

Abstract
Raf kinases are downstream effectors of small GTPase Ras. Mutations in Ras and Raf are associated with a variety of cancers and genetic disorders. Of the three Raf isoforms, cRaf is most frequently involved in tumor initiation by Ras. Cytosolic Raf is auto-inhibited and becomes active upon recruitment to the plasma membrane. Since the catalytic domain of Raf is its kinase domain, we ask the following: does the kinase domain of Raf has potential to interact with membrane and if yes, what role does the membrane interaction play? We present a model of cRaf kinase domain in complex with a heterogeneous membrane bilayer using atomistic molecular dynamics simulation. We show that the kinase domain of cRaf has three distinct membrane-interacting regions: a polybasic motif (R.RKTR) from the regulatory αC-helix, an aromatic/hydrophobic cluster from the N-terminal acidic region (NtA) and positively charged/aromatic cluster from the activation segment (AS). We show that residues from these regions form an extended membrane-interacting surface that resembles the membrane-interacting residues from known membrane-binding domains. Activating phosphorylatable regions (NtA and AS), make direct contact with the membrane whereas R.RKTR forms specific multivalent salt bridges with PA. PA lipids dwell for longer times around the R.RKTR motif. Our results suggest that membrane interaction of monomeric cRaf kinase domain likely orchestrates the Raf activation process and modulates its function. We show that R.RKTR is a hotspot that interacts with membrane when cRaf is monomeric and becomes part of the interface upon Raf dimerization. We propose that in terms of utilizing a specific hotspot to form membrane interaction and dimer formation, both Raf and its upstream binding partner KRas, are similar.
Introduction
Raf kinases are downstream binding partners of Ras1. Ras-Raf activates MAPK signaling pathway involved in a variety of cell signaling processes2. Ras shuttles between GTP-bound or active and GDP-bound or inactive states, is prenylated, and binds with membrane for its normal function3–5. Mammalian Raf kinases exist in three isoforms, a-, b- and cRaf1. cRaf is the most important isoform involved in the tumor initiation by Ras6,7. GTP-bound Ras recruits Raf to the plasma membrane8,9. Raf is a multi-domain protein made up of a Ras-binding domain (RBD), a cysteine-rich domain (CRD), a linker region, and a catalytic kinase domain (KinaseD) (Fig. 1). The functional unit of Raf is a dimer formed by the kinase domain of two Raf protomers10. Either a homo- or hetero-dimer is formed by almost all possible combinations of the three isoforms with heterodimers specifically between cRaf and bRaf being the most common11,12. The N-lobe of Raf KinaseD consists of a regulatory helix, αC-helix, and functional modulation of Raf kinase activity by the spatial location of αC-helix is well-known13,14. αC-helix in cRaf:KinaseD harbors an RKTR motif that is directly involved in the Raf dimer interface formation10. RBD and CRD together form an N-terminal regulatory region15. Cytosolic Raf is inactive, and exists in an auto-inhibited state wherein the N-terminal regulatory region engages the C-terminal kinase domain, thereby masking and rendering it inactive15. The auto-inhibition is released upon a Ras-dependent recruitment of Raf to the plasma membrane evoked by interaction of Raf-RBD with Ras-GTP, leading to unmasking of the kinase domain8,9.
Figure 1.

The domain architecture of cRaf (A) and mTOR (B) kinases. Raf has a Ras-binding domain (RBD), a cysteine-rich domain (CRD) and a catalytic kinase domain (KinaseD) containing an αC-helix in the N-lobe of the KinaseD. The N-terminus of mTOR is made up of FAT domain followed by a kinase domain, N-lobe of which has a FRB (FKBP12-Rapamycin-binding) domain. The membrane-bound peptides and protein domains which are subjected to MD simulations are shown by numbers (1), (2) and (3).
Studies show that tethering cRaf with the minimal membrane anchor of K-Ras containing prenylation motif, CAAX, henceforth referred to as tK-CAAX, results in the recruitment of Raf to the plasma membrane in a Ras-independent manner8,9. Tethering tK-CAAX to an isolated kinase domain of Raf alleviates the need for release of auto-inhibition, possibly because the N-terminal regulatory region is no longer present, and this construct mimics a Ras-induced state of Raf16. The crystal structure of RBD17, CRD18 and kinaseD11 of cRaf have been solved in isolation; however, the long linker region (~200 residues long) connecting the N-terminal regulatory region with the kinaseD is not yet known. While RBD’s primary role is to interact with membrane-bound GTP-Ras19, the CRD, which is structurally and functionally similar to a membrane-interacting C1 domain, also interacts with lipids18,20. Recently, complexes of Ras and Ras:Raf-RBD-CRD with membrane have been investigated using a number of methods including computational, spectroscopic and biochemical approaches21–28. This raises an intriguing question: does the kinase domain of Raf also interact with the membrane?
Previous studies suggested the presence of a PA (phosphatidic acid) interacting region in the cRaf:kinaseD29,30. A lipid-interacting region has also been observed in another kinase, mTOR (mammalian target of Rapamycin)31. mTOR is a downstream effector of the Ras homologue in brain (Rheb) and, like Raf, it has a catalytic kinase domain (Fig. 1)32. The αC helix in cRaf and the FRB domain in mTOR, which lie in the N-lobe of kinase domain, harbors this potential PA-interacting region (Fig. 1). While the spatial location of αC-helix is a well-known functional modulator of Raf kinase activity13,14, mutation in the PA-interacting region within the FRB domain correlated with a ~60% reduction in the mTOR downstream signaling activity31. This is suggestive of an important role of membrane in modulating the function of cRaf and mTOR.
