

Abstract
COP1 is a highly conserved ubiquitin ligase that regulates diverse cellular processes in plants and metazoans. Tribbles pseudokinases, which only exist in metazoans, act as scaffolds that interact with COP1 and its substrates to facilitate ubiquitination. Here, we report that, in addition to this scaffolding role, TRIB1 promotes nuclear localization of COP1 by disrupting an intramolecular interaction between the WD40 domain and a previously uncharacterized regulatory site within COP1. This site, which we have termed the pseudosubstrate latch (PSL), resembles the consensus COP1‐binding motif present in known COP1 substrates. Our findings support a model in which binding of the PSL to the WD40 domain stabilizes a conformation of COP1 that is conducive to CRM1‐mediated nuclear export, and TRIB1 displaces this intramolecular interaction to induce nuclear retention of COP1. Coevolution of Tribbles and the PSL in metazoans further underscores the importance of this role of Tribbles in regulating COP1 function.
Keywords: COP1, E3 ubiquitin ligase, nuclear export, pseudokinase, Tribbles
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Constitutive photomorphogenic 1 (COP1), also known as RFWD2, is a RING domain‐containing E3 ubiquitin ligase that is essential for mammalian embryonic development. COP1 regulates diverse cellular processes, including cell proliferation and the DNA damage response, by promoting the degradation of key transcription factors (Dornan et al, 2006; Migliorini et al, 2011). Overexpression of COP1 is observed in breast and ovarian adenocarcinomas, as well as hepatocellular carcinoma and pancreatic cancer (Dornan et al, 2004a; Lee et al, 2010; Su et al, 2011). Deletion of the COP1 locus has also been reported in many cancer types, such as prostate cancer and melanoma (Migliorini et al, 2011; Vitari et al, 2011). These findings indicate that COP1 can have pro‐oncogenic or tumor‐suppressing functions depending on the cellular context. The opposing effects of COP1 in cancer are likely reflective of the ability of COP1 to target for degradation both transcription factors that function as tumor suppressors, such as C/EBPα and p53, as well as proto‐oncogenes, such as c‐Jun and ETS family members (Dornan et al, 2004b; Wertz et al, 2004; Keeshan et al, 2010; Vitari et al, 2011).
COP1 is highly conserved in metazoans, as well as in plants. The domain organization of COP1 is the same in these multicellular eukaryotes and, in addition to the N‐terminal RING domain, includes a putative coiled coil domain and a C‐terminal WD40 domain, consisting of seven WD40 repeats. First identified as a key repressor of photomorphogenesis in plants (Deng et al, 1991), COP1 influences the expression of the majority of light‐controlled genes in Arabidopsis, which accounts for > 20% of the genome (Ma et al, 2002). COP1 achieves this by ubiquitinating and promoting the degradation of transcription factors, such as HY5, LAF1, and HFR, that positively regulate light signaling (Lau & Deng, 2012). This activity of COP1 is itself regulated by light, which controls trafficking of COP1 between the nucleus and cytoplasm. In dark‐grown plants, COP1 localizes to the nucleus where it binds to and ubiquitinates transcription factors. Light inhibits COP1 activity by triggering its nuclear export, away from its substrates (Von Arnim & Deng, 1994; Pacín et al, 2014). Thus, shuttling between the cytosol and nucleus is an essential mechanism for regulation of COP1 activity in plants.
In mammals, COP1 acts as a component of the CRL4COP1/DET1 ubiquitin ligase complex where it binds to the adaptor protein De‐etiolated‐1 (DET1), which also mediates interactions with the DDB1‐CUL4A‐RBX1 core complex (Wertz et al, 2004; Zhang et al, 2017). Like plant COP1, mammalian COP1 localizes to both the nucleus and cytoplasm. However, the mechanisms governing COP1 localization in mammalian cells are not well understood, and in the absence of the light/dark cycle, they would need to be different than those in plants. In contrast to the light‐controlled binary pattern of either cytosolic or nuclear localization of COP1 in plant cells, the partitioning of COP1 in mammalian cells under steady‐state growth conditions varies quite markedly from cell to cell, ranging from primarily nuclear to primarily cytoplasmic (Yi et al, 2002; Bianchi et al, 2003). Since many substrates of mammalian COP1 are also transcription factors, partitioning of COP1 between the nucleus and cytoplasm is likely important for regulation of its activity as it is in plants.
Pseudokinases in the Tribbles family (TRIB1, TRIB2, TRIB3) have been identified as critical regulators of COP1 function in mammals and are not present in plants. In particular, TRIB1 and TRIB2 are essential for COP1‐mediated ubiquitination of the tumor suppressor C/EBPα (Dedhia et al, 2010; Keeshan et al, 2010; Yoshida et al, 2013). Dysregulation of Tribbles expression can be pathogenic in a COP1‐dependent manner. In acute myeloid leukemia, overexpression of TRIB1 or TRIB2 has been shown to induce leukemogenesis in mice through excessive COP1‐dependent degradation of C/EBPα (Dedhia et al, 2010; Keeshan et al, 2010; Yoshida et al, 2013). Loss of TRIB1 expression is also detrimental. Mice that have TRIB1‐deficient hematopoietic cells experience defects in myeloid cell differentiation due to aberrant levels of C/EBPα and develop metabolic disorders, such as hypertriglyceridemia and insulin resistance, when fed a high‐fat diet (Satoh et al, 2013).
All three Tribbles family members possess a highly conserved pseudokinase domain flanked by an N‐terminal extension and a C‐terminal tail. Tribbles interact directly with the WD40 domain of COP1 via a conserved COP1‐binding motif in their C‐terminal tails that conforms to the consensus sequence (D/E)(D/E)(X)XXVP(D/E), present in numerous COP1 substrates (Holm et al, 2001; Dedhia et al, 2010; Uljon et al, 2016). Through this motif, Tribbles recruit substrates to COP1 lacking a canonical COP1‐binding motif, such as C/EBPα. Thus, Tribbles pseudokinases have been proposed to regulate COP1 by functioning as scaffolds that interact directly with both COP1 and its substrates to facilitate ubiquitination (Qi et al, 2006; Keeshan et al, 2010; Uljon et al, 2016).
In this study, we show that, in addition to this scaffolding role, TRIB1 emulates the regulatory control that light exerts on COP1 localization in plants and modulates the extent of COP1 nuclear localization. We demonstrate that TRIB1 blocks nuclear export of COP1 through disruption of an intramolecular interaction within COP1. We identify a regulatory site in COP1, which we have termed the “pseudosubstrate latch,” that closely resembles the canonical COP1‐binding motif used by COP1‐binding partners. The pseudosubstrate latch in COP1 appears to have coevolved with Tribbles pseudokinases in animals and, notably, is missing in plants, which use light to control COP1 nucleocytoplasmic shuttling. Our work highlights how two orthogonal mechanisms have evolved in different kingdoms to regulate the same process.
Results
TRIB1 promotes nuclear accumulation of COP1
To investigate the effects of TRIB1 expression on COP1 localization, we transiently expressed GFP‐tagged human COP1 in COS7, Huh7, and HEK293 cells and examined its subcellular distribution in the presence and absence of co‐transfected human TRIB1 via confocal microscopy. In the absence of TRIB1, COP1 localized to both the nucleus and cytoplasm, while TRIB1 localized primarily to the nucleus (Kiss‐Toth et al, 2006; Soubeyrand et al, 2016; Appendix Fig S1A). Consistent with previous studies, the distribution of COP1 between these two compartments varied considerably within the cell population (Yi et al, 2002; Bianchi et al, 2003; Fig EV1A and B). Strikingly, when TRIB1 and COP1 were coexpressed, COP1 shifted to being predominantly nuclear (Figs 1A, and EV1C and E). To quantify this effect, we measured the ratio of nuclear to cytoplasmic COP1 fluorescence and found that coexpression of TRIB1 markedly increased the ratio of nuclear to cytoplasmic COP1 across the entire cell population (Figs 1B, and EV1D and F). TRIB1 also increased the nuclear population of endogenous COP1 when expressed in NIH3T3 cells (Fig EV1G and H).
Figure EV1. TRIB1 promotes nuclear localization of COP1.
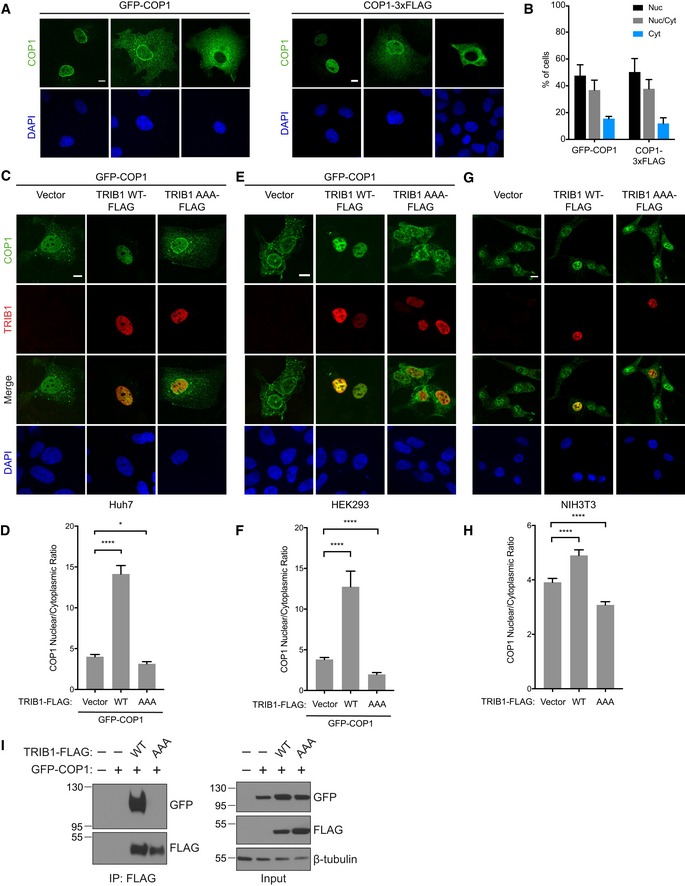
-
ARepresentative immunofluorescence images of COS7 cells expressing GFP‐COP1 or COP1‐3xFLAG showing the different patterns of localization (nuclear, nuclear and cytoplasmic, and cytoplasmic) typically observed within a cell population. For visualizing COP1‐3xFLAG, the cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
-
BQuantification of percentage of cells observed in (A) with the indicated COP1 construct exhibiting nuclear (Nuc), nuclear and cytoplasmic (Nuc/Cyt), or cytoplasmic (Cyt) localization. Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed.
-
C–FRepresentative images of Huh7 cells (C) and HEK293 cells (E) expressing GFP‐COP1 (green) co‐transfected with empty vector or the indicated TRIB1‐FLAG construct (red). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. See also Appendix Fig S1C and D for Western blots showing the expression levels of the constructs used. (D, F) Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 in (C) and (E). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 with empty vector was calculated using the Student's t‐test (*P < 0.01, ****P < 0.00001).
-
G, HRepresentative images of NIH3T3 cells (G) transfected with empty vector or the indicated TRIB1‐FLAG construct (red). Cells were stained with anti‐FLAG (Cell Signaling), anti‐COP1 (Abcam), anti‐mouse Alexa Fluor 647, and anti‐rabbit Alexa Fluor 488. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. See also Appendix Fig S1E for Western blots showing the expression levels of the constructs used. (H) Quantification of the average ratio of nuclear/cytoplasmic fluorescence of endogenous COP1 in (G). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to the empty vector control was calculated using the Student's t‐test (****P < 0.00001).
