

Abstract
Key points
The TASK‐1 channel gene (KCNK3) has been identified as a possible disease‐causing gene in heritable pulmonary arterial hypertension (PAH).
In the present study, we show that novel mutated TASK‐1 channels, seen in PAH patients, have a substantially reduced current compared to wild‐type TASK‐1 channels.
These mutated TASK‐1 channels are located at the plasma membrane to the same degree as wild‐type TASK‐1 channels.
ONO‐RS‐082 and alkaline pH 8.4 both activate TASK‐1 channels but do not recover current through mutant TASK‐1 channels.
We show that the guanylate cyclase activator, riociguat, a novel treatment for PAH, enhances current through TASK‐1 channels but does not recover current through mutant TASK‐1 channels.
Abstract
Pulmonary arterial hypertension (PAH) affects ∼15–50 people per million. KCNK3, the gene that encodes the two pore domain potassium channel TASK‐1 (K2P3.1), has been identified as a possible disease‐causing gene in heritable PAH. Recently, two new mutations have been identified in KCNK3 in PAH patients: G106R and L214R. The present study aimed to characterize the functional properties and regulation of wild‐type (WT) and mutated TASK‐1 channels and determine how these might contribute to PAH and its treatment. Currents through WT and mutated human TASK‐1 channels transiently expressed in tsA201 cells were measured using whole‐cell patch clamp electrophysiology. Localization of fluorescence‐tagged channels was visualized using confocal microscopy and quantified with in‐cell and on‐cell westerns. G106R or L214R mutated channels were located at the plasma membrane to the same degree as WT channels; however, their current was markedly reduced compared to WT TASK‐1 channels. Functional current through these mutated channels could not be restored using activators of WT TASK‐1 channels (pH 8.4, ONO‐RS‐082). The guanylate cyclase activator, riociguat, enhanced current through WT TASK‐1 channels; however, similar to the other activators investigated, riociguat did not have any effect on current through mutated TASK‐1 channels. Thus, novel mutations in TASK‐1 seen in PAH substantially alter the functional properties of these channels. Current through these channels could not be restored by activators of TASK‐1 channels. Riociguat enhancement of current through TASK‐1 channels could contribute to its therapeutic benefit in the treatment of PAH.
Keywords: Pulmonary arterial hypertension, KCNK3 (TASK‐1) potassium channel, riociguat
Key points
The TASK‐1 channel gene (KCNK3) has been identified as a possible disease‐causing gene in heritable pulmonary arterial hypertension (PAH).
In the present study, we show that novel mutated TASK‐1 channels, seen in PAH patients, have a substantially reduced current compared to wild‐type TASK‐1 channels.
These mutated TASK‐1 channels are located at the plasma membrane to the same degree as wild‐type TASK‐1 channels.
ONO‐RS‐082 and alkaline pH 8.4 both activate TASK‐1 channels but do not recover current through mutant TASK‐1 channels.
We show that the guanylate cyclase activator, riociguat, a novel treatment for PAH, enhances current through TASK‐1 channels but does not recover current through mutant TASK‐1 channels.
Introduction
Pulmonary arterial hypertension (PAH, Group 1 pulmonary hypertension) (Simonneau et al. 2013, Galie et al. 2015) is a progressive and incurable disease that occurs when the pulmonary arteries undergo pathological remodelling, causing the blood vessels to vasoconstrict, with a consequent increase in pulmonary vascular resistance. Continued high pressure in the pulmonary artery leads to right ventricular heart failure and death if left untreated (Gaine & McLaughlin, 2017). Although PAH is a rare disease with an incidence of 15–50 people per million in the population (Humbert et al. 2014), the prognosis for patients is poor, with a 5 year survival rate of 34% (Tang et al. 2016). PAH can be either idiopathic (iPAH), heritable/familial (hPAH), or associated with other pathological conditions such as connective tissue diseases (e.g. scleroderma), human immunodeficiency virus (HIV), advanced liver disease, congenital heart disease and Schistosomiasis infection (Butrous 2015, Garg et al. 2017, Ghofrani et al. 2017). Despite being an incurable disease, there are now treatments and medication that can delay disease progression and increase life expectancy. Currently, there are three known pathways that contribute to cell proliferation and vasoconstriction in the pulmonary arteries: the prostacyclin, endothelin and nitric oxide pathways. Clinical treatments are aimed at appropriate up‐ or down‐regulation of each of these pathways to slow disease progression (Hill et al. 2016). Recently, riociguat (BAY 63–2521), a guanylate cyclase stimulator, acting downstream of nitric oxide through increased cGMP production, has been approved for use in PAH (Hill et al. 2016, Ghofrani et al. 2017). The therapeutic benefits of riociguat include improvement in pulmonary vascular haemodynamics and increased exercise ability in patients with PAH.
Advances in DNA sequencing and wide availability of whole exome sequencing has resulted in a large number of genetic defects being identified in both hPAH and iPAH sufferers, enhancing the molecular understanding of the pathogenic mechanism(s) underlying the disease. Both hPAH and iPAH have been linked to a defective copy of the bone morphogenic protein receptor type II (BRPM2) gene, which regulates vascular cell proliferation (Machado et al. 2015). Other, less frequent, mutations associated with PAH include ALK1, ENG, TBX4, EIF2AK4, SMAD, CAV1 and NOTCH3 (Tang et al. 2016). Additionally, a number of ion channels have been identified as risk factors for hPAH and iPAH. In particular, two potassium ion channels, Kv1.5 (KCNA5) and TASK‐1 (KCNK3, K2P3.1), which are implicated in hPAH and iPAH development, play a crucial role in regulating pulmonary vascular tone (Yuan et al. 1998, Boucherat et al. 2015, Hemnes & Humbert 2017, Olschewski et al. 2017). Down‐regulation, inhibition and mutations of these channels resulting in loss of channel function have been shown to contribute to cell proliferation, resistance to apoptosis and vasoconstriction in pulmonary arterial smooth muscle cells (PASMCs) (Remillard et al. 2007, Antigny et al. 2016). Furthermore, a reduction in TASK‐1 channel function has been observed in right ventricular cardiomyoctes prior to the development of right ventricular hypertrophy related to pulmonary hypertension (Lambert et al. 2018).
TASK‐1 channels belong to the two pore domain family of potassium ion channels (Enyedi & Czirjak 2010). Six heterozygous TASK‐1 mutations were first described in 2013, for both hPAH and iPAH patients (Ma et al. 2013). Characterization of these mutations using patch clamp electrophysiology showed that, when expressed homozygously, these mutations resulted in loss of channel function. However, the application of a phospholipase A2 inhibitor (ONO‐RS‐O82) was able to partially restore channel function for two of the homozygous mutant channels, thus opening a new avenue for PAH‐directed therapeutic development (Girerd et al. 2014). This report was followed by the identification of two further mutations, this time homozygous mutations, from a Spanish cohort of PAH patients, with an aggressive form of the disease (Navas et al. 2016).
In the present study, we characterize the functional properties and membrane localization of the two new homozygous mutations (G106R and L214R) identified by Navas et al. (2016). We show that current through these channels is considerably reduced compared to WT TASK‐1 channels and that functional current through these mutated channels cannot be restored using activators of WT TASK‐1 channels (pH 8.4, ONO‐RS‐082). In addition, we show for the first time that the guanylate cyclase activator, riociguat, enhances current through WT TASK‐1 channels. Similar to the other activators, however, riociguat does not have any effect on current through G106R or L214R mutated TASK‐1 channels. A preliminary account of some of these results has been reported previously (Cunningham et al. 2017).
Methods
Molecular biology
Human wild‐type (WT) TASK‐1 (KCNK9) cDNA was cloned into pcDNA3.1 vector (Invitrogen, Carlsbad, CA, USA) and was a kind gift from Helen Meadows (GlaxoSmithKline, Harlow, UK). WT TASK‐1 was cut out of pcDNA3.1 vector and cloned into the MCS of pAcGFP1‐N1 vector (Clontech‐Takara Bio Europe, Saint‐Germain‐en‐Laye, France) to create a fusion construct with the N‐terminus of AcGFP1. The terminating stop codon of TASK‐1 was removed by site‐directed mutagenesis and maintained in the same reading frame with the start codon of AcGFP1. Constructs were fully sequencing to ensure correct sequence incorporation (Eurofins MWG Operon, Huntsville, AL, USA).