Based upon these observations, we hypothesize that the kinase domain of cRaf as well as mTOR harbors a membrane-interacting region that is distinct from their previously characterized membrane interacting regions/motifs. To test this hypothesis, we first performed molecular dynamics (MD) simulations of the putative PA-interacting regions of cRaf and mTOR, and the full-length kinase domain of cRaf attached to tK-CAAX, in a binary lipid bilayer containing 12% PA and 88% PC. The latter construct ensures membrane localization in a Ras-independent manner8,9,16. Our results show that a polybasic R.RKTR motif from the regulatory αC-helix, an aromatic/hydrophobic cluster from the N-terminal acidic region (NtA), and aromatic/positively charged residues from the activation segment (AS) in the kinase domain of cRaf make direct contact with the membrane. R391 and 398-RKTR (R.RKTR) is PA-specific, and PA lipids dwell for long near this cluster. Polybasic cluster-PA interactions are stabilized by multivalent salt bridges. αC-helix and activating phosphorylatable regions (NtA and AS)1 are in direct contact with the membrane throughout the simulations. Together, these three regions project an extended membrane interacting surface containing positively charged, aromatic, and hydrophobic residues, that resembles the membrane-interacting residues from other established membrane-binding domains (e.g.33). The functional implication of KinaseD-membrane interaction likely lies in priming Raf for activation by properly positioning phosphorylatable regions and αC-helix for phosphorylation and dimerization, considering that membrane targeting enhances phosphorylation and that phosphorylation precedes dimerization34,35. To our knowledge, this is the first study that presents a detailed atomistic model of the complex between cRaf kinase domain and a heterogeneous membrane.
Methods
We constructed a membrane bilayer patch containing 366 POPC (88%) and 50 POPA (12%) lipids using CHARMM-GUI36 and subjected it to equilibration as described below. Using this bilayer, molecular dynamics (MD) simulations were carried out on (1) the potential PA-interacting regions of cRaf and mTOR kinase domains (cRaf αC-Pept and mTORFRB-pept), (2) full-length cRaf kinase domain (cRaf KinaseD), and (3) full-length cRaf KinaseD glued to a modeled C-terminus of cRaf that in turn is linked to the prenylated C-terminus of K-Ras () (Table S1).
Preparation of initial models
cRaf αC-Pept and mTORFRB-pept: The potential PA-interacting regions of cRaf (residues 389–426) and mTOR (residues 2101–2137) were taken from 3OMV37 and 4JSV32, respectively (Fig. 1). Each peptide was simulated in a PC/PA bilayer. cRaf αC-Pept simulation contained four cRaf peptides (two each in the top and bottom leaflets) and mTORFRB-pept simulation contained two mTOR peptides (one each in the top and bottom leaflets). For cRaf, the peptides were placed with their centers of mass approximately 60 Å apart in a box of length 120 × 120 Å in the x-y dimension. In each simulation, the peptides were positioned near the interface of a PC/PA bilayer model with their hydrophilic residues in close proximity to the lipids.
cRaf KinaseD: Chain A of the kinase domain of cRaf from 3OMV37 was used as the starting structure (residues 340–615). This and the majority of other crystal structures of kinases have missing activation segment (AS) (residues 492–503), which was modeled using Modeller 9.1138. While inactive Raf kinase has an ordered AS39, here, our primary goal is to probe the membrane interaction of an active monomer that will likely undergo dimerization. Therefore, we selected an AS model with a completely disordered conformation. The position of ATP molecule and magnesium ions was determined by a structural alignment of the cRAF kinase domain and cyclic AMP dependent protein kinase (PDB: 4DFX40). The starting structure for the cRaf KinaseD - membrane complex was obtained by docking the kinase domain onto one of the membrane-bound peptides in the last snapshot of the cRaf αC-Pept simulation.
: Studies have shown that the kinase domain of cRaf is recruited to the plasma membrane in a Ras-independent manner if a CAAX motif of K-Ras (tK-CAAX) is attached at its C-terminus8,9,16. Therefore, first we modeled the C-terminus of cRaf (Fig. 1) using PEPFOLD41 (residues 615–648). We obtained five low energy models with a full or partial helical content. We picked the first lowest energy model, which was completely helical. Also, a protein blast search of the C-terminus sequence against protein-data bank yielded a hit with an alpha-helical structure having 50% sequence similarity with the query. Additionally, other studies have shown that the C-terminus of other kinases, including mTOR (FATC, Fig. 1), adopts a helical structure upon binding to membrane-mimetic micelles42. A farnesylated K-Ras lipid anchor made up of 17 C-terminal amino acids (tk-CAAX) obtained from our previous study of membrane-bound K-Ras23 was added onto the modeled C-terminus of cRaf. The KinaseD from this construct and the last snapshot obtained from cRaf KinaseD simulation were structurally aligned, and farnesyl was pulled inside the PA/PC membrane. This system is referred as (Fig. 1, Table S1).
Molecular dynamics simulation
The systems were energy minimized for 2000 steps with lipids and proteins fixed and equilibrated for 200 ps with the lipid phosphate and heavy atoms of proteins harmonically restrained with a force constant k = 4 kcal/mol/Å2. The k was gradually scaled by 0.75, 0.50, 0.25 and 0 with a time step of 1 fs. Production run was performed using a time step of 2 fs using SHAKE to restrain all bonds involving hydrogen atoms43. PME was used for electrostatics with 12 and 14 Å cutoffs for non-bonded interactions44. NPT ensemble was used with constant pressure maintained at 1 bar by the Nose-Hoover Langevin piston method and temperature of 310 K controlled by Langevin thermostat. The CHARMM36 force field for proteins45 and lipids46 was used with CMAP dihedral correction for proteins47,48. Simulations were run with the NAMD2.11 program49. cRaf αC-Pept and mTORFRB-pept were simulated for ~1 μs. Each of cRaf KinaseD and was simulated for 500 ns (Table S1).
Analysis
Encounter between the peptide and lipid is defined as a contact when heavy atoms of proteins are within a distance, d, of 4 Å of any heavy atom of the lipid headgroups. Residence time of PA (tres) around those protein residues that are in contact with the membrane for >50% of simulation time is calculated as follows:
| 1 |
where, ti and tf are the initial and final time with Δt = 10 ps. Aggregate residence time is defined as Σtres.
Results
Sequence and structural analysis predicts a membrane-interacting region in the kinase domain of cRaf
There are several protein domains that have a high propensity of interacting with the membranes33,50. Some examples include C1, C2, PH and FYVE domains50. Some of these have a defined lipid-binding pocket that facilitates interaction with the membrane by recognizing specific membrane components such as PS, phosphoinositides, phorbol acetate, or even ions (e.g. PKCδ51, Fig. 2A)33,50. Others have no well-defined lipid-binding pockets but rather contain a positively charged surface patch (e.g. FERM, PDZ, PTB, ANTH, Spectrin52; Fig. 2B)33,50. Even the same domain type in different multi-domain proteins can have either a well-defined lipid binding pocket or a positively charged surface patch. For example, PH domain forms a lipid-binding pocket in SOS53 and a relatively solvent exposed surface patch of positively charged residues in Spectrin52. Note that a surface patch or pocket can be formed by the spatial proximity of residues from distal secondary structure elements (separated by double dots in Table 1).