-
ICoimmunoprecipitation of GFP‐COP1 with TRIB1‐FLAG in COS7 cells showing that the AAA mutation ablates the TRIB1/COP1 interaction.
Figure 1. TRIB1 promotes nuclear accumulation of COP1.
- Representative images of COS7 cells expressing GFP‐COP1 (green) co‐transfected with empty vector or the indicated TRIB1‐FLAG construct (red). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. See also Appendix Fig S1B for Western blots showing the expression levels of the constructs used.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 in (A). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 with empty vector was calculated using the Student's t‐test (****P < 0.00001).
- Representative images of COS7 cells showing GFP‐COP1 (green) and TRIB1‐FLAG (red) colocalizing in punctate nuclear structures. Insets show enlarged images of these nuclear puncta. Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Representative images of COS7 cells showing that GFP‐COP1 (green) and TRIB1‐HA (red) are diffuse in the nucleoplasm when coexpressed with C/EBPα (cyan). Cells were stained with anti‐HA (Santa Cruz), anti‐C/EBPα (Cell Signaling), anti‐mouse Alexa Fluor 405, and anti‐rabbit Alexa Fluor 680. Scale bar, 10 μm. See also Fig EV2C for images of cells expressing each construct alone or in pairs.
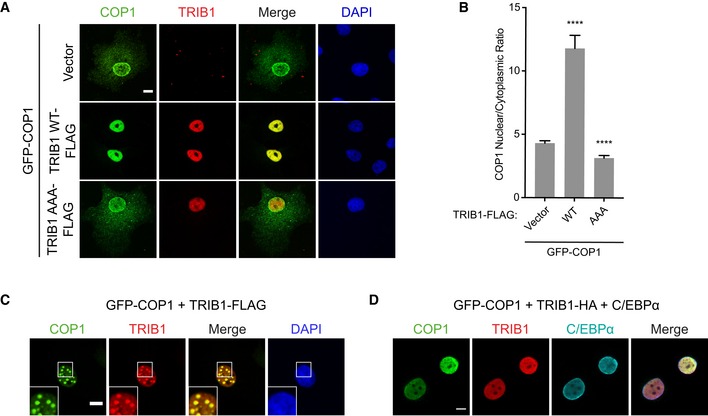
Notably, we found that in approximately half of the cells co‐transfected with COP1 and TRIB1, both proteins colocalized in punctate structures in the nucleus (Fig 1C), a phenomenon previously observed for Arabidopsis COP1 (Von Arnim & Deng, 1994). These structures colocalized with DAXX, a component of PML bodies (Fig EV2A), but not with markers of other types of subnuclear compartments (Fig EV2B). Arabidopsis COP1 recruits its substrates and binding partners to nuclear puncta (Ang et al, 1998; Wang et al, 2001; Seo et al, 2003). Thus, we tested whether the COP1 substrate, C/EBPα, is also recruited to the COP1‐ and TRIB1‐positive nuclear puncta in mammalian cells by co‐transfecting TRIB1, COP1, and C/EBPα in COS7 cells and examining their localization via immunofluorescence. C/EBPα localized exclusively to the nucleus, and its coexpression with either TRIB1 or COP1 alone had no effect on their localization (Fig EV2C). Surprisingly, however, upon C/EBPα coexpression, TRIB1 and COP1 no longer localized in nuclear puncta (Fig 1D). These results demonstrate a potential difference in the composition of the punctate nuclear structures that COP1 localizes to in plants versus mammals and show that binding of C/EBPα to the TRIB1/COP1 complex results in the redistribution of this complex between subnuclear compartments.
Figure EV2. COP1 and TRIB1 colocalize in punctate nuclear structures.
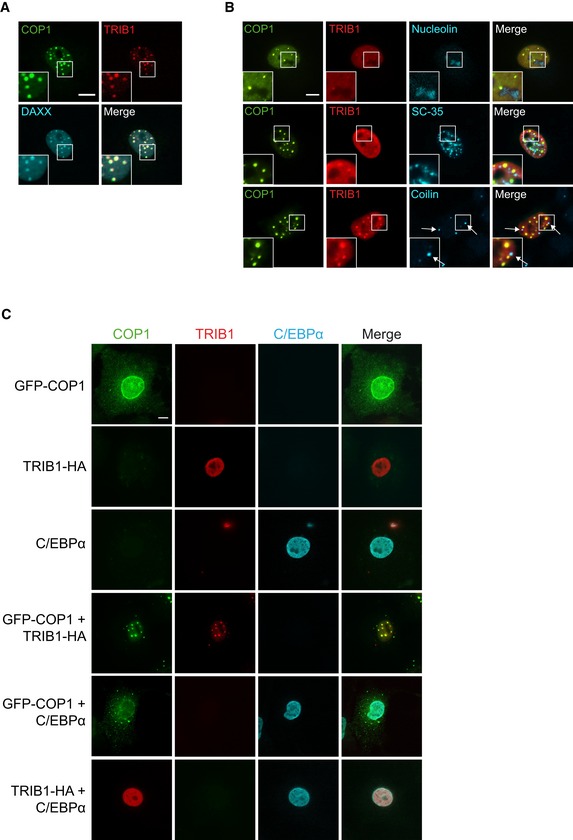
- Representative images of COS7 cells showing that COP1‐3xFLAG (green) and TRIB1‐HA (red) colocalize with GFP‐DAXX (cyan). Insets show enlarged images of nuclear puncta. Cells were stained with anti‐FLAG (Cell Signaling), anti‐HA (Santa Cruz), anti‐mouse Alexa Fluor 405, and anti‐rabbit Alexa Fluor 680. Scale bar, 10 μm.
- Representative images of COS7 cells showing that GFP‐COP1 (green) and TRIB1‐FLAG (red) do not colocalize with markers (cyan) for nucleoli (nucleolin), nuclear speckles (SC‐35), or Cajal bodies (coilin). Cells were stained with anti‐FLAG (Cell Signaling), anti‐mouse Alexa Fluor 405, anti‐rabbit Alexa Fluor 680, and either anti‐nucleolin, anti‐SC‐35, or anti‐coilin, as indicated. Scale bar, 10 μm.
- Representative images of COS7 cells expressing the indicated combinations of GFP‐COP1 (green), TRIB1‐HA (red), and C/EBPα (cyan). Images for cells coexpressing all three constructs are shown in Fig 1D. Cells were stained with anti‐HA (Santa Cruz), anti‐C/EBPα (Cell Signaling), anti‐mouse Alexa Fluor 405, and anti‐rabbit Alexa Fluor 680. Scale bar, 10 μm.
The TRIB1 C‐terminal tail is sufficient to promote nuclear localization of COP1
To test whether a direct interaction between TRIB1 and COP1 is required for promoting nuclear localization of COP1, we generated a mutant, TRIB1 AAA, in which the COP1‐binding motif in the TRIB1 C‐terminal tail is mutated from DQIVPE to AQIAAE. An analogous set of mutations abolishes the interaction between murine TRIB2 and COP1 (Keeshan et al, 2006), and we confirmed through coimmunoprecipitation that TRIB1 AAA also failed to interact with COP1 (Fig EV1I). Although TRIB1 AAA localized to the nucleus like TRIB1 WT (Appendix Fig S1A), this mutant failed to increase COP1 nuclear localization (Fig 1A and B, and EV1C–H). Hence, a direct interaction between TRIB1 and COP1 via the COP1‐binding motif in the TRIB1 C‐terminal tail is required for TRIB1‐mediated COP1 nuclear accumulation.
To determine whether the tail of TRIB1 is sufficient for regulation of COP1 localization, we fused the C‐terminal tail of TRIB1 (residues 343–372) to GFP and tested the ability of this construct to modulate the localization of a FLAG‐tagged COP1 construct. For comparison, we also fused full‐length TRIB1 to GFP and created AAA mutants of both the full‐length and tail fusion constructs. Both GFP‐TRIB1 WT and GFP‐TRIB1 AAA localized exclusively to the nucleus. In contrast, GFP‐TRIB1 tail WT and GFP‐TRIB1 tail AAA, which lack the NLS present in the N‐terminal extension of TRIB1, although enriched in the nucleus, were also found in the cytoplasm. In accordance with our earlier results, GFP‐TRIB1 WT potentiated COP1 nuclear localization, whereas GFP alone and GFP‐TRIB1 AAA did not. Remarkably, however, GFP‐TRIB1 tail was sufficient to promote COP1 nuclear localization. This effect was abrogated by the AAA mutation, which eliminates COP1 binding (Fig 2A and B). These findings reveal that the TRIB1 C‐terminal tail is both necessary and sufficient to enhance COP1 nuclear localization.
Figure 2. The TRIB1 C‐terminal tail is sufficient to promote COP1 nuclear localization.
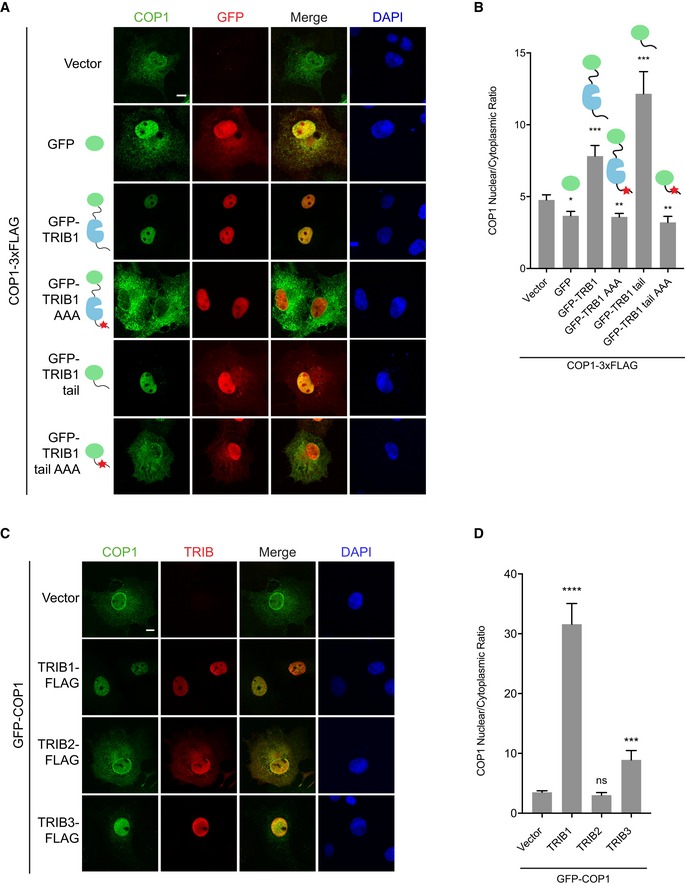
- Representative images of COS7 cells expressing COP1‐3xFLAG (green) co‐transfected with empty vector, GFP, or the indicated GFP‐TRIB1 constructs (red). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. See also Appendix Fig S2A for images of GFP‐tagged constructs in the absence of COP1 coexpression and Appendix Fig S2B for Western blots showing the expression levels of the constructs used.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of COP1‐3xFLAG in (A). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 with empty vector was calculated using the Student's t‐test (*P < 0.01; **P < 0.001; ***P < 0.0001).
- Representative images of COS7 cells expressing GFP‐COP1 (green) co‐transfected with empty vector, TRIB1‐FLAG, TRIB2‐FLAG, or TRIB3‐FLAG (red). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 in (C). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 with empty vector was calculated using the Student's t‐test (ns, not significant; ***P < 0.0001; ****P < 0.00001).