Mutations
Point mutations were introduced into both pcDNA3.1 and pAcGFP1‐N1 encoding TASK‐1 cDNA by site‐directed mutagenesis using a QuikChange site‐directed mutagenesis kit and procedure (Stratagene, La Jolla, CA, USA). A pair of complementary oligonucleotide primers (25–35 bases) incorporating the intended mutation (either G106R or L214R) were synthesized (Eurofins MWG Operon, Ebersberg, Germany). All constructs were fully sequenced to ensure correct mutation incorporation.
Cell culture
tsA201 cells, which are modified human embryonic kidney 293 cells stably transfected with the SV40 large T antigen (ECACC; Sigma‐Aldrich, Gillingham, Dorset, UK), were grown in a monolayer tissue culture flask maintained in growth medium that was composed of 88% minimum essential media with Earle's salts, 2 mm l‐glutamine, 10% heat‐inactivated foetal bovine serum, 1% penicillin (10,000 units mL−1) and streptomycin (10 mg mL−1), and 1% nonessential amino acids (Sigma‐Aldrich; Pan Biotech, Aidenbach, Germany; Fisher Scientific, Pittsburgh, PA, USA). The cells were stored in an incubator at 37 °C with a humidified atmosphere of 95% O2 and 5% CO2. When the cells reached 80% confluency, they were were split and resuspended in a four‐well plate containing glass coverslips (diameter 13 mm) coated with poly‐d‐lysine (1 mg mL–1) at a concentration of 7 × 104 cells in 0.5 mL of media, ready for transfection the next day.
Transfection
For the electrophysiological experiments, cells were transiently transfected using a modified calcium‐phosphate protocol described by Chen & Okayama (1987). Next, 500 ng of pcDNA3.1 vector (Invitrogen) encoding human WT or mutant human TASK‐1 (KCNK3) (Genbank AF006823) and 500 ng of cDNA encoding for green fluorescent protein (GFP) was added into each well. The cells were incubated for 6–8 h at 37 °C in 95% O2 and 5% CO2. Following incubation, cells were washed twice with a 1 × PBS, and incubated in 0.5 mL of fresh growth media overnight. The cells were used for electrophysiological recordings the next day.
For confocal microscopy, cells were transfected using TurboFect transfection reagent (ThermoFisher, Loughborough, UK). Then, 1 μg of pAcGFP1‐N1 vector (Clontech‐Takara Bio Europe) encoding human WT or mutant human TASK‐1 was diluted in 100 μL of serum‐free cell media. To this, 1.5 μL of the Turbofect transfection reagent was added and incubated for 15–20 min. After incubation, this mixture was added to one well of the plate and incubated for 24–48 h at 37 °C in 95% O2 and 5% CO2, prior to membrane staining and/or fixation.
Membrane staining
The plasma membranes of cells were stained using CellMask Deep Red (Thermofisher). A freshly prepared 1:1000 dilution of CellMask Deep Red plasma membrane stain (1×) was prepared in 1 × PBS. Transfected cells were then washed three times with 1 × PBS and submerged into 1 mL of prepared CellMask membrane stain for 5–10 min at 37 °C. The staining solution was removed by washing the coverslips twice in PBS, ready for fixation.
Cell fixation
Prior to fixation cells were washed twice with room temperature 1 × PBS and then fixed in 1 mL of 2% paraformaldehyde (PFA) solution and incubated at 4 °C for 20 min. Following incubation, the cells were washed twice in 1 × PBS. For nuclei staining, 500 μL of freshly prepared Hoechst 33528 (1:500 dilution in PBS; Sigma‐Aldrich) was added to each well and incubated at 37 °C for up to 10 min. Cells were then washed twice with PBS and rinsed once with ddH2O to remove any salt crystals. Coverslips were mounted face down onto slides containing a drop of Vectashield anti‐fade mounting medium (Vector Laboratories, Peterborough UK).
Confocal microscopy
Images were taken using a LSM 880 confocal microscope (Carl Zeiss, Oberkochen, Germany), placed upon an anti‐vibration table and processed using Zen Black software (Carl Zeiss). Cells were imaged using oil immersion under a Plan‐Apochromat 63×/1.4 oil DIC M27 objective (Carl Zeiss). The cells were excited with an argon laser at either 561, 488 or 405 nm for the CellMask Deep Red plasma membrane stain, pAcGFP‐channel and Hoechst 33528, respectively.
Co‐localization analysis
Zen Black software was used to determine the degree of co‐localization. Regions of interest around cells with detectable green fluorescence were selected for analysis. Control images for pAcGFP‐only cells and Deep Red membrane stain‐only cells were taken with each set of images to ensure co‐localized quadrants were not set arbitrarily. Pearson's correlation coefficient (PCC) was used to represent the degree of co‐localization.
In‐cell and on‐cell western
Cells were seeded onto a poly‐d‐lysine coated 96‐well plate at a density of 2 × 104 cells/well (in 100 μL). The cultures were incubated for 24 h at 37 °C in 5% CO2 until a confluence of 80–95% was reached. Each well was transfected with 200 ng of DNA for HA‐tagged WT or mutated TASK1 and 0.4 μL of Turbofect and incubated for 24 h at 37 °C in 5% CO2. Cell culture media was removed and cells were immediately fixed with 40 μL of 2% PFA and incubated at room temperature for 20 min on the bench. Fixing solution was removed and cells washed.
For staining, cells were incubated 40 μL of a 1:500 dilution of 2 μg μL–1 monoclonal anti‐HA antibody (mouse) in blocking solution. A 1:1000 dilution IRDye 800CW (green) goat anti‐mouse secondary antibody and DRAQ5 (1:10,000; 5 mm) was added to each well. For whole cell staining (in‐cell), the membrane was permeablized with 40 μL of 0.1% Triton X‐100. Plates were scanned with detection in both the 700 and 800 nm channels using a Odyssey SA near‐infrared fluorescence imager (Li‐Cor, Lincoln, NE, USA). Integrated intensities were recorded and analysed. For each experiment, each condition was repeated in triplicate (three coverslips) and each experiment was repeated on three separate independent occasions.
Whole‐cell patch clamp electrophysiology
Currents were recorded from tsA201 cells transiently transfected with the channel of interest using whole‐cell patch clamp in a voltage clamp configuration. A coverslip with transfected tsA201 cells was transferred into a recording chamber filled with an external solution composed of (in mm) 145 NaCl, 2.5 KCL, 3 MgCl2, 1 CaCl2 and 10 Hepes (pH to either 7.4 or 8.4, using NaOH), mounted under an inverted microscope (Diaphot; Nikon, Tokyo, Japan) with epifluorescence. External solution and modulatory compounds were superfused at a rate of 4–5 mL min−1. Complete exchange of the bath solution occurred within 100–120 s. Only cells that were transfected with GFP as evident from green fluorescence (excitation 395–440 nm; emission 470–600 nm) were selected for electrophysiological recordings. Patch pipettes were pulled from thin walled borosilicate glass (GC150TF; Harvard Apparatus, Edenbridge, UK) and had resistances of 3–6 MΩ when filled with pipette solution. The pipette solution contained (in mm) 150 KCL, 3 MgCl2, 5 EGTA and 10 Hepes (pH adjusted to 7.4 with KOH). Whole‐cell currents were recorded at a holding potential of –60 mV at 20–24 °C (room temperature). Cells were hyperpolarized to –80 mV for 100 ms and then subjected to a step to –40 mV for 500 ms, followed by a a step to –120 mV for 100 ms. These step changes were followed by a 500 ms voltage ramp to +20 mV and a step back to –80 mV for another 100 ms before returning to the holding potential of –60 mV. This protocol was composed of sweeps lasting 1.5 s, including sampling at the holding voltage and was repeated once every 5 s. Currents were recorded using an Axopatch 200 or 1D patch clamp amplifier (Molecular Devices, Sunnyvale, CA, USA) and analysed using pCLAMP, version 10.2 (Molecular Devices), Excel (Microsoft Corp., Redmond, WA, USA) and Prism, version 6 or 7 (GraphPad Software Inc., San Diego, CA, USA). For analysis of outward current, we measured the current amplitude during the step to −40 mV.
Chemicals
ONO‐RS‐082 was purchased from Abcam (Cambridge, UK) and dissolved in dimethyl sulphoxide (DMSO) to create a 10 mm stock solution. This was diluted in external solution to the desired concentration just before use. Riociguat was purchased from MedChemExpress (Shanghai, China), diluted in DMSO to create a 10 mm stock. All other chemicals were purchased from Sigma‐Aldrich.