Figure 2.
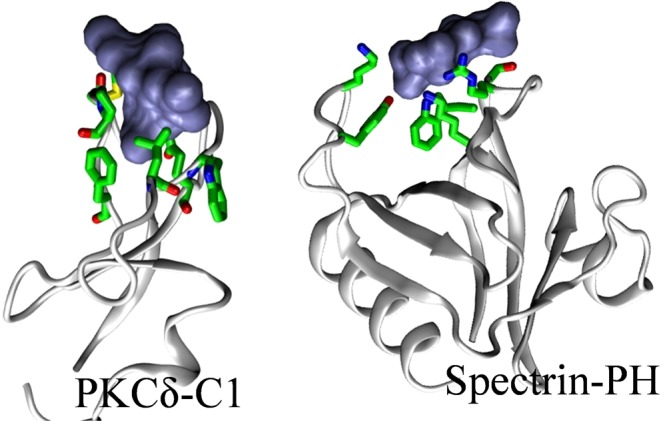
Membrane-interacting residues (green sticks) in C1 (A) and PH (B) domains of PKCδ (1PTR) and Spectrin (1BTN), respectively, in complex with phorbol acetate and inositol phosphate (blue surface). Color scheme: protein (gray), nitrogen (blue), oxygen (red), carbon (green).
Table 1.
Known and predicted membrane interacting residues from membrane-binding protein domains.
| Protein | Domain | Membrane-interacting residues* |
|---|---|---|
| PKCα | C2 | K.K..KY..W..N |
| Syntaxin1A | BAR-like | KK.KY.K.RRKK |
| SOS | PH | K..R..K..R..FF..Y |
| PKCδ | C1 | M.P.F.L.W.Q# |
| Kcc4p (yeast) | KA | K..K..K..K..ILFL..KK |
| MARK/PAR1 kinase | KA1 | R.R..R..KR |
| Spectrin | PH | K..R.W..Y.K |
| cRaf | Kinase | R.RK.R& |
| mTOR | FRB-kinase | R.R.K& |
*Single dot = residues from same secondary structure element; double dot = residues from different secondary structure elements; No dot = residues are contiguous.
#An exception containing hydrophobic and aromatic residues only.
&Predicted from our study.
We observe that, for the selected known membrane-binding domains, the membrane-interacting residues can be classified roughly into three groups: (1) largely positively charged (e.g. syntaxin 1A, MARK/PAR1), (2) positive and aromatic/hydrophobic (e.g. PKCα, SOS, spectrin) and (3) polar, aromatic and hydrophobic (e.g. PKCδ) (Table 1). In line with this, we observed a positively charged cluster, 391R-398-RKTR and 2108-RRISK, in the suggested PA-interacting region of cRaf and mTOR, respectively (Table 1). Aromatic and/or hydrophobic residues either alone or in combination with positively charged residues are not unexpected given that the aromatic residues prefer lipid-water interfaces while hydrophobic residues can insert into the membrane, as suggested recently for a newly identified membrane-interacting kinase associated domain-1 (KA1)54,55.
Furthermore, subjecting the PA-interacting region of cRaf (390-FRNEVAVLRKTRHVNILLFMGYMTKDNLAIVTQWCEG) to a blast search against the non-redundant protein database identified other serine/threonine kinases (STK) and STK- or Raf-like kinases with a similar region, highlighting a conserved sequence motif containing a positively charged residue cluster, RxEx4R(K)K(R)T(L)R, where x is any amino acid (Fig. 3). All Raf isoforms contain the motif R(K)xEx4RKTR (Fig. 3). The sequences analyzed here are from diverse organisms ranging from plants, drosophila to humans (Table S2). Direct interaction of the catalytic kinase domain of aRaf and cRaf with the membrane has been suggested29,56 and evidence exists for membrane localization of two other kinases: KSR and CTR157,58. However, the kinase domains of KSR and CTR1 are not shown to interact with the membrane directly but rather via their N-terminal region either directly (like Raf-CRD) or indirectly (like Raf-RBD). Based upon this and together with the presence of a putative membrane-interacting sequence motif in most kinase domains (Fig. 3), one can speculate that direct membrane interaction of kinase domains is generalizable.
Figure 3.

Multiple sequence alignment of selected sequences. A conserved putative membrane-interacting sequence motif, RxEx4R(K)K(R)T(L)R, is observed among all Raf kinases. (see Supplementary Information).
In addition to the involvement of RxEx4R(K)K(R)T(L)R in the formation of dimer interface in the Raf kinases10 and likely in other kinases as well, the combined observation from our sequence and structure-based analysis is suggestive of the presence of a putative membrane-interacting region in the catalytic domain of kinases in general, and specifically in cRaf and mTOR kinases (Table 1). Focusing on the polybasic residues of cRaf, henceforth, we write RxEx4RKTR as R.RKTR.
R.RKTR in cRaf and RRISK in mTOR are membrane-interacting motifs
We performed MD simulations on the potential PA-interacting segments of cRaf and mTOR, cRaf αC-Pept and mTORFRB-pept, respectively, in a PA/PC bilayer model membrane. We ensured that the individual peptides in cRaf αC-Pept and mTORFRB-pept do not interact amongst themselves and thus can be considered as four independent peptide-membrane systems (Fig. S1). Since the lipid distribution is also symmetric (each leaflet contains 183 randomly placed POPC and 25 POPA lipids), the addition of four or two peptides did not perturb the global structure of the bilayer. For example, the average area per lipid was ~62.4 Å2, which is in agreement with POPC-dominated binary lipid mixtures59. Therefore, we performed our analysis by taking an average over the four (cRaf) and the two (mTOR) peptides, unless mentioned otherwise.
