

Abstract
Exquisite control of catalytic metathesis reactivity is possible through ligand‐based variation of ruthenium carbene complexes. Sterically hindered alkenes, however, remain a generally recalcitrant class of substrates for intermolecular cross‐metathesis. Allylic chalcogenides (sulfides and selenides) have emerged as “privileged” substrates that exhibit enhanced turnover rates with the commercially available second‐generation ruthenium catalyst. Increased turnover rates are advantageous when competing catalyst degradation is limiting, although specific mechanisms have not been defined. Herein, we describe facile cross‐metathesis of allylic sulfone reagents with sterically hindered isoprenoid alkene substrates. Furthermore, we demonstrate the first example of intermolecular cross‐metathesis of ruthenium carbenes with a tetrasubstituted alkene. Computational analysis by combined coupled cluster/DFT calculations exposes a favorable energetic profile for metallacyclobutane formation from chelating ruthenium β‐chalcogenide carbene intermediates. These results establish allylic sulfones as privileged reagents for a substrate‐based strategy of cross‐metathesis derivatization.
Keywords: tetrasubstituted, chalcogenide, terpenoid, squalene, coupled cluster
The ligand‐based design of ruthenium metathesis catalysts has achieved remarkable efficiency with broad tolerance of functional groups under a wide range of reaction conditions. The versatile second generation Hoveyda−Grubbs(II) precatalyst [HG(II)] incorporates imidazolin‐2‐ylidene and chelating benzylidene‐ether ligands.1 Intermolecular cross‐metathesis is most successful with matched alkene pairs where one exhibits high reactivity, characterized by rapid homodimerization, and the second substrate is less reactive and dimerizes with reluctance due to electronic or steric factors.2 Recently, trisubstituted prenyl derivatives have been employed as partners for ring‐closing and cross‐metathesis with challenging 1,1‐disubstituted alkenes, by promoting catalyst turnover through the avoidance of unstable methylidene complexes.3,4 Within the interplay of steric and electronic factors, substituted alkenes remain reluctant partners for cross‐metathesis and impeded turnover kinetics can result in catalyst degradation and competing secondary processes.5 Modification of aryl ligand substituents in ruthenium and molybdenum catalysts opens the coordination environment to accommodate sterically hindered alkene substrates, while substitution of the N‐heterocyclic carbene backbone provides catalysts with improved efficiency for ring‐closing metathesis to yield tetrasubstituted cycloalkenes.6,7 Problems associated with this approach include reduced catalyst stability and limited scope of effective substrates, highlighting the need for alternative strategies.
Allylic sulfides and selenides have been identified as “privileged substrates” that promote efficient cross‐metathesis coupling with the HG(II) catalyst.8, 9, 10, 11 Enhanced relative cross‐metathesis rates are particularly advantageous under challenging conditions such as aqueous media where high turnover frequency overcomes competitive catalyst decomposition.8 A mechanistic rationale for this enhanced reactivity was suggested to involve a relay process in which sulfur coordinates and positions the alkene proximal to the alkylidene complex.10 A subtle structural balance between stability and reactivity was evident from observations that homologous butenyl and pentenyl sulfides were inactive as metathesis substrates.8 The stabilizing effects of chelating benzylidene sulfide ligands are evident in “latent” ruthenium carbene catalysts that require thermal or photochemical activation to initiate metathesis propagation.12 In this work, we investigated HG(II)‐catalyzed cross‐metathesis of allylic sulfide and sulfone reagents with a panel of severely hindered alkene substrates. Product identification and quantitation of metathesis products was facilitated using derivatives possessing a triazaborolopyridinium chromophore, and unique features of reactivity and regioselectivity were revealed. We report the unprecedented intermolecular cross‐metathesis of allylic chalcogenides with tetramethylethylene. Extensive computer simulation with combined coupled cluster/density functional theory calculations support a mechanistic pathway that involves chelate‐stabilized ruthenium β‐chalcogenide carbene complexes that provide an energetically accessible pathway to π‐complexes with highly substituted alkenes, followed by rate‐determining formation of metallacyclobutane intermediates.
The three fluorescent metathesis probes shown in Figure 1 with vinyl (1 b), allylic sulfide (1 c), and sulfone (1 d) groups were synthesized (Supplemental Scheme 1). Sulfoxides were not included in this study because they are reported to be poor substrates in cross−metathesis reactions, although they readily participate in ring closing metathesis.13,14 Complete experimental procedures, compound characterization data, photophysical properties of (1 b–1 d), and computational details are provided in the Supplementary Information.