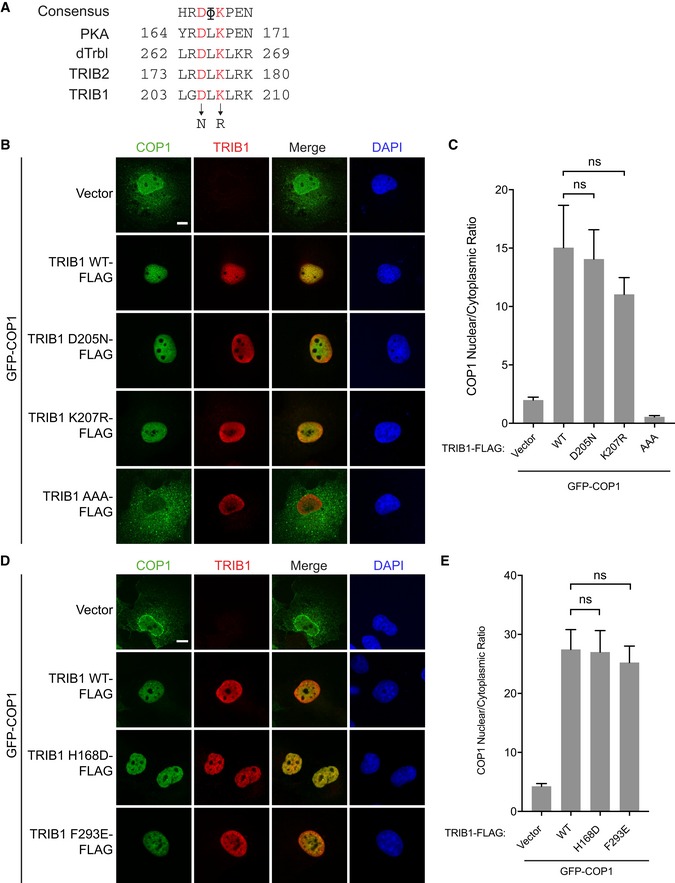
Though the C‐terminal tail of TRIB1 was sufficient for promoting COP1 nuclear localization, we next tested whether the pseudokinase domain of TRIB1 could play an additional regulatory role in this process. Several studies have shown that mutations in the pseudokinase domain affect interactions between Tribbles and its binding partners. The pseudokinase domain retains several conserved motifs important for catalysis in catalytically competent kinases, including the HRD motif in the catalytic loop. A mutation within the HRD motif in the Drosophila Tribbles homolog ablates its interactions with Akt and the C/EBP homolog Slbo (Masoner et al, 2013; Das et al, 2014). Similarly, mutations in the catalytic loop of murine TRIB2 impair binding of C/EBPα (Keeshan et al, 2010). Although TRIB1 is catalytically inactive (Murphy et al, 2015), these mutations might bias the pseudokinase domain to adopt different conformations and thereby affect the ability of TRIB1 to interact with its binding partners. Recently, a crystal structure of TRIB1 in complex with a C/EBPα peptide revealed the binding site for C/EBPα within the C‐lobe of the TRIB1 pseudokinase domain (Jamieson et al, 2018). We generated point mutations in the pseudoactive site of TRIB1 corresponding to those analyzed in Drosophila Tribbles (D205N) and in murine TRIB2 (K207R) (Fig EV3A). We also introduced two point mutations in TRIB1 that had been shown to ablate interaction with C/EBPα (H168D, F293E) seen in the crystal structure (Jamieson et al, 2018). None of these mutations, however, caused a significant change in the ability of TRIB1 to promote COP1 nuclear localization (Fig EV3B–E).
Figure EV3. Mutations that disrupt binding to C/EBPα do not affect the ability of TRIB1 to promote nuclear localization of COP1.
-
ASequence alignment of the catalytic loop in TRIB1, TRIB2, Drosophila Tribbles (dTrbl), and PKA.
-
B–ERepresentative images of COS7 cells showing GFP‐COP1 (green) (B, D) co‐transfected with empty vector or the indicated TRIB1‐FLAG constructs (red). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. (C, E) Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 in (B) and (D). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 coexpressed with TRIB1 WT was calculated using the Student's t‐test (ns, not significant).
Other members of the Tribbles family, TRIB2 and TRIB3, also contain a COP1‐binding motif in their C‐terminal tails. We examined whether they are likewise capable of promoting COP1 nuclear localization. Like TRIB1, TRIB3 localized predominantly to the nucleus and induced an increase in COP1 nuclear localization, albeit to a lesser extent than TRIB1 (Fig 2C and D). In contrast, expression of TRIB2 did not have a significant effect on COP1 localization (Fig 2C and D), possibly because TRIB2 localized throughout the nucleus and cytoplasm in COS7 cells. In Bel‐7402 liver cancer cells, in which TRIB2 is predominantly nuclear, TRIB2 was shown to increase nuclear localization of endogenous COP1 (Xu et al, 2014). Hence, all Tribbles family members have an intrinsic ability, encoded in their C‐terminal tails, to modulate the extent of nuclear accumulation of COP1.
Nuclear localization of COP1 is regulated by the WD40 domain
The TRIB1 tail binds to the WD40 domain of COP1 (Uljon et al, 2016). Therefore, we expected that mutations in the TRIB1‐binding site in the COP1 WD40 domain would prevent TRIB1 from promoting COP1 nuclear localization. Based on the recently published crystal structure of the TRIB1 tail in complex with the COP1 WD40 domain (Uljon et al, 2016), we generated several COP1 WD40 point mutants (K472E, R515E, W517A, K600E, E642R) to test this hypothesis. Each of these residues is highly conserved, and analogous mutations in Arabidopsis COP1 affect binding of substrates containing a COP1‐binding motif (Holm et al, 2001). Four of these residues (K472, R515, W517, K600) directly engage the TRIB1 tail and their mutation should therefore prevent TRIB1 binding, while E642 is not involved in the interaction (Fig 3A).
Figure 3. Nuclear localization of COP1 is regulated by the WD40 domain.
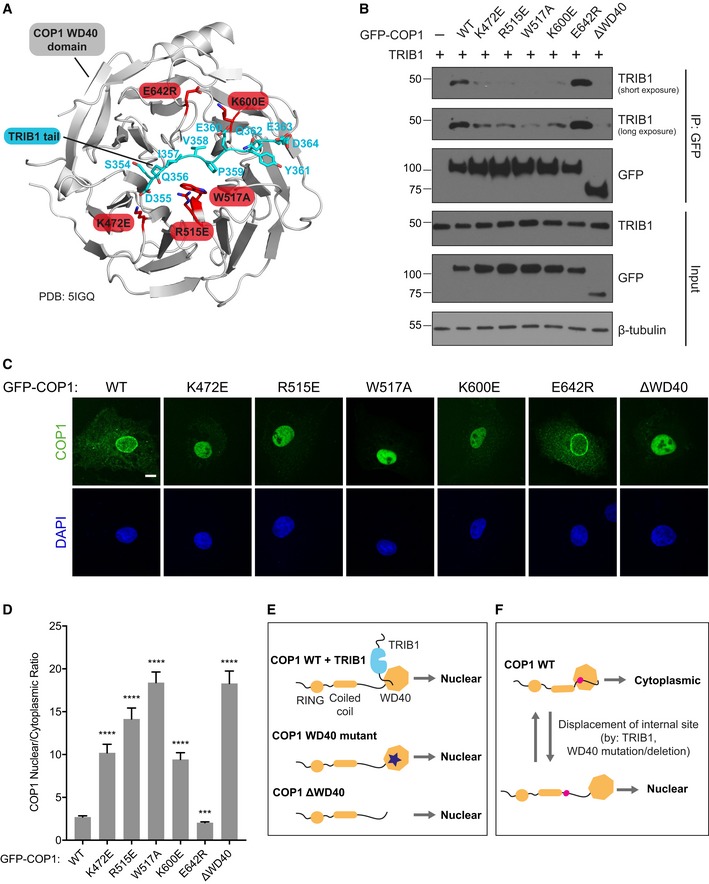
- A cartoon representation of a crystal structure of the COP1 WD40 domain (gray) in complex with the TRIB1 tail (cyan) (PDB: 5IGQ). Point mutations introduced in the WD40 domain are shown in red.
- Coimmunoprecipitation of COP1 WD40 mutants with TRIB1 from COS7 cells transiently transfected with the indicated GFP‐COP1 constructs and untagged TRIB1.
- Representative images of COS7 cells transiently transfected with the indicated GFP‐COP1 constructs (green). Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. See also Fig EV4A for corresponding images of these GFP‐COP1 constructs coexpressed with TRIB1‐FLAG.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 constructs in (C). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 WT was calculated using the Student's t‐test (***P < 0.0001; ****P < 0.00001).
- Cartoons summarizing the effects of TRIB1 binding and WD40 domain mutation/deletion on COP1 localization.
- Proposed model for regulation of COP1 subcellular localization by an intramolecular regulatory site that competes with TRIB1 for binding to the WD40 domain.
We confirmed through coimmunoprecipitation that K472E, R515E, W517A, and K600E mutations indeed markedly weaken the interaction between TRIB1 and COP1, while E642R did not (Fig 3B). Counterintuitively, however, all four COP1 WD40 mutants that exhibited impaired TRIB1 binding localized primarily to the nucleus even in the absence of TRIB1 (Fig 3C and D). Coexpression with TRIB1 did not result in a significant change in the localization of these mutants (Fig EV4A and B). COP1 E642R, on the other hand, behaved similarly to COP1 WT and accumulated exclusively in the nucleus only in the presence of TRIB1 coexpression (Fig 3C and D, and EV4A and B). The WD40 domain mutations that interfere with TRIB1 binding mimicked the effect of deleting the entire WD40 domain (Fig 3C and D), which was previously shown to cause mammalian COP1 to localize exclusively to the nucleus (Yi et al, 2002; Bianchi et al, 2003). These findings reveal that an intact TRIB1‐binding site is necessary for proper nucleocytoplasmic shuttling of COP1.
Figure EV4. Localization of TRIB1‐binding deficient COP1 WD40 mutants is unaffected by TRIB1 coexpression.
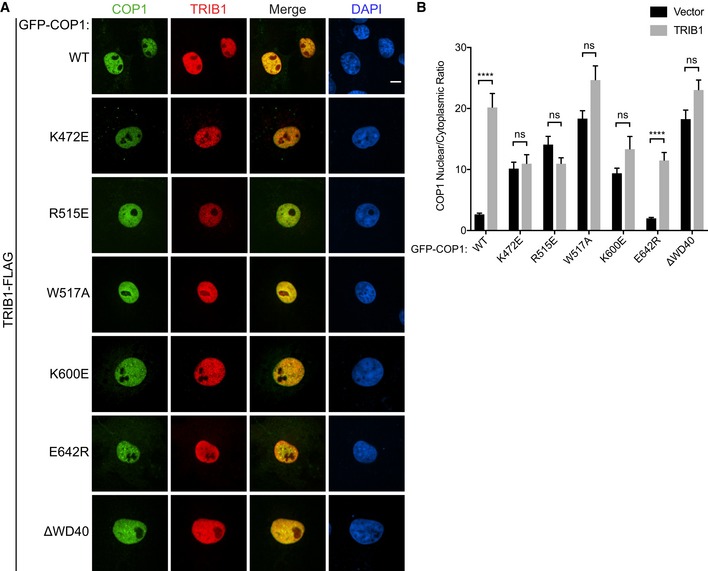
- Representative images of COS7 cells co‐transfected with TRIB1‐FLAG (red) and the indicated GFP‐COP1 constructs (green). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence for each of the indicated GFP‐COP1 constructs co‐transfected with empty vector (Fig 3C) or with TRIB1‐FLAG (Fig EV4A). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance was calculated using the Student's t‐test (ns, not significant; ****P < 0.00001).