Both cRaf αC-Pept and mTORFRB-pept form stable interactions with the membrane throughout the simulation, with 6 to 12 residues being in direct contact with the PC or PA lipids (Fig. S2). In the beginning of the simulations, the peptides were placed with their hydrophilic residues facing the membrane. The peptides were further drawn towards the membrane during the initial simulation stage. Therefore, though we did not expect an increase in the number of residues that are in contact with lipids with time, we wanted to assess how stable the peptide-membrane interaction is, and if it occurs via specific residues. To do so, we focused on the helical region of both cRaf αC-Pept and mTORFRB-pept that contains a polar face made up of positively charged residues that may interact with the negatively charged environment provided by the anionic lipids (Fig. 4, left). All peptides in cRaf αC-Pept and mTORFRB-pept remain bound to membrane throughout the simulation via this charged surface (Fig. 4, middle). One of the peptides in mTORFRB-pept dissociated and re-associated with the membrane (and hence the lower average probability of contact in Fig. 4, middle), but it remained stably bound for the last ~500 ns. While re-associating, it made interactions via the same charged surface as before (Fig. 4, left and middle).
Figure 4.

Peptide-lipid interactions in cRaf (A) and mTOR. (B) The helices in both cRaf αC-Pept and mTORFRB-pept are amphipathic (left). Average probability of contact between protein and lipids (middle). Contact is defined when any protein heavy atoms is within 4Å of heavy atoms of lipid headgroups. A snapshot showing residues in contact with membrane for >50% of the simulation length (right). Color scheme: (left)-charged surface (blue transparent ovals), basic residues (blue), acidic residues (red), hydrophobic (white), others (green); (right)-helical region (orange), carbon (yellow), residues in contact with membrane are highlighted by blue points. The membrane is shown in gray sticks.
Charged residues from the helical region involved in stable protein-lipid contacts are R391, R398, K399 and R401 in cRaf and H2106, R2109, R2110 and K2113 in mTOR (Fig. 4, middle). The former corresponds to our sequence motif, R.RKTR, and the latter, to the RRISK of mTOR. In the latter, R2134, a non-helical residue, also interacts with membrane but here we focus primarily on the helical region. This is because non-helical regions, owing to their higher flexibility, may show different dynamics when the full-length protein is considered. The helical residues made stable interactions with the POPA and POPC phospholipids for >50% of the simulation time (Fig. 4, middle and right). Henceforth, we refer to R391 and 398-RKTR (R.RKTR) either as a polybasic cluster or membrane-interacting sequence motif of cRaf and use these two terms interchangeably throughout the text. Similarly, R2109, R2110 and K2113 of mTOR is referred to as mTOR’s polybasic cluster (Fig. 4, middle). The interaction pattern resembles a perfect amphipathic helix where hydrophobic residues L2103, Y2104, V2107, F2108, I2111, L2115 do not interact with the membrane and are involved in hydrophobic packing interaction in the four-helix bundle of the FRB domain.
Recently, studies suggesting a role of membrane in modulating the function of membrane-bound small GTPases have emerged. This concept is termed as membrane orientation dynamics (or membrane reorientation) according to which specific protein surfaces, often containing positively charged residues, interact directly and transiently with the membrane21,60. As a result, the proteins adopt distinct and specific orientation with respect to the membrane (e.g.60). These distinct orientations in turn have been related to variable functional outputs. Membrane orientation dynamics has been observed in Ras (N-, H- and K-Ras)21–24, other members of the Ras superfamily such as Arf and Rheb61,62, and the membrane-interacting CRD domain of Raf kinases25,28. The protein-membrane interaction during membrane reorientation serves as a secondary interaction since the specific membrane-anchoring motifs (such as CAAX motif in K-Ras) provides the primary interaction with membrane. The helical region harboring the polybasic cluster in both cRaf αC-Pept and mTORFRB-pept adopts a perpendicular orientation with respect to the membrane normal (Fig. S3). The orientation of the helical region is found conserved in the full-length kinase domain, , and possibly modulates the activation of Raf (section 4). We believe that under physiological conditions, the observed interaction between αC-helix of cRaf and FRB domain of mTOR with the membrane is secondary since membrane-interacting motifs provide the primary attachment in both mTOR42,63,64 and cRaf5,17,29,65,66.
Our results show that the polybasic cluster in cRaf and mTOR acts as membrane-interacting motif. This strengthens our hypothesis that kinase domain has a membrane-interacting region. In subsequent sections, we focus our attention on cRaf.
R.RKTR motif is PA-specific
PA clusters around the R.RKTR motif of cRaf αC-Pept as observed from a 2D radial pair distribution function that shows a primary peak around ~4Å and secondary and tertiary peaks around ~4.8Å and ~5.7Å (Fig. 5A,B). Though only 12% of PA is present in the simulation, nearly 20% of the PA phospholipids are found near the R.RKTR motif (Fig. S4). Similar results are obtained for mTORFRB-pept(Fig. S5). Overall, we observe that the polybasic cluster interacts specifically with PA (Fig. 5). We observed surface “grooves” formed by multivalent interactions between PA and positively charged residues (Fig. 5C). These surface grooves are formed by the polybasic residues that engage the phosphate group of PA stably and for much longer times (Fig. 5C, Table S3). This is not completely unexpected given the complementarity between the R.RKTR motif and anionic phospholipid, PA.
Figure 5.

Two dimensional radial distribution function, g2D(r), of phosphorous atoms of PA (A) and PC (B) around specific atoms of the basic cluster residues. Nξ atom of K399 and Nδ atom of R391, R398 and R401 were used. The g2D(r) is averaged over all four peptides in the cRaf simulation, cRaf αC-Pept. The different colors show convergence using time-blocks 600–700 ns (black), 700–800 ns (red), 800–900 ns (blue), and 900 to 1000 ns (green). (C) Histogram of tres (residence time of PA around individual residues of the cRaf basic cluster). tres is calculated using Eq. 1. A surface “groove” formed by basic cluster residues e.g. R391 and R398 is shown in surface representation with the following color scheme: blue (+), red (−) and white (all hydrophobic) and green (all other amino acids). The phosphate of POPA is shown in red (oxygen) and tan (phosphorous) vdW spheres. (D) Multivalent salt-bridge interaction between a single PA lipid and two residues (R391 and R398) from the basic cluster.