Figure 1.
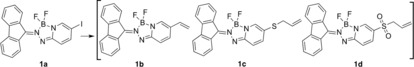
Structures of spectrophotometric probes
Comparative homodimerization reactions of the alkenyl‐probes (vinyl 1 b, allylic sulfide 1 c, and sulfone 1 d were investigated to establish their relative metathesis reactivity using the HG(II) catalyst (5 mol %), in dichloromethane at 40 °C (see SI for details). The allylic sulfide 1c and sulfone probes 1 d produced the homodimers 2 c and 2 d in high yields, respectively. In contrast, the vinyl substrate 1 b produced only a trace quantity of homodimer 2 b even after extended reaction time (12 hours), consistent with expectations for substituted styrenes.15
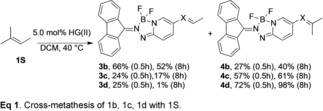
The alkene 2‐methyl‐2‐butene (1 S) is a convenient substrate for the preparation of isoprenoid derivatives due to its volatility (35–38 °C), that enables the use of excess reagent to drive a cross‐metathesis event towards complete conversion.16,17 Comparative reactions of the alkenyl probes 1 b, 1 c, and 1 d were conducted using 100 equivalents of 2‐methyl‐2‐butene and 5 mol % of the HG(II) catalyst in dichloromethane at 40 °C (Equation 1). The cross‐metathesis of 1 b yielded a product ratio favoring disubstituted/trisubstituted alkene 3 b/4 b=2.4/1.0 and trace amounts of homodimer 2 b after 0.5 hours. Extension of the reaction period to 8 hours increased the amount of trisubstituted alkene 3 b/4 b=1.3/1.0. In contrast, allylic sulfide 1 c reacted rapidly with regioselectivity for the trisubstituted prenylsulfide 3 c/4 c=1.0/2.4 at 0.5 hours. This product distribution and the amount of homodimer 2 c varied little after an extended reaction time (8 hours), although small amounts of oxidized compounds (sulfoxide and sulfone) were detected and isolated yields for preparative‐scale reactions decreased. Sulfone 1 d reacted rapidly and regioselectively to preferentially produce the prenylsulfone 3 d/4 d=1.0/2.9 at 0.5 hours. Extension of the reaction period of 1 d to 8 hours resulted in complete conversion to the trisubstituted prenyl sulfone.
These results demonstrate that the disubstituted sulfone‐functionalized alkene 3 d remained a competent substrate in the catalytic cycle with 1 S to produce the thermodynamically favored trisubstituted prenyl product 4 d. Only trace amounts of the allylsulfone homodimerization product 2 d were observed under these conditions that involved a large excess of 1 S. Control experiments using homodimer 2 d and excess 1 S in the presence of HG(II) catalyst proceeded slowly to form the expected di‐ and trisubstituted products 3 d/4 d (8 % and 23 %, respectively) after 8 hours (see SI for details Figure S11). While it is clear that homodimer 2 d re‐entered the catalytic cross‐metathesis reaction, poor solubility limited conversion under these reaction conditions. Additional control experiments with the isolated prenylsulfide 4 c and prenylsulfone 4 d demonstrated that these trisubstituted alkenes were unable to initiate metallacyclobutane formation with the HG(II) catalyst precursor (data not shown), consistent with expectations based on steric influences.3,6, 19
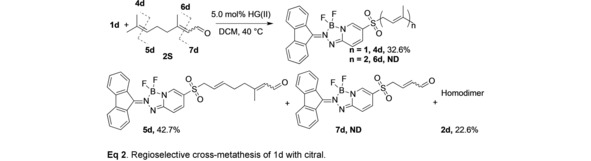
Catalytic metathesis of terpenoids is of interest for the production of fine chemicals and polymers.18 In light of the efficient labeling and high isolated yields of prenyl products from sulfone 1 d, we investigated the possible electronic effects of the substrate on the relative reactivity of this probe. The terpenoid substrate citral (2 S), which consists of an E/Z isomeric mixture of citral A/B (61/31), was selected as a model system to evaluate possible electronic factors that control regioselectivity of 1 d for different trisubstituted alkene groups. For this experiment the stoichiometry was modified by using a 1 : 1 ratio of allyl sulfone 1 d to citral in the presence of the catalyst HG(II) (5 mol %) in dichloromethane at 40 °C (Equation 2). After 1 h cross‐metathesis occurred exclusively at the isolated isoprenyl tail to yield disubstituted alkene 5 d (43 %) and trisubstituted prenyl product 4 d (33 %), accompanied by the homodimerization product 2 d (24 %) and trace amounts of unreacted 1 d (<1 %). No products from cross‐metathesis of the electron deficient conjugated enal group (6 d, 7 d) were observed, which illustrates the preferential reactivity of 1 d with electron‐rich alkene substrates. Authentic samples of compound 7 d were synthesized by an alternative route (see SI) and used to verify that 7 d was undetectable as a product under these conditions.
