetal Neonatal Ed 81, F162–F167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden LP, Hobbs BD, Cohen RT, Wise RA, Checkley W, Crapo JD & Hersh CP; COPDGene Investigators (2015). Childhood pneumonia increases risk for chronic obstructive pulmonary disease: the COPDGene study. Respir Res 16, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes D Jr, Tobias JD, Mansour HM, Kirkby S, McCoy KS, Daniels CJ & Whitson BA (2014). Pulmonary hypertension in cystic fibrosis with advanced lung disease. Am J Respir Crit Care Med 190, 898–905. [DOI] [PubMed] [Google Scholar]
- Hibbs AM, Johnson NL, Rosen CL, Kirchner HL, Martin R, Storfer‐Isser A & Redline S (2008). Prenatal and neonatal risk factors for sleep disordered breathing in school‐aged children born preterm. J Pediatr 153, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman N, Kallapur SG & Jobe A (2012). Physiology of transition from intrauterine to extrauterine life. Clin Perinatol 39, 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian CG, Gonzales M, Rodriguez A, Bellido D, Salmon CS, Ladenburger A, Reardon L, Vargas E & Moore LG (2015). Perinatal hypoxia increases susceptibility to high‐altitude polycythemia and attendant pulmonary vascular dysfunction. Am J Physiol Heart Circ Physiol 309, H565–H573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaner RJ, Ladetto JV, Singh R, Fukuda N, Matthay MA & Crystal RG (2000). Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am J Respir Cell Mol Biol 22, 657–664. [DOI] [PubMed] [Google Scholar]
- Kardon G, Ackerman KG, McCulley DJ, Shen Y, Wynn J, Shang L, Bogenschutz E, Sun X & Chung WK (2017). Congenital diaphragmatic hernias: from genes to mechanisms to therapies. Dis Model Mech 10, 955–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara Y, Tuder RM, Taraseviciene‐Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J & Voelkel NF (2000). Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106, 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha SJ, Watkins WJ, Heron J, Henderson J, Dunstan FD & Kotecha S (2010). Spirometric lung function in school‐age children: effect of intrauterine growth retardation and catch‐up growth. Am J Respir Crit Care Med 181, 969–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer BW, Kallapur S, Newnham J & Jobe AH (2009). Prenatal inflammation and lung development. Semin Fetal Neonatal Med 14, 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie SS, Elliott JE, Beasley KM, Mangum TS, Goodman RD, Duke JW, Gladstone IM & Lovering AT (2018). Exaggerated increase in pulmonary artery pressure during exercise in adults born preterm. Am J Respir Crit Care Med 197, 821–823. [DOI] [PubMed] [Google Scholar]
- Levine RJ, Maynard SE, Qian C, Lim K‐H, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP & Karumanchi SA (2004). Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med 350, 672–683. [DOI] [PubMed] [Google Scholar]
- Lewandowski AJ, Augustine D, Lamata P, Davis EF, Lazdam M, Francis J, McCormick K, Wilkinson AR, Singhal A, Lucas A, Smith NP, Neubauer S & Leeson P (2013a). Preterm heart in adult life: cardiovascular magnetic resonance reveals distinct differences in left ventricular mass, geometry, and function. Circulation 127, 197–206. [DOI] [PubMed] [Google Scholar]
- Lewandowski AJ, Bradlow WM, Augustine D, Davis EF, Francis J, Singhal A, Lucas A, Neubauer S, McCormick K & Leeson P (2013b). Right ventricular systolic dysfunction in young adults born preterm. Circulation 128, 713–720. [DOI] [PubMed] [Google Scholar]
