

Summary
Nuclear receptor peroxisome proliferator‐activated receptor γ (PPAR‐γ) activation can prevent immunoinflammatory disorders and diabetes. B cells play protective roles during inflammation as well. However, the roles of endogenous PPAR‐γ in the regulatory properties of B cells to relieve inflammation remain unknown. Here, we developed B‐cell‐specific PPAR‐γ knockout (B‐PPAR‐γ −/−) mice and found that the conditional deletion of PPAR‐γ in B cells resulted in exaggerated contact hypersensitivity (CHS). Meanwhile, interferon‐γ (IFN‐γ) of CD4+ CD8+ T cells was up‐regulated in B‐PPAR‐γ −/− mice in CHS. This showed that the regulatory function of B cells in B‐PPAR‐γ −/− mice declined in vivo. Whereas splenic CD5+ CD1dhi regulatory B‐cell numbers and peripheral regulatory T‐cell numbers were not changed in naive B‐PPAR‐γ −/− mice. Loss of PPAR‐γ in B cells also did not affect either CD86 or FasL expression in splenic CD5+ CD1dhi regulatory B cells after activation. Notably, interleukin‐10 (IL‐10) production in CD5+ CD1dhi regulatory B cells reduced in B‐PPAR‐γ‐deficient mice. In addition, functional IL‐10‐producing CD5+ CD1dhi regulatory B cells decreased in B‐PPAR‐γ −/− mice in the CHS model. These findings were in accordance with augmented CHS. The current work indicated the involvement of endogenous PPAR‐γ in the regulatory function of B cells by disturbing the expansion of IL‐10‐positive regulatory B cells.
Keywords: contact hypersensitivity, interleukin‐10, peroxisome proliferator‐activated receptor γ, regulatory B cell
Abbreviations
- B‐PPAR‐γ−/−
B‐cell‐specific PPAR‐γ knockout
- Breg
regulatory B cell
- CFSE
carboxyfluorescein succinimidyl ester
- CHS
contact hypersensitivity
- Ct
control
- EAE
experimental allergic encephalomyelitis
- IFN‐γ
interferon‐γ
- IL‐10
interleukin‐10
- LPS
lipopolysaccharide
- PIM
PMA, ionomycin, and monensin
- PMA
phorbol 12‐myristate 13‐acetate
- PPAR‐γ
peroxisome proliferator‐activated receptor γ
- Treg
regulatory T cell
Introduction
B cells play an important role in the innate and adaptive immune responses, they differentiate into effector antibody‐secreting plasma cells and memory B cells to ensure an efficient response during adaptive immunity.1 Recently, B cells with regulatory functions, named regulatory B (Breg) cells, were also described.2, 3, 4, 5, 6, 7 Breg cells can play protective roles to negatively regulate immune and inflammatory diseases.8, 9, 10 Until now, two well‐characterized functional Breg cells were identified in murine splenocytes, namely, T2‐MZP (CD19+ CD23+ CD21hi CD1dhi) and B10 (CD19+ CD5+ CD1dhi).3, 4, 11 First defined in 2008, CD5+ CD1dhi Breg cells suppressed T‐cell‐mediated responses and inhibited contact hypersensitivity (CHS), experimental allergic encephalomyelitis (EAE), lupus erythematosus, inflammatory bowel disease and collagen‐induced arthritis.4, 10, 12, 13, 14
Peroxisome proliferator‐activated receptor γ (PPAR‐γ) is a member of the nuclear hormone receptor superfamily.15 Both natural and synthesized ligands such as rosiglitazone can bind to PPAR‐γ.15, 16 PPAR‐γ activation was first noted to treat diabetes; its additional anti‐inflammatory and immunosuppressive functions were recently demonstrated through its ability to ameliorate CHS, EAE, arthritis etc.17, 18, 19, 20 Whole‐body PPAR‐γ +/− mice were more susceptible to autoimmune and inflammatory diseases such as immune arthritis and EAE,21, 22 which further indicated that endogenous PPAR‐γ activation also limited inflammation and immune responses.
Most efforts have been towards directly understanding how PPAR‐γ activation suppresses the effector function of T cells and B cells to elicit anti‐inflammatory functions.23, 24, 25 To affect effector T cells, PPAR‐γ agonists or ligands decreased T helper type 1 cytokines such as interferon‐γ (IFN‐γ), tumor necrosis factor‐α (TNF‐α) and interleukin‐1β (IL‐1β), and promoted T helper type 2 cytokines such as IL‐4 and IL‐10.19, 25 T helper type 17 differentiation strongly increased in T‐cell‐specific PPAR‐γ knockout mice.26 For effector B cells, there have been paradoxical results reported for PPAR‐γ.21, 27, 28 Among them, PPAR‐γ activation inhibited lipopolysaccharide (LPS)‐induced B‐cell proliferation in an asthma model and reduced IgE synthesis in peripheral blood mononuclear cells of atopic dermatitis patients, findings that showed the suppression of effector B cells and that were in line with the anti‐inflammatory functions of PPAR‐γ activation.25, 29, 30
In contrast to effector cells, regulatory cells are crucial in maintaining peripheral tolerance and controlling immune responses. Of interest, PPAR‐γ deficiency in T cells decreased the numbers and function of CD4+ Foxp3+ regulatory T (Treg) cells, PPAR‐γ activation enhanced the inducible Treg cells.31, 32 PPAR‐γ was also a major driver of accumulation and phenotypes of adipose tissue Treg cells.33 Reports and our previous study revealed that Breg cells showed similar regulatory functions to Treg cells in immune diseases.9, 10, 34, 35 To date, the intrinsic role of PPAR‐γ in Breg cells is attractive but remains unclear.
Notably, B‐cell‐deficient mice elicited exacerbated CHS,4 which indicated that B cells possessed protective regulatory roles to suppress CHS. It also showed the value of Breg cells in CHS. To address whether endogenous PPAR‐γ expression affects CHS and the regulatory property of B cells, in this study B‐cell‐specific PPAR‐γ‐knockout mice were generated, and it was first observed that conditional deletion of PPAR‐γ in B cells resulted in an enhanced CHS response. This indicated the reduced regulatory function of B cells in vivo after endogenous PPAR‐γ deficiency, and then the underlying mechanisms mainly targeting CD5+ CD1dhi Breg cells were further explored.
Materials and methods
Mouse generation and breeding
Strain C.129P2(C)‐Cd19tm1(cre)Cgn/J with the Cre recombinase under the control of the CD19 promoter on the BALB/c genetic background and Strain B6.129‐Pparg tm2Rev /J, which possesses loxP sites on either side of exons 1 and 2 of the PPAR‐γ gene, were from The Jackson Laboratory (Bar Harbor, ME). The generation and breeding program of mice was previously described.28 The female progeny with the genotype CD19‐Cre+/− PPAR‐γ fl/fl (B‐PPAR‐γ −/−) were B‐cell‐specific PPAR‐γ knockout mice, and the progeny with the genotype CD19‐Cre−/− PPAR‐γ fl/fl (normal B‐cell littermate controls, Ct) were used as the control mice. Progeny were genotyped with a polymerase chain reaction (PCR) method provided by the Jackson Laboratory. Wild‐type mice on C57BL/6 background were from the Guangdong Medical Laboratory Animal Center (Foshan City, Guangdong Province, China). All animals were bred in an animal facility with a 12‐hr light : 12‐hr dark cycle and used at 8–12 weeks of age. All animal experiments were carried out under the approval of the Southern Medical University Animal Care and Use Committee in accordance with the guidelines for the ethical treatment of animals.
Cell isolation and preparation
Freshly isolated spleens were passed through a nylon cell strainer to create a single‐cell suspension, and splenic lymphocytes were collected using Mouse 1 × Lymphocyte Separation Medium (DKW33‐R0100; Dakewe, Biotech Company, Shenzhen City, China). CD19+ B cells were purified using a Mouse B‐Cell Negative Isolation kit (19854A; Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer's instructions, with purity of > 95%. For sorting CD19+ CD5+ CD1dhi Breg cells, 5 × 106 bead‐separated splenic CD19+ B cells pooled from CHS mice in each group at 48 hr after challenge were cultured in 2 ml complete medium in 24‐well flat‐bottom plates and stimulated with LPS (10 μg/ml, Escherichia coli serotype O111:B4, L4391; Sigma‐Aldrich, St Louis, MO) for 24 hr, then stained with phycoerythrin‐Cychrome 7 (PE‐Cy7) ‐conjugated CD19 (clone 1D3), fluorescein isothiocyanate (FITC) ‐conjugated CD5 (clone 53‐7.3), or PE‐conjugated CD1d (clone 1B1; BD Pharmingen, San Diego, CA). Next, the CD19+ CD5+ CD1dhi B cells were selected by a BD FACS Aria III flow cytometer (BD Biosciences, San Jose, CA) with purity of ~90–95%. CD4+ T cells from splenocytes of wild‐type C57BL/6, for use in a functional study of Breg cells in vitro, were purified with the Mouse CD4+ T Cell Negative Isolation kit (19812A; Stem Cell Technologies), and the purity was > 90%.
Quantitative real‐time PCR
Total RNA of CD5+ CD1dhi B cells, CD5+ CD1dlow B cells and CD5− CD1dlow B cells of wild‐type mice were extracted using the Total RNA Extraction Kit (LS1040; Promega, Beijing City, China) according to the manufacturer' s instructions. Total RNA was reverse‐transcribed using PrimeScript™ Master Mix (RR036A; TaKaRa, Shiga, Japan). Quantitative real‐time PCR was performed using qPCR Master Mix (A6001; Promega) according to the manufacturer's directions. All data were normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) values and the results were analyzed using the comparative ∆∆Ct method. The relative gene expression is presented as the fold‐change. Quantitative PCR was performed using primers (Invitrogen, Carlsbad, CA) for PPAR‐γ (forward, 5′‐CGG AAG CCC TTT GGT GAC TTT ATG‐3′ and reverse, 5′‐GCA GCA GGT TGT CTT GGA TGT C‐3′), GAPDH (forward, 5′‐AAC TTT GGC ATT GTG GAA GG‐3′ and reverse, 5′‐ACA CAT TGG GGG TAG GAA CA‐3′).
Breg markers and CD86, FasL measurement
For detecting the surface markers CD5 and CD1d on Breg cells, single‐cell suspensions were prepared from mouse spleen. For CD86 and FasL expression of Breg cells, 5 × 105 splenocytes were cultured in 0·2 ml complete medium in a 96‐well flat‐bottom plate and stimulated with LPS for 24 hr, then stained with peridinin chlorophyll protein‐conjugated CD19 (clone 1D3), FITC‐conjugated CD5, PE‐conjugated CD1d, allophycocyanin (APC)‐conjugated CD86 (cloneGL‐1), and APC‐conjugated CD178 (FasL, clone MFL3) purchased from BD Pharmingen. These surface molecules were measured by BD Canto II flow cytometer (BD Biosciences) and analyzed by flowjo V10 (version 10.0; LLC, Ashland, OR).
Foxp3+ Treg cells assay
A 200‐μl sample of peripheral blood was collected, and 10 μl EDTA (0·5 m, 15575‐038; Invitrogen) was added to prevent the blood from clotting. Erythrocytes were lyzed in red blood cell lysis buffer (420301; Biolegend, San Diego, CA) under the basic manufacturer's instructions. Then, cells were stained with Fc‐block anti‐antibody CD16/32. Next, cells were stained with the FITC‐conjugated CD4 (clone GK1.5; BD Pharmingen) and APC‐conjugated CD25 (clone PC61.5; eBioscience, San Diego, CA). Then, cells were fixed and permeabilized with the Foxp3/Transcription Factor Straining Buffer Set Kit (005523‐00; eBioscience) according to the manufacturer's instructions. The permeabilized cells were stained with PE‐conjugated Foxp3 (clone FJK‐16s; eBioscience). The CD4+ CD25+ Foxp3+ T cells in peripheral blood were detected with the BD Canto II flow cytometer and analyzed by flowjo V10.
Intracellular IL‐10 detection
First, 1 × 106 splenocytes or 5 × 105 CD19+ B cells from naive or CHS mice were cultured in 0·2 ml complete medium in a 96‐well flat‐bottom plate. For analysis of IL‐10, cells were stimulated with LPS for 5 hr or 24 hr, and phorbol 12‐myristate 13‐acetate (PMA), ionomycin, and monensin (PMA 50 ng/ml, P8139; Sigma‐Aldrich; ionomycin, 500 ng/ml, I0634; Sigma‐Aldrich; monensin, 2 μm, 00‐4505‐51; eBioscience) were added for the last 5 hr. For intracellular staining of IL‐10, cells were stained for the surface molecule and then fixed and permeabilized using a Cytofix/Cytoperm kit (54714; BD Biosciences) according to the manufacturer's instructions. The permeabilized cells were stained with APC‐conjugated IL‐10 and analyzed using a BD Canto II flow cytometer.
Intracellular IFN‐γ detection
A total of 1 × 106 splenocytes pooled from CHS mice in each group at 48 hr after challenge were plated in triplicate in 0·2 ml complete medium. For analysis of IFN‐γ, cells were stimulated with anti‐CD3e (clone 145‐2C11; eBioscience) and anti‐CD28 (clone 37.51; eBioscience) for 24 hr; then PMA, 50 ng/ml, ionomycin, 500 ng/ml, and brefeldin A, 3 μg/ml (004506‐51; eBioscience) were added for the last 5 hr. Then, cells were harvested and washed, followed by surface staining with PE‐conjugated CD4 (clone GK1.5; BD Pharmingen) and FITC‐conjugated CD8 (clone 53‐6.7; eBioscience); then fixed and permeabilized using a Cytofix/Cytoperm kit (54714; BD Biosciences) according to the manufacturer's instructions. The permeabilized cells were stained with APC‐conjugated IFN‐γ (clone XMG1.2; BD Pharmingen) and analyzed by BD Canto II flow cytometer.
Functional study of Breg cells to inhibit CD4+ T‐cell proliferation in vitro
The sorted CD19+ CD5+ CD1dhi B cells from CHS mice were resuspended at a density of 1·5 × 106/ml in complete medium after stimulation with LPS for 24 hr. CD4+ T cells separated after negative selection were resuspended at a density of 1 × 107 cells/ml in PBS containing 4 μm carboxyfluorescein succinimidyl ester (CFSE; Molecular Probes and C34554; Invitrogen) for 10 min at 37°, protected from light. Labeling was terminated by addition of complete culture medium. Then, 0·1 ml CFSE‐labeled CD4+ T cells were added into 96‐well flat‐bottom plates pre‐coated with anti‐CD3e (clone 145‐2C11; eBioscience) and anti‐CD28 (clone 37.51; eBioscience), and then cocultured with 1·5 × 105 CD19+ CD5+ CD1dhi B cells in 0·1 ml complete medium at a 1 : 1 ratio for 72 hr. Next, cells were harvested and stained with APC‐conjugated CD4. The proliferation of CD4+ T cells was analyzed by BD Canto II flow cytometry.
Contact hypersensitivity reaction
The CHS reactions were induced with oxazolone as described previously.4 Briefly, B‐cell‐specific PPAR‐γ knockout mice and control mice (n = 5 per group) were sensitized with 25 μl of oxazolone (100 mg/ml; Sigma‐Aldrich) in acetone/olive oil (4 : 1, v/v) for 2 consecutive days on a shaved right abdomen. On day 5, 10 μl of oxazolone solution (10 mg/ml) in acetone/olive oil (4 : 1) was applied to the right ear (5 μl on the dorsal side and 5 μl on the ventral side) to challenge the response. The thickness of each ear was measured at 24, 48, 72 and 96 hr after challenge with a constant‐force, calibrated digital thickness gauge. To observe ear swelling by hematoxylin & eosin stain, mice were killed after ear swelling measurement at 48 hr after challenge, ears were collected and fixed in 4% paraformaldehyde and embedded in paraffin to be cut into slices and stained with hematoxylin & eosin later; to evaluate Breg cells in CHS meanwhile, splenocytes and the purified CD19+ B cells pooled from B‐PPAR‐γ −/− mice and control mice were activated with LPS for 5 or 24 hr to measure cell surface markers and intracellular IL‐10. The sorted CD19+ CD5+ CD1dhi Breg cells from B‐PPAR‐γ −/− mice and control mice were co‐cultured with CD4+ T cells to detect their inhibitory effects on CD4+ T‐cell proliferation.
Statistical analysis
Data are expressed as the means ± SEM. Significance was determined by a two‐tailed unpaired Student's t‐test. Statistical analyses were performed using graphpad prism 5·0 (GraphPad Software, San Diego, CA). Probability values of P < 0·05 were considered statistically significant; *P < 0·05, ** P < 0·01, ***P < 0·001 were applied.
Results
B‐PPAR‐γ−/− mice had enhanced CHS response and T‐cell IFN‐γ production
Whole‐body PPAR‐γ knockout in mice is embryonically lethal.28 To determine whether endogenous PPAR‐γ affects the regulatory function of B cells in CHS in vivo, we generated conditional knockout mice in which only B cells harbored a Cre‐dependent deletion of PPAR‐γ (B‐PPAR‐γ‐deficient). B‐cell‐deficient mice have an exacerbated CHS response, showing the decreased regulatory function of B cells to control inflammation in CHS in vivo;4 hence, the CHS model was first employed. We observed that CD19‐Cre+/− mice had normal CHS responses compared with wild‐type (CD19‐Cre−/−) mice, which excluded the affect of CD19‐Cre on CHS (Fig. S1). We found that the oxazolone‐induced ear swelling was significantly higher in B‐PPAR‐γ −/− mice than in control mice at 48 hr after challenge (P < 0·05, Fig. 1a). At 72 and 96 hr after challenge, no difference was apparent between B‐PPAR‐γ −/− and control mice (Fig. 1a). This curve was in line with the previous reports that PPAR‐γ +/− mice exhibited a pronounced increase of methylated bovine serum albumin‐induced arthritis at day 1 after methylated bovine serum albumin challenge and gradually lost the difference compared with the control mice after that.21 Because B‐cell‐deficient mice had increased CHS responses even at 72 and 96 hr after challenge,4 it seemed that the dampening of the B‐cell regulatory function might gradually recover 48 hr after challenge in B‐PPAR‐γ −/− mice. At 48 hr after challenge, B‐PPAR‐γ −/− mice also showed more serious infiltration of cells in comparison with control mice in hematoxylin & eosin staining assays (Fig. 1b). The intraepidermal abscess and the surrounding epidermis hyperplasia were more evident in B‐PPAR‐γ −/− mice. In addition, the IFN‐γ production of splenic CD4+ and CD8+ T cells after anti‐CD3e and anti‐CD28 stimulation was up‐regulated in B‐PPAR‐γ −/− mice at 48 hr after challenge (Fig. 1c). In summary, the B‐PPAR‐γ −/− mice displayed an enhanced CHS response compared with the control mice at 48 hr after challenge (Fig. 1a,b); this indicated that the regulatory function of B cells was impaired in B‐PPAR‐γ −/− mice due to the loss of PPAR‐γ in CD19+ B cells, especially at 48 hr after challenge in CHS.
Figure 1.
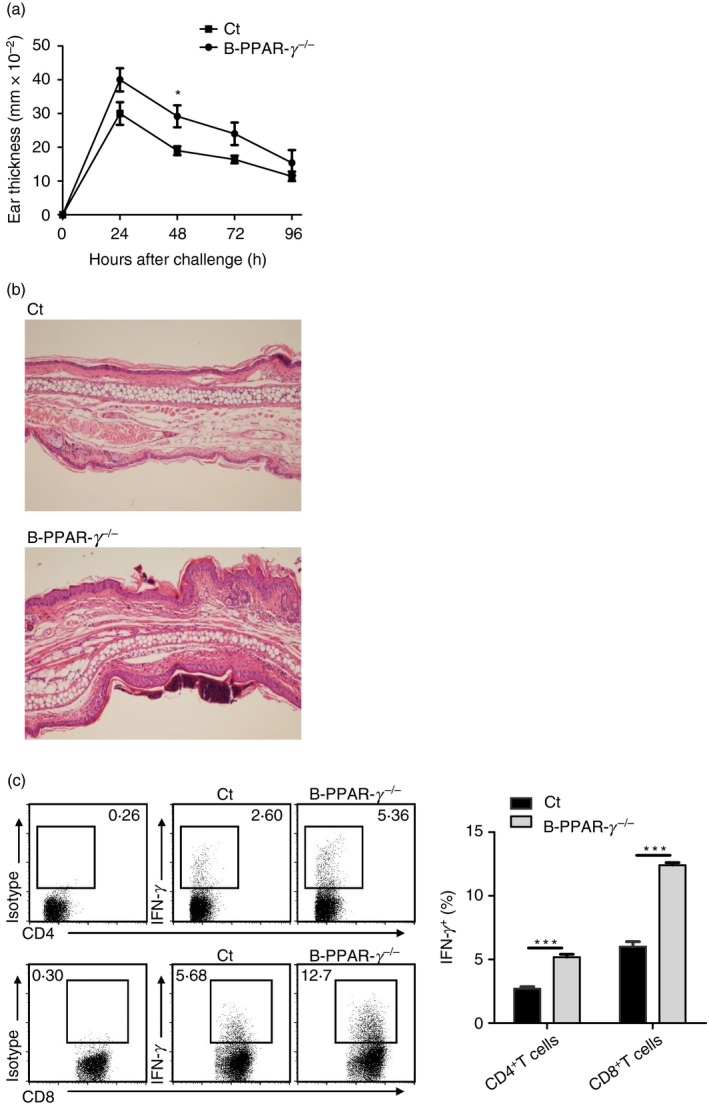
B‐cell‐specific peroxisome proliferator‐activated receptor γ knockout (B‐PPAR‐γ −/−) mice have increased contact hypersensitivity (CHS) responses and interferon‐γ (IFN‐γ) production of T cells. (a) CHS responses in control mice and B‐PPAR‐γ −/− mice were indicated by ear thickness after oxazolone challenge. Values represent means (± SEM) from n = 5 per group, *P < 0·05. (b) Histopathology assessment of hematoxylin & eosin‐stained ear lobe sections from B‐PPAR‐γ −/− and control CHS mice at 48 hr after oxazolone challenge (original magnification × 100), n ≥ 3 per group. Similar results were obtained from two independent experiments. (c) Representative flow cytometry plots and bar graph show IFN‐γ expression of splenic CD4+ T cells and CD8+ T cells from CHS mice at 48 hr after challenge. Representative flow cytometry plots are gated for CD4+ or CD8+ cells. Values are mean (± SEM) of three triplicates in each group, which represent two independent experiments (**P < 0·01, ***P < 0·001).
Naive B‐PPAR‐γ−/− mice had normal levels of splenic CD5+ CD1dhi Breg cells and CD4+ CD25+ Foxp3+ Treg cells
Splenic CD19+ CD5+ CD1dhi Breg cells can inhibit the CHS response and other immune responses both in vitro and in vivo.4, 6, 9 PPAR‐γ expression in splenic CD19+ CD5+ CD1dhi Breg cells is higher than other B‐cell subsets by qPCR analysis (Fig. S2). To investigate whether loss of PPAR‐γ in B cells affected splenic CD19+ CD5+ CD1dhi Breg cell generation in naive B‐PPAR‐γ −/− mice, we measured the CD19+ CD5+ CD1dhi Breg cells of spleen in naive B‐PPAR‐γ −/− mice and control mice first. Naive B‐PPAR‐γ −/− mice had normal levels of splenic CD19+ B cells and CD19+ CD5+ CD1dhi Breg cells (Fig. 2a,b). The Breg cells can induce Foxp3+ Treg cells to perform inhibitory functions.36, 37 Therefore, we examined the CD4+ CD25+ Foxp3+ Treg cell frequencies in peripheral blood of naive B‐PPAR‐γ −/− mice and control mice (Fig. 2c). As shown in Fig. 2(c), the proportion of CD4+ T cells in peripheral blood, CD25+ Foxp3+ Treg cells in CD4+ T cells from peripheral blood, did not change in B‐PPAR‐γ −/− mice compared with control mice. Therefore, the development of CD5+ CD1dhi Breg cells in spleen and Foxp3+ Treg cells in peripheral blood was normal in naive B‐PPAR‐γ‐deficient mice. Loss of PPAR‐γ did not affect the number or the generation of splenic CD5+ CD1dhi Breg cells and peripheral Foxp3+ Treg cells, which could contribute to the regulatory function of B cells.
Figure 2.
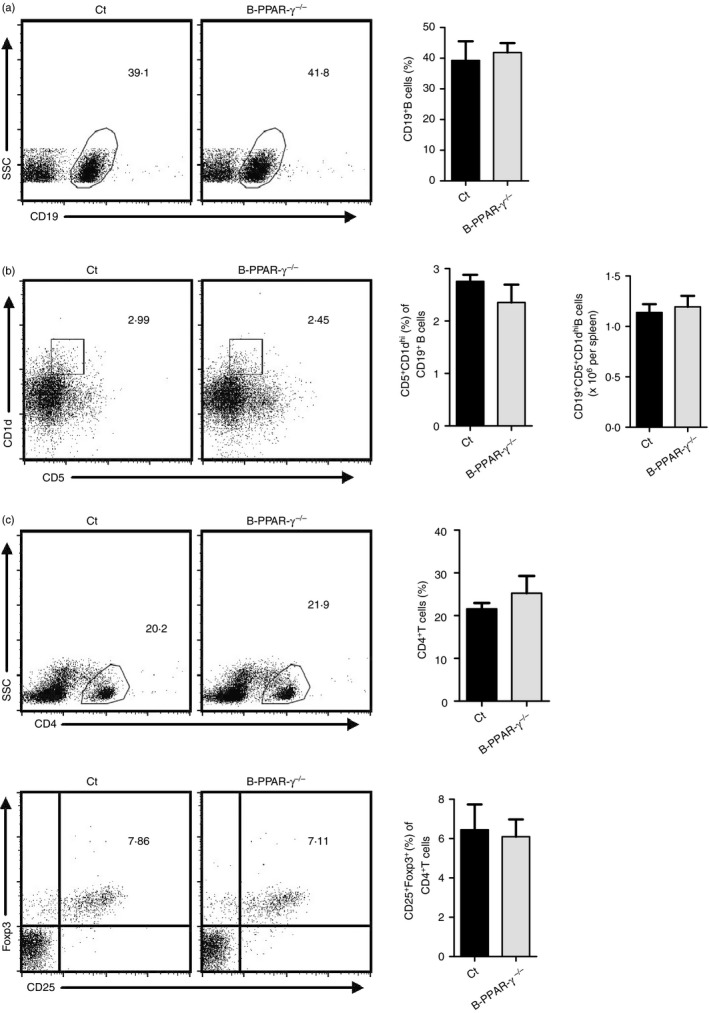
Naive B‐cell‐specific peroxisome proliferator‐activated receptor γ knockout (B‐PPAR‐γ −/−) mice have normal splenic CD5+ CD1dhi regulatory B (Breg) and peripheral CD4+ CD25+ Foxp3+ regulatory T (Treg) cells. Splenocytes and peripheral blood were collected from naive control mice and B‐PPAR‐γ −/− mice. Representative flow cytometry plots and bar chart show (a) the frequency of CD19+ B cells in splenic lymphocytes. Representative flow cytometry plots are gated for lymphocytes. (b) The frequency of CD5+ CD1dhi Breg cells in CD19+ B cells and the number of CD5+ CD1dhi Breg cells in spleen. Representative flow cytometry plots are gated for CD19+ cells. (c) CD25+ Foxp3+ Treg cells among the total CD4+ T cells in peripheral blood were analyzed. Representative flow cytometry plots are gated for lymphocytes or CD4+ cells. Bar graphs indicate mean (± SEM), n = 3, similar results were obtained from two independent experiments.
CD86 and FasL expression in CD5+ CD1dhi Breg cells did not change in naive B‐PPAR‐γ‐deficient mice after activation
Regulatory B cells achieve immune inhibitory functions after activation.8 LPS can potently activate CD5+ CD1dhi Breg cells after Toll‐like receptor 4 ligation.38 CD86 is a surface molecule related not only to B‐cell activation but also to the regulatory function of B cells.8, 39, 40 Anti‐CD86 antibody can block the immune suppressive effects of Breg cells in humans and exacerbate EAE in mice.5, 41 Hence, we further investigated the effects of B‐cell‐specific PPAR‐γ knockout on the CD86 expression of CD5+ CD1dhi Breg cells after activation by LPS for 24 hr in vitro. The percentage of CD19+ B cells and CD5+ CD1dhi Breg cells and the number of CD19+ CD5+ CD1dhi B cells were not obviously different between the B‐PPAR‐γ −/− and control mice (Fig. 3a,b). In addition, CD86 expression in CD19+ B cells and CD19+ CD5+ CD1dhi Breg cells was unchanged in B‐PPAR‐γ −/− mice compared with control mice after LPS activation (Fig. 3c).
Figure 3.
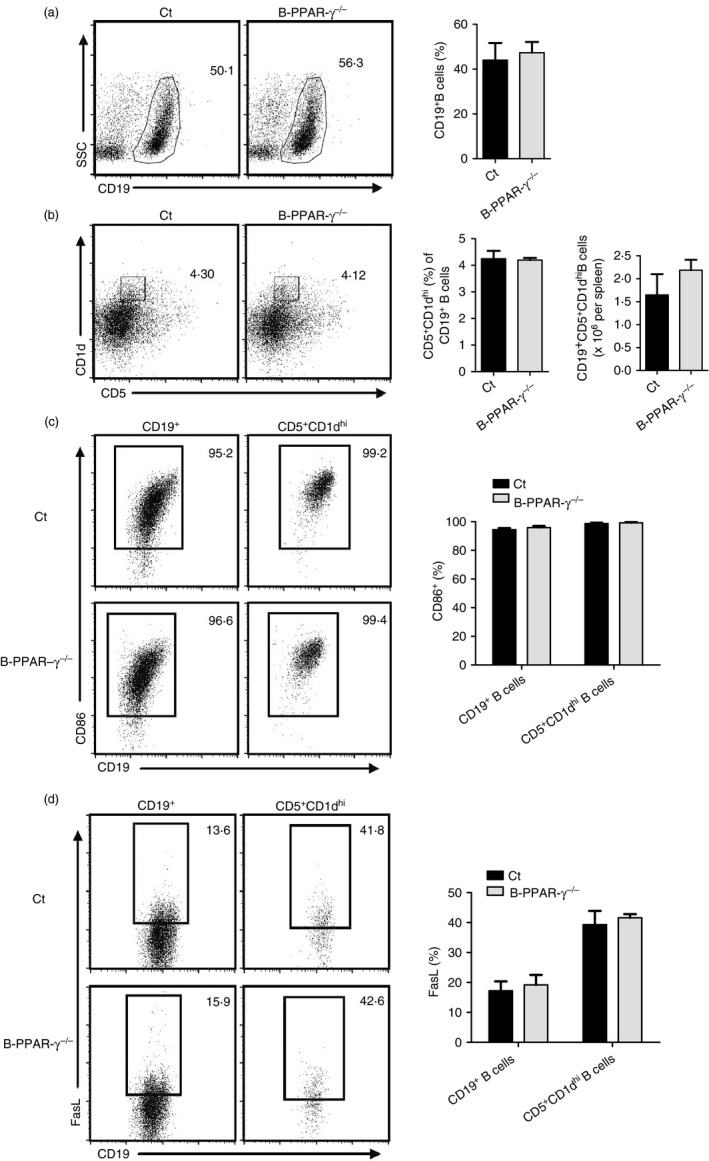
Naive B‐cell‐specific peroxisome proliferator‐activated receptor γ knockout (B‐PPAR‐γ −/−) mice have normal CD86 and FasL expression in CD5+ CD1dhi regulatory B (Breg) cells after activation. Splenic lymphocytes collected from control and B‐PPAR‐γ −/− mice were stimulated with lipopolysaccharide (LPS) for 24 hr in vitro. Representative flow cytometry plots and bar chart show (a) the percentage of CD19+ B cells of lymphocytes, (b) the frequency of CD5+ CD1dhi Breg cells in the total CD19+ B cells and the number of CD5+ CD1dhi Breg cells in spleen, (c) CD86, and (d) FasL expression in CD19+ and CD5+ CD1dhi Breg cells. Representative flow cytometry plots are gated for lymphocytes or CD19+ cells. Bar graphs indicate mean (± SEM), n = 3, data represented two independent experiments.
FasL can bind to Fas to induce apoptosis of immune cells and harbor an IL‐10‐independent suppression of CD4+ T‐cell activation.42 Our previous study revealed that an anti‐FasL antibody partly blocked the inhibitory effects of CD19+ CD5+ CD1dhi Breg cells on CD4+ T‐cell proliferation.43 Therefore, we next evaluated FasL expression in CD19+ CD5+ CD1dhi Breg cells after activation. FasL expression was not obviously affected in CD19+ CD5+ CD1dhi Breg cells and CD19+ B cells in B‐PPAR‐γ −/− mice in comparison with control mice after LPS activation (Fig. 3d).
IL‐10 production decreased in CD5+ CD1dhi Breg cells in B‐PPAR‐γ‐deficient mice
Interleukin‐10 is the pivotal molecule by which CD5+ CD1dhi Breg cells exhibit their regulatory functions.4 Anti‐IL‐10 antibody impaired the inhibitory effects of CD5+ CD1dhi Breg cells in vitro.12 Hence, we asked if IL‐10 production in CD5+ CD1dhi Breg cells changed after B‐cell‐specific PPAR‐γ knockout. First, we excluded the affect of CD19‐Cre on CD5+ CD1dhi Breg cell expansion and IL‐10 expression after LPS stimulation by using CD19‐Cre+/− mice and wild‐type (CD19‐Cre−/−) mice. CD19‐Cre+/− mice showed no different percentages of CD19, CD5+ CD1dhi Breg cells and IL‐10 expression in comparison with wild‐type mice (Fig. S1). We then found that after 5 hr incubation with LPS+PIM (PMA, ionomycin, and monensin), the percentage of CD5+ CD1dhi Breg cells and the number of CD19+ CD5+ CD1dhi B cells were not obviously different between the naive B‐PPAR‐γ −/− and control mice (Fig. 4a), but IL‐10 expression was significantly decreased in CD19+ B cells and CD5+ CD1dhi Breg cells from B‐PPAR‐γ −/− mice compared with control mice after LPS activation for 5 hr (Fig. 4b). Meanwhile, the IL‐10 production in CD5+ CD1dlow and CD5− CD1dlow B‐cell populations was not obviously reduced (Fig. 4b). On the whole, the proportion of IL‐10+ CD5+ CD1dhi Breg cells in CD19+ B cells and the number of IL‐10+ CD19+ CD5+ CD1dhi B cells in spleen after activation were also obviously down‐regulated in naive B‐PPAR‐γ −/− mice (Fig. 4c). This might contribute to the declined regulatory function of B cells in B‐PPAR‐γ −/− mice in vivo.
Figure 4.
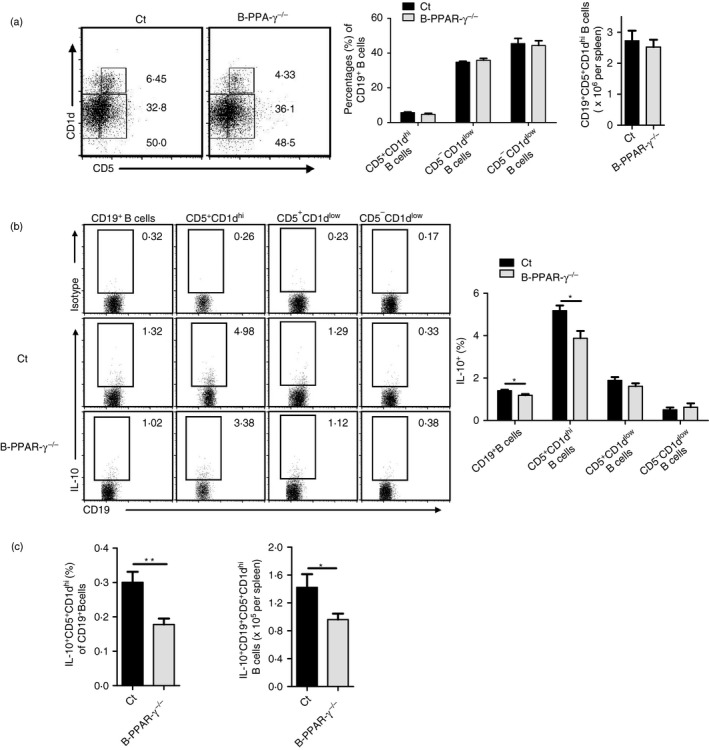
Interleukin‐10 (IL‐10) production of CD5+ CD1dhi regulatory B (Breg) cells decreases in naive B‐cell‐specific peroxisome proliferator‐activated receptor γ knockout (B‐PPAR‐γ −/−) mice. Splenic lymphocytes from naive control mice and B‐PPAR‐γ −/− mice were cultured with lipopolysaccharide (LPS) plus PIM for 5 hr. Representative flow cytometry plots and bar chart show (a) the percentage of CD5+ CD1dhi Breg cells, CD5+ CD1dlow B cells, and CD5− CD1dlow B cells and the number of CD19+ CD5+ CD1dhi Breg cells in the spleen. (b) IL‐10 expression of CD19+ B cells, CD5+ CD1dhi B cells and other B‐cell subsets. Representative flow cytometry plots are gated for CD19+, CD5+ CD1dhi, CD5+ CD1dlow, or CD5− CD1dlow B cells. (c) The proportion of IL‐10+ CD5+ CD1dhi B cells in CD19+ B cells and the number of IL‐10+ CD19+ CD5+ CD1dhi B cells in spleen. Unpaired Student's t‐test showed significant differences between groups, bar graphs indicate mean (± SEM), n = 6, *P < 0·05.
Functional IL‐10‐producing CD5+ CD1dhi Breg cells reduced in B‐PPAR‐γ‐deficient mice in CHS
Next, we focused on IL‐10‐producing CD5+ CD1dhi Breg cells in CHS to address their relationship with the regulatory function defect of B cells in B‐PPAR‐γ −/− mice. Splenocytes and splenic CD19+ B cells isolated from the B‐PPAR‐γ −/− mice and control mice at 48 hr after oxazolone challenge were pooled and stimulated with LPS for 5 hr or 24 hr to detect IL‐10 in CD19+ CD5+ CD1dhi Breg cells. Of note, CD5+ CD1dhi Breg cell frequency decreased in B‐PPAR‐γ −/− mice after LPS activation for 5 hr or 24 hr in purified CD19+ B cells and splenocytes (Fig. 5a). Furthermore, there was an obvious reduction of IL‐10 production of CD19+ B cells and CD5+ CD1dhi Breg cells in purified CD19+ B cells after LPS activation for 24 hr (Fig. 5b,c). Here, different results on CD5+ CD1dhi Breg cell population and IL‐10 production were observed in purified CD19+ B cells and splenocytes (Fig. 5a–c). This might be a result of the different signals for Breg cell expansion and activation. In purified B cells, Breg cells were only activated by LPS with ligation by Toll‐like receptor 4; whereas in splenocytes from CHS mice, there might be other signals from other cells such as primed T cells, which could also provide activation signals to Breg cells.5, 9 On the whole, the proportion of IL‐10‐producing CD5+ CD1dhi Breg cells of CD19+ B cells obviously decreased in both purified CD19+ B cells and splenocytes from B‐PPAR‐γ −/− mice (Fig. 5d). At the same time, the IFN‐γ production of CD4+ and CD8+ T cells after anti‐CD3e and anti‐CD28 activation was obviously up‐regulated in vitro (Fig. 1c), which was in accordance with the increased CHS reaction in vivo. Therefore, B‐PPAR‐γ −/− mice had a decreased IL‐10‐producing CD5+ CD1dhi Breg cell population in B cells after activation in CHS, which should contribute to the inhibition of activated effector T cells in CHS.
Figure 5.
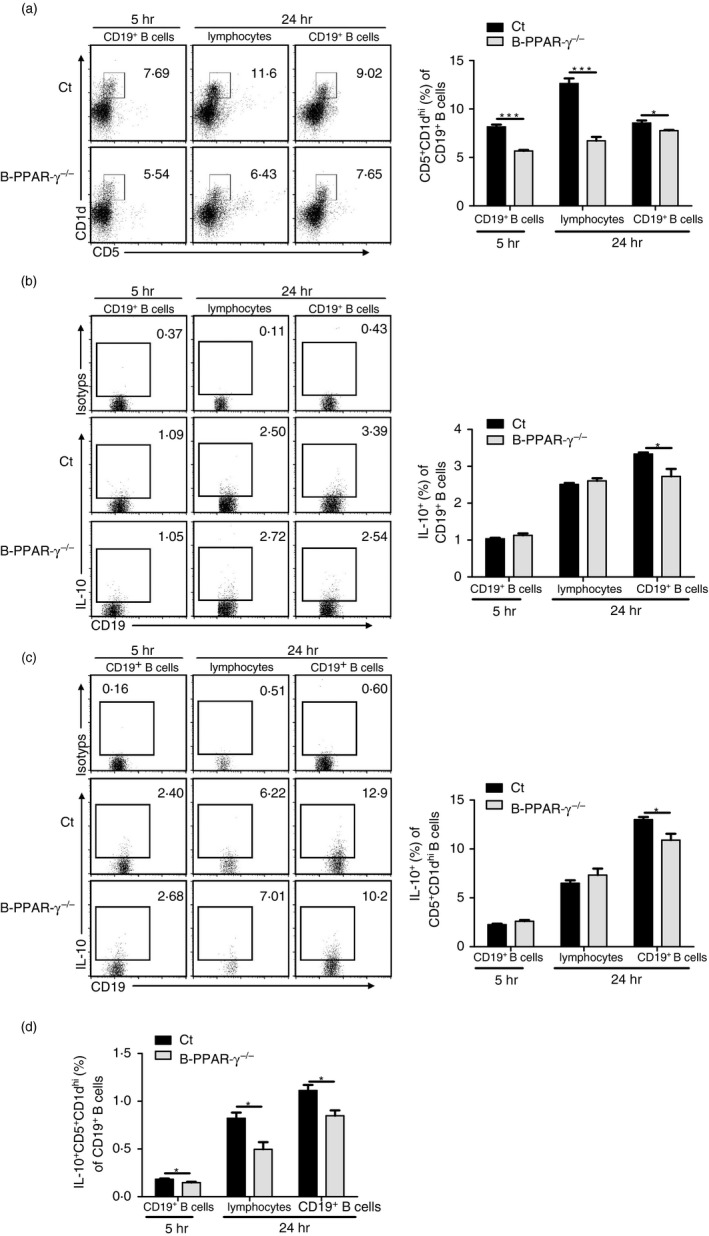
B‐cell‐specific peroxisome proliferator‐activated receptor γ knockout (B‐PPAR‐γ −/−) mice have decreased interleukin‐10 (IL‐10) ‐producing‐CD5+ CD1dhi regulatory B (Breg) cells in contact hypersensitivity (CHS). The splenic lymphocytes and splenic CD19+ B cells were isolated from control mice and B‐PPAR‐γ −/− mice at 48 hr after the oxazolone challenge in CHS. Cells were cultured with lipopolysaccharide (LPS) activation for 5 or 24 hr and PIM was added for the last 5 hr. Representative flow cytometry plots and bar chart show (a) the percentage of CD5+ CD1dhi Breg cells, Representative flow cytometry plots are gated for CD19+ cells. (b) IL‐10 expression of CD19+ B cells. (c) IL‐10 expression of CD19+ CD5+ CD1dhi Breg cells. Representative flow cytometry plots are gated for CD19+ cells or CD19+ CD5+ CD1dhi cells. (d) The percentage of IL‐10+ CD5+ CD1dhi Breg cells in B cells was analyzed. Bar graphs indicate mean (± SEM), n = 3, *P < 0·05, **P < 0·01, ***P < 0·001. data represented two independent experiments.
To confirm that these CD19+ CD5+ CD1dhi Breg cells were functional after activation in CHS, we separated splenic CD19+ B cells using beads from B‐PPAR‐γ −/− mice and control mice 48 hr after challenge, stimulated them with LPS for 24 hr, sorted the CD19+ CD5+ CD1dhi Breg cells and co‐cultured them with the CFSE‐labeled CD4+ T cells from naive C57BL/6 mice for 72 hr. The divided CD4+ T cells during proliferation showed decreased CFSE fluorescence after each division. As expected, the CD19+ CD5+ CD1dhi Breg cells of both control mice and B‐PPAR‐γ −/− mice exhibited regulatory function by inhibiting the proliferation of CD4+ T cells (Fig. 6). The inhibitory function of CD19+ CD5+ CD1dhi Breg cells was seen in B‐PPAR‐γ −/− mice as well as in control mice (Fig. 6). In combination with the results in Fig. 5, these data showed that the number of functional CD19+ CD5+ CD1dhi IL‐10+ Breg cells in B‐PPAR‐γ −/− mice decreased after activation. This could cause the decline in regulatory function of B cells with enhanced T‐cell‐mediated inflammation, leading to differences in CHS in vivo between B‐PPAR‐γ −/− mice and control mice.
Figure 6.
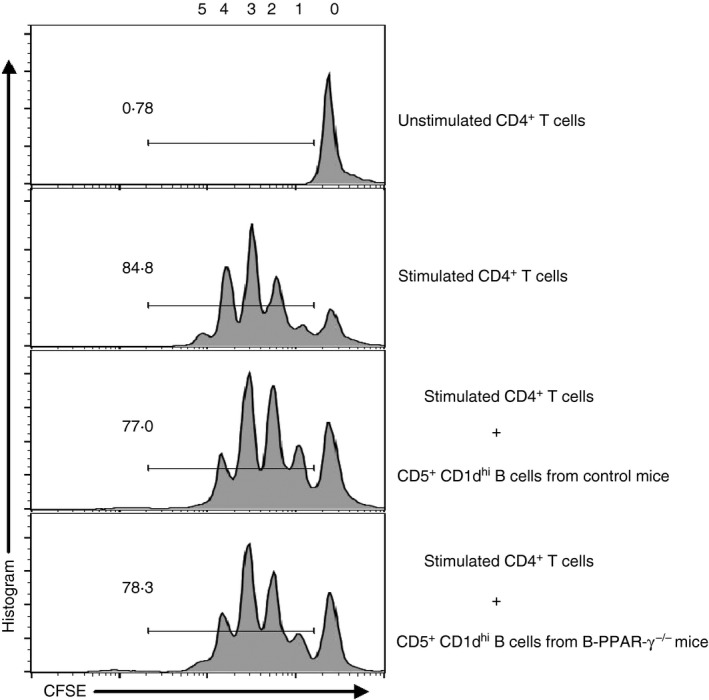
CD5+ CD1dhi regulatory B (Breg) cells from contact hypersensitivity (CHS) mice functionally inhibit CD4+ T‐cell proliferation. Splenic CD19+ B cells were isolated from control mice and B‐cell‐specific peroxisome proliferator‐activated receptor γ knockout (B‐PPAR‐γ −/−) mice at 48 hr after the oxazolone challenge. CD19+ CD5+ CD1dhi B cells were sorted after lipopolysaccharide (LPS) stimulation for 24 hr and co‐cultured with CD4+ T cells at a 1 : 1 ratio for 72 hr in the presence of anti‐CD3e and anti‐CD28. The proliferation of CFSE‐labeled CD4+ T cells was analyzed after the co‐culture by flow cytometry. Representative flow cytometry plots are gated for CD4+ cells. Data represented two independent experiments.
Discussion
Regulatory B cells were recently discovered to be negative regulators of inflammatory and immune disorders.2, 4, 9, 10 The emerging role of PPAR‐γ activation to control inflammation and immune disorders has been documented for the past few decades.25, 44 Of note, PPAR‐γ is widely expressed, including in B cells and T cells, whereas the involvement of PPAR‐γ in B cells is rare and a paradox.21, 27, 28 In addition, it remains unclear whether PPAR‐γ can affect the regulatory ability of B cells to down‐regulate T‐cell‐mediated inflammation such as CHS. Because whole‐body PPAR‐γ knockout is embryonically lethal in mice,28 and PPAR‐γ agonists also elicit PPAR‐γ‐independent effects,31 we generated B‐cell‐specific PPAR‐γ knockout mice to evaluate the intrinsic role of PPAR‐γ on CHS and regulatory B cells. The results first denote the participation of endogenous PPAR‐γ in the regulatory function of B cells in vivo by using a CHS model.
CD19‐deficient mice have increased and prolonged CHS response reactions, indicating an inhibitory role of B cells.4 CHS is a model for human allergic contact dermatitis and is mainly mediated by antigen‐specific effector T cells.4, 38 The skin Langerhans cell is the exclusive antigen‐presenting cell in CHS4. CHS was thought to have a minimized stimulatory role for B cells in the immune responses, which allowed the regulatory effect of B cells to be observed.4 In the current study, we bred B‐cell‐specific PPAR‐γ‐deficient mice to investigate the role of PPAR‐γ on Breg cells using the CHS model. We found that B‐cell‐specific PPAR‐γ‐deficient mice had an enhanced CHS response compared with the control mice, especially at 48 hr after challenge, showing that loss of PPAR‐γ in B cells caused the reduced regulatory function of B cells in vivo.
To investigate why PPAR‐γ deficiency lowered the regulatory function of B cells in vivo, we first asked if the number of naive Breg cells decreased. As previously described, splenic CD19+ CD5+ CD1dhi Breg cells are the recognized important subset of Breg cells responsible for suppressing CHS.4, 6 To adoptively transfer CD19+ CD5+ CD1dhi Breg cells to B‐cell‐deficient mice could rescue the regulatory function defect of B cells in the CHS model.4, 6 We then detected splenic CD5+ CD1dhi Breg cells in CD19+ B cells from naive mice and observed that the frequency and number of CD5+ CD1dhi Breg cells was not changed between the control mice and the B‐PPAR‐γ knockout mice. This indicated that the generation of splenic CD19+ CD5+ CD1dhi B cells was not affected after B‐cell‐specific deficiency of PPAR‐γ. This finding was in line with the previous report that loss of PPAR‐γ in B cells did not affect the development of B cells or B‐cell populations, such as pro‐B cells, pre‐B cells, follicular B cells, marginal zone precursor cells, marginal zone cells, among others, in spleen or bone marrow.28 As reported, mice lacking Breg cells presented with reduced Treg cell frequency.36, 37 We next investigated the CD4+ CD25+ Foxp3+ Treg cells in peripheral blood and observed no significant change in B‐PPAR‐γ −/− mice. This finding suggested that the generation of splenic CD5+ CD1dhi Breg cells and peripheral Treg cells was normal in naive B‐PPAR‐γ −/− mice, which could not contribute to the reduced regulatory function of B cells in vivo after PPAR‐γ deficiency.
Regulatory B cells can efficiently suppress CHS, especially after activation.4, 6 Next, we asked if Breg cell activation was affected by the loss of PPAR‐γ in the B‐cell lineage. CD86 (B7‐2) and FasL are the important surface molecules for Breg cells to perform their regulatory function after activation. Anti‐CD86 antibody can reverse the suppressive function of human CD19+ CD24+ CD38+ Breg cells on Th1 cells.5 During EAE, reconstitution with B cells from wild‐type mice, but not B7‐deficient mice, resulted in disease recovery.40 We tested the CD86 expression in CD19+ B cells and CD5+ CD1dhi Breg cells after LPS activation and found no difference between B‐PPAR‐γ −/− and control mice. Of interest, B‐PPAR‐γ −/− mice had normal CD86 expression in B cells in B‐PPAR‐γ −/− mice after anti‐CD40 and IL‐4 activation,28 consistent with our results here. For CD86 expressed in both activated effector B cells and Breg cells, our results and previous reports suggested that CD86 might not be involved in PPAR‐γ‐mediated regulatory function and effector function of B cells. B lymphocytes and CD5+ CD1dhi Breg cells also expressed elevated FasL after LPS stimulation, which could mediate the death of immune cells, including CD4+ T cells, and then inhibit the inflammatory response or diseases.43 We observed that FasL expression was not affected in CD19+ B cells and CD5+ CD1dhi Breg cells in B‐PPAR‐γ −/− mice after LPS activation, suggesting that other factors might contribute to the reduced regulatory function of B cells in B‐cell‐specific PPAR‐γ‐deficient mice.
Interleukin‐10 is considered a pivotal inhibitory cytokine for the suppressive activity of Breg cells.2, 4 Interleukin‐10‐deficient mice have enhanced inflammatory reactions and diseases.4, 45 In addition, IL‐10‐ and FasL‐expressing Breg cells could be different populations among CD5+ CD1dhi B cells.43, 46 Interleukin‐10 is not constitutively expressed in CD5+ CD1dhi Breg cells, it can be well detected after antigen, LPS, CpG, anti‐CD40 and PIM stimulation in mice.4, 38 We found that IL‐10 secretion statistically decreased in CD19+ B cells and CD5+ CD1dhi Breg cells after LPS+PIM activation in naive B‐PPAR‐γ −/− mice. The expansion of IL‐10‐expressing CD5+ CD1dhi Breg cells dramatically deceased in B cells from B‐cell‐specific PPAR‐γ‐deficient mice. In the previous study, CD138+ plasma B cells decreased obviously after ovalbumin immunization in B‐cell‐specific PPAR‐γ‐deficient mice compared with control mice.28 For CD138+, plasma cells could secrete IL‐10 and be regarded as a mouse Breg cell population in addition to producing antibodies,9, 47 this suggested that CD138+ Breg cells might also be impaired in B‐cell‐specific‐PPAR‐γ‐deficient mice, which was consistent with the decreased IL‐10+ CD5+ CD1dhi Breg cells here. It was reported that the PPAR response element present in the human IL‐10 promoter region and the PPAR‐γ agonist rosiglitazone could specifically induce the production of IL‐10 from T‐cell‐receptor‐activated human CD4+ T cells.48 Together with our results, these findings indicated that the intrinsic PPAR‐γ deficiency in B cells might affect the production of IL‐10 in Breg cells or the IL‐10+ CD5+ CD1dhi Breg cell differentiation after activation, dampening the immune suppressive function of B cells.
These IL‐10‐expressing Breg cells were then focused upon in CHS to address the intrinsic role of PPAR‐γ deficiency in B cells. During CHS, the percentage of CD5+ CD1dhi Breg cells and IL‐10 production of CD5+ CD1dhi Breg cells were decreased in purified B cells after activation in B‐PPAR‐γ −/− mice. As a whole, the IL‐10+ CD5+ CD1dhi Breg cell proportion was particularly reduced in B cells from CHS in B‐PPAR‐γ −/− mice. Simultaneously, the sorted splenic CD5+ CD1dhi Breg cells after activation from CHS mice here showed regulatory functions to inhibit CD4+ T‐cell proliferation. Collectively, the functional IL‐10‐expressing CD5+ CD1dhi Breg cells decreased in B‐PPAR‐γ −/− mice, which was consistent with the increase of ear swelling and IFN‐γ production by T cells in B‐PPAR‐γ −/− mice in CHS. In line with this, the PPAR‐γ agonist pioglitazone was recently reported to increase the Breg cells to ameliorate liver injury in mice given a high‐fat diet.49 Based on the results above, we concluded that decrease of functional IL‐10‐producing CD19+ CD5+ CD1dhi Breg cells among B cells after activation led to an exacerbated contact hypersensitivity after the loss of PPAR‐γ in B cells. The endogenous PPAR‐γ deficiency in B cells might impair IL‐10‐producing Breg cell differentiation after activation to aggravate the CHS response.
In conclusion, we generated B‐cell‐specific PPAR‐γ‐deficient mice and found that endogenous PPAR‐γ deficiency impacted the regulatory function of B cells in CHS. The impaired regulatory function after knockout of PPAR‐γ in B cells was not due to reduced naive splenic CD5+ CD1dhi Breg cell generation, but instead to the activation‐induced IL‐10+ CD5+ CD1dhi Breg cell expansion. Our group and other groups recently described that nuclear factor‐κB and nuclear factor of activated T‐cell signal inhibition, which had been observed after PPAR‐γ activation in effector cells,20, 50 could dampen Breg cell activation.13, 43, 51 We cannot exclude the possibility that paradoxical effects of PPAR‐γ activation on Breg cells initiated by a different immune balance might sometimes occur during immune responses. The effects of PPAR‐γ‐related signals on Breg cells need to be further elucidated in the future.
Disclosure
The authors declare no conflicts of interest.
Supporting information
Figure S1. CD19‐Cre+/− mice have normal CD5+CD1dhi Breg cells and contact hypersensitivity (CHS) response.
Figure S2. The mRNA expression of peroxisome proliferator‐activated receptor γ (PPAR‐γ) is higher in CD5+CD1dhi B cells than other two B cell subsets.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81373123, 81611130078). Jianbing Su and Keng Wang performed experiments, analyzed and/or interpreted data. Xuan Zhou, Yiyuan Wang, Jialan Xu, Lei Tao, Xiangzhou Zeng and Nana Chen performed experiments. Xiaojuan Li designed the study, Jianbing Su and Xiaojuan Li wrote the paper. Xiaochun Bai and Xiaojuan Li revised, finally approved and have overall responsibility for the published work.
J. Su and K. Wang contributed equally to this work.
Contributor Information
Xiaochun Bai, Email: baixc15@smu.edu.cn.
Xiaojuan Li, Email: lixiaoj@smu.edu.cn.
References
- 1. Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev 2004; 197:179–191. [DOI] [PubMed] [Google Scholar]
- 2. Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM. B cells regulate autoimmunity by provision of IL‐10. Nat Immunol 2002; 3:944–950. [DOI] [PubMed] [Google Scholar]
- 3. Evans JG, Chavez‐Rueda KA, Eddaoudi A, Meyer‐Bahlburg A, Rawlings DJ, Ehrenstein MR et al Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol 2007; 178:7868–7878. [DOI] [PubMed] [Google Scholar]
- 4. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhi CD5+ phenotype controls T cell‐dependent inflammatory responses. Immunity 2008; 28:639–650. [DOI] [PubMed] [Google Scholar]
- 5. Blair PA, Norena LY, Flores‐Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR et al CD19+CD24hiCD38hi B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 2010; 32:129–140. [DOI] [PubMed] [Google Scholar]
- 6. Matsushita T, Le Huu D, Kobayashi T, Hamaguchi Y, Hasegawa M, Naka K et al A novel splenic B1 regulatory cell subset suppresses allergic disease through phosphatidylinositol 3‐kinase‐Akt pathway activation. J Allergy Clin Immunol 2016; 138:1170–1182 e1179. [DOI] [PubMed] [Google Scholar]
- 7. Dambuza IM, He C, Choi JK, Yu CR, Wang R, Mattapallil MJ et al IL‐12p35 induces expansion of IL‐10 and IL‐35‐expressing regulatory B cells and ameliorates autoimmune disease. Nat Commun 2017; 8:719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol 2012; 30:221–241. [DOI] [PubMed] [Google Scholar]
- 9. Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42:607–612. [DOI] [PubMed] [Google Scholar]
- 10. Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol 2015; 194:1395–1401. [DOI] [PubMed] [Google Scholar]
- 11. Yang M, Rui K, Wang S, Lu L. Regulatory B cells in autoimmune diseases. Cell Mol Immunol 2013; 10:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang M, Sun L, Wang S, Ko KH, Xu H, Zheng BJ et al Novel function of B cell‐activating factor in the induction of IL‐10‐producing regulatory B cells. J Immunol 2010; 184:3321–3325. [DOI] [PubMed] [Google Scholar]
- 13. Jin G, Hamaguchi Y, Matsushita T, Hasegawa M, Le Huu D, Ishiura N et al B‐cell linker protein expression contributes to controlling allergic and autoimmune diseases by mediating IL‐10 production in regulatory B cells. J Allergy Clin Immunol 2013; 131:1674–1682. [DOI] [PubMed] [Google Scholar]
- 14. Meng X, Grotsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX et al Hypoxia‐inducible factor‐1α is a critical transcription factor for IL‐10‐producing B cells in autoimmune disease. Nat Commun 2018; 9:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vamecq J, Latruffe N. Medical significance of peroxisome proliferator‐activated receptors. Lancet 1999; 354:141–148. [DOI] [PubMed] [Google Scholar]
- 16. Janani C, Ranjitha Kumari BD. PPAR γ gene – a review. Diabetes Metab Syndr 2015; 9:46–50. [DOI] [PubMed] [Google Scholar]
- 17. Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett‐Racke AE et al Peroxisome proliferator‐activated receptor‐gamma agonist 15‐deoxy‐δ(12,14)‐prostaglandin J2 ameliorates experimental autoimmune encephalomyelitis. J Immunol 2002; 168:2508–2515. [DOI] [PubMed] [Google Scholar]
- 18. Li T, Chen H, Yang Z, Wei N, Zhang S, Mei X et al Topical application of Pseudolaric acid B improve DNFB‐induced contact hypersensitivity via regulating the balance of Th1/Th17/Treg cell subsets. Eur J Pharm Sci 2012; 45:668–676. [DOI] [PubMed] [Google Scholar]
- 19. Tomita T, Kakiuchi Y, Tsao PS. THR0921, a novel peroxisome proliferator‐activated receptor γ agonist, reduces the severity of collagen‐induced arthritis. Arthritis Res Ther 2006; 8:R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choo J, Lee Y, Yan XJ, Noh TH, Kim SJ, Son S et al A novel peroxisome proliferator‐activated receptor (PPAR) γ agonist 2‐hydroxyethyl 5‐chloro‐4,5‐didehydrojasmonate exerts anti‐inflammatory effects in colitis. J Biol Chem 2015; 290:25609–25619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Setoguchi K, Misaki Y, Terauchi Y, Yamauchi T, Kawahata K, Kadowaki T et al Peroxisome proliferator‐activated receptor‐γ haploinsufficiency enhances B cell proliferative responses and exacerbates experimentally induced arthritis. J Clin Invest 2001; 108:1667–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Natarajan C, Muthian G, Barak Y, Evans RM, Bright JJ. Peroxisome proliferator‐activated receptor‐γ‐deficient heterozygous mice develop an exacerbated neural antigen‐induced Th1 response and experimental allergic encephalomyelitis. J Immunol 2003; 171:5743–5750. [DOI] [PubMed] [Google Scholar]
- 23. Ray DM, Akbiyik F, Bernstein SH, Phipps RP. CD40 engagement prevents peroxisome proliferator‐activated receptor γ agonist‐induced apoptosis of B lymphocytes and B lymphoma cells by an NF‐κB‐dependent mechanism. J Immunol 2005; 174:4060–4069. [DOI] [PubMed] [Google Scholar]
- 24. Hontecillas R, Bassaganya‐Riera J. Peroxisome proliferator‐activated receptor is required for regulatory CD4+ T cell‐mediated protection against colitis. J Immunol 2007; 178:2940–2949. [DOI] [PubMed] [Google Scholar]
- 25. da Rocha Junior LF, Dantas AT, Duarte AL, de Melo Rego MJ, Pitta Ida R, Pitta MG. PPARγ agonists in adaptive immunity: what do immune disorders and their models have to tell us? PPAR Res 2013; 2013:519724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S et al The nuclear receptor PPAR γ selectively inhibits Th17 differentiation in a T cell‐intrinsic fashion and suppresses CNS autoimmunity. J Exp Med 2009; 206:2079–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia‐Bates TM, Baglole CJ, Bernard MP, Murant TI, Simpson‐Haidaris PJ, Phipps RP. Peroxisome proliferator‐activated receptor γ ligands enhance human B cell antibody production and differentiation. J Immunol 2009; 183:6903–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramon S, Bancos S, Thatcher TH, Murant TI, Moshkani S, Sahler JM et al Peroxisome proliferator‐activated receptor γ B cell‐specific‐deficient mice have an impaired antibody response. J Immunol 2012; 189:4740–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Farnesi‐de‐Assuncao TS, Alves CF, Carregaro V, de Oliveira JR, da Silva CA, Cheraim AB et al PPAR‐γ agonists, mainly 15d‐PGJ2, reduce eosinophil recruitment following allergen challenge. Cell Immunol 2012; 273:23–29. [DOI] [PubMed] [Google Scholar]
- 30. Ruhl R, Dahten A, Schweigert FJ, Herz U, Worm M. Inhibition of IgE‐production by peroxisome proliferator‐activated receptor ligands. J Invest Dermatol 2003; 121:757–764. [DOI] [PubMed] [Google Scholar]
- 31. Wohlfert EA, Nichols FC, Nevius E, Clark RB. Peroxisome proliferator‐activated receptor (PPAR) and Immunoregulation: enhancement of regulatory T cells through PPAR ‐dependent and ‐independent mechanisms. J Immunol 2007; 178:4129–4135. [DOI] [PubMed] [Google Scholar]
- 32. Guri AJ, Mohapatra SK, Horne WT 2nd, Hontecillas R, Bassaganya‐Riera J. The role of T cell PPAR γ in mice with experimental inflammatory bowel disease. BMC Gastroenterol 2010; 10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE et al PPAR‐γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012; 486:549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li X, Zhong H, Bao W, Boulad N, Evangelista J, Haider MA et al Defective regulatory B‐cell compartment in patients with immune thrombocytopenia. Blood 2012; 120:3318–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Semple JW. Bregging rights in ITP. Blood 2012; 120:3169. [DOI] [PubMed] [Google Scholar]
- 36. Carter NA, Vasconcellos R, Rosser EC, Tulone C, Munoz‐Suano A, Kamanaka M et al Mice lacking endogenous IL‐10‐producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol 2011; 186:5569–5579. [DOI] [PubMed] [Google Scholar]
- 37. Liu Y, Cheng LS, Wu SD, Wang SQ, Li L, She WM et al IL‐10‐producing regulatory B‐cells suppressed effector T‐cells but enhanced regulatory T‐cells in chronic HBV infection. Clin Sci (Lond) 2016; 130:907–919. [DOI] [PubMed] [Google Scholar]
- 38. Yanaba K, Bouaziz JD, Matsushita T, Tsubata T, Tedder TF. The development and function of regulatory B cells expressing IL‐10 (B10 cells) requires antigen receptor diversity and TLR signals. J Immunol 2009; 182:7459–7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suvas S, Singh V, Sahdev S, Vohra H, Agrewala JN. Distinct role of CD80 and CD86 in the regulation of the activation of B cell and B cell lymphoma. J Biol Chem 2002; 277:7766–7775. [DOI] [PubMed] [Google Scholar]
- 40. Mann MK, Maresz K, Shriver LP, Tan Y, Dittel BN. B cell regulation of CD4+ CD25+ T regulatory cells and IL‐10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol 2007; 178:3447–3456. [DOI] [PubMed] [Google Scholar]
- 41. Racke MK, Scott DE, Quigley L, Gray GS, Abe R, June CH et al Distinct roles for B7‐1 (CD‐80) and B7‐2 (CD‐86) in the initiation of experimental allergic encephalomyelitis. J Clin Invest 1995; 96:2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lundy SK, Fox DA. Reduced fas ligand‐expressing splenic CD5+ B lymphocytes in severe collagen‐induced arthritis. Arthritis Res Ther 2009; 11:R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang K, Tao L, Su J, Zhang Y, Zou B, Wang Y et al TLR4 supports the expansion of FasL+CD5+CD1dhi regulatory B cells, which decreases in contact hypersensitivity. Mol Immunol 2017; 87:188–199. [DOI] [PubMed] [Google Scholar]
- 44. Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2002; 2:748–759. [DOI] [PubMed] [Google Scholar]
- 45. Carter NA, Rosser EC, Mauri C. Interleukin‐10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen‐induced arthritis. Arthritis Res Ther 2012; 14:R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klinker MW, Reed TJ, Fox DA, Lundy SK. Interleukin‐5 supports the expansion of fas ligand‐expressing killer B cells that induce antigen‐specific apoptosis of CD4+ T cells and secrete interleukin‐10. PLoS ONE 2013; 8:e70131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mauri C, Menon M. The expanding family of regulatory B cells. Int Immmunol 2015; 27:479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thompson PW, Bayliffe AI, Warren AP, Lamb JR. Interleukin‐10 is upregulated by nanomolar rosiglitazone treatment of mature dendritic cells and human CD4+ T cells. Cytokine 2007; 39:184–191. [DOI] [PubMed] [Google Scholar]
- 49. Xu Z, Wang G, Zhu Y, Liu R, Song J, Ni Y et al PPAR‐γ agonist ameliorates liver pathology accompanied by increasing regulatory B and T cells in high‐fat‐diet mice. Obesity (Silver Spring) 2017; 25:581–590. [DOI] [PubMed] [Google Scholar]
- 50. Bao Y, Li R, Jiang J, Cai B, Gao J, Le K et al Activation of peroxisome proliferator‐activated receptor γ inhibits endothelin‐1‐induced cardiac hypertrophy via the calcineurin/NFAT signaling pathway. Mol Cell Biochem 2008; 317:189–196. [DOI] [PubMed] [Google Scholar]
- 51. Baba Y, Matsumoto M, Kurosaki T. Signals controlling the development and activity of regulatory B‐lineage cells. Int Immunol 2015; 27:487–493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. CD19‐Cre+/− mice have normal CD5+CD1dhi Breg cells and contact hypersensitivity (CHS) response.
Figure S2. The mRNA expression of peroxisome proliferator‐activated receptor γ (PPAR‐γ) is higher in CD5+CD1dhi B cells than other two B cell subsets.