To further evaluate steric influences, we investigated the cross‐metathesis of 1 d with the triterpene squalene (3 S). This symmetrical polyisoprenoid substrate consists of two head‐to‐head linked farnesyl units, and can produce six different products of intermolecular cross‐metathesis. The reaction stoichiometry was modified to a 5‐fold excess of squalene so that 1 d was the limiting reactant, while maintaining standard conditions and reaction time (1 hour). The product distribution was analyzed by HPLC‐ PDA‐HRMS, and quantified results are shown in Figure 2. The product mixture contained all of the expected trisubstituted (4 d, 6 d, 10 d) and disubstituted (8 d, 9 d, 11 d) products. The predominant products resulted from cross‐metathesis at the terminal alkene that produced the disubstituted alkene 8 d (20 %) and the regioisomeric trisubstituted C5 prenyl compound 4 d (45 %). Similar ratios (∼1 : 2) of di‐/trisubstituted products were observed for each of the other internal alkene positions, which resulted in similar amounts of the C10 geranyl 6 d (6 %) and C15 farnesyl 10 d (9 %) analogs. These results demonstrate efficient catalyst turnover for cross‐metathesis of 1 d with electron‐rich trisubstituted alkenes and suggest the operation of sulfone‐associated structural features that poise the incipient ruthenium β‐sulfonyl carbene complexes for enhanced reactivity with sterically hindered substrates.
Figure 2.
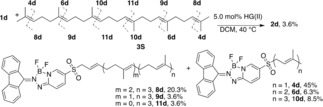
Regioselective cross‐metathesis of 1 d with polyisoprenoid squalene.
A review of the literature was unable to identify examples of metathesis events for ruthenium carbene complexes incorporating a tetrasubstituted alkene, and previously reported attempts to engage 2,3‐dimethyl‐2‐butene (4 S) in cross‐metathesis were unsuccessful.16
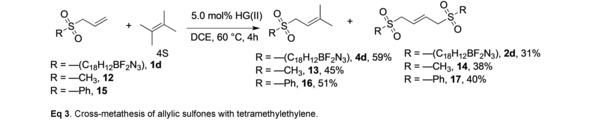
The facile reactivity of allylic sulfone 1 d with isoprenoid substrates encouraged us to investigate the possibility of cross‐metathesis with 4 S. Remarkably, the allylic sulfone 1 d efficiently produced the cross‐metathesis product 4 d (59 %), accompanied by homodimer 2 d (31 %), see Equation 3. The allyl sulfide 1 c was also converted to the cross‐metathesis product 4 c (24 %), although the yield was lower relative to the sulfone, and minor amounts of unreacted 1 c, and oxidized products were also detected (SI Figure S15). Additional allylic sulfone substrates were evaluated to assess the scope of this surprising reaction and to determine if any specific features of the reactivity were associated with the triazaborolopyridinium dye. The HG(II) catalyzed cross metathesis of tetrasubstituted 4 S was successful with aliphatic substrate methylallylsulfone 12, producing the trisubstituted prenyl derivative 13 (45 %) and homodimer 14 (38 %). In a similar way, the phenylallylsulfone analog 15 underwent efficient cross‐metathesis with 4 S to produce the alkene 16 (51 %) and homodimer 17 (40 %). Additional cross‐metathesis reactions of 4 S with the electron deficient vinyl sulfone 18, and the allylic chalcogenide allyl p‐anisyl ether 19 were attempted (see SI). No cross metathesis products of the vinylsulfone 18 with 4 S were detected under these conditions over 12 h. Previous reports have shown cross metathesis of 18 with type I alkene substrates.20 Only trace amounts of cross metathesis products from the allylic ether 19 with 4 S were detected, similar to published reports demonstrating increased reactivity of allylic sulfides compared to ethers.9 These results demonstrate that unique structural features of the allylic sulfone and sulfide moieties are associated with the enhanced cross‐metathesis reactivity of sterically obstructed alkenes. With experimental evidence documenting cross metathesis of allylic chalcogenides with tetrasubstituted alkene substrates, computational studies were initiated to elucidate potential structural features and the energetics of the interaction with activated ruthenium N‐heterocyclic carbene catalysts.