Statistical analysis
Data are expressed as the mean ± 95% confidence intervals (CI) or the mean ± SEM, where n represents the number of individual cells, and ‘days’ represents the number of different recording days. On any given day, recordings were randomized to include both control and treatment recordings, for both electrophysiological and imaging experiments. Statistical analysis was performed using Prism, version 6 or 7 (GraphPad Software Inc.). Statistical analysis used one‐way ANOVA with a post hoc Dunnett's test for multiple comparison tests, as well as paired or unpaired Student's t tests. P < 0.05 was considered statistically significant. Where appropriate, the 95% CI for the difference in means is also given.
Results
Structural analysis of the human TASK‐1 mutations
TASK‐1, which is encoded by the gene KCNK3, belongs to the two‐pore domain potassium (K2P) superfamily of potassium ion channels. The K2P family of ion channels is characterized by a unique molecular topology consisting of two α‐subunits that dimerize together to produce a single central pore, with each α‐subunit comprising two pore (P) loop forming domains and four transmembrane domains (Enyedi & Czirjak 2010). Two novel pathogenic missense TASK‐1 mutations were identified by Navas et al. (2016) from three Spanish patients with an aggressive form of iPAH and hPAH. These mutations c.614 T>G (L214R) and c.316 G>C G106R were identified on exon 2 of KCNK3 and were the first homozygous mutations to be reported in PAH (Navas et al. 2016). Using a generic TASK channel homology model, based on a crystal structure of the K2P channel TRAAK (Brohawn et al. 2012), the positions of the two mutations (on the two subunits in the TASK channel dimer) are indicated in Fig. 1 A. The glycine (G) 106 [mutated to an arginine (R)] is located extracellularly between the first pore domain and the second transmembrane domain, whereas the leucine (L) 214 (also mutated to R) is located extracellularly between the second pore domain and the fourth transmembrane domain. Alignment of TASK‐1 with other members of the acid‐sensitive TASK subfamily (TASK‐3 and TASK‐5) shows that these are highly conserved residues and are probably critical for channel function (Fig. 1 B).
Figure 1. Homology model of TASK‐1 variants.
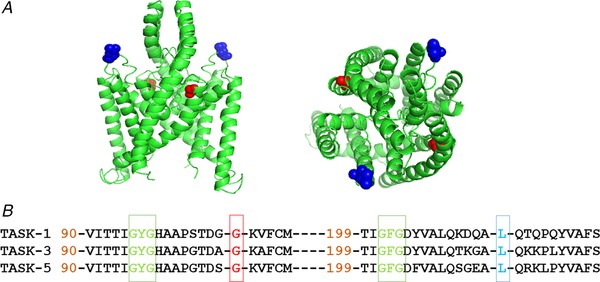
A, homology model of TASK channels based on TRAAK crystal structure (PDB ID: 3UM7; Brohawn et al. 2012) depicting the location of the two PAH TASK‐1 mutations: G106R and L214R. TASK‐1_G106 amino acids are shown in red and TASK‐1_L214 are shown in blue. Left: side view of the channel. Right: view from above the channel. B, amino acid sequence alignment of TASK‐1 with the two other members of the TASK subfamily: TASK‐3 and TASK‐5. Gaps are indicated by dashes and numbers indicate where sequence begins relative to the full length channel. The amino acids mutated in PAH patients, glycine (G) 106 and leucine (L) 214 are in red and blue, respectively. The selectivity filter regions are shown in green. [Color figure can be viewed at wileyonlinelibrary.com]
Characterization of current through G106R and L214R mutated human TASK‐1 channels
Previously identified mutations of TASK‐1 from PAH patients were found to cause loss of function of the channel at physiological pH when expressed transiently as homodimer mutants (Ma et al. 2013). Whole‐cell patch clamp techniques were utilized to determine functionality of the two novel homodimer TASK‐1 mutants, G106R and L214R. Whole‐cell recordings from tsA201 cells transiently transfected with human TASK‐1 cDNA gave whole cell currents of 7.8 pA pF–1 (n = 44 cells from 17 days, 95% CI = 5.8–9.7) (measured as current density at –40 mV; see Methods) (Fig. 2 A), using a physiological extracellular solution of 2.5 mm [K+]o. Ramp changes in holding potential from –120 to +20 mV demonstrate that the current is outwardly rectifying (Fig. 2 B and C) with a mean zero current potential of –78 mV (n = 44, 95% CI = –74 to –82) close to the equilibrium potential for potassium ions under these recording conditions. We have shown previously that TASK‐1 channels expressed in tsA201 cells are enhanced by alkaline pH, blocked by acidic pH and blocked by methanandamide (Aller et al 2005, Veale et al 2007).
Figure 2. Electrophysiological profiling of currents through WT TASK‐1 and TASK‐1 mutant channels.
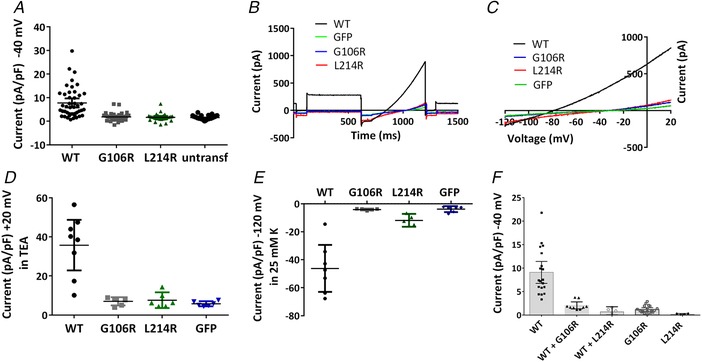
A, current density (pA pF–1) measured at –40 mV from individual cells transiently expressing WT TASK‐1, TASK‐1_G106R, TASK‐1_L214R channels or untransfected cells. Error bars represent the 95% CI. B, raw data trace from exemplar human TASK‐1, TASK‐1_G106R, TASK‐1_L214R channels and GFP alone transfected cells in 10 mm TEA using a step‐ramp voltage protocol as detailed in the Methods. C, current–voltage relationship for the cells in (B) evoked by ramp changes in voltage from –120 to –20 mV. D, current density (pA pF–1) measured at +20 mV in the presence of 10 mm TEA for cells expressing WT TASK‐1, TASK‐1_G106R, TASK‐1_L214R channels or GFP alone. E, current density (pA pF–1) measured at –120 mV in 25 mm K external for cells expressing WT TASK‐1, TASK‐1_G106R, TASK‐1_L214R channels or GFP alone. F, current density (pA pF–1) measured at –40 mV for cells expressing WT TASK‐1, TASK‐1_G106R, TASK‐1_L214R channels or co‐expression of WT TASK_1 and TASK‐1_G106R or WT TASK_1 and TASK‐1_L214R. [Color figure can be viewed at wileyonlinelibrary.com]
By contrast, mutation of a small uncharged glycine (G) residue at position 106 to a large positively charged arginine (R) residue, resulted in a poorly functioning channel with significantly reduced current, when expressed alone as a homozygous mutant channel in tsA201 cells (Fig. 2 A–C). The average whole‐cell current measured at –40 mV was 1.8 pA pF–1 (n = 38 cells from 14 days, 95% CI = 1.3–2.4). Similarly, mutation of a small hydrophobic leucine (L) residue at position 214 to a large positively charged arginine (R) residue was found to have substantially reduced current when expressed alone as a homozygous mutant channel in tsA201 cells (Fig. 2 A–C). The average whole‐cell current measured at –40 mV was 1.6 pA pF–1 (n = 27 cells from 10 days, 95% CI = 1.0–2.3). The outward currents recorded from TASK‐1_G106R and TASK‐1_L214R channels were statistically significantly reduced (P < 0.05, one‐way ANOVA followed by a Dunnett's multiple comparisons test) from WT TASK‐1 and not significantly different from untransfected cells. Untransfected tsA201 cells had an average whole‐cell current measured at –40 mV of 1.5 pA pF–1 (n = 36 cells from 9 days, 95% CI = 1.2–1.8) (Fig. 2 A–C).
Outward currents at +20 mV were recorded in the presence of tetraethylammonium chloride (TEA) (10 mm) to minimize the contribution of currents through endogenous KV channels in tsA201 cells. As for outward currents at –40 mV, currents through both mutated channels at +20 mV were significantly smaller than current through WT TASK‐1 channels and not significantly different from cells transfected with GFP alone (Fig. 2 D). Inward currents though WT and mutated TASK‐1 channels were measured at –120 mV, when the external solution was raised to 25 mm K to identify inward current through WT channels (Fig. 3). Again, inward currents through both mutated channels at –120 mV were significantly smaller than current through WT TASK‐1 channels and not significantly different from cells transfected with GFP alone (Fig. 2 E)
Figure 3. Ion permeability of TASK‐1 and mutated TASK‐1 channels.