Identification of an intramolecular WD40‐binding site that regulates the subcellular distribution of COP1
Our findings demonstrate that association of TRIB1 with the WD40 domain and disruption of the TRIB1‐binding site on the WD40 domain paradoxically both cause COP1 to be retained in the nucleus (Fig 3E). These seemingly contradictory observations could be explained by a model in which the TRIB1‐binding site on the WD40 domain participates in an intramolecular interaction that promotes COP1 nuclear export such that the TRIB1 C‐terminal tail competes with this interaction, causing nuclear retention (Fig 3F). We hypothesized that this intramolecular site resembles the consensus COP1‐binding motif found in numerous COP1 binding partners. By examining the sequence of COP1, we identified a sequence, EDSTVPQ, which contains key structural features of the COP1‐binding motif, including the “VP” motif and acidic residues a few positions N‐terminal to the VP motif (Fig 4A). This site is located within the linker between the coiled coil and WD40 domains and spans residues 308–314 (Fig 3F).
Figure 4. TRIB1 competes with an intramolecular site for binding to the WD40 domain to modulate COP1 localization.
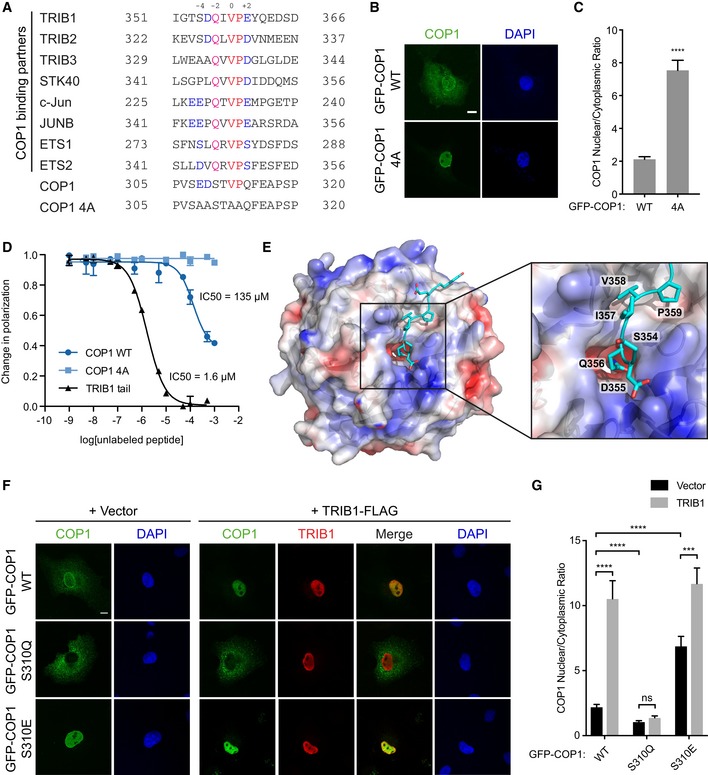
- Sequence alignment of COP1 pseudosubstrate latch with the COP1 4A mutant and COP1‐binding motifs from previously identified COP1‐binding partners. Residues in the sequences are colored to indicate residues conserved in the COP1‐binding motif: Val‐Pro dipeptide (red), acidic residues (blue), conserved Gln (magenta).
- Representative images of COS7 cells transiently transfected with GFP‐COP1 WT or GFP‐COP1 4A (green). Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 in (B). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 WT was calculated using the Student's t‐test (****P < 0.00001).
- Fluorescence polarization competition assay measuring the ability of unlabeled COP1 PSL, COP1 PSL 4A, and TRIB1 tail peptides to compete with FITC‐TRIB1 tail for binding to the COP1 WD40 domain (376–731). The COP1 WD40 domain was pre‐incubated with FITC‐TRIB1 tail before addition of the indicated concentrations of unlabeled peptide. The change in polarization is plotted as a function of unlabeled peptide concentration. Curves were fit to a sigmoidal dose–response curve. Data are presented as mean values ± s.e.m. for three independent experiments where each measurement was made in duplicate.
- Surface representation of the crystal structure (PDB: 5IGQ) of the COP1 WD40 domain colored according to electrostatic surface potential, calculated by APBS (Baker et al, 2001), in complex with the TRIB1 tail (cyan). Inset shows zoomed view of TRIB1 Q356 inserting its side chain into the central channel of the COP1 WD40 domain.
- Representative images of COS7 cells transiently transfected with the indicated GFP‐COP1 constructs (green) and empty vector or TRIB1‐FLAG (red). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 constructs in (F). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance was calculated using the Student's t‐test (ns, not significant, ***P < 0.0001; ****P < 0.00001).
If this putative intramolecular site is responsible for interacting with the WD40 domain to enable COP1 nuclear export, then its mutation should result in increased COP1 nuclear localization, mimicking the effect of TRIB1 binding or mutating/deleting the WD40 domain. Indeed, mutation of this site (COP1 4A) resulted in enhanced nuclear localization of COP1 (Fig 4A–C). In addition, deletions in the linker between the coiled coil and WD40 domains resulted in increased nuclear localization only if they included the EDSTVPQ sequence (Fig EV5A–C). Together, these data support a role for this intramolecular WD40‐binding site in regulation of COP1 nuclear export. Because of the similarity between the intramolecular site and the COP1‐binding motifs in COP1 substrates, we have termed this site the pseudosubstrate latch (PSL).
Figure EV5. Identification of an intramolecular WD40‐binding site that regulates the subcellular distribution of COP1.
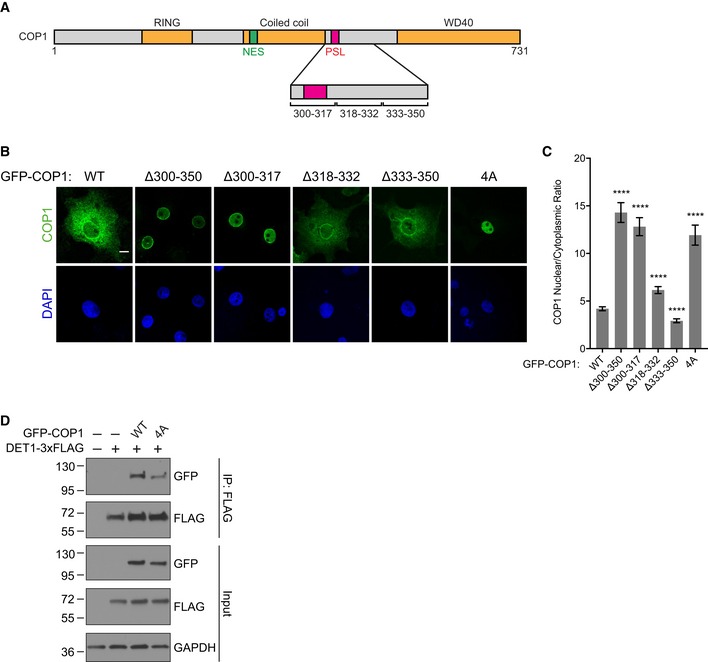
- Schematic of COP1 illustrating locations of the coiled coil–WD40 domain linker deletions and PSL.
- Representative images of COS7 cells transfected with the indicated GFP‐COP1 constructs (green). Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Quantification of the average ratio for nuclear/cytoplasmic fluorescence for each of the indicated GFP‐COP1 constructs. Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance relative to GFP‐COP1 WT was calculated using the Student's t‐test (****P < 0.00001).
- Coimmunoprecipitation of GFP‐COP1 with DET1‐3xFLAG in COS7 cells showing that the 4A mutation weakens the DET1/COP1 interaction.
We next used a fluorescence polarization competition assay to directly measure whether the PSL binds to the same interface on the WD40 domain as the TRIB1 tail. An unlabeled COP1 PSL peptide was able to compete with a FITC‐labeled TRIB1 tail peptide for binding to recombinant human COP1 WD40 domain with an IC50 of 135 μM, whereas a COP1 PSL 4A peptide did not displace FITC‐TRIB1 tail even at millimolar concentrations (Fig 4D). These results demonstrate that the COP1 PSL binds to the WD40 domain in a manner dependent on the COP1‐binding motif within the PSL. An unlabeled TRIB1 tail peptide competed with FITC‐TRIB1 tail with an IC50 of 1.6 μM. Although the PSL bound to the WD40 domain with ~ 100‐fold lower affinity than the TRIB1 tail, the PSL/WD40 interaction can occur in cis in the context of full‐length COP1, which would increase its effective affinity in a cellular environment.
The lower affinity of the intramolecular PSL/WD40 interaction could be in place to allow the TRIB1 tail to outcompete it. To test this model, we sought to reengineer the PSL into a stronger WD40‐binding site, which would be expected to decrease the ability of TRIB1 to promote COP1 nuclear localization. We compared the COP1‐binding motif in the PSL to those in known COP1 binding partners and found that a unique feature of the PSL is its lack of a glutamine residue (Q356 in TRIB1) in the −2 position relative to the VP motif (Fig 4A). Examination of the electrostatic surface potential of the WD40 domain in the crystal structure of the TRIB1 tail/COP1 WD40 domain complex revealed that the side chain of TRIB1 Q356 inserts into the negatively charged central channel of the COP1 WD40 domain and forms a hydrogen bond with Y491 (Fig 4E). In the COP1 PSL, the corresponding residue is a serine (S310), which is expected not to engage as well in hydrogen bonding with Y491 due to its shorter side chain and lower polarity. We therefore reasoned that mutating S310 to glutamine (S310Q) would strengthen the interaction between the PSL and WD40 domain and, consequently, enhance the ability of the PSL to compete with TRIB1 for binding to the WD40 domain. Indeed, COP1 S310Q exhibited reduced nuclear localization compared to COP1 WT and was largely indifferent to TRIB1 (Fig 4F and G).
In contrast to glutamine substitution, replacement of S310 with a negatively charged residue should weaken the binding of the PSL to the WD40 domain and, consequently, enhance nuclear localization, even without TRIB1 present. In agreement with this prediction, a S310E mutation resulted in increased nuclear COP1, and addition of TRIB1 further potentiated this effect by less than twofold (Fig 4F and G). Due to the phosphomimetic potential of the S310E mutation, these data suggest that binding of the PSL to the WD40 domain could be regulated through phosphorylation of S310, which, like TRIB1 binding, would be expected to promote COP1 nuclear localization. This could be another reason why the S310Q mutation, which removes the putative phosphorylation site, enhances PSL binding and results in efficient nuclear export of COP1.
The PSL site encompasses residues 308–314, which closely follow the C‐terminal portion of the putative coiled coil domain (residues 280–299) that has been implicated in the interaction of COP1 with the adaptor protein DET1. Splice variants of COP1 lacking this region of the coiled coil domain fail to interact with DET1, preventing assembly of the CRL4COP1/DET1 ubiquitin ligase complex (Wertz et al, 2004; Savio et al, 2008). Using coimmunoprecipitation, we tested whether mutations within the PSL affect the interaction between COP1 and DET1. While COP1 4A still interacted with DET1, the interaction was notably weaker than that observed for COP1 WT (Fig EV5D), suggesting a role for the PSL/WD40 interaction in regulation of DET1 binding.
TRIB1 inhibits CRM1‐mediated nuclear export of COP1
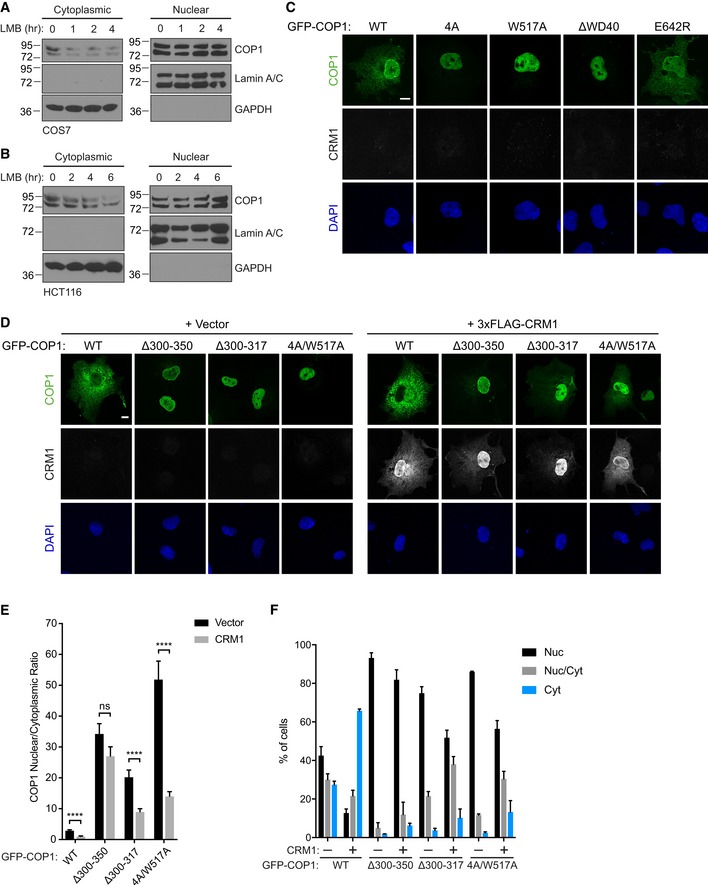
TRIB1 could increase COP1 nuclear localization by promoting nuclear import and/or inhibiting nuclear export of COP1. Since TRIB1 is found primarily in the nucleus (Kiss‐Toth et al, 2006; Soubeyrand et al, 2016), TRIB1 is more likely to regulate COP1 nuclear export after COP1 has already undergone nuclear import. A leucine‐rich nuclear export signal (NES) at the N‐terminus of the COP1 coiled coil domain is recognized by the nuclear export receptor CRM1 (Yi et al, 2002; Kırlı et al, 2015). To confirm that CRM1 regulates COP1 localization, COS7 and HCT116 cells were treated with leptomycin B, a specific inhibitor of CRM1, and localization of endogenous COP1 was examined via subcellular fractionation. In both cases, treatment with leptomycin B reduced the amount of COP1 in the cytoplasmic fraction (Fig EV6A and B). Furthermore, mutation of the NES (COP1 NESmut) resulted in constitutively nuclear localization of COP1 (Fig 5A and C).
Figure EV6. TRIB1 inhibits CRM1‐mediated nuclear export of COP1.
-
A, BSubcellular fractionation of COS7 cells (A) and HCT116 cells (B) treated with 20 nM leptomycin B (LMB) for the indicated times, showing a reduction of endogenous COP1 levels in the cytoplasmic fraction over time.
-
CRepresentative images of COS7 cells expressing the indicated GFP‐COP1 (green) constructs co‐transfected with empty vector. Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
-
DRepresentative images of COS7 cells expressing the indicated GFP‐COP1 (green) constructs co‐transfected with empty vector or 3xFLAG‐CRM1 (grayscale). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
-
EQuantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 constructs in (D). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance was calculated using the Student's t‐test (ns, not significant; ****P < 0.00001).
-
FQuantification of percentage of cells observed in (D) with the indicated GFP‐COP1 construct exhibiting nuclear (Nuc), nuclear and cytoplasmic (Nuc/Cyt), or cytoplasmic (Cyt) localization. Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed.
Figure 5. TRIB1 inhibits CRM1‐mediated nuclear export of COP1.
- Representative images of COS7 cells transiently transfected with GFP‐COP1 WT or GFP‐COP1 NESmut (V238A/L242A) (green) and empty vector or 3xFLAG‐CRM1 (grayscale). Cells were stained with anti‐FLAG (Cell Signaling) and anti‐rabbit Alexa Fluor 680. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Representative images of COS7 cells transfected with GFP‐COP1 WT (green), the indicated TRIB1‐HA constructs (red), and empty vector or 3xFLAG‐CRM1 (grayscale). Cells were stained with anti‐FLAG (Cell Signaling), anti‐HA (Sigma), anti‐rabbit Alexa Fluor 680, and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 constructs in (A) and (B). Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance was calculated using the Student's t‐test (ns, not significant; *P < 0.01; ****P < 0.00001).
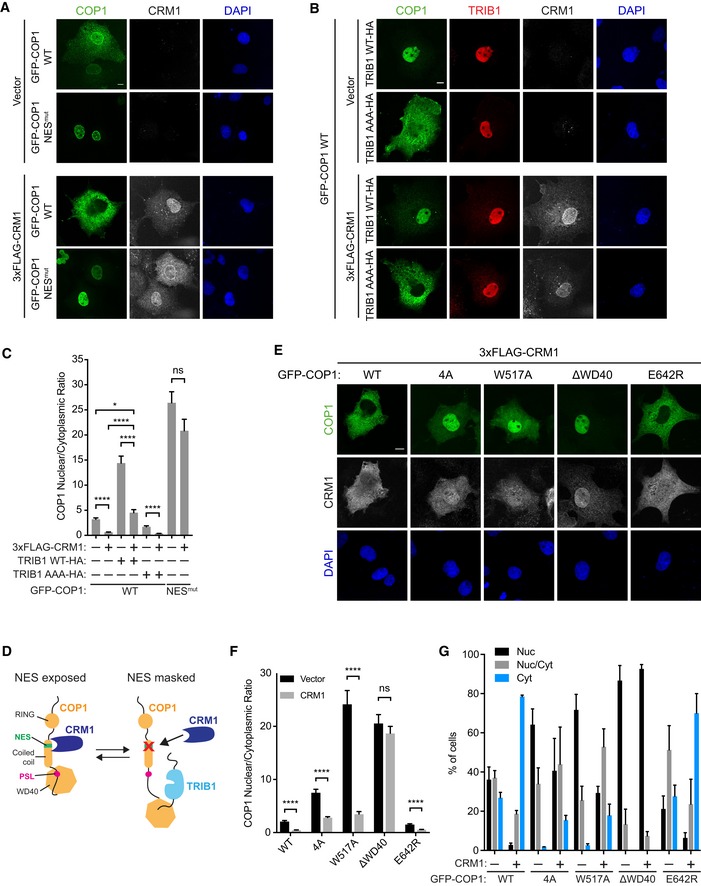
- A hypothesized model for inhibition of COP1 nuclear export by TRIB1 through NES masking. In this model, binding of the PSL to the WD40 domain stabilizes a conformation in which the NES, located in the distal putative coiled coil domain, is exposed. This could be achieved by an effective reduction of the distance between the WD40 domain and the putative coiled coil domain, resulting from PSL binding. Loss of this proximity upon TRIB1 binding could then induce a conformational change in the coiled coil domain that results in NES masking.
- Representative images of COS7 cells transiently transfected with the indicated GFP‐COP1 constructs (green) and 3xFLAG‐CRM1 (grayscale). Cells were stained with anti‐FLAG (Sigma) and anti‐mouse Alexa Fluor 568. Nuclei were counterstained with DAPI (blue). Scale bar, 10 μm. See also Fig EV6C for corresponding images of these GFP‐COP1 constructs co‐transfected with empty vector.
- Quantification of the average ratio of nuclear/cytoplasmic fluorescence of GFP‐COP1 constructs in (E) and Fig EV6C. Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed. Significance was calculated using the Student's t‐test (ns, not significant; ****P < 0.00001).
- Quantification of percentage of cells observed in (E) and Fig EV6C with the indicated GFP‐COP1 construct exhibiting nuclear (Nuc), nuclear and cytoplasmic (Nuc/Cyt), or cytoplasmic (Cyt) localization. Mean values ± s.e.m. are shown for three independent experiments where 50 individual cells per experiment were analyzed.
To test whether TRIB1 influences COP1 localization through a CRM1‐dependent pathway, we coexpressed COP1 and CRM1 with or without TRIB1 and examined their localization via immunofluorescence. Overexpression of CRM1 shifted COP1 WT to predominantly cytoplasmic localization in a NES‐dependent manner (Fig 5A and C). However, this shift was markedly reduced when COP1 was coexpressed with both CRM1 and TRIB1 WT, but not TRIB1 AAA (Fig 5A–C), suggesting that TRIB1 binding to COP1 directly competes with CRM1‐dependent nuclear export.
Recognition of cargo proteins by CRM1 is often regulated through masking or occlusion of the NES in the cargo. This can be achieved through intermolecular interactions that involve changes in oligomerization states, as observed for p53 (Stommel et al, 1999), or through conformational changes, as shown for NFAT1 (Okamura et al, 2000). Binding of the COP1 PSL to the WD40 domain could enable COP1 nuclear export by unmasking the NES and making it accessible to CRM1 (Fig 5D). Therefore, loss of the PSL/WD40 interaction should mask the NES and render COP1 less responsive to CRM1. To test this hypothesis, we coexpressed CRM1 with mutants of COP1 in which the PSL/WD40 interaction is disrupted (COP1 4A, COP1 W517A, and COP1 ΔWD40), as well as COP1 WT and COP1 E642R, in which this interaction is maintained. As expected, COP1 4A and COP1 W517A were less affected by the presence of CRM1 than COP1 WT and COP1 E624R (Fig 5E and F). Although the average nuclear to cytoplasmic ratio for COP1 4A and COP1 W517A still markedly decreased when CRM1 was coexpressed, both of these mutants remained substantially more nuclear than COP1 WT and COP1 E642R in the presence of CRM1 (Fig 5F). These differences were more striking when we considered how many cells showed exclusively cytosolic localization in the presence of CRM1, with ~ 80% of cells showing this phenotype for wild‐type COP1 and only ~ 20% for COP1 4A and COP1 W517A (Fig 5G). In contrast, COP1 ΔWD40 was completely unaffected by CRM1 overexpression and remained primarily nuclear under all conditions (Figs 5E–G and EV6C), in agreement with this deletion being more potent than single WD40 point mutations in blocking binding of the COP1‐binding motif (Fig 3B). Since COP1 4A and COP1 W517A could still be exported to a certain degree by CRM1 unlike COP1 ΔWD40, these mutations are likely less effective in abolishing the PSL/WD40 interaction. Thus, we generated a COP1 construct in which both the PSL and WD40 domain are mutated (COP1 4A/W517A). We also evaluated the localization of mutants in which the entire PSL is deleted (COP1 Δ300–350, COP1 Δ300–317) in the presence of CRM1 and found that, as predicted, they were more refractory to CRM1‐mediated nuclear export than COP1 4A and COP1 W517A (Fig EV6D–F). Collectively, these findings show that the PSL/WD40 interaction modulates COP1 responsiveness to CRM1.
Coevolution of Tribbles and the COP1 pseudosubstrate latch
The TRIB1‐dependent control of the nuclear localization of human COP1 that we describe here is mechanistically very different from the light‐exerted control of COP1 described in plants. COP1 is a highly conserved protein with homologs present in most animal and plant species, as well as some fungi. In contrast, Tribbles homologs have only been identified thus far in metazoans (Eyers et al, 2016). This evolutionary divergence led us to wonder whether appearance of the PSL site in COP1 coincides with appearance of Tribbles in evolution.
To ascertain the degree of conservation of the PSL in COP1, sequences of COP1 homologs from 100 diverse species were aligned and used to construct a phylogenetic tree (Fig 6). The sequence of each of these homologs was examined for the presence of a putative PSL, based on two criteria: (i) The site had to be present between the coiled coil and WD40 domains and (ii) had to contain the VP dipeptide characteristic of the COP1‐binding motif. We then determined whether each of the species examined also possesses at least one Tribbles homolog, as well as whether these Tribbles homologs contain a COP1‐binding motif in their C‐terminal tail.
Figure 6. Coevolution of the COP1 pseudosubstrate latch and Tribbles.
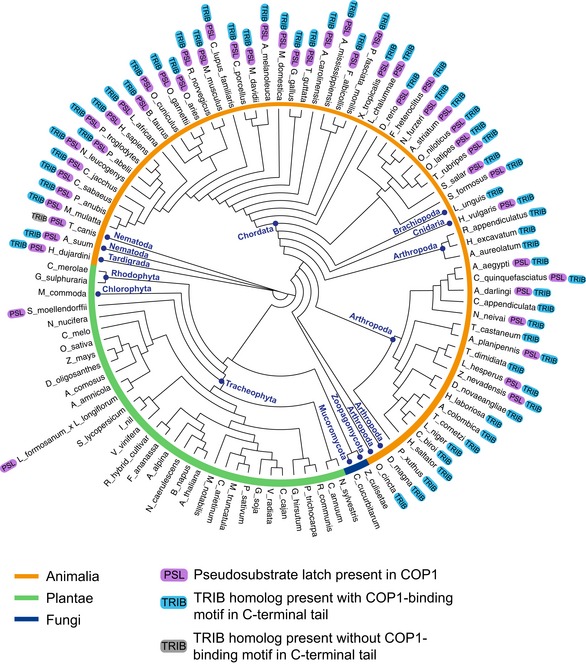
Phylogenetic tree of COP1 homologs from 100 species showing the presence of the pseudosubstrate latch (PSL) in COP1 and the presence of Tribbles homologs in the indicated species.
We found that, with only a few exceptions, the PSL in COP1 was present only in animal species that have at least one Tribbles homolog (Fig 6). In these species, Tribbles almost always had a COP1‐binding motif, a feature that is remarkably well conserved in Tribbles homologs. The coevolution of Tribbles and the PSL site in COP1 supports the hypothesis that they are engaged in a conserved regulatory interaction. Notably, the sequence alignment also revealed that, in species where the PSL is present, the sequence of the entire linker connecting the coiled coil and WD40 domains is more conserved than in species where the PSL is absent, emphasizing the importance of this region in regulation of COP1 function in species in which Tribbles is also present.
Several substrates of COP1, such as ETS transcription factors and c‐Jun, are also known to possess a COP1‐binding motif, and theoretically, they could utilize the mechanism described here for TRIB1 to promote COP1 nuclear localization. To determine whether the COP1‐binding motifs in these substrates also coevolved with the COP1 PSL, we examined whether each of the species included in the phylogenetic tree of COP1 sequences shown in Fig 6 possessed ETS or c‐Jun homologs that also have a COP1‐binding motif. Like Tribbles, ETS transcription factors and c‐Jun are highly conserved and possess a COP1‐binding motif in most metazoan species but are absent in plants (Appendix Table S1). This suggests that control of COP1 nuclear export by the PSL could be a broader regulatory mechanism adopted by not only Tribbles but also a wider range of COP1 substrates and binding partners.
Discussion
Regulation of COP1 activity through nucleocytoplasmic shuttling was first characterized for Arabidopsis COP1. In plants, which do not have any Tribbles homologs, this process is controlled by light and involves several photoreceptors, including CRY1, phyA, and phyB (Osterlund & Deng, 1998). Like mammalian COP1, Arabidopsis COP1 has a cytoplasmic localization signal within its coiled coil domain that is necessary for its nuclear export and resembles the leucine‐rich NESs recognized by CRM1 (Stacey et al, 1999; Subramanian et al, 2004). The export of mammalian COP1 is primarily regulated by association with CRM1, as well as by 14‐3‐3σ under conditions of cell stress in response to DNA damage (Yi et al, 2002; Su et al, 2010; Kırlı et al, 2015). In plants, binding partners, such as SPA proteins and CSN1, that have been reported to modulate COP1 localization, associate with the putative coiled coil domain of COP1 (Wang et al, 2009; Balcerowicz et al, 2017). For Arabidopsis COP1, control of COP1 subcellular localization seems to be exerted solely through interactions mediated by the putative coiled coil domain, and the WD40 domain is not essential for light‐regulated nucleocytoplasmic trafficking (Torii et al, 1998; Stacey et al, 1999). In stark contrast to Arabidopsis COP1, deletion of the WD40 domain prevents nuclear export of mammalian COP1, underscoring the importance of this domain for proper shuttling between the nucleus and cytoplasm (Yi et al, 2002; Bianchi et al, 2003).
In this study, we elucidate the mechanism by which the WD40 domain controls nucleocytoplasmic shuttling of mammalian COP1. We show that the WD40 domain engages in an intramolecular interaction that promotes nuclear export, and hence, its deletion results in nuclear enrichment of COP1. At this point, we can only speculate about the nature of the mechanism underlying this effect. The NES in human COP1 is located at the N‐terminus of the putative coiled coil domain, while the intramolecular WD40‐binding element, PSL, is located immediately following the C‐terminal end of the putative coiled coil domain and separated by a ~ 70 residue‐long linker from the WD40 domain. Binding of the WD40 domain to the PSL would effectively shorten the linker region and bring the WD40 domain in close proximity to the putative coiled coil domain. Our data are consistent with a model in which this binding event is coupled to a conformational rearrangement that would change the ability of the NES to interact with CRM1 and result in its “unmasking” when the PSL is bound to the WD40 domain (Fig 5D). Due to our lack of structural information for full‐length COP1, the nature of this conformational change remains unknown. Currently, structural information for COP1 is limited to the WD40 domain (Uljon et al, 2016). Structural studies of the putative coiled coil domain and, ideally, full‐length COP1 are needed to reveal the conformational changes induced by the PSL/WD40 interaction and by binding of TRIB1 to the WD40 domain, as well as how the accessibility of the COP1 NES is affected by these changes.
The location of the PSL in COP1 places this site in close proximity to the C‐terminal portion of the putative coiled coil domain that is required for engaging the adaptor protein DET1 (Wertz et al, 2004; Savio et al, 2008). Our finding that mutation of the PSL weakens the interaction of COP1 with DET1 suggests that binding of the PSL to the WD40 domain could be involved in regulation of DET1 binding and, consequently, the recruitment of the DDB1‐CUL4A‐RBX1 ubiquitination complex to COP1 (Wertz et al, 2004; Zhang et al, 2017). At the same time, this interaction of COP1 with DET1 and/or the DDB1‐CUL4A‐RBX1 complex might play a role in controlling the accessibility of the COP1 NES. Interestingly, we have observed that the COP1 WD40 mutants that disrupt the PSL/WD40 interaction, and in consequence, localize predominantly to the nucleus, exhibit higher expression levels than COP1 WT or the E642R mutants, which retain the ability to bind to the PSL and are more cytosolic. This correlation between nuclear localization of COP1 and its stability is consistent with previous work showing that, upon nuclear export, COP1 undergoes autoubiquitination (Dornan et al, 2006). Hence, retention of COP1 in the nucleus through disruption of the PSL/WD40 “lock” by Tribbles or other COP1 binding partners could play a role in potentiating COP1 activity by increasing its stability.
One of the major implications of our data is that, aside from TRIB1, other proteins containing a COP1‐binding motif that localize to the nucleus should also control nuclear export of COP1. Indeed, we found that TRIB3 also potentiated COP1 nuclear localization. While we did not observe the same effect for TRIB2, this is likely due to its propensity to localize to both the nucleus and cytoplasm in COS7 cells, as TRIB2 has been reported to increase COP1 nuclear localization in cells where it localizes primarily in the nucleus (Xu et al, 2014). Another Tribbles homolog, STK40 (SgK495), also interacts with COP1 through a COP1‐binding motif in its C‐terminal tail and thus has the potential to inhibit COP1 nuclear export (Durzynska et al, 2017). Several COP1 substrates, such as ETS family transcription factors and c‐Jun, are also known to bind directly to COP1 through a consensus COP1‐binding motif (Wertz et al, 2004; Vitari et al, 2011). Their COP1‐binding motifs engage the COP1 WD40 domain with affinities that are higher than those of the COP1 PSL (Uljon et al, 2016). Thus, these substrates, which by and large, are localized to the nucleus, have the potential to compete with the PSL and thereby promote COP1 nuclear localization. In further support of this general mechanism for regulation of COP1 nuclear export, our evolutionary analysis of Tribbles, ETS family members, and c‐Jun demonstrates a high degree of conservation of their COP1‐binding motifs and their coevolution with the PSL site in COP1.
Arabidopsis COP1 binding partners like HY5 and UVR8, which bind to the WD40 domain via a COP1‐binding motif similar to the one present in Tribbles, do not alter COP1 localization. Instead, Arabidopsis COP1, which spontaneously localizes to nuclear puncta denoted as nuclear speckles, alters the localization of its substrates and recruits them to the speckles (Ang et al, 1998; Favory et al, 2009). This speckle targeting has been speculated to regulate the function of Arabidopsis COP1 since loss of COP1 function correlates with loss of its localization to speckles (Stacey & Von Arnim, 1999). We found that mammalian COP1 localizes to similar punctate nuclear structures, which are positive for a PML body marker, only when coexpressed with TRIB1. Although it is not certain that the TRIB1/COP1 nuclear puncta we observe represent the same compartment occupied by Arabidopsis COP1, these observations indicate that subnuclear localization might be important for regulating the function of TRIB1 and COP1 in mammalian cells. Notably, we no longer observe TRIB1 and COP1 in nuclear puncta upon coexpression of C/EBPα, indicating that binding of C/EBPα changes subnuclear localization of the TRIB1/COP1 complex. This could potentially be due to competition between C/EBPα and unknown factors that organize TRIB1 and/or COP1 in punctate structures in PML bodies. Alternatively, a conformational change in the TRIB1 pseudokinase domain induced by C/EBPα binding (Jamieson et al, 2018) could alter the ability of TRIB1 to form interactions necessary for its localization to PML bodies.
Like other pseudokinases, Tribbles have evolved to perform non‐catalytic functions that often are unique, difficult to anticipate a priori, and challenging to understand mechanistically (Reiterer et al, 2014; Kung & Jura, 2016; Jacobsen & Murphy, 2017). Sometimes, these functions are heavily influenced by adjacent domains, contributing to the challenges in characterizing the molecular mechanisms by which pseudokinases signal. We found that the pseudokinase TRIB1 uses its C‐terminal tail to control the nuclear export of COP1, in addition to its role as a scaffold that facilitates COP1 interaction with its substrates, which is primarily orchestrated by the pseudokinase domain (Keeshan et al, 2010). It is unclear at present whether the pseudokinase domain contributes to regulation of COP1 nuclear export. The crystal structure of the TRIB1 pseudokinase domain and the C‐terminal tail, however, offers a potential mechanism for this regulation. In the structure, the TRIB1 C‐terminal tail binds to a pocket formed by the αC helix in the N‐lobe of the pseudokinase domain in a manner reminiscent of binding of a conserved hydrophobic motif to the PIF pocket in AGC kinases (Pearce et al, 2010; Murphy et al, 2015). Since the residues within the TRIB1 C‐terminal tail used to engage both the αC pocket in the TRIB1 pseudokinase domain and the COP1 WD40 domain partially overlap, these two interactions should be mutually exclusive. Thus, binding of the C‐terminal tail to the TRIB1 pseudokinase domain could serve as an autoinhibitory mechanism restricting COP1 binding. Indeed, we observed that the TRIB1 tail by itself potentiated COP1 nuclear localization to a greater extent than did full‐length TRIB1, indicating that the pseudokinase domain might have an inhibitory role in this process. The ability of the TRIB1 pseudokinase domain to directly bind its C‐terminal tail, which contains the COP1‐binding motif, also opens up the possibility that, upon binding of the TRIB1 tail to the COP1 WD40 domain and displacement of the COP1 PSL, the PSL site could in turn dock into the pocket in the pseudokinase domain where the TRIB1 tail binds. The length of the linker between the WD40 domain and PSL makes such an interaction possible and could serve as another stabilizing interaction within the COP1/TRIB1 complex.