The energetic profiles of the reaction of 2,3‐dimethyl‐2‐butene with HG(II) and the incipient carbenes derived from methylallyl sulfide 12 (that is, [Ru]=CH−CH2−S−CH3) and sulfone 15 (that is, [Ru]=CH−CH2−SO2−CH3), were determined using electronic structure calculations. We analyzed the intermediates and transition states following the established three‐step reaction course according to the Chauvin mechanism that involves formation of the ruthenium carbene π‐complex, followed by the formation of metallacyclobutane, and finally dissociation to the product π‐complex.21 Previous computational studies on the mechanisms of HG(II) initiation reveal that an interchange mechanism involving synchronous departure of the chelating ether ligand with incoming alkene was the most common route leading to π‐complex formation with α‐olefins, compared with the alternative dissociative or associative mechanisms.22 The extreme steric parameters associated with a tetrasubstituted alkene component have not previously been explored by computation. Electronic structure calculations were performed using a state‐of‐the‐art combined coupled cluster/DFT approach, in which the geometry structures of interest were optimized at the M06‐L/def2‐SV(P)+PCM(dichloromethane) level of theory [see Figure S18 and Table S3 in the Supporting Information for computational details and comparison of the computed and X‐ray structures of HG(II)], and the electronic energies were further refined by the single‐point calculations at the DLPNO‐CCSD(T)/def2‐TZVP level of theory.23
The calculated free energy profile for the interaction of 2,3‐dimethyl‐2‐butene 4 S with HG(II) showed that formation of the corresponding metallocyclobutane is a prohibitively high endothermic step (ΔG=100 kJ/mol, see Figure 3 (red lines). These results suggest that a dissociative mechanism would be required with HG(II) due to steric constraints of the tetrasubstituted substrate. In contrast, the free energy diagram for the intermolecular cross‐metathesis of 4 S with the β‐sulfonyl‐ruthenium carbene complex derived from allylsulfone reveals that an efficient route is available with a net free energy of activation ΔG ≠=42 kJ/mol as shown in Figure 3 (green lines). The free energy diagram for the β‐sulfidyl‐ruthenium carbene complex shown in Figure 3 (black lines) exposes a significantly lower reaction profile than that for the HG(II) catalyst, but still higher than the corresponding potential energy surface of the β‐sulfonyl‐ruthenium carbene complex. Chelation of the sulfone and sulfide appendages are important structural features involved in both of the computed lowest energy ruthenium carbene complexes derived from the allylic chalcogenides, but in presence of alkene 4 S each of these intermediates proceeds to form the non‐chelated π‐complexes. These results suggest that chelation of the β‐chalcogenide groups provides anchimeric stabilization of the activated ruthenium carbene intermediates and may thereby avoid competitive catalyst degradation that often accompanies stalled catalytic cycles. The coordination sphere then opens to accommodate ligation of tetramethylethylene that initiates the cross‐metathesis process. The fine balance involved in the energetics of chelate stabilization in this case contrasts with the well‐known examples of latent metathesis catalysts in which the stability of the chelated carbene requires significant thermal or photochemical initiation of metathesis. Strategies that involve exogenous addition of Lewis acids have been implemented to overcome impeded metathesis with ester‐functionalized substrates, but this approach does not extend the steric tolerance for hindered substrates.24 The increased energetic cost of the sulfide‐based Ru catalyst as compared with the sulfone‐based Ru catalyst develops steadily as the reaction progresses and is therefore likely related to the total endothermicity of the reaction (i. e. ΔG=54 and 30 kJ/mol for the profile with the sulfide‐ and sulfone‐based catalyst). The larger energetic cost associated with the β‐sulfidyl‐ruthenium carbene complex in the reaction with 4 S is consistent with the experimental observations of reduced product yields (SI Figure S15), and the detection of oxidized byproducts that likely result from catalyst degradation in the corresponding reactions of 1 c that were discussed previously.
Figure 3.