Next, we focused our attention on the dynamics of specific PA molecules that interact with the residues in R.RKTR. A recent study reported a range of 50 to 100 ns as the residence time of POPA near a charged motif in an amphipathic helix of a protein67. We observe ~15 binding events with tres ≥ 40 ns, with the maximum residence time of a PA molecule, , is 125 ns near R398 (Figs 5C and S6). This suggests that PA’s dynamics is slowed down upon binding to R.RKTR. We must note that the values of the residence time are sensitive to the Δt (10 ps in this case) and the actual residence time in some cases will be >100 ns. As an example, for one of the peptides in cRaf αC-Pept, a single PA phospholipid remained bound to R398 for >300 ns with a Δt = 50 ps (Fig. S6). Note, the aggregate residence time of PA near R398 is >500 ns (Fig. S6).
Owing to the suggested PA-specificity of the αC-helix that harbors the R.RKTR motif, combined with the available knowledge that the αC-helix modulates Raf kinase function14 and that the dimerization of Raf involves the RKTR motif68, we reasoned that the membrane-interacting sequence motif of cRaf may play a functional role. We investigated this in the next section.
αC-helix, N-terminal acidic region (NtA) and activation segment (AS) are membrane-interacting regions of cRaf kinase domain
We studied the membrane interaction properties of the full-length kinase domain of cRaf with and without tK-CAAX, and cRafKinaseD, respectively. The former construct is biologically relevant as it localizes Raf on the plasma membrane in a Ras-independent manner16 (see introduction & methods). Despite the absence of tK-CAAX, once placed on the membrane surface, Raf remains bound to the bilayer throughout the simulation in cRaf KinaseD. Therefore, we merged the cRaf KinaseD and trajectories for further analysis.
Residues from the three regions of the catalytic kinase domain of cRaf made direct contact with the membrane for >80% of the simulation length. These are: (1) Y340, Y341, W342 at the NtA, (2) the R.RKTR motif of the αC-helix, and (3) R462, N463, S494, R495, W496 and S497 at the AS (Fig. 6). We observe that a combination of hydrophobic and aromatic residues from NtA and positive and aromatic/hydrophobic residues from AS contribute towards the membrane binding. These residues resemble the membrane-interacting residues from known membrane-binding domains (Table 1). Specifically, the NtA residues resemble PKCδ’s and AS’s resemble PKCα’s, while the R.RKTR motif resembles the KA1 domain from MARK/PAR1 kinase (Table 1, section 1). Together, the three lipid-binding regions of cRaf:KinaseD give rise to an extended membrane-interacting surface (Fig. 6C, brown boxes). Quite interestingly, R401 of the R.RKTR motif interacted only for ~35% time with the membrane in the full-length KinaseD and remained out of contact for the remaining 65%. This is interesting because R401 of RKTR motif plays a key role in Raf dimerization10.
Figure 6.

(A) Last snapshot of cRaf kinase domain (orange ribbons) bound to the PA/PC membrane (blue surface) from the simulation . Only the kinase domain is shown. Water is shown as green dots. αC-helix from the N-lobe of cRaf kinase domain is labeled. (B) Probability of the residue contact with the lipids in cRaf KinaseD plus . Cutoff criteria are same as in Fig. 4. Three regions are in direct contact with membrane phospholipids, N-terminal acidic region (NtA, yellow); the basic cluster (blue); and the activation segment (AS, green). (C) A snapshot from the simulation with membrane-interacting residues shown as sticks (C: green, N: blue; O: red). Protein is in orange and membrane in white surface representation. An extended membrane-interacting sequence code is highlighted in the brown box.
The αC-helix sits in an orientation perpendicular to the membrane normal, as observed in cRaf αC-Pept(Fig. S3). In kinases, a set of specific hydrophobic residues aligned together results in the formation of a regulatory or R-spine. In cRaf, F408, L397, F487 and H466 are involved in the formation of the R-spine. In our simulation, we observe R-spine residues aligned in a linear manner throughout the simulation. Additionally, we observe that W342, an NtA residue which is conserved among all Raf isoforms, extends the cRaf’s R-spine. The extended R-spine remains stable as well. A previous study suggested a similar Trp-mediated R-spine extension in bRaf dimers, though it was in the absence of membrane35. Since W342 interacts with the membrane throughout the simulation this suggests that the R-spine in cRaf is likely to adopt a preferred orientation with respect to the membrane plane.
PA lipids around the R.RKTR motif show a high residence of tres > 100 ns (Fig. S6). This is similar to our results from cRaf αC-Pept where the PA lipids showed longer dwell time near the polybasic cluster. In addition, residues from NtA (Y340; = 21 ns) and AS (R462; = 15 ns) also hold onto PAs with relatively longer dwell times (Fig. S6). In fact, a single PA lipid is shared among the residues belonging to the three distinct regions (Fig. 7). For example, the residues R398 and K399 (polybasic cluster) and Y340 (NtA) interact with a PA for ~152 ns (Fig. 7). Afterwards, while maintaining the interaction with the polybasic cluster residue(s), the same PA lipid interacts with the activation segment (R426).
Figure 7.
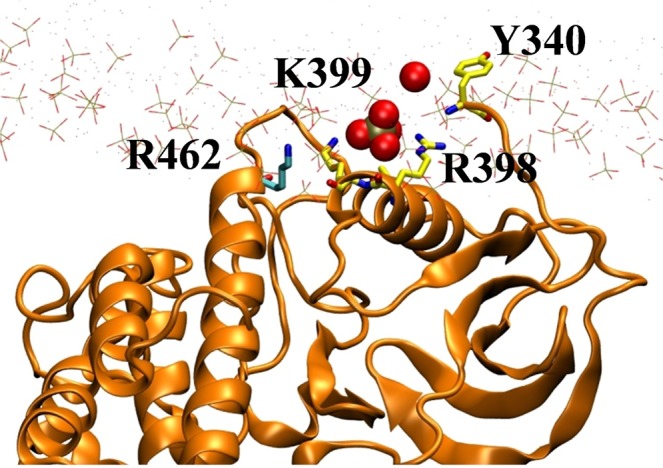
PA lipid shown in vdW spheres (red:oxygen and tan:phosphorous) with = 125 ns for R398 forms multivalent salt bridges with protein residues from three different regions of the cRaf:KinaseD including residues Y340 (NtA), R398 and K399 (basic cluster) and R462 (AS). The kinase domain is in orange ribbon, membrane is shown as lines with O: red, P: tan, N: blue and selected residues shown as sticks (C:cyan).
Therefore, we demonstrate that an extended membrane-interacting surface on the kinase domain of cRaf is formed by positively charged, aromatic and hydrophobic residues contributed by the three distinct regions (NtA, αC and AS). A previous study29 proposed that lipid-interaction of cRaf’s PA-interacting region is driven not just by electrostatics. Our observation of additional regions, NtA and AS, making direct contact with the membrane in addition to the αC-helix (R.RKTR motif) may provide a possible explanation for this observation. This is because interaction with multiple regions especially those containing key phosphorylation sites (NtA and AS, see discussion) makes the KinaseD-membrane interaction non-random.
Discussion
Some studies suggested a putative interaction between the phosphatidic acid (PA) and the kinase domain of the cRaf and mTOR kinases29–31. Such an interaction may either involve sequestration of PA by the protein or a direct interaction between protein and membrane. Studies indicated a possibility for the latter29–31. Based upon this, we first performed a comparative sequence and structural analysis on the known membrane-interacting domains such as C1, C2, PH and the putative PA-interacting regions of cRaf and mTOR (Figs 2 and 3). We observe a putative membrane-interacting sequence motif, RxEx4R(K)K(R)T(L)(Q)R conserved in Raf-like kinases across diverse organisms (Fig. 3). Our results showed the presence of a polybasic cluster in the suggested PA-interacting region of cRaf (R.RKTR) and mTOR (RRISK) kinase domains, that resembles the membrane-interacting residues from known membrane-binding domains (Fig. 3, Table 1). We thus hypothesized that the kinase domain of cRaf and mTOR has membrane-interacting regions. To investigate this, we performed MD simulations on the potential PA-interacting region of cRaf and mTOR (cRafαC-Pept and mTORFRB-pept, respectively) in an explicit PA (12%) and PC (88%) membrane bilayer model as a proof-of-principle (Fig. 1). cRaf αC-Pept and mTORFRB-pept remain stably bound to the PA/PC membrane throughout the simulation, and the polybasic clusters, R.RKTR of cRaf and RRISK in mTOR were found to be the membrane-binding residues (Fig. 4). In cRaf, PA clustered around the R.RKTR motif and PA lipids interacted with the polybasic cluster with larger dwell times (Fig. 5). These interactions were stabilized by multivalent salt bridges between a PA lipid and multiple charged residues (Fig. 5C,D, Table S3). Our results from cRaf αC-Pept simulations thus support our hypothesis that cRaf:KinaseD interacts stably with membrane via its R.RKTR motif.
In order to gain insight into kinaseD-lipid interaction in a more physiologically relevant context, we performed MD simulations of the full-length kinase domain of cRaf with and without tK-CAAX embedded in the same PA/PC membrane ( and cRaf KinaseD; see methods). Attaching tK-CAAX to cRaf mimics an auto-inhibition released state of Raf on the membrane8,9,16. Our results revealed that three distinct regions, namely NtA, αC-helix, and AS interact directly and stably with the membrane. Together, the three regions contributed towards the formation of an extended membrane-interacting surface involving a combination of aromatic, hydrophobic and positively charged residues: YYW..R.RKTR..RN.RW. We termed this extended surface because it is comprised of three different groups of the membrane-interacting residues in known membrane-binding domains (section 1, Table 1).
The disordered AS and the NtA regions, which are engaged in the protein-membrane interaction, contain activating phosphorylatable residues that are occluded by the membrane in our simulation. And yet membrane targeting has been shown to facilitate the phosphorylation of NtA residues34. It is possible that upon release of auto-inhibition and recruitment of Raf to the plasma membrane, the kinase domain interacts transiently with the membrane and thereby occlude its phosphorylatable residues and αC-helix that is required for dimerization. Then, tumbling of the kinase domain on the membrane would expose some of the activating phosphorylatable residues (e.g. NtA’s) to membrane-localized kinases. Even if slight, the repulsion between the anionic membrane and negative charges introduced by phosphorylation may expose the αC-helix for dimer formation. Upon completion of the phosphorylation, the NtA and AS regions likely experience conformational changes that prime Raf protomers for dimerization. Differential conformational dynamics of a membrane interacting phosphorylated peptide versus its non-phosphorylated counterpart has been observed69. Quite interestingly, R401, a key residue involved in Raf dimerization10 and whose mutation results in dimer disruption, interacts less efficiently with the membrane in the and cRaf KinaseD simulation (only 35%). Thus, we speculate that while R391, R398 and K399 act as anchor points to the membrane, R401 that lies in a flexible loop preceding the C-terminus of αC-helix, is free to participate in dimer formation.
Ras, upstream of Raf, is a single domain small GTPase having two lobes: lobe 1 or effector-lobe and lobe 2 or allosteric-lobe. We and others22,23, using computations and experiments, have shown that K-Ras adopts two distinct orientations with respect to membrane surface, one where lobe 2 directly interacts with membrane (OS1) and another where lobe 1 does so (OS2). Since lobe 1 is effector-interacting, occlusion of this surface by membrane in OS2 can affect its function. On the other hand, in OS1 helices 3 and 4 are in direct contact with the membrane, and the effector-lobe is free to interact with the downstream binding partners (Fig. 8). We also showed that the interface of one of the dominant K-Ras dimers, I1 dimer, is formed by helices 3 and 470 (Fig. 8). Our results from the current work show that the same RKTR motif in cRaf is involved in membrane interaction and in dimer formation (Fig. 8). This suggests that RKTR is a hotspot similar to the helices 3 and 4 of K-Ras, and interacts with membrane when cRaf is in the monomeric state and forms an interface upon Raf dimerization.
Figure 8.

An illustration showing similarities between KRAS and cRaf. Both contain hostpot regions, helix 3 (H3) and helix 4 (H4) in KRAS and RKTR motif in cRaf kinase domain (cRaf:KinaseD). In both, the hotspot interacts with the membrane when they are monomers. Both undergo dimerization with their hotspot regions lying at the dimer interface.
The interaction of monomeric cRaf with membrane, therefore, appears to be an important step in the activation of Raf. The formation of this complex possibly acts as an intermediate in the activation process and (1) reduces the search space of monomeric cRaf proteins to find other protomers for dimerization, (2) facilitates the interaction of phosphorylatable regions (NtA and AS) with membrane thereby positioning them to possibly get phosphorylated by membrane-localized kinases and (3) regulates the exposure of the αC-helix via which Raf dimerizes. These are suggested by a recent study that shows membrane targeting of cRaf enhances the phosphorylation of residues in NtA34. An interdependence of phosphorylation and activation of kinases by membrane binding has also been shown for another protein kinase, AKT71. Studies have also shown that phosphorylation precedes Raf dimerization34,35. Interaction of cRaf:KinaseD with specific membrane phospholipids may also serve as one of the factors responsible for unmasking the kinase domain after its Ras-dependent recruitment to the plasma membrane. To our knowledge, this is the first study explicitly showing the interaction between the catalytic kinase domain of cRaf and membrane, pinpointing the key regions with possible functional implications.
Conclusions
Previous studies suggested a possibility of interaction between the cRaf kinase domain and the PA lipids29,30. Molecular and atomistic details of such an interaction are missing. We present a modeled complex of the cRaf kinase domain bound to a PA/PC bilayer membrane. Our study shows R.RKTR motif is PA-specific and three distinct regions of the kinase domain of cRaf, the regulatory αC-helix, N-terminal acidic region (NtA) and activation segment (AS) interact directly with the membrane. The three regions, together, form an extended membrane-interacting surface that resembles the membrane-interacting residues from well-known membrane-binding domains. Activating phosphorylatable residues of Raf form stable contact with the membrane. We propose that while Raf is recruited to the plasma membrane primarily via its N-terminal domains, the catalytic kinase domain may transiently interact with the membrane and possibly modulate Raf activation process.
Supplementary information
Acknowledgements
AAG is thankful to NIH grant #R01GM100078, Extreme Science and Engineering Discovery Environment (XSEDE) grant #MCB150054, Cancer Prevention and Research Institute of Texas (CPRIT) grant #DP150093. J.F.H. is thankful to CPRIT grant #RP170233. P.P. is thankful to Texas Advanced Computing Center (TACC).
Author Contributions
P.P. conceived the idea, designed the experiment, analyzed data and wrote the initial draft of the manuscript. J.F.H. and A.A.G. provided conceptual advice. A.A.G. provided resources for the project. All authors provided critical review and final approval of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-38770-w.
References
- 1.Matallanas D, et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer. 2011;2:232–260. doi: 10.1177/1947601911407323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vetter IR, Wittinghofer A. The Guanine Nucleotide-Binding Switch in Three Dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 4.Wittinghofer A, Vetter IR. Structure-function relationships of the G domain, a canonical switch motif. Annu Rev Biochem. 2011;80:943–971. doi: 10.1146/annurev-biochem-062708-134043. [DOI] [PubMed] [Google Scholar]
- 5.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177. doi: 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- 6.Blasco RB, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–663. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-Ras(G12D) Cancer Discov. 2011;1:128–136. doi: 10.1158/2159-8290.CD-10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stokoe D, Macdonald SG, Cadwallader K, Symons M, Hancock JF. Activation of Raf as a result of recruitment to the plasma membrane. Science. 1994;264:1463–1467. doi: 10.1126/science.7811320. [DOI] [PubMed] [Google Scholar]
- 9.Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369:411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- 10.Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- 11.Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- 12.Rushworth LK, Hindley AD, O’Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–2272. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS. Kinases and pseudokinases: lessons from RAF. Mol Cell Biol. 2014;34:1538–1546. doi: 10.1128/MCB.00057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roskoski RJ. RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun. 2010;399:313–317. doi: 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- 15.Cutler RE, Jr., Stephens RM, Saracino MR, Morrison DK. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci USA. 1998;95:9214–9219. doi: 10.1073/pnas.95.16.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavoie H, et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol. 2013;9(428–436):1257. doi: 10.1038/nchembio.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fetics SK, et al. Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure. 2015;23:505–516. doi: 10.1016/j.str.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mott HR, et al. The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc Natl Acad Sci USA. 1996;6:8312–8317. doi: 10.1073/pnas.93.16.8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrmann C, Martin GA, Wittinghofer A. Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J Biol Chem. 1995;270:2901–2905. doi: 10.1074/jbc.270.7.2901. [DOI] [PubMed] [Google Scholar]
- 20.Medkova M, Cho W. Interplay of C1 and C2 domains of protein kinase C-alpha in its membrane binding and activation. J Biol Chem. 1999;274:19852–19861. doi: 10.1074/jbc.274.28.19852. [DOI] [PubMed] [Google Scholar]
- 21.Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF, McCammon JA. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J Med Chem. 2007;50:674–684. doi: 10.1021/jm061053f. [DOI] [PubMed] [Google Scholar]
- 22.Mazhab-Jafari MT, et al. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc Natl Acad Sci USA. 2015;112:6. doi: 10.1073/pnas.1419895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prakash P, Zhou Y, Liang H, Hancock JF, Gorfe AA. Oncogenic K-Ras Binds to an Anionic Membrane in Two Distinct Orientations: A Molecular Dynamics Analysis. Biophys J. 2016;110:1125–1138. doi: 10.1016/j.bpj.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapoor S, et al. Revealing conformational substates of lipidated N-Ras protein by pressure modulation. Proc Natl Acad Sci USA. 2012;109:460–465. doi: 10.1073/pnas.1110553109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Srivastava PP, Buck MA. “Tug of War” maintains a dynamic protein-membrane complex: MD simulations of C-Raf RBD-CRD bound to K-Ras4B at an anionic membrane. ACS Cent Sci. 2017;4:298–305. doi: 10.1021/acscentsci.7b00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Cao S, Buck M. K-Ras at Anionic Membranes: Orientation, Orientation…Orientation. Recent Simulations and Experiments. Biophys J. 2016;110:1033–1035. doi: 10.1016/j.bpj.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li ZL, Buck M. Computational Modeling Reveals that Signaling Lipids Modulate the Orientation of K-Ras4A at the Membrane Reflecting Protein Topology. Structure. 2017;25:679–689 e672. doi: 10.1016/j.str.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Travers T, et al. Molecular recognition of RAS/RAF complex at the membrane: Role of RAF cysteine-rich domain. Sci Rep. 2018;8:8461. doi: 10.1038/s41598-018-26832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J Biol Chem. 1996;271:8472–8480. doi: 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]
- 30.Baljuls A, et al. Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J Biol Chem. 2011;286:16491–16503. doi: 10.1074/jbc.M110.194167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, et al. mTOR kinase structure, mechanism and regulation. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moravcevic K, Oxley CL, Lemmon MA. Conditional peripheral membrane proteins: facing up to limited specificity. Structure. 2012;20:15–27. doi: 10.1016/j.str.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi, M., Li, Y., Dillon, T. J., Kariya, Y. & Stork, P. J. S. Phosphorylation of the C-Raf N-region promotes Raf dimerization. Mol Cell Biol (2017). [DOI] [PMC free article] [PubMed]
- 35.Jambrina PG, et al. Phosphorylation of RAF Kinase Dimers Drives Conformational Changes that Facilitate Transactivation. Angew Chem Int Ed Engl. 2016;55:983–986. doi: 10.1002/anie.201509272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 37.Hatzivassiliou G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 38.Eswar N, et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;Chapter 5:Unit–5 6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thevakumaran N, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22:37–43. doi: 10.1038/nsmb.2924. [DOI] [PubMed] [Google Scholar]
- 40.Bastidas AC, et al. Role of N-terminal myristylation in the structure and regulation of cAMP-dependent protein kinase. J Mol Biol. 2012;422:215–229. doi: 10.1016/j.jmb.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maupetit J, Derreumaux P, Tuffery P. A fast method for large-scale de novo peptide and miniprotein structure prediction. J Comput Chem. 2010;31:726–738. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
- 42.Dames SA. Structural basis for the association of the redox-sensitive target of rapamycin FATC domain with membrane-mimetic micelles. J Biol Chem. 2010;285:7766–7775. doi: 10.1074/jbc.M109.058404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J Comput Phys. 1977;23:15. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 44.Darden T, York D, Pedersen L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 45.Best RB, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klauda JB, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buck M, Bouguet-Bonnet S, Pastor RW, MacKerell AD., Jr. Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of hen lysozyme. Biophys J. 2006;90:L36–38. doi: 10.1529/biophysj.105.078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 49.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 51.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-X. [DOI] [PubMed] [Google Scholar]
- 52.Hyvonen M, et al. Structure of the binding site for inositol phosphates in a PH domain. EMBO J. 1995;14:4676–4685. doi: 10.1002/j.1460-2075.1995.tb00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng J, et al. The solution structure of the pleckstrin homology domain of human SOS1. A possible structural role for the sequential association of diffuse B cell lymphoma and pleckstrin homology domains. J Biol Chem. 1997;272:30340–30344. doi: 10.1074/jbc.272.48.30340. [DOI] [PubMed] [Google Scholar]
- 54.Hadders MA, Williams RL. Kinases charging to the membrane. Cell. 2010;143:865–867. doi: 10.1016/j.cell.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 55.Moravcevic K, et al. Kinase associated-1 domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell. 2010;143:966–977. doi: 10.1016/j.cell.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson LM, James KM, Chamberlain MD, Anderson DH. Identification of key residues in the A-Raf kinase important for phosphoinositide lipid binding specificity. Biochemistry. 2005;44:3432–3440. doi: 10.1021/bi0487692. [DOI] [PubMed] [Google Scholar]
- 57.Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13:1356–1364. doi: 10.1016/S0960-9822(03)00535-9. [DOI] [PubMed] [Google Scholar]
- 58.Gao Z, et al. Localization of the Raf-like kinase CTR1 to the endoplasmic reticulum of Arabidopsis through participation in ethylene receptor signaling complexes. J Biol Chem. 2003;278:34725–34732. doi: 10.1074/jbc.M305548200. [DOI] [PubMed] [Google Scholar]
- 59.Chiu SW, Jakobsson E, Subramaniam S, Scott HL. Combined Monte Carlo and molecular dynamics simulation of fully hydrated dioleyl and palmitoyl-oleyl phosphatidylcholine lipid bilayers. Biophys J. 1999;77:2462–2469. doi: 10.1016/S0006-3495(99)77082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prakash, P. & Gorfe, A. A. Membrane orientation dynamics of lipid-modified small GTPases. Small GTPases, 1–10 (2016). [DOI] [PMC free article] [PubMed]
- 61.Liu Y, Kahn RA, Prestegard JH. Dynamic structure of membrane-anchored Arf*GTP. Nat Struct Mol Biol. 2010;17:876–881. doi: 10.1038/nsmb.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mazhab-Jafari MT, et al. Membrane-dependent modulation of the mTOR activator Rheb: NMR observations of a GTPase tethered to a lipid-bilayer nanodisc. J Am Chem Soc. 2013;135:3367–3370. doi: 10.1021/ja312508w. [DOI] [PubMed] [Google Scholar]
- 63.Chen J, Fang Y. A novel pathway regulating the mammalian target of rapamycin (mTOR) signaling. Biochem Pharmacol. 2002;64:1071–1077. doi: 10.1016/S0006-2952(02)01263-7. [DOI] [PubMed] [Google Scholar]
- 64.Foster DA. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007;67:1–4. doi: 10.1158/0008-5472.CAN-06-3016. [DOI] [PubMed] [Google Scholar]
- 65.Brtva TR, et al. Two distinct Raf domains mediate interaction with Ras. J Biol Chem. 1995;270:9809–9812. doi: 10.1074/jbc.270.17.9809. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh S, et al. The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J Biol Chem. 1994;269:10000–10007. [PubMed] [Google Scholar]
- 67.Hofbauer HF, et al. The molecular recognition of phosphatidic acid by an amphipathic helix in Opi1. J Cell Biol. 2018;217:3109–3126. doi: 10.1083/jcb.201802027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Durrant DE, Morrison DK. Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br J Cancer. 2018;118:3–8. doi: 10.1038/bjc.2017.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou Y, et al. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell. 2016;168:13. doi: 10.1016/j.cell.2016.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prakash P, et al. Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci Rep. 2017;7:40109. doi: 10.1038/srep40109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lucic I, et al. Conformational sampling of membranes by Akt controls its activation and inactivation. Proc Natl Acad Sci USA. 2018;115:E3940–E3949. doi: 10.1073/pnas.1716109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.