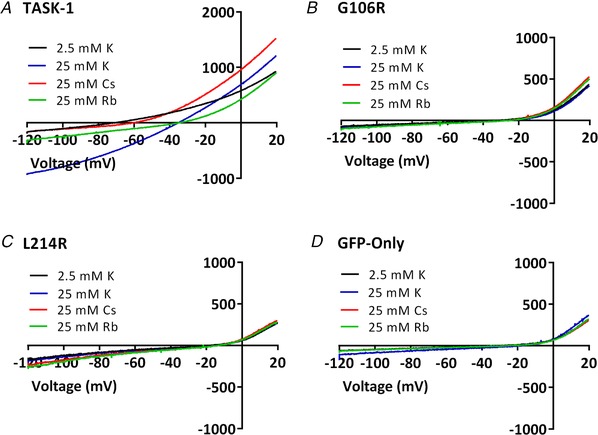
A–D, current–voltage relationships from exemplar human TASK‐1, TASK‐1_G106R, TASK‐1_L214R channels and GFP alone transfected cells in 2.5 mm K, 25 mm K, 25 mm Rb or 25 mm Cs. [Color figure can be viewed at wileyonlinelibrary.com]
Heterodimeric channels
Co‐expression of either of the mutated TASK‐1 channels with WT TASK‐1 channels to allow the formation of heterodimeric channels gave substantially and significantly reduced currents from WT TASK‐1 channels alone (P < 0.05, one‐way ANOVA) that were not significantly different from mutated channels alone (Fig. 2 F). This suggests that the mutated channels are dominant negative, when expressed heterologously with WT TASK‐1 channels.
Ion selectivity of WT and mutated TASK‐1 channels
Ramp changes in voltage from –120 to +20 mV were applied to cells expressing WT TASK_1 channels, both of the mutant channels or GFP alone in normal K external solution (2.5 mm). The reversal potential of the current was obtained from these ramps and compared with that obtained with an external solution containing 25 mm K (Fig. 2). The external solution was then changed again to one where 25 mm of Rb or Cs replaced the 25 mm K. For WT TASK‐1 channels, clear shifts in V rev were seen compared to recordings in 25 mm K (Fig. 3 A and Table 1). By contrast, no changes in current reversal potential were seen for either mutant channel in any condition and the reversal potentials seen were not significantly different from that seen for cells transfected with GFP alone (Fig. 3 B–D and Table 1).
Table 1.
The effect of permeating ion on TASK‐1 channel current reversal potential
| 2.5 mm K | 25 mm K | 25 mm Cs | 25 mm Rb | |
|---|---|---|---|---|
| TASK‐1 | –73 ± 2 (n = 13) | –38 ± 1 (n = 10) | –64 ± 2 (n = 8) | –41 ± 3 (n = 7) |
| TASK‐1_G106R | –33 ± 8 (n = 5) | –28 ± 4 (n = 5) | –29 ± 5 (n = 5) | –25 ± 3 (n = 5) |
| TASK‐1_L214R | –19 ± 3 (n = 5) | –17 ± 2 (n = 5) | –18 ± 4 (n = 5) | –15 ± 3 (n = 5) |
| GFP alone | –32 ± 2 (n = 13) | –25 ± 3 (n = 6) | –31 ± 4 (n = 5) | –27 ± 3 (n = 5) |
Reversal potentials (in mV) obtained for TASK‐1, TASK‐1_G106R, TASK‐1_L214R and GFP alone transfected cells when the external solution contained 2.5 mm K, 25 mm K, 25 mm Cs or 25 mm Rb.
Cellular localization of fluorescence‐labelled TASK‐1 variants
To determine whether the reduced current levels recorded through TASK‐1_G106R and TASK‐1_L214R variants were a consequence of reduced trafficking to the plasma membrane, fluorescence‐tagged channels were used to examine cellular localization. GFP was fused to the C terminus of TASK‐1 and the two TASK‐1 variants (see Methods).
Electrophysiological recordings showed that neither the GFP tag (9.7 pA pF–1, n = 9 cells, 95% CI = 6.8–12.7 for WT_TASK‐1 channels vs. 8.2 pA pF–1, n = 8 cells, 95% CI = 4.9–11.5 for pACGFP_TASK‐1 channels), nor the different transfection agent (8.0 pA pF–1, n = 5 cells, 95% CI = 5.8–10.1 for CaCl2 transfection vs. 9.5 pA pF–1, n = 5 cells, 95% CI = 8.3–10.7 for turbofect transfection), altered the magnitude of functional current through WT‐TASK‐1 channels, as measured in temporally matched experiments.
Cellular localization of WT TASK‐1‐GFP in tsA201 cells was examined using confocal microscopy. GFP fluorescence was observed at the plasma membrane of cells transiently transfected with WT TASK‐1‐GFP but there was also expression of WT TASK‐1 intracellularly. Figure 4 Aa represents a typical cell expressing TASK‐1‐GFP fused channels (green), excited at 480 nm. Fluorescence was also observed for the plasma membrane specific stain, CellMask Deep Red. Figure 4 Ab shows the fluorescence signal (red) for the same cell excited at 561 nm. Expression of the channel at the membrane was confirmed by co‐localization of the green signal of the channel with the red signal emitted from the plasma membrane specific stain (Fig. 4 Ac). The nuclei are stained blue. Co‐localization of WT TASK‐1 with the plasma membrane was quantified using PCC from 12 cells, from eight different plates from four different cultures. A strong linear correlation of 0.65 (n = 12 cells, 95% CI = 0.57–0.73) was observed for WT TASK‐1 at the membrane (Fig. 4 E). The same analysis for the two TASK‐1 variants gave results similar to that seen for WT TASK‐1 channels. Strong green fluorescence was observed at the membrane of cells transiently transfected with a GFP fused TASK‐1_G106R or TASK‐1_L214R mutant channels (Fig. 4 Ba and Ca), which showed some overlap with the red fluorescence from the membrane specific stain (Fig. 4 Bb, Bc, Cb and Cc). PCC values of 0.75 (n = 12 cells, 95% CI = 0.69–0.81 and 0.72 (n = 12 cells, 95% CI = 0.64–0.80 were calculated for TASK‐1_G106R and TASK‐1_L214R, respectively (Fig. 4 E), which was not significantly different from WT TASK‐1 (P > 0.05, one‐way ANOVA) suggesting that the two variants were both translated and trafficked to the membrane to the same degree as WT TASK‐1 under these experimental conditions. In comparison, untransfected cells showed no green fluorescence (Fig. 4 Da–Dc).
Figure 4. Evaluation of cellular localization of labelled TASK‐1 variants.
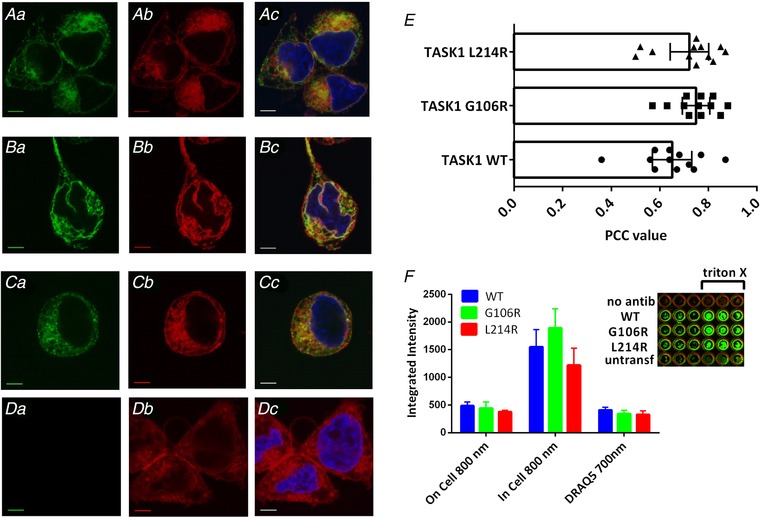
Aa and Ab, photomicrograph taken using confocal microscopy showing cellular localization of WT TASK‐1 channels C terminally fused with GFP, relative to the location of the plasma membrane stained with CellMask Deep Red. Ac, overlay of (Aa) and (Ab) indicating co‐localization of TASK‐1‐GFP with the plasma membrane in yellow. Nuclei were stained with the blue fluorescent dye Hoechst 33258. Ba, cellular localization of TASK‐1_G106R fused with GFP, relative to the location of the membrane (Bb). Bc, co‐localization of the G106R variant at the membrane in yellow. Ca, localization of TASK‐1_L214R in relation to the plasma membrane (Cb). Cc, overlap of the GFP‐fused variant with the plasma membrane, stained red. Da, cells untransfected with TASK‐1; (Db) and (Dc) as above. All scale bars are 5 μm. E, quantification of the co‐localization observed in experiments, as shown in (Ac), (Bc) and (Cc), using Pearson's correlation coefficient. A correlation coefficient of 1 represents 100% correlation. F, integrated fluorescence intensity for HA‐tagged WT and mutant TASK‐1 channels in non‐permeabilized (membrane) and permeabilized (whole‐cell) cells detected at 800 nm and for DRAQ5 (whole cell) detected at 700 nm using a Li‐Cor Odyssey SA fluorescence imager. Each column represents the mean ± SEM from three independent experiments performed in triplicate. The mean integrated intensity obtained from untransfected cells on the same plate, treated in the same way, was subtracted from each value. Inset: exemplar coverslips from a single plate, for ‘no antib’ (WT TASK‐1 with no primary antibody), WT TASK‐1, TASK‐1_G106R, TASK‐1_L214 transfected cells and untransfected cells ‘untransf’. Each row has three unpermeabilized coverslips and three permeabilized (with Triton X‐100) coverslips. [Color figure can be viewed at wileyonlinelibrary.com]
In‐cell and on‐cell westerns
To quantify the expression of WT TASK‐1 and mutated channel protein in the cells and at the membrane, we used in‐cell and on‐cell westerns. An HA tag was engineered between positions alanine (50) and arginine (51) in the extracellular loop between the first transmembrane domain and the first pore region (M1/P1 loop) of WT TASK‐1 channels and the two mutant TASK‐1 channels. Thus, the signal seen in unpermeabilized cells is proportional to the amount of channel expressed at the cell membrane, whereas the signal in cells permeabilized with Triton X‐100 is proportional to the total number of channels expressed in the cells. The number of cells in each coverslip was quantified using DRAQ5, a far‐red DNA stain. Figure 4 F shows that there was no significant difference between the total number cells in coverslips for WT and mutated channels (DRAQ5 700 nm), the total amount of TASK channel protein expressed in the cells (in‐cell 800 nm) nor the amount of channel expression at the cell membrane (on‐cell 800 nm) (P > 0.05, two‐way ANOVA). Consistent with previous work (Renigunta et al. 2006, Mathie et al. 2010), the data show that the majority of TASK‐1 channel expression is intracellular, although the distributions of WT and mutated channels are similar in transfected cells.
Functional characterization of TASK‐1 variants under conditions of alkalosis
The next step was to characterize the functionality of these variants using a variety of modulators and pharmacological tools. TASK‐1 channels have a pK a of 7.3 and can be efficiently inhibited and activated by acidosis or alkalosis, respectively (Duprat et al. 1997). We investigated whether the TASK‐1 variants were still sensitive to pH changes, as well as whether it was possible to rescue functionality of these channels, by increasing extracellular pH from 7.4 to 8.4. As expected for WT TASK‐1 a change in pHo from 7.4 to 8.4 caused a significant increase in outward current density measured at –40 mV (P < 0.05, paired t test, 95% CI of difference = 11.6–20.0) (Fig. 5 A–C). For the TASK‐1_G106R variant, there was no significant effect of external alkalosis (P > 0.05, paired t test, 95% CI of difference = –0.2 to 3.1]) (Fig. 5 D–F). Similarly, for TASK‐1_L214R, there was also no significant effect of external alkalosis (P > 0.05, paired t test, 95% CI of difference = –1.0 to 1.0]) (Fig. 5 G–I).
Figure 5. Effect of extracellular alkalosis on WT TASK‐1 and TASK‐1 variants.
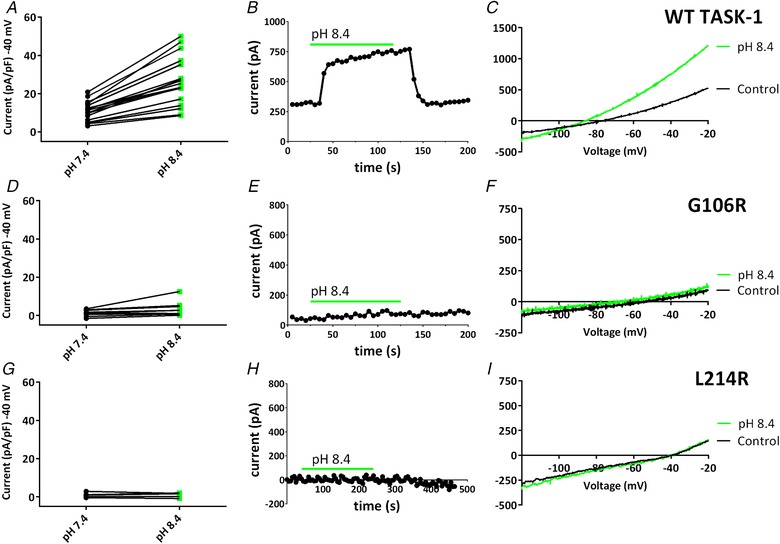
A, plot of currents (pA pF–1) measured at –40 mV, recorded through WT TASK‐1 channels in either pH 7.4 (black dots) or pH 8.4 (green squares). A black line links the same cell in each condition. B, time course plot showing the effect of pH 8.4 on WT TASK‐1 currents. Each point is a 5 s average of the current at –40 mV. Application of pH 8.4 is indicated by the green bar. Current prior to and after the green bar is measured at pH 7.4. C, representative currents recorded through WT TASK‐1 in a single cell, evoked by ramp changes in voltage from –120 to –20 mV in pH 7.4 (black line) or pH 8.4 (green line). D–F, as shown for (A) to (C), but for TASK‐1_G106R channels. G–I, as shown for (A) to (C), but for TASK‐1_L214R channels. [Color figure can be viewed at wileyonlinelibrary.com]
Effect of ONO‐RS‐082 on novel TASK‐1 variants
It has previously been reported that the phospholipase A2 inhibitor, ONO‐RS‐082, was able to rescue current through two of six heterozygous TASK‐1 mutations identified in patients with PAH (Ma et al. 2013). As reported by Ma et al. 2013, acute application of 10 μm ONO‐RS‐082 to WT TASK‐1 resulted in a significant increase in current density (P < 0.05, paired t test, 95% CI of difference = 2.3–14.3) (Fig. 6 A and B). This effect could be reversed in wash (Fig. 6 B). By contrast, current density through the two mutant TASK‐1 channels could not be recovered by ONO‐RS‐082 using the same experimental conditions. For TASK‐1_G106R, P > 0.05, paired t test, 95% CI of difference = –0.7 to 0.5] and for TASK‐1_L214R, P > 0.05, paired t test, 95% CI of difference = –1.5 to 1.0] (Fig. 6 C–F).
Figure 6. Effect of ONO‐RS‐082 (10 μM) on WT TASK‐1 and TASK‐1 variants.
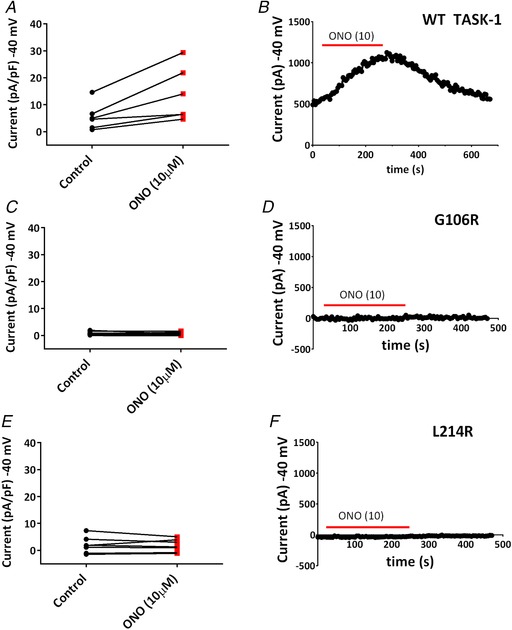
A, a plot of currents (pA pF–1) measured at –40 mV, recorded through WT TASK‐1 channels in either extracellular recording solution (control) (black dots) or control solution containing 10 μm ONO‐RS‐082 (red squares). A black line links the same cell in each condition. B, time course plot showing the acute application of ONO‐RS‐082 (10 μm) on WT TASK‐1 currents. Each point is a 5 s average of the current at –40 mV. Application of ONO‐RS‐082 is indicated by the red bar. Current prior to and after the red bar is measured in control solution. C and D, as shown for (A) and (B), but for TASK‐1_G106R channels. E and F, as shown for (A) and (B), but for TASK‐1_L214R channels. [Color figure can be viewed at wileyonlinelibrary.com]
Riociguat‐induced enhancement of TASK‐1 current
A relatively recent drug licensed for the treatment of PAH, riociguat belongs to a novel class of pharmacological agents that directly stimulate soluble guanylate cyclase (sGC), a key enzyme in the nitric oxide–cGMP signalling pathway, a major player in the pathophysiology of PAH. Riociguat stimulates the production of cGMP, which acts to regulate vascular tone by controlling dilatation and cellular proliferation of the vascular wall. We aimed to investigate the action of the sGC stimulator, riociguat, on WT TASK‐1 and the TASK‐1 variants. Incubation of WT TASK‐1 channels in either extracellular solution containing 10 μm riociguat or extracellular solution minus drug for 20 min before recording from cells resulted in a significant increase in current measured at –40 mV (P < 0.05, unpaired t test, 95% CI of difference = 1.6–8.5) from 5.8 pA pF–1 (n = 19, 5 days, 95% CI = 3.8–7.8) for control cells to 10.9 pA pF–1 (n = 17, 5 days, 95% CI = 7.8–14.0) for cells incubated in riociguat (Fig. 7 A and B). The increased current through TASK‐1 channels in riociguat led to a significant hyperpolarization of the zero current level (V M) (P < 0.05, unpaired t test, 95% CI of difference = 7.8–23.0) from –77 mV (95% CI = –72 to –82) to –93 mV (95% CI = –87 to –99).
Figure 7. Effect of Riociguat (10 μm) on WT TASK‐1 and TASK‐1 variants.
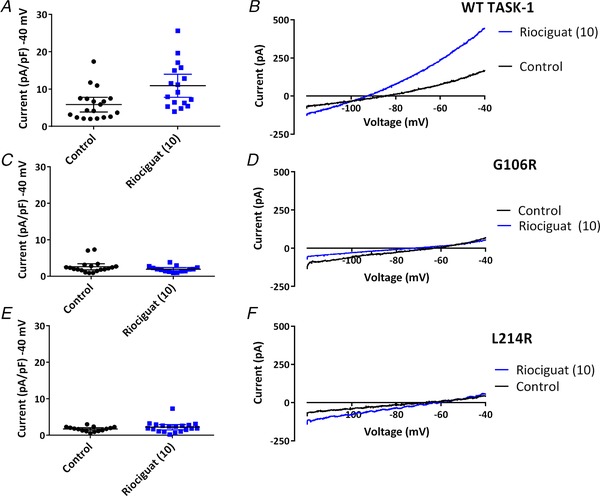
A, plot of current (pA pF–1) measured at –40 mV from individual cells transiently expressing WT TASK‐1 either incubated in extracellular solution minus riociguat (control: black dots) or incubated in extracellular solution containing 10 μm riociguat (blue squares). Error bars represent the 95% CI. B, representative currents recorded through WT TASK‐1, evoked by ramp changes in voltage from –120 mV to –40 mV under control conditions (2.5 mm [K+]o), (black line) and after incubation in 10 μm riociguat (blue line). C and D, as shown for (A) and (B), but for TASK‐1_G106R channels. E and F, as shown for (A) and (B), but for TASK‐1_L214R channels. [Color figure can be viewed at wileyonlinelibrary.com]
Using the same experimental procedure as described for WT TASK‐1, we investigated the effect of riociguat (10 μm) on the two TASK‐1 variants aiming to determine whether current could be rescued through these mutant channels. As seen for ONO‐RS‐082 and pH 8.4, no increase in functional current by riociguat was observed for either TASK‐1_G106R or TASK‐1_L214R. Incubation of TASK‐1_G106R channels in either extracellular solution containing 10 μm riociguat or extracellular solution minus drug for 20 min before recording from cells resulted in no significant difference in current measured at –40 mV (P > 0.05, unpaired t test, 95% CI of difference = –1.7 to 0.3) (Fig. 7 C and D). Similarly, incubation of TASK‐1_L214R channels in either extracellular solution containing 10 μm riociguat or extracellular solution minus drug for 20 min before recording from cells resulted in no significant difference in current measured at –40 mV (P > 0.05, unpaired t test, 95% CI of difference = –0.3 to 1.4) (Fig. 7 E and F). By contrast to cells expressing WT channels, riociguat had no significant effect on the zero current level (V M) of cells expressing TASK‐1_G106R channels (P > 0.05, 95% CI of difference = –12.6 to 5.7) or TASK‐1_L214R channels (P > 0.05, 95% CI of difference = –9.8 to 8.5).
Discussion
In the present study, we report for the first time the functional consequences of two mutations recently reported in PAH patients. We show that current through two new homozygous mutations (G106R, L214R) of TASK‐1 channels is considerably reduced compared to WT TASK‐1 channels. Although the two mutations of TASK‐1 considerably reduce channel current, this is not the result of any trafficking defects because WT and mutant channels appear to be equally distributed at the cell membrane. Instead, the two mutations must alter the functional properties of TASK‐1 channels to reduce current. In both cases, G106R and L214R, two large positively‐charged arginine residues, are substituted into the channel, close to the selectivity filter (Fig. 1), which might be hypothesized to interfere with flow of potassium ions through the channel.
TASK‐1 is expressed in rabbit, mouse, human and rat PASMCs (Gurney et al. 2003, Gardener et al. 2004, Olschewski et al. 2006, Manoury et al. 2009). The non‐inactivating K current in rat, rabbit and human PASMCs has functional characteristics of TASK‐1 channels (Olschewski et al. 2017), although this may not be the case in mouse PASMCs (Manoury et al. 2009, 2011). TASK‐1 channels are important regulators of PASMC resting membrane potential and excitability. In rat models of PAH, TASK‐1 current decreased progressively during development of the disease and this was associated with a membrane depolarization (Antigny et al. 2016). Indeed, the results of the present study suggest that reduced function of TASK‐1 channel activity is important in both hPAH and iPAH.
Pharmacological enhancement of WT TASK‐1 current is observed with both alkaline pH (8.4) and application of the phospholipase A2 inhibitor, ONO‐RS‐082. In rat models of PAH, treatment with ONO‐RS‐082 reversed the proliferation, vasoconstriction and inflammation that was observed (Antigny et al. 2016), indicating the importance of TASK‐1 channels in PAH.
Importantly, in some of the TASK‐1 mutations (T8K, E182K), but not all (e.g. G203D) described by Ma et al. (2013), ONO‐RS‐082 was able to restore current through mutant channels. However, this only occurred at membrane potentials more positive to the resting membrane potential of the cells and more positive to that seen for enhancement of WT TASK‐1 current. Similarly, we have shown previously that current through the mutated TASK‐3 channel (G236R) that underlies KCNK9 imprinting syndrome (Graham et al. 2016) can be recovered both pharmacologically through application of fenamate compounds and genetically through further gain of function channel mutations (Veale et al. 2014). These pharmacological interventions restore both current amplitude and the apparent K selectivity in the channel (Veale et al. 2014; Bohnen et al. 2017) because, without the latter, enhancement of a current with reduced K selectivity would exacerbate any pathophysiological responses resulting from cellular depolarization, as would be the case for PASMCs in PAH (Olschewski et al. 2017). However, neither of the two mutant channels in the present study showed any recovery of current following exposure to either alkaline pH or ONO‐RS‐082.
Although the two mutations in the present study have been observed as homozygous mutations in patients, previously identified mutations were heterozygous and heterozygous channel dimers did display functional TASK‐1 current, although this was considerably reduced compared to WT TASK‐1 (Ma et al. 2013; Bohnen et al. 2017). Current through these heterozygous mutations could be restored by ONO‐RS‐082 (Bohnen et al. 2017). For the two mutations in the present study, current through heterozygous channels was no larger than that seen for the homozygous mutants. Furthermore, mutant TASK‐1 channels (such as WT TASK‐1 channels) (Czirjak & Enyedi 2002, Berg et al. 2004, Kang et al. 2004) could form functional heteromeric channels with TASK‐3 channels (Bohnen et al. 2017). Homomeric TASK‐1 channels are the predominant TASK channels in PASMCs, particularly human PASMCs (Olschewski et al. 2006), and the role of these channels in regulating pulmonary vascular tone and controlling the resting membrane potential of these cells is well established in humans (Boucherat et al. 2015). However, in other tissues, such as atrial cardiomyoctes (Rinne et al. 2015), carotid body glomus cells (Kim et al. 2009) and cerebellar granule neurons (Aller et al. 2005), the effect of TASK‐1 channel mutations could be somewhat mitigated through the formation of functional heterodimers with TASK‐3 channel subunits in these cells.
The guanylate cyclase activator, riociguat, is approved for the treatment of PAH. Riociguat catalyses the synthesis of cGMP, and activates protein kinase G (PKG), which acts through several putative mechanisms to reduce intracellular Ca and inhibit smooth muscle contraction (Ghofrani et al. 2017).
Previous studies have shown that cGMP can enhance current through both recombinant TASK‐1 channels in expression systems and native TASK‐1 currents in basal forebrain neurons (Kang et al. 2007, Toyoda et al. 2008, 2010). Similarly, TNFα activation of TASK‐3 channels (El Hachmane et al. 2014) is considered to be mediated through cGMP. It has been proposed that cGMP activates PKG, which then tunes the pH sensitivity of TASK‐1 channels, through PKG‐mediated phosphorylation, such that the phosphorylated channels are less inhibited by H ions at physiological pH values (Toyoda et al. 2010).
In the present study, we show for the first time that riociguat enhances current through WT TASK‐1 channels, which could contribute to its therapeutic benefit in PAH. However, riociguat does not have any effect on current through G106R or L214R mutated TASK‐1 channels. Whether this is important in terms of the therapeutic strategy for patients with these mutations will depend on whether the primary action of riociguat in PAH is mediated through activation of TASK‐1 channels or whether this is an additional (but beneficial) action of riociguat (Ghofrani et al. 2017). In this regard, it is of interest that other regulatory pathways contributing to cell proliferation and vasoconstriction in the pulmonary arteries alter the function of TASK‐1 channels in PASMCs. For example, endothelin‐1, a potent vasoconstrictor that stimulates vascular remodelling, inhibits TASK‐1 channels via both a protein kinase C‐dependent pathway (Tang et al. 2009, Schiekel et al. 2013) and Rho kinase (Seyler et al. 2012). Similarly, treprostinil, a stable analogue of prostacyclin, acts via PKA to up‐regulate TASK‐1 current in human PASMCs (Olschewski et al. 2006).
Down‐regulation or inhibition of TASK‐1 channel activity has been proposed to contribute to vascular remodelling in PAH (Olschewski et al. 2006, Tang et al. 2009, Antigny et al. 2016) and a loss of TASK‐1 channel function and expression is a characteristic of right ventricular hypertrophy associated with pulmonary hypertension (Lambert et al. 2018). Guanylate cyclase activators, endothelin receptor antagonists and prostacyclin analogues are established vascular therapeutic strategies in the treatment of PAH, acting to reduce vasoconstriction and smooth muscle proliferation, and all three will stimulate activation of TASK‐1 channels in PASMCs, which would contribute to their therapeutic benefit.
In summary, novel mutations in TASK‐1 channels recently seen in PAH are associated with a loss of function. Current through these channels could not be restored by activators of TASK‐1 channels. Riociguat enhancement of current through TASK‐1 channels could contribute to its therapeutic benefit in the treatment of PAH.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
ALC, AM, PE‐S and ELV participated in the research design. KPC, RGH and ELV conducted the experiments. KPC, RGH, ELV and AM performed the data analysis. AM and ELV wrote the manuscript. KPC, RGH, ALC, PE‐S, ELV and AM revised the final manuscript and approved the version submitted for publication.
Funding
This study was supported by the Biotechnology and Biological Sciences Research Council (UK) [BB/J000930/1] (AM, ELV); the Spanish Ministerio de Economia y Competitividad (SAF2016‐77222‐R to AC) with funds co‐financed by ERDF (FEDER) Funds from the European Commission, ‘A way of making Europe’; by Comunidad de Madrid en Biomedicina (B2017/BMD‐3727) (AC); and by the Cardiovascular Medical Research and Education Fund (USA) (AC, AM).
Acknowledgements
KPC and RGH are supported by the award of University of Kent VC scholarships.
Biography
Kevin Cunningham studied for his BSc (with honours) in Pharmacology at Kingston University and followed this with an MSc in Cancer Biology from the University of Kent, where he received a Kent Cancer Trust Scholarship. He is currently a final year PhD student in receipt of a prestigious 3 year University of Kent, Vice Chancellor's Research Scholarship under the supervision of Professor Alistair Mathie and Dr Emma Veale. Kevin's research investigates the role of two pore domain potassium (K2P) channels and their role in pulmonary arterial hypertension. Post‐PhD, Kevin hopes to continue exploring ion channel regulation within cardiovascular disease.

Edited by: Harold Schultz & Larissa Shimoda
References
- Aller MI, Veale EL, Linden AM, Sandu C, Schwaninger M, Evans LJ, Korpi ER, Mathie A, Wisden W & Brickley SG (2005). Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar granule neurons. J Neurosci 25, 11455–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antigny F, Hautefort A, Meloche J, Belacel‐Ouari M, Manoury B, Rucker‐Martin C, Péchoux C, Potus F, Nadeau V, Tremblay E, Ruffenach G, Bourgeois A, Dorfmüller P, Breuils‐Bonnet S, Fadel E, Ranchoux B, Jourdon P, Girerd B, Montani D, Provencher S, Bonnet S, Simonneau G, Humbert M & Perros F (2016). Potassium channel subfamily K member 3 (KCNK3) contributes to the development of pulmonary arterial hypertension. Circulation 133, 1371–1385. [DOI] [PubMed] [Google Scholar]
- Berg AP, Talley EM, Manger JP & Bayliss DA (2004). Motoneurons express heteromeric TWIK‐related acid‐sensitive K+ (TASK) channels containing TASK‐1 (KCNK3) and TASK‐3 (KCNK9) subunits. J Neurosci 24, 6693–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen MS, Roman‐Campos D, Terrenoire C, Jnani J, Sampson KJ, Chung WK & Kass RS (2017). The impact of heterozygous KCNK3 mutations associated with pulmonary arterial hypertension on channel function and pharmacological recovery. J Am Heart Assoc 6, e006465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucherat O, Chabot S, Antigny F, Perros F, Provencher S & Bonnet S (2015). Potassium channels in pulmonary arterial hypertension. Eur Respir J 46, 1167–1177. [DOI] [PubMed] [Google Scholar]
- Brohawn SG, del Mármol J & MacKinnon R (2012). Crystal structure of the human K2P TRAAK, a lipid‐ and mechano‐sensitive K+ ion channel. Science 335, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butrous G (2015). Human immunodeficiency virus‐associated pulmonary arterial hypertension: considerations for pulmonary vascular diseases in the developing world. Circulation 131, 1361–1370. [DOI] [PubMed] [Google Scholar]
- Chen C & Okayama H (1987). High‐efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham KP, Veale EL, Escribano‐Subias P, Cogolludo AL & Mathie A (2017). Electrophysiological characterisation of two novel KCNK3 (TASK‐1) mutations indicated in pulmonary arterial hypertension. Basic Clin Pharmacol Toxicol 121 (Suppl 2), P59. [Google Scholar]
- Czirják G & Enyedi P (2002). Formation of functional heterodimers between the TASK‐1 and TASK‐3 two‐pore domain potassium channel subunits. J Biol Chem 277, 5426–5432. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C & Lazdunski M (1997). TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J 16, 5464–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hachmane MF, Rees KA, Veale EL, Sumbayev VV & Mathie A (2014). Enhancement of TWIK‐related acid‐sensitive potassium channel 3 (TASK3) two‐pore domain potassium channel activity by tumor necrosis factor α. J Biol Chem 289, 1388–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P & Czirják G (2010). Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev 90, 559–605. [DOI] [PubMed] [Google Scholar]
- Gaine S & McLaughlin V (2017). Pulmonary arterial hypertension: tailoring treatment to risk in the current era. Eur Respir Rev 26, 170095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M & Hoeper M (2015). 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46, 903–975. [DOI] [PubMed] [Google Scholar]
- Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM & Weston AH (2004). Functional evidence of a role for two‐pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol 142, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg L, Akbar G, Agrawal S, Agarwal M, Khaddour L, Handa R, Garg A, Shah M, Patel B & Dalal BD (2017). Drug‐induced pulmonary arterial hypertension: a review. Heart Fail Rev 22, 289–297. [DOI] [PubMed] [Google Scholar]
- Ghofrani HA, Humbert M, Langleben D, Schermuly R, Stasch JP, Wilkins MR & Klinger JR (2017). Riociguat: mode of action and clinical development in pulmonary hypertension. Chest 151, 468–480. [DOI] [PubMed] [Google Scholar]
- Girerd B, Perros F, Antigny F, Humbert M & Montani D (2014). KCNK3: new gene target for pulmonary hypertension? Expert Rev Respir Med 8, 385–387. [DOI] [PubMed] [Google Scholar]
- Graham JM Jr, N Zadeh, Kelley M, Tan ES, Liew W, Tan V, Deardorff MA, Wilson GN, Sagi‐Dain L & Shalev SA (2016). KCNK9 imprinting syndrome‐further delineation of a possible treatable disorder. Am J Med Genet A 170, 2632–2637. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ & Kempsill FE (2003). Two‐pore domain K channel, TASK‐1, in pulmonary artery smooth muscle cells. Circ Res 93, 957–964. [DOI] [PubMed] [Google Scholar]
- Hemnes AR & Humbert M (2017). Pathobiology of pulmonary arterial hypertension: understanding the roads less travelled. Eur Respir Rev 26, 170093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill NS, Cawley MJ & Heggen‐Peay CL (2016). New therapeutic paradigms and guidelines in the management of pulmonary arterial hypertension. J Manag Care Spec Pharm 22 (3 Suppl A), S3‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Lau EM, Montani D, Jaïs X, Sitbon O & Simonneau G (2014). Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 130, 2189–2208. [DOI] [PubMed] [Google Scholar]
- Kang D, Han J, Talley EM, Bayliss DA & Kim D (2004). Functional expression of TASK‐1/TASK‐3 heteromers in cerebellar granule cells. J Physiol 554, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Dempo Y, Ohashi A, Saito M, Toyoda H, Sato H, Koshino H, Maeda Y & Hirai T (2007). Nitric oxide activates leak K+ currents in the presumed cholinergic neuron of basal forebrain. J Neurophysiol 98, 3397–3410. [DOI] [PubMed] [Google Scholar]
- Kim D, Cavanaugh EJ, Kim I & Carroll JL (2009). Heteromeric TASK‐1/TASK‐3 is the major oxygen‐sensitive background K+ channel in rat carotid body glomus cells. J Physiol 587, 2963–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M, Boet A, Rucker‐Martin C, Mendes‐Ferreira P, Capuano V, Hatem S, Adão R, Brás‐Silva C, Hautefort A, Michel JB, Dorfmuller P, Fadel E, Kotsimbos T, Price L, Jourdon P, Montani D, Humbert M, Perros F & Antigny F (2018). Loss of KCNK3 is a hallmark of RV hypertrophy/dysfunction associated with pulmonary hypertension. Cardiovasc Res 114, 880–893. [DOI] [PubMed] [Google Scholar]
- Ma L, Roman‐Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Trégouët DA, Borczuk A, Rosenzweig EB, Girerd B, Montani D, Humbert M, Loyd JE, Kass RS & Chung WK (2013). A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 369, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado RD, Southgate L, Eichstaedt CA, Aldred MA, Austin ED, Best DH, Chung WK, Benjamin N, Elliott CG, Eyries M, Fischer C, Gräf S, Hinderhofer K, Humbert M, Keiles SB, Loyd JE, Morrell NW, Newman JH, Soubrier F, Trembath RC, Viales RR & Grünig E (2015). Pulmonary arterial hypertension: a current perspective on established and emerging molecular genetic defects. Hum Mutat 36, 1113–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoury B, Etheridge SL, Reid J & Gurney AM (2009). Organ culture mimics the effects of hypoxia on membrane potential, K(+) channels and vessel tone in pulmonary artery. Br J Pharmacol 158, 848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoury B, Lamalle C, Oliveira R, Reid J & Gurney AM (2011). Contractile and electrophysiological properties of pulmonary artery smooth muscle are not altered in TASK‐1 knockout mice. J Physiol 589, 3231–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Rees KA, El Hachmane MF & Veale EL (2010). Trafficking of neuronal two pore domain potassium channels. Curr Neuropharmacol 8, 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas P, Tenorio J, Quezada CA, Barrios E, Gordo G, Arias P, López Meseguer M, Santos‐Lozano A, Palomino Doza J, Lapunzina P & Escribano Subías P (2016). Molecular analysis of BMPR2, TBX4, and KCNK3 and genotype‐phenotype correlations in Spanish patients and families with idiopathic and hereditary pulmonary arterial hypertension. Rev Esp Cardiol (Engl Ed) 69, 1011–1019. [DOI] [PubMed] [Google Scholar]
- Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, Kwapiszewska G, Klepetko W, Seeger W & Olschewski H (2006). Impact of TASK‐1 in human pulmonary artery smooth muscle cells. Circ Res 98, 1072–1080. [DOI] [PubMed] [Google Scholar]
- Olschewski A, Veale EL, Nagy BM, Nagaraj C, Kwapiszewska G, Antigny F, Lambert M, Humbert M, Czirják G, Enyedi P & Mathie A (2017). TASK‐1 (KCNK3) channels in the lung: from cell biology to clinical implications. Eur Respir J 50, 1700754. [DOI] [PubMed] [Google Scholar]
- Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O'connor DT & Yuan JX (2007). Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292, C1837–C1853 [DOI] [PubMed] [Google Scholar]
- Renigunta V, Yuan H, Zuzarte M, Rinne S, Koch A, Wischmeyer E, Schlichthorl G, Gao Y, Karschin A, Jacob R, Schwappach B, Daut J & Preisig‐Muller R (2006). The retention factor p11 confers an endoplasmic reticulum‐localization signal to the potassium channel TASK‐1. Traffic 7, 168–181. [DOI] [PubMed] [Google Scholar]
- Rinné S, Kiper AK, Schlichthörl G, Dittmann S, Netter MF, Limberg SH, Silbernagel N, Zuzarte M, Moosdorf R, Wulf H, Schulze‐Bahr E, Rolfes C & Decher N (2015). TASK‐1 and TASK‐3 may form heterodimers in human atrial cardiomyocytes. J Mol Cell Cardiol 81, 71–80. [DOI] [PubMed] [Google Scholar]
- Schiekel J, Lindner M, Hetzel A, Wemhöner K, Renigunta V, Schlichthörl G, Decher N, Oliver D & Daut J (2013). The inhibition of the potassium channel TASK‐1 in rat cardiac muscle by endothelin‐1 is mediated by phospholipase C. Cardiovasc Res 97, 97–105. [DOI] [PubMed] [Google Scholar]
- Seyler C, Duthil‐Straub E, Zitron E, Gierten J, Scholz EP, Fink RH, Karle CA, Becker R, Katus HA & Thomas D (2012). TASK1 (K(2P)3.1) K(+) channel inhibition by endothelin‐1 is mediated through Rho kinase‐dependent phosphorylation. Br J Pharmacol 165, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM & Souza R (2013). Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62(25 Suppl), D34–41. [DOI] [PubMed] [Google Scholar]
- Tang B, Li Y, Nagaraj C, Morty RE, Gabor S, Stacher E, Voswinckel R, Weissmann N, Leithner K, Olschewski H & Olschewski A (2009). Endothelin‐1 inhibits background two‐pore domain channel TASK‐1 in primary human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 41, 476–483. [DOI] [PubMed] [Google Scholar]
- Tang H, Desai AA & Yuan JX (2016). Genetic insights into pulmonary arterial hypertension. Application of whole‐exome sequencing to the study of pathogenic mechanisms. Am J Respir Crit Care Med 194, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Saito M, Okazawa M, Hirao K, Sato H, Abe H, Takada K, Funabiki K, Takada M, Kaneko T & Kang Y (2010). Protein kinase G dynamically modulates TASK1‐mediated leak K+ currents in cholinergic neurons of the basal forebrain. J Neurosci 30, 5677–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda H, Saito M, Sato H, Dempo Y, Ohashi A, Hirai T, Maeda Y, Kaneko T & Kang Y (2008). cGMP activates a pH‐sensitive leak K+ current in the presumed cholinergic neuron of basal forebrain. J Neurophysiol 99, 2126–2133. [DOI] [PubMed] [Google Scholar]
- Veale EL, Buswell R, Clarke CE & Mathie A (2007). Identification of a region in the TASK3 two pore domain potassium channel that is critical for its blockade by methanandamide. Br J Pharmacol 152, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veale EL, Hassan M, Walsh Y, Al‐Moubarak E & Mathie A (2014). Recovery of current through mutated TASK3 potassium channels underlying Birk Barel syndrome. Mol Pharmacol 85, 397–407. [DOI] [PubMed] [Google Scholar]
- Yuan JX, Aldinger AM, Juhaszova M, Wang J, Jr Conte JV, Gaine SP, Orens JB & Rubin LJ (1998). Dysfunctional voltage‐gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 98, 1400–1406. [DOI] [PubMed] [Google Scholar]