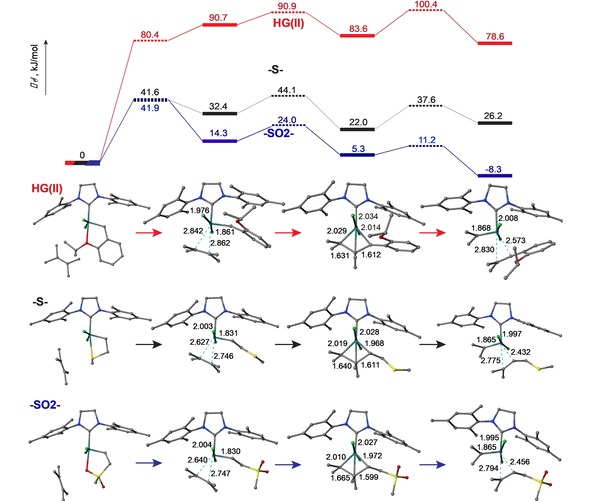
Top: Free energy profile of interaction of 2,3‐dimethyl‐2‐butene 4 S with HG(II), [Ru]=CH−CH2−S−CH3 and [Ru]=CH−CH2−SO2−CH3, shown using red, black, and blue colors, respectively [DLPNO‐CCSD(T)/def2‐TZVP//M06‐L/def2‐SV(P)+PCM(MeCN)]. Bottom: Calculated initial chelate structures of the HG(II) precatalyst and ruthenium β‐chalcogenide carbene complexes are aligned along the left edge, the corresponding key intermediates in the reaction coordinate pathways are correlated within columns directly below the composite free energy profiles, and selected bond lengths are associated with the computed structures. It is noted that the transition state free energy of the π‐complex formation involving HG(II) lies below the free energy of the resulting π‐complex due to the entropy correction (no such effect is present on the corresponding potential energy surface diagram).
These results reveal favorable energetics for the production of π‐complex and metallocyclobutane intermediates from ruthenium carbene complexes that possesses β‐chalcogenide ligands, and provide a rationale for the observed intermolecular cross‐metathesis reactions of allylic sulfones with sterically hindered substrates, including the first example involving a tetrasubstituted alkene. The β‐sulfonyl chelate structure achieves a functional balance between a stabilized resting state for the intermediate carbene catalyst to promote turnover and avoid the energetic traps that result in latent or stalled catalysts. Additionally, the β‐sulfonyl chelate structure provides access to coordination by alkenes with severe steric constraints. These results support the classification allylic sulfone reagents as privileged substrates for intermolecular cross‐metathesis, and we anticipate that this method will have broad utility for the derivatization of isoprenoids, recycling of polyisoprenoid rubber materials,25 and for bioconjugation and bioorthogonal labeling applications.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was supported by the National Science Foundation (NSF CMI 1507070) and mass spectrometry data was recorded on instrumentation obtained through the National Science Foundation Major Research Instrumentation Program (NSF MRI 1626468). Computational resources were provided by ICT Supercomputing of NMSU and by the Extreme Science and Engineering Discovery Environment (XSEDE) award TG‐CHE170004.
R. R. Sapkota, J. M. Jarvis, T. M. Schaub, M. R. Talipov, J. B. Arterburn, ChemistryOpen 2019, 8, 201.
References
- 1.
- 1a. Garber S., Kingsbury J., Gray B., Hoveyda A., J. Am. Chem. Soc. 2000, 122, 8168–8179; [Google Scholar]
- 1b. Hoveyda A., Zhugralin A., Nature 2007, 450, 243–251. [DOI] [PubMed] [Google Scholar]
- 2. Chatterjee A., Choi T., Sanders D., Grubbs R., J. Am. Chem. Soc. 2003, 125, 11360–11370. [DOI] [PubMed] [Google Scholar]
- 3. Wang Z., Jackson W., Robinson A., Org. Lett. 2013, 15, 3006–3009. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Bahou K., Braddock D., Meyer A., Savage G., Org. Lett. 2017, 19, 5332–5335; [DOI] [PubMed] [Google Scholar]
- 4b. Burnley J., Wang Z. J., Jackson W. R., Robinson A. J., J. Org. Chem. 2017, 82, 8497–8505. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Yu B., Hamad F., Sels B., Van Hecke K., Verpoort F., Dalton Trans. 2015, 44, 11835–11842; [DOI] [PubMed] [Google Scholar]