- McEvoy CT, Milner KF, Scherman AJ, Schilling DG, Tiller CJ, Vuylsteke B, Shorey‐Kendrick LE, Spindel ER, Schuff R, Mitchell J, Peters D, Metz J, Haas D, Jackson K, Tepper RS & Morris CD (2017). Vitamin C to Decrease the Effects of Smoking in Pregnancy on Infant Lung Function (VCSIP): Rationale, design, and methods of a randomized, controlled trial of vitamin C supplementation in pregnancy for the primary prevention of effects of in utero tobacco smoke exposure on infant lung function and respiratory health. Contemp Clin Trials 58, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath‐Morrow SA, Lauer T, Collaco JM, Yee M, O'Reilly M, Mitzner W, Neptune E, Wise R & Biswal S (2011). Neonatal hyperoxia contributes additively to cigarette smoke‐induced chronic obstructive pulmonary disease changes in adult mice. Am J Respir Cell Mol Biol 45, 610–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BA & Abman SH (2017). Translational advances in the field of pulmonary hypertension. focusing on developmental origins and disease inception for the prevention of pulmonary hypertension. Am J Respir Crit Care Med 195, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestan KK, Check J, Minturn L, Yallapragada S, Farrow KN, Liu X, Su E, Porta N, Gotteiner N & Ernst LM (2014). Placental pathologic changes of maternal vascular underperfusion in bronchopulmonary dysplasia and pulmonary hypertension. Placenta 35, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M & Bland RD (2010). Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298, L23–L35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery‐Downs HE, Young ME, Ross MA, Polak MJ, Ritchie SK & Lynch SK (2010). Sleep‐disordered breathing symptoms frequency and growth among prematurely born infants. Sleep Med 11, 263–267. [DOI] [PubMed] [Google Scholar]
- Mourani PM, Sontag MK, Younoszai A, Miller JI, Kinsella JP, Baker CD, Poindexter BB, Ingram DA & Abman SH (2015). Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med 191, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JD, Rabinovitch M, Goldstein JD & Reid LM (1981). The structural basis of persistent pulmonary hypertension of the newborn infant. J Pediatr 98, 962–967. [DOI] [PubMed] [Google Scholar]
- Naumburg E, Axelsson I, Huber D & Soderstrom L (2015). Some neonatal risk factors for adult pulmonary arterial hypertension remain unknown. Acta Paediatr 104, 1104–1108. [DOI] [PubMed] [Google Scholar]
- Naumburg E, Söderström L, Huber D & Axelsson I (2017). Risk factors for pulmonary arterial hypertension in children and young adults. Pediatr Pulmonol 52, 636–641. [DOI] [PubMed] [Google Scholar]
- Papamatheakis DG, Blood AB, Kim JH & Wilson SM (2013). Antenatal hypoxia and pulmonary vascular function and remodeling. Curr Vasc Pharmacol 11, 616–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson DL, Dawling S, Walsh WF, Haines JL, Christman BW, Bazyk A, Scott N & Summar ML (2001). Neonatal pulmonary hypertension – urea‐cycle intermediates, nitric oxide production, and carbamoyl‐phosphate synthetase function. N Engl J Med 344, 1832–1838. [DOI] [PubMed] [Google Scholar]
- Peng T & Morrisey EE (2013). Development of the pulmonary vasculature: Current understanding and concepts for the future. Pulm Circ 3, 176–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Tian Y, Boogerd CJ, Lu MM, Kadzik RS, Stewart KM, Evans SM & Morrisey EE (2013). Coordination of heart and lung co‐development by a multipotent cardiopulmonary progenitor. Nature 500, 589–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyter A‐C, Delhaes F, Diaceri G, Menétrey S, Tolsa J‐F (2014). Perinatal nitric oxide therapy prevents adverse effects of perinatal hypoxia on the adult pulmonary circulation. BioMed Res Int 2014, 949361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U & Sartori C (2011). Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol 301, H247–H252. [DOI] [PubMed] [Google Scholar]
- Rimoldi SF, Sartori C, Rexhaj E, Bailey DM, de Marchi SF, McEneny J, Arx R, Cerny D, Duplain H, Germond M, Allemann Y & Scherrer U (2015). Antioxidants improve vascular function in children conceived by assisted reproductive technologies: A randomized double‐blind placebo‐controlled trial. Eur J Prev Cardiol 22, 1399–1407. [DOI] [PubMed] [Google Scholar]
- Rozance PJ, Seedorf GJ, Brown A, Roe G, O'Meara MC, Gien J, Tang JR & Abman SH (2011). Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol 301, L860–L871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez‐Roisin R & Barbera JA (2002). Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 19, 632–638. [DOI] [PubMed] [Google Scholar]
- Sartori C, Allemann Y, Trueb L, Delabays A, Nicod P & Scherrer U (1999). Augmented vasoreactivity in adult life associated with perinatal vascular insult. Lancet 353, 2205–2207. [DOI] [PubMed] [Google Scholar]
- Scherrer U, Rimoldi SF, Rexhaj E, Stuber T, Duplain H, Garcin S, de Marchi SF, Nicod P, Germond M, Allemann Y & Sartori C (2012). Systemic and pulmonary vascular dysfunction in children conceived by assisted reproductive technologies. Circulation 125, 1890–1896. [DOI] [PubMed] [Google Scholar]
- Shimoya K, Taniguchi T, Matsuzaki N, Moriyama A, Murata Y, Kitajima H, Fujimura M & Nakayama M (2000). Chorioamnionitis decreased incidence of respiratory distress syndrome by elevating fetal interleukin‐6 serum concentration. Hum Reprod 15, 2234–2240. [DOI] [PubMed] [Google Scholar]
- Suresh S, O'Callaghan M, Sly PD & Mamun AA (2015). Impact of childhood anthropometry trends on adult lung function. Chest 147, 1118–1126. [DOI] [PubMed] [Google Scholar]
- Thebaud B & Abman SH (2007). Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 175, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Marter LJ, Dammann O, Allred EN, Leviton A, Pagano M, Moore M & Martin C (2002). Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr 140, 171–176. [DOI] [PubMed] [Google Scholar]
- Vollsaeter M, Roksund OD, Eide GE, Markestad T & Halvorsen T (2013). Lung function after preterm birth: development from mid‐childhood to adulthood. Thorax 68, 767–776. [DOI] [PubMed] [Google Scholar]
- von Arx R, Allemann Y, Sartori C, Rexhaj E, Cerny D, de Marchi SF, Soria R, Germond M, Scherrer U & Rimoldi SF (2015). Right ventricular dysfunction in children and adolescents conceived by assisted reproductive technologies. J Appl Physiol (1985) 118, 1200–1206. [DOI] [PubMed] [Google Scholar]
- Wadhwa PD, Buss C, Entringer S & Swanson JM (2009). Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med 27, 358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterberg KL, Demers LM, Scott SM & Murphy S (1996). Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 97, 210–215. [PubMed] [Google Scholar]
- Weir EK, Lopez‐Barneo J, Buckler KJ & Archer SL (2005). Acute oxygen‐sensing mechanisms. N Engl J Med 353, 2042–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann N, Grimminger F & Seeger W (2012). Smoking: Is it a risk factor for pulmonary vascular diseases? Pulm Circ 2, 395–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee M, White RJ, Awad HA, Bates WA, McGrath‐Morrow SA & O'Reilly MA (2011). Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol 178, 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GZ, Aye CY, Lewandowski AJ, Davis EF, Khoo CP, Newton L, Yang CT, Al Haj Zen A, Simpson LJ, O'Brien K, Cook DA, Granne I, Kyriakou T, Channon KM, Watt SM & Leeson P (2016). Association of maternal antiangiogenic profile at birth with early postnatal loss of microvascular density in offspring of hypertensive pregnancies. Hypertension 68, 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian RT, Hedlin H, Greuenwald P, Wilson DM, Segal JI, Jorden M, Kudelko K, Liu J, Hsi A, Rupp A, Sweatt AJ, Tuder R, Berry GJ, Rabinovitch M, Doyle RL, De Jesus Perez V & Kawut SM (2018). Features and outcomes of methamphetamine associated pulmonary arterial hypertension. Am J Respir Crit Care Med 197, 788–800. [DOI] [PMC free article] [PubMed] [Google Scholar]