Another regulatory step in COP1/TRIB1 binding could involve phosphorylation. It remains to be seen whether S310, located in the PSL, is a bona fide phosphorylation site. Our data suggest that phosphorylation of this site has the potential to regulate binding of COP1 to the TRIB1 C‐terminal tail. Phosphorylation of S387, which is located in the linker connecting the PSL to the WD40 domain, has previously been shown to promote nuclear export of COP1 (Dornan et al, 2006). This phosphorylation event catalyzed by the ATM kinase could regulate COP1 localization by modulating the PSL/WD40 interaction.
The differences in the nature of the nucleocytoplasmic shuttling mechanisms of COP1 in plants versus the one described here is remarkable given the high degree of structural similarity between plant and mammalian COP1. The net output of this regulation is modulation of the amount of COP1 in the nucleus and hence the efficiency with which it can degrade transcription factors. This process is likely to be important in human leukemias where upregulation of TRIB1 drives excessive COP1‐dependent degradation of transcription factors and might be important to consider in efforts toward pharmacological targeting of this pseudokinase.
Materials and Methods
Plasmids, cell culture, and antibodies
The human TRIB1 coding sequence was synthesized by GenScript. Human cDNAs for COP1, C/EBPα, and DET1 were purchased from transOMIC (BC094728, BC160133, BC109060). Constructs for each of these proteins were cloned into pcDNA3.1(+). 3xFLAG‐CRM1 was a gift from Xin Wang (Addgene plasmid #17647). GFP‐DAXX was a gift from Michael Rosen (UT Southwestern). Mutations and deletions were introduced using QuikChange Site‐Directed Mutagenesis Kit (Agilent). All constructs were verified by DNA sequencing. COS7, HEK293, Huh7, NIH3T3, HCT116, and U2OS cells were maintained in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. H1299 and C4‐2 cells were maintained in RPMI supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. U87MG cells were maintained in MEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Huh7 cells were obtained from Kevan Shokat (UCSF). HCT116 cells were obtained from Frank McCormick (UCSF). U2OS and U87MG cells were obtained from Jim Wells (UCSF). H1299 cells were obtained from Hani Goodarzi (UCSF). C4‐2 cells were obtained from Jason Gestwicki (UCSF). Transfections for COS7, HEK293, and Huh7 cells were carried out using Lipofectamine 3000 (Invitrogen), while those for NIH3T3 cells were performed using Lipofectamine LTX (Invitrogen). The following primary antibodies were used: anti‐FLAG (Sigma, F1804), anti‐COP1 (Bethyl, A300‐894A), anti‐COP1 (Abcam, ab56400), anti‐TRIB1 (Abcam, ab137717), anti‐GFP (Santa Cruz, sc‐9996), anti‐FLAG (Cell Signaling, 2368), anti‐HA (Sigma, H9658), anti‐HA (Santa Cruz, sc‐7392), anti‐C/EBPα (Cell Signaling, 2295), anti‐nucleolin (Invitrogen, 39‐6400), anti‐SC‐35 (Abcam, ab11826), anti‐coilin (Abcam, ab11822), anti‐TRIB2 (Sigma, WH0028951M4), anti‐TRIB3 (Abcam, ab75846), anti‐CRM1 (Santa Cruz, sc‐5595), anti‐Lamin A/C (Abcam, ab8984), anti‐GAPDH (Cell Signaling, 97166), anti‐β‐tubulin (Cell Signaling, 2128). For Western blotting and immunoprecipitation, the following secondary antibodies were used: anti‐mouse‐HRP (GE Healthcare, NA931), anti‐rabbit‐HRP (Cell Signaling, 7074), anti‐mouse IgG Veriblot (Abcam, ab131368), and Veriblot for IP secondary (ab131366). GFP‐tagged constructs were immunoprecipitated using GFP‐trap beads (ChromoTek). For immunofluorescence, anti‐mouse Alexa Fluor 405, anti‐mouse Alexa Fluor 568, anti‐mouse Alexa Fluor 647, and anti‐rabbit Alexa Fluor 680 secondary antibodies (Molecular Probes) were used.
Immunofluorescence
Cells were seeded onto glass coverslips in 6‐well plates and transfected with the indicated constructs. Approximately 24 h post‐transfection, cells were fixed in 3.7% formaldehyde, permeabilized in 0.1% Triton X‐100, and blocked with 1% BSA. Cells were then incubated with the indicated primary antibodies for 1 h at 37°C, washed with PBS, and incubated with the indicated secondary antibodies for 1 h at 37°C. Nuclei were visualized using DAPI where indicated. Coverslips were mounted onto glass slides using ProLong Anti‐Fade mounting medium (Thermo). Coverslips were imaged using a Nikon spinning disk confocal microscope.
To calculate nuclear/cytoplasmic ratios of COP1 fluorescence, confocal images were analyzed using FIJI (ImageJ). Binary image masks were created of DAPI‐positive staining to define nuclear regions of interest (ROI) for analysis. This was done by applying a Gaussian Blur filter (2 × 2 pixel radius) to reduce noise followed by automatic thresholding using the Otsu algorithm to convert the image into a binary mask that included all fluorescence above background. This mask was then used to calculate the average fluorescence intensity for COP1 in the nucleus of each cell. For COS7 and Huh7 cells, the average fluorescence intensity of COP1 was then measured in a cytoplasmic ROI of equal size to the corresponding nuclear ROI for each cell. For HEK293 and NIH3T3 cells, the cytoplasmic ROI was defined by manually tracing the boundaries of each cell. The average nuclear fluorescence intensity was then divided by the average cytoplasmic fluorescence intensity to determine the ratio of nuclear to cytoplasmic COP1 fluorescence in each cell. Cells exhibiting punctate and diffuse nuclear localization of COP1 were combined into one population when quantifying the nuclear/cytoplasmic ratio. Data presented are the means ± s.e.m. determined from at least three independent experiments. In each experiment, 50 cells per sample were randomly selected for analysis. Student t‐tests were used to compare samples. Statistical analyses were performed using GraphPad Prism 7 (GraphPad).
Immunoprecipitation and Western blotting
COS7 cells were seeded onto 60‐mm dishes at 3.5 × 105 cells/dish and transfected with the indicated constructs. Approximately 24 h post‐transfection, cells were lysed in IP lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP‐40, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4) supplemented with Roche complete mini EDTA‐free protease inhibitor cocktail. The lysates were centrifuged for 10 min at 21,000× g. The supernatants were then incubated with the indicated primary antibodies and Protein A Sepharose beads (Invitrogen) or with GFP‐trap beads (ChromoTek) overnight rotating at 4°C. Beads were washed three times with wash buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4) supplemented with protease inhibitors. Proteins were eluted using SDS–PAGE loading buffer, detected using standard Western blotting protocols with the indicated antibodies, and visualized using enhanced chemiluminescence (ECL) reagent (GE Healthcare, Amersham). To reduce detection of IgG bands, Veriblot secondary antibodies (Abcam) were used to detect immunoprecipitated proteins. For Western blotting of endogenous protein expression levels and for detection of exogenous protein expression in imaging experiments, cells were lysed in RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% NP‐40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO4) supplemented with Roche complete mini EDTA‐free protease inhibitor cocktail. Subcellular fractionation was performed using the NE‐PER kit (Thermo).
Protein purification
His‐tagged COP1 WD40 domain (residues 376–731) was expressed in Sf9 cells using the Bac‐to‐Bac expression system (Invitrogen) and purified with Talon beads (Clontech) followed by anion‐exchange chromatography on a Mono Q column (GE Healthcare). Purified proteins were then buffer exchanged into storage buffer (20 mM Tris pH 8.0, 250 mM NaCl, 5% glycerol, 2 mM TCEP), flash frozen, and stored at −80°C.
Fluorescence polarization
Fluorescence polarization assays were performed using purified COP1 WD40 domain in FP assay buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 2 mM TCEP, 0.1% Triton X‐100). FITC‐labeled and unlabeled peptides were synthesized by Elim Bio and purified to > 90% purity, as assessed by HPLC. 25 nM FITC‐TRIB1 tail (residues 338–366) was pre‐incubated with 1 μM COP1 WD40 domain before addition of indicated concentrations of the unlabeled COP1 PSL peptide (residues 302–320) or unlabeled TRIB1 tail peptide. Measurements were performed in duplicate in 20 μl well volumes in black 384‐well plates (Corning) on an Analyst AD plate reader (Molecular Devices) with excitation and emission wavelengths of 485 and 530 nm, respectively. Data presented are the means ± s.e.m. determined from three independent experiments.
| Peptide | Sequence |
|---|---|
| FITC‐TRIB1 tail | FITC‐DSEIGTSDQIVPEYQEDSD |
| Unlabeled TRIB1 tail | DSEIGTSDQIVPEYQEDSD |
| Unlabeled COP1 PSL | LYSPVSEDSTVPQFEAPSP |
| Unlabeled COP1 PSL 4A | LYSPVSAASTAAQFEAPSP |
Phylogenetic analysis
Sequences of COP1 homologs were collected from the UniProt and NCBI databases. These sequences were aligned using MUSCLE 3.8 (Edgar, 2004), and this alignment was used to generate a phylogenetic tree using the phylogeny software PhyML 3.0, which built the tree based on the maximum‐likelihood principle (Guindon et al, 2010). The tree was visualized using Dendroscope 3 (Huson & Scornavacca, 2012).
Author contributions
JEK performed all experiments. JEK and NJ designed the experiments, analyzed data, and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank all members of the Jura Lab for helpful discussions. J.E.K. was supported by a National Science Foundation Graduate Research Fellowship and a UCSF Discovery Fellowship.
The EMBO Journal (2019) 38: e99708
References
- Ang LH, Chattopadhyay S, Wei N, Oyama T, Okada K, Batschauer A, Deng XW (1998) Molecular interaction between COP1 and HY5 defines a regulatory switch for light control of Arabidopsis development. Mol Cell 1: 213–222 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcerowicz M, Kerner K, Schenkel C, Hoecker U (2017) SPA proteins affect the subcellular localization of COP1 in the COP1/SPA ubiquitin ligase complex during photomorphogenesis. Plant Physiol 174: 1314–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi E, Denti S, Catena R, Rossetti G, Polo S, Gasparian S, Putignano S, Rogge L, Pardi R (2003) Characterization of human constitutive photomorphogenesis protein 1, a RING finger ubiquitin ligase that interacts with Jun transcription factors and modulates their transcriptional activity. J Biol Chem 278: 19682–19690 [DOI] [PubMed] [Google Scholar]
- Das R, Sebo Z, Pence L, Dobens LL (2014) Drosophila Tribbles antagonizes insulin signaling‐mediated growth and metabolism via interactions with Akt kinase. PLoS One 9: e109530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedhia PH, Keeshan K, Uljon S, Xu L, Vega ME, Shestova O, Zaks‐Zilberman M, Romany C, Blacklow SC, Pear WS (2010) Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood 116: 1321–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng XW, Caspar T, Quail PH (1991) cop1: a regulatory locus involved in light‐controlled development and gene expression in Arabidopsis . Genes Dev 5: 1172–1182 [DOI] [PubMed] [Google Scholar]
- Dornan D, Bheddah S, Newton K, Ince W, Frantz GD, Dowd P, Koeppen H, Dixit VM, French DM (2004a) COP1, the negative regulator of p53, is overexpressed in breast and ovarian adenocarcinomas. Cancer Res 64: 7226–7230 [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004b) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Dornan D, Shimizu H, Mah A, Dudhela T, Eby M, O'Rourke K, Seshagiri S, Dixit VM (2006) ATM engages autodegradation of the E3 ubiquitin ligase COP1 after DNA damage. Science 313: 1122–1126 [DOI] [PubMed] [Google Scholar]
- Durzynska I, Xu X, Adelmant G, Ficarro SB, Marto JA, Sliz P, Uljon S, Blacklow SC (2017) STK40 is a pseudokinase that binds the E3 ubiquitin ligase COP1. Structure 25: 287–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers PA, Keeshan K, Kannan N (2016) Tribbles in the 21st century: the evolving roles of tribbles pseudokinases in biology and disease. Trends Cell Biol 27: 284–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favory J‐J, Stec A, Gruber H, Rizzini L, Oravecz A, Funk M, Albert A, Cloix C, Jenkins GI, Oakeley EJ, Seidlitz HK, Nagy F, Ulm R (2009) Interaction of COP1 and UVR8 regulates UV‐B‐induced photomorphogenesis and stress acclimation in Arabidopsis . EMBO J 28: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J‐F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321 [DOI] [PubMed] [Google Scholar]
- Holm M, Hardtke CS, Gaudet R, Deng XW (2001) Identification of a structural motif that confers specific interaction with the WD40 repeat domain of Arabidopsis COP1. EMBO J 20: 118–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Scornavacca C (2012) Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol 61: 1061–1067 [DOI] [PubMed] [Google Scholar]
- Jacobsen AV, Murphy JM (2017) The secret life of kinases: insights into non‐catalytic signalling functions from pseudokinases. Biochem Soc Trans 45: 665–681 [DOI] [PubMed] [Google Scholar]
- Jamieson SA, Ruan Z, Burgess AE, Curry JR, McMillan HD, Brewster JL, Dunbier AK, Axtman AD, Kannan N, Mace PD (2018) Substrate binding allosterically relieves autoinhibition of the pseudokinase TRIB1. Sci Signal 11: eaau0597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K, He Y, Wouters BJ, Shestova O, Xu L, Sai H, Rodriguez CG, Maillard I, Tobias JW, Valk P, Carroll M, Aster JC, Delwel R, Pear WS (2006) Tribbles homolog 2 inactivates C/EBPα and causes acute myelogenous leukemia. Cancer Cell 10: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeshan K, Bailis W, Dedhia PH, Vega ME, Shestova O, Xu L, Toscano K, Uljon SN, Blacklow SC, Pear WS (2010) Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood 116: 4948–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kırlı K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, Urlaub H, Görlich D (2015) A deep proteomics perspective on CRM1‐mediated nuclear export and nucleocytoplasmic partitioning. Elife 4: 1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss‐Toth E, Wyllie DH, Holland K, Marsden L, Jozsa V, Oxley KM, Polgar T, Qwarnstrom EE, Dower SK (2006) Functional mapping and identification of novel regulators for the Toll/Interleukin‐1 signalling network by transcription expression cloning. Cell Signal 18: 202–214 [DOI] [PubMed] [Google Scholar]
- Kung JE, Jura N (2016) Structural basis for the non‐catalytic functions of protein kinases. Structure 24: 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau OS, Deng XW (2012) The photomorphogenic repressors COP1 and DET1: 20 years later. Trends Plant Sci 17: 584–593 [DOI] [PubMed] [Google Scholar]
- Lee Y‐H, Andersen JB, Song H‐T, Judge AD, Seo D, Ishikawa T, Marquardt JU, Kitade M, Durkin ME, Raggi C, Woo HG, Conner EA, Avital I, Maclachlan I, Factor VM, Thorgeirsson SS (2010) Definition of ubiquitination modulator COP1 as a novel therapeutic target in human hepatocellular carcinoma. Cancer Res 70: 8264–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Gao Y, Qu L, Chen Z, Li J, Zhao H, Deng XW (2002) Genomic evidence for COP1 as a repressor of light‐regulated gene expression and development in Arabidopsis . Plant Cell 14: 2383–2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoner V, Das R, Pence L, Anand G, LaFerriere H, Zars T, Bouyain S, Dobens LL (2013) The kinase domain of Drosophila Tribbles is required for turnover of fly C/EBP during cell migration. Dev Biol 375: 33–44 [DOI] [PubMed] [Google Scholar]
- Migliorini D, Bogaerts S, Defever D, Vyas R, Denecker G, Radaelli E, Zwolinska A, Depaepe V, Hochepied T, Skarnes WC, Marine J‐C (2011) Cop1 constitutively regulates c‐Jun protein stability and functions as a tumor suppressor in mice. J Clin Invest 121: 1329–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Nakatani Y, Jamieson SA, Dai W, Lucet IS, Mace PD (2015) Molecular mechanism of CCAAT‐enhancer binding protein recruitment by the TRIB1 pseudokinase. Structure 23: 2111–2121 [DOI] [PubMed] [Google Scholar]
- Okamura H, Aramburu J, García‐Rodríguez C, Viola JP, Raghavan A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A (2000) Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell 6: 539–550 [DOI] [PubMed] [Google Scholar]
- Osterlund MT, Deng XW (1998) Multiple photoreceptors mediate the light‐induced reduction of GUS‐COP1 from Arabidopsis hypocotyl nuclei. Plant J 16: 201–208 [DOI] [PubMed] [Google Scholar]
- Pacín M, Legris M, Casal JJ (2014) Rapid decline in nuclear costitutive photomorphogenesis1 abundance anticipates the stabilization of its target elongated hypocotyl5 in the light. Plant Physiol 164: 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR (2010) The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11: 9–22 [DOI] [PubMed] [Google Scholar]
- Qi L, Heredia JE, Altarejos JY, Screaton R, Goebel N, Niessen S, Macleod IX, Liew CW, Kulkarni RN, Bain J, Newgard C, Nelson M, Evans RM, Yates J, Montminy M (2006) TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science 312: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Reiterer V, Eyers PA, Farhan H (2014) Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol 24: 489–505 [DOI] [PubMed] [Google Scholar]
- Satoh T, Kidoya H, Naito H, Yamamoto M, Takemura N, Nakagawa K, Yoshioka Y, Morii E, Takakura N, Takeuchi O, Akira S (2013) Critical role of Trib1 in differentiation of tissue‐resident M2‐like macrophages. Nature 495: 524–528 [DOI] [PubMed] [Google Scholar]
- Savio MG, Rotondo G, Maglie S, Rossetti G, Bender JR, Pardi R (2008) COP1D, an alternatively spliced constitutive photomorphogenic‐1 (COP1) product, stabilizes UV stress‐induced c‐Jun through inhibition of full‐length COP1. Oncogene 27: 2401–2411 [DOI] [PubMed] [Google Scholar]
- Seo HS, Yang J‐Y, Ishikawa M, Bolle C, Ballesteros ML, Chua N‐H (2003) LAF1 ubiquitination by COP1 controls photomorphogenesis and is stimulated by SPA1. Nature 423: 995–999 [DOI] [PubMed] [Google Scholar]
- Soubeyrand S, Martinuk A, Lau P, McPherson R (2016) TRIB1 is regulated post‐transcriptionally by proteasomal and non‐proteasomal pathways. PLoS One 11: e0152346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey MG, Hicks SN, Von Arnim AG (1999) Discrete domains mediate the light‐responsive nuclear and cytoplasmic localization of Arabidopsis COP1. Plant Cell 11: 349–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey MG, Von Arnim AG (1999) A novel motif mediates the targeting of the Arabidopsis COP1 protein to subnuclear foci. J Biol Chem 274: 27231–27236 [DOI] [PubMed] [Google Scholar]
- Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM (1999) A leucine‐rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 18: 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C‐H, Zhao R, Velazquez‐Torres G, Chen J, Gully C, Yeung S‐CJ, Lee M‐H (2010) Nuclear export regulation of COP1 by 14‐3‐3σ in response to DNA damage. Mol Cancer 9: 243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C‐H, Zhao R, Zhang F, Qu C, Chen B, Feng Y‐H, Phan L, Chen J, Wang H, Wang H, Yeung S‐CJ, Lee M‐H (2011) 14‐3‐3sigma exerts tumor‐suppressor activity mediated by regulation of COP1 stability. Cancer Res 71: 884–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian C, Kim B‐H, Lyssenko NN, Xu X, Johnson CH, von Arnim AG (2004) The Arabidopsis repressor of light signaling, COP1, is regulated by nuclear exclusion: mutational analysis by bioluminescence resonance energy transfer. Proc Natl Acad Sci USA 101: 6798–6802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii KU, McNellis TW, Deng XW (1998) Functional dissection of Arabidopsis COP1 reveals specific roles of its three structural modules in light control of seedling development. EMBO J 17: 5577–5587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uljon S, Xu X, Durzynska I, Stein S, Adelmant G, Marto JA, Pear WS, Blacklow SC (2016) Structural basis for substrate selectivity of the E3 ligase COP1. Structure 24: 687–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Leong KG, Newton K, Yee C, O'Rourke K, Liu J, Phu L, Vij R, Ferrando R, Couto SS, Mohan S, Pandita A, Hongo J‐A, Arnott D, Wertz IE, Gao W‐Q, French DM, Dixit VM (2011) COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature 474: 403–406 [DOI] [PubMed] [Google Scholar]
- Von Arnim AG, Deng XW (1994) Light inactivation of Arabidopsis photomorphogenic repressor COP1 involves a cell‐specific regulation of its nucleocytoplasmic partitioning. Cell 79: 1035–1045 [DOI] [PubMed] [Google Scholar]
- Wang H, Ma LG, Li JM, Zhao HY, Deng XW (2001) Direct interaction of Arabidopsis cryptochromes with COP1 in light control development. Science 294: 154–158 [DOI] [PubMed] [Google Scholar]
- Wang X, Li W, Piqueras R, Cao K, Deng XW, Wei N (2009) Regulation of COP1 nuclear localization by the COP9 signalosome via direct interaction with CSN1. Plant J 58: 655–667 [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, Dixit VM (2004) Human De‐etiolated‐1 regulates c‐Jun by assembling a CUL4A ubiquitin ligase. Science 303: 1371–1374 [DOI] [PubMed] [Google Scholar]
- Xu S, Tong M, Huang J, Zhang Y, Qiao Y, Weng W, Liu W, Wang J, Sun F (2014) TRIB2 inhibits Wnt/β‐Catenin/TCF4 signaling through its associated ubiquitin E3 ligases, β‐TrCP, COP1 and Smurf1, in liver cancer cells. FEBS Lett 588: 4334–4341 [DOI] [PubMed] [Google Scholar]
- Yi C, Wang H, Wei N, Deng XW (2002) An initial biochemical and cell biological characterization of the mammalian homologue of a central plant developmental switch, COP1. BMC Cell Biol 3: 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kato J‐Y, Nakamae I, Yoneda‐Kato N (2013) COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1. Blood 122: 1750–1760 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Newton K, Kummerfeld SK, Webster J, Kirkpatrick DS, Phu L, Eastham‐Anderson J, Liu J, Lee WP, Wu J, Li H, Junttila MR, Dixit VM (2017) Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc Natl Acad Sci USA 114: 3903–3908 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File