- 5b. Engel J., Smit W., Foscato M., Occhipinti G., Tornroos K., Jensen V., J. Am. Chem. Soc. 2017, 139, 16609–16619. [DOI] [PubMed] [Google Scholar]
- 6. Stewart I., Douglas C., Grubbs R., Org. Lett. 2008, 10, 441–444. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Nguyen T., Koh M., Mann T., Schrock R., Hoveyda A., Nature 2017, 552, 347–354; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Cesar V., Zhang Y., Kosnik W., Zielinski A., Rajkiewicz A., Ruamps M., Bastin S., Lugan N., Lavigne G., Grela K., Chem. Eur. J. 2017, 23, 1950–1955. [DOI] [PubMed] [Google Scholar]
- 8. Lin Y., Chalker J., Floyd N., Bernardes G., Davis B., J. Am. Chem. Soc. 2008, 130, 9642–9643. [DOI] [PubMed] [Google Scholar]
- 9. Lin Y., Chalker J., Davis B., J. Am. Chem. Soc. 2010, 132, 16805–16811. [DOI] [PubMed] [Google Scholar]
- 10. Chalker J. M., Aust. J. Chem. 2015, 68,1801–1809. [Google Scholar]
- 11. Lin Y., Davis B., Beilstein J. Org. Chem. 2010, 6, 1219–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Ben-Asuly A., Tzur E., Diesendruck C., Sigalov M., Goldberg I., Lemcoff N., Organometallics 2008, 27, 811–813; [Google Scholar]
- 12b. Tzur E., Szadkowska A., Ben-Asuly A., Makal A., Goldberg I., Wozniak K., Grela K., Lemcoff N., Chem. Eur. J. 2010, 16, 8726–8737; [DOI] [PubMed] [Google Scholar]
- 12c. Vougioukalakis G., Grubbs R., Chem. Rev. 2010, 110, 1746–1787. [DOI] [PubMed] [Google Scholar]
- 13. Grela K., Bieniek M., Kim M., Klajn R., Tetrahedron 2003, 59, 4525. [Google Scholar]
- 14. Cezary S., Grela K., Arkivoc, 2011, iv, 71–81. [Google Scholar]
- 15. Crowe W., Zhang Z., J. Am. Chem. Soc. 1993, 115, 10998–10999. [Google Scholar]
- 16. Chatterjee A., Sanders D., Grubbs R., Org. Lett. 2002, 4, 1939–1942. [DOI] [PubMed] [Google Scholar]
- 17. Chatterjee A., Grubbs R., Org. Lett. 1999, 1, 1751–1753. [DOI] [PubMed] [Google Scholar]
- 18. Bruneau C., Fischmeister C., Mandelli D., Carvalho W., dos Santos E., Dixneuf P., Fernandes L., Catal. Sci. Technol. 2018, 8, 3989–4004 [Google Scholar]
- 19.R. H. Grubbs, A. G. Wenzel, D. J. O′Leary, E. Khosravi, Second edition. ed., Wiley-VCH, Weinheim, Germany, 2015, p. 1 online resource (3 volumes).
- 20. Grela K., Bieniek M., Tet. Lett. 2001, 42, 6425–6428. [Google Scholar]
- 21.
- 21a. Herisson J., Chauvin Y., Makromol. Chem. 1971, 141, 161–162; [Google Scholar]
- 21b. Connon S., Blechert S., Angew. Chem. 2003, 42, 1900–1923. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Vorfalt T., Wannowius K., Plenio H., Angew. Chem. 2010, 49, 5533–5536; [DOI] [PubMed] [Google Scholar]
- 22b. Thiel V., Hendann M., Wannowius K., Plenio H., J. Am. Chem. Soc. 2012, 134, 1104–1114; [DOI] [PubMed] [Google Scholar]
- 22c. Ashworth I., Hillier I., Nelson D., Percy J., Vincent M., ACS Catal. 2013, 3, 1929–1939. [Google Scholar]
- 23.
- 23a. Neese F., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78; [Google Scholar]
- 23b. Riplinger C., Neese F., J. Chem. Phys. 2013, 138; [DOI] [PubMed] [Google Scholar]
- 23c. Mondal B., Neese F., Ye S., Inorg. Chem. 2015, 54, 7192–7198; [DOI] [PubMed] [Google Scholar]
- 23d. Li F., Talipov M., Dong C., Bali S., Ding K., J. Biol. Inorg. Chem. 2018, 23, 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Furstner A., Langemann K., J. Am. Chem. Soc. 1997, 119, 9130–9136; [Google Scholar]
- 24b. Moise J., Arseniyadis S., Cossy J., Org. Lett. 2007, 9, 1695–1698. [DOI] [PubMed] [Google Scholar]
- 25. Wolf S., Plenio H., Green Chem. 2011, 13, 2008–20012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary