

Abstract
Chromosome segregation requires the centromere, the site on chromosomes where kinetochores assemble in mitosis to attach chromosomes to the mitotic spindle. Centromere identity is defined epigenetically by the presence of nucleosomes containing the histone H3 variant CENP‐A. New CENP‐A nucleosome assembly occurs at the centromere every cell cycle during G1, but how CENP‐A nucleosome assembly is spatially and temporally restricted remains poorly understood. Centromere recruitment of factors required for CENP‐A assembly is mediated in part by the three‐protein Mis18 complex (Mis18α, Mis18β, M18BP1). Here, we show that Xenopus M18BP1 localizes to centromeres during metaphase—prior to CENP‐A assembly—by binding to CENP‐C using a highly conserved SANTA domain. We find that Cdk phosphorylation of M18BP1 is necessary for M18BP1 to bind CENP‐C and localize to centromeres in metaphase. Surprisingly, mutations which disrupt the metaphase M18BP1/CENP‐C interaction cause defective nuclear localization of M18BP1 in interphase, resulting in defective CENP‐A nucleosome assembly. We propose that M18BP1 may identify centromeric sites in metaphase for subsequent CENP‐A nucleosome assembly in interphase.
Keywords: CENP‐A, CENP‐C, centromere, epigenetics
Subject Categories: Cell Cycle; Chromatin, Epigenetics, Genomics & Functional Genomics
Introduction
During cell division, eukaryotes segregate their genomes by attaching chromosomes to the microtubules of the mitotic spindle through the kinetochore (Westhorpe & Straight, 2015). Kinetochores assemble in mitosis on a specialized region of the chromosome termed the centromere (Musacchio & Desai, 2017). Mutations that disrupt the functions of centromere and kinetochore proteins cause chromosome missegregation, genome instability, and cell death (Stoler et al, 1995; Fachinetti et al, 2013). Centromeres are distinguished by the incorporation of a histone H3 variant termed CENP‐A (CENtromere Protein A) into centromeric nucleosomes (Palmer et al, 1987; Yoda et al, 2000; Van Hooser et al, 2001; Guse et al, 2011; Mendiburo et al, 2011). CENP‐A nucleosomes epigenetically determine the function of centromeres, and loss of CENP‐A results in defective kinetochore assembly and improper chromosome segregation (Karpen & Allshire, 1997; McKinley & Cheeseman, 2016). Thus, a central question in chromosome segregation is how cells selectively maintain CENP‐A chromatin at the centromere.
CENP‐A nucleosomes are equally distributed to each sister chromatid during DNA replication (Jansen et al, 2007). To prevent the replication‐coupled dilution of CENP‐A chromatin, new CENP‐A nucleosomes are assembled once per cell cycle in vertebrates during G1. New CENP‐A assembly is mediated by HJURP (Holliday junction‐recognizing protein), the histone chaperone that binds soluble CENP‐A/H4 dimers (Dunleavy et al, 2009; Foltz et al, 2009). HJURP localization is sufficient to promote local CENP‐A deposition so its localization must be restricted to centromeres (Barnhart et al, 2011; Shono et al, 2015; French et al, 2017). Centromere‐specific HJURP targeting requires a CENP‐A nucleosome‐binding protein, CENP‐C (Carroll et al, 2010; Kato et al, 2013), and a three‐protein complex termed the Mis18 complex (Barnhart et al, 2011; Moree et al, 2011; Tachiwana et al, 2015; Nardi et al, 2016; French et al, 2017; Pan et al, 2017; Spiller et al, 2017). The Mis18 complex recognizes centromeric chromatin either directly by binding to CENP‐A nucleosomes (Kral, 2015; French et al, 2017; Hori et al, 2017; Sandmann et al, 2017) or indirectly by binding CCAN components (Moree et al, 2011; Dambacher et al, 2012; Shono et al, 2015; Stellfox Madison et al, 2016). Together, CENP‐C and the Mis18 complex restrict HJURP localization to the pre‐existing centromere.
The process of CENP‐A assembly is restricted to G1 in vertebrates in part by Cdk1 activity (Jansen et al, 2007; Bernad et al, 2011; Moree et al, 2011; Silva et al, 2012). When Cdk activity is inhibited, HJURP and the Mis18 complex localize to centromeres prior to G1 resulting in CENP‐A assembly during G2 or even S phase (Silva et al, 2012; Muller et al, 2014). Cdk phosphorylation of HJURP inhibits HJURP binding to Mis18β, preventing its localization (Muller et al, 2014; Wang et al, 2014; Stankovic et al, 2017). Cdk phosphorylation of M18BP1 also inhibits M18BP1 binding to Mis18α/Mis18β and prevents M18BP1 centromere localization in human cells (Silva et al, 2012; McKinley & Cheeseman, 2014; Pan et al, 2017; Spiller et al, 2017; Stankovic et al, 2017). In addition, Cdk1 phosphorylation of CENP‐A has been proposed to restrict CENP‐A assembly by inhibiting CENP‐A association with the HJURP chaperone, though conflicting data for this model exist (Hu et al, 2011; Yu et al, 2015; Fachinetti et al, 2017).
While the Mis18 complex plays a clear role in localizing HJURP to centromeres in G1, whether it has additional roles in regulating CENP‐A assembly remains unclear. Notably, M18BP1/KNL2 localizes throughout the cell cycle in Xenopus (Moree et al, 2011), Caenorhabditis elegans (Maddox et al, 2007), and chicken (Perpelescu et al, 2015; Hori et al, 2017), including all stages of mitosis. Metaphase M18BP1 localization has also been reported in human (McKinley & Cheeseman, 2014). The Mis18 complex has been proposed to “prime” centromeric chromatin for CENP‐A assembly, perhaps by regulating centromeric histone acetylation (Hayashi et al, 2004; Fujita et al, 2007; Kim et al, 2012; Ohzeki et al, 2012, 2016; Shang et al, 2016). However, directly tethering HJURP to chromatin is sufficient to drive CENP‐A assembly even in the absence of the Mis18 complex (Barnhart et al, 2011; French et al, 2017), suggesting that the primary role of the Mis18 complex in CENP‐A assembly is HJURP localization in G1.
To understand how M18BP1 controls CENP‐A assembly, we identified the requirements for M18BP1 localization to metaphase and interphase centromeres using a cell‐free CENP‐A assembly system in Xenopus laevis egg extracts. We previously demonstrated that M18BP1 localization to metaphase centromeres in Xenopus requires CENP‐C (Moree et al, 2011). CENP‐C binding by the Mis18 complex is conserved in human, Xenopus, and mouse (Moree et al, 2011; Dambacher et al, 2012; Stellfox Madison et al, 2016). Here, we show that the interaction between M18BP1 and CENP‐C is mediated by the highly conserved SANTA domain in M18BP1. Their interaction requires M18BP1 phosphorylation at threonine 166, a Cdk site, and is therefore restricted to mitosis. Surprisingly, mutations that disrupt the interaction between M18BP1 and CENP‐C not only prevent proper metaphase localization, but also interphase localization. This results in part from defective nuclear localization. We propose that M18BP1 may identify centromeric sites in metaphase for subsequent CENP‐A nucleosome assembly in interphase.
Results
Metaphase localization of M18BP1 requires the conserved SANTA domain
Xenopus laevis expresses two M18BP1 isoforms that are both capable of supporting interphase CENP‐A assembly but differ in the timing of their localization to centromeres (Moree et al, 2011; Session et al, 2016). We confirmed that Myc‐tagged versions of M18BP1‐1 and M18BP1‐2, made by in vitro translation and added to Xenopus egg extract, recapitulated this localization to sperm chromatin. Consistent with previous results, we found that M18BP1‐1 localized to metaphase centromeres while M18BP1‐2 was only weakly detectable (Moree et al, 2011; Fig EV1A).
Figure EV1. Comparison of M18BP1‐1 and M18BP1‐2 metaphase localization.
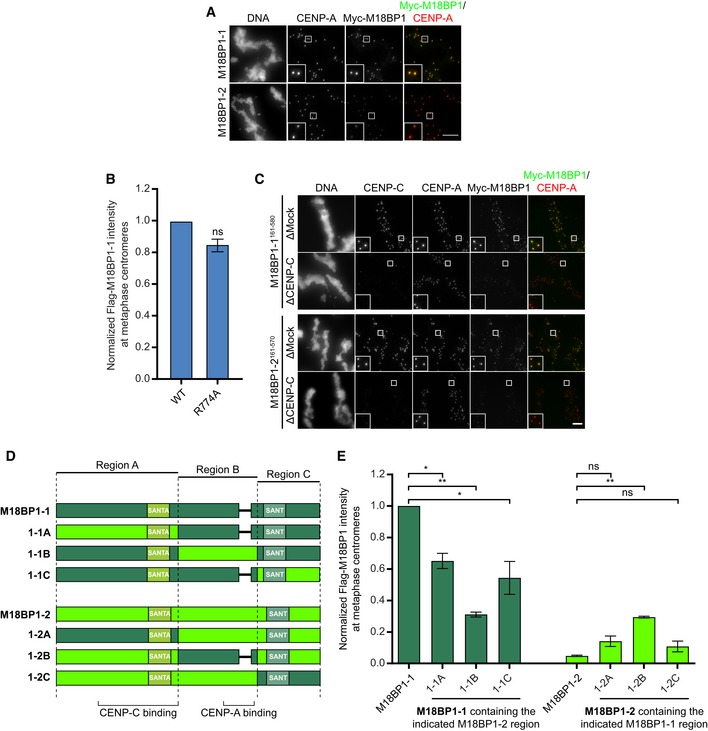
- Full‐length M18BP1‐1 and M18BP1‐2 differ in their metaphase localization. Immunofluorescence images of M18BP1 isoform localization to metaphase sperm centromeres. The M18BP1 isoform is indicated to the left; immunolocalized protein is indicated above. Myc‐tagged, in vitro translated M18BP1‐1 or M18BP1‐2 was incubated with sperm chromatin in metaphase‐arrested Xenopus egg extract depleted of endogenous M18BP1. M18BP1‐1 localizes robustly to metaphase centromeres whereas M18BP1‐2 was only weakly detectable. This image represents the maximum amount of metaphase M18BP1‐2 localization observed. Scale bar, 10 μm. Insets are magnified 3×.
- CENP‐A nucleosome binding‐deficient mutant of M18BP1‐1 remains at metaphase centromeres. Quantification of immunofluorescence intensity of full‐length Flag‐M18BP1‐1 (mutant species indicated at bottom) at metaphase centromeres normalized to WT. R774A shows 85 ± 2% of WT localization. Error bars represent SEM from two independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test.
- M18BP1‐2161–570 exhibits CENP‐C‐dependent localization to metaphase centromeres similar to M18BP1‐1161–580. Representative immunofluorescence images showing M18BP1 truncation localization (indicated at left) in metaphase extract depleted of endogenous M18BP1. In addition, extract was mock‐depleted or depleted of CENP‐C as indicated at left. Immunolocalized protein indicated above. Scale bar, 10 μm. Insets are magnified 3×. M18BP1‐1 images are the same as those in Fig 3E.
- Schematic showing M18BP1 chimeras in which the indicated domains from M18BP1‐1 (dark green) were substituted into M18BP1‐2 (light green) and vice versa for assessing their role in metaphase centromere localization.
- Quantification of metaphase centromere localization of M18BP1 chimeras in (D). M18BP1 species indicated at bottom. Graph shows mean centromere intensity normalized to M18BP1‐1WT. Error bars represent SEM from three independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005.
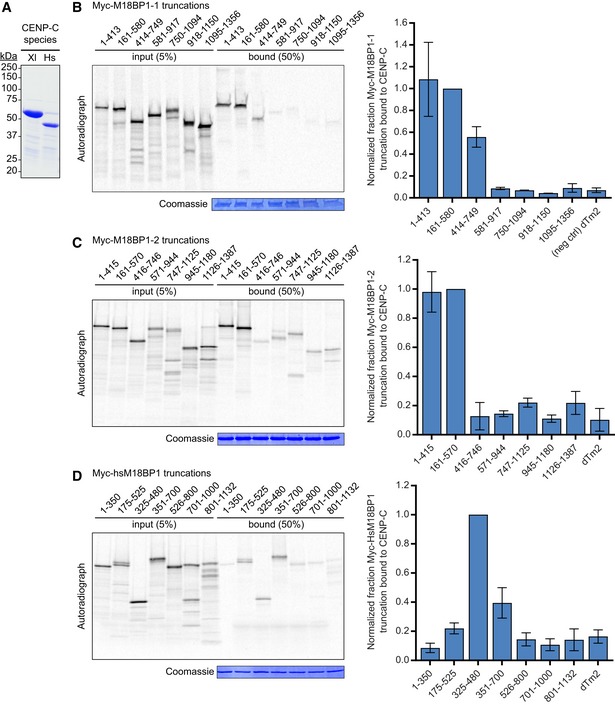
To determine which regions of the M18BP1‐1 protein were important for metaphase localization, we measured the extent of localization of Myc‐tagged M18BP1‐1 truncations at sperm centromeres. In metaphase extracts depleted of both M18BP1 isoforms, a truncated version of M18BP1‐1 containing only the first 580 amino acids (M18BP1‐11–580) was sufficient for centromeric localization. This result demonstrates that residues 581–1356 of M18BP1‐1—which contain both the CENP‐A nucleosome‐binding domain (French et al, 2017) and the DNA‐binding SANT/Myb domain (Maddox et al, 2007)—are dispensable for metaphase localization (Fig 1A–C). Consistent with this, mutation of the CENP‐A nucleosome‐binding domain in M18BP1‐1 had little effect on metaphase centromere localization (Fig EV1B). A fragment of M18BP1‐1 containing amino acids 161–580 retained 67 ± 4% of the protein at centromeres compared to the wild‐type protein (Fig 1A–C); however, further truncation of this region abolished metaphase localization (Fig 1A–C). Thus, M18BP1‐1161–580 is capable of localizing to metaphase centromeres.
Figure 1. M18BP1‐1161–580 is sufficient for centromere localization in metaphase egg extract.
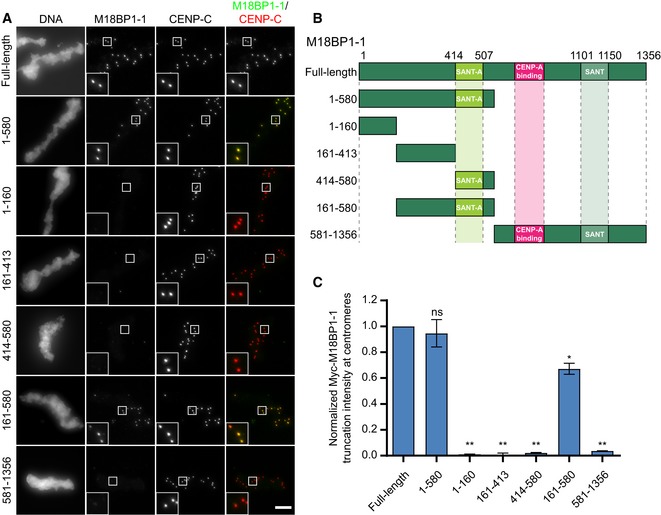
- Representative immunofluorescence images showing localization of Myc‐tagged M18BP1‐1 truncations schematized in (B) to metaphase sperm centromeres in M18BP1‐depleted extract. Truncations analyzed are indicated on the left; immunolocalized proteins are indicated above. Scale bar, 10 μm. Insets are magnified 3×.
- Schematic of M18BP1‐1 truncations used to define the metaphase targeting domain in (A) and (C).
- Quantification of the average Myc intensity at centromeres from (A). Values are normalized to centromeric signal of full‐length M18BP1‐1. Graph shows the mean ± SEM of at least three experiments. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005.
We hypothesized that amino acid differences between M18BP1‐1 and M18BP1‐2 within this region (161–580 in M18BP1‐1; 161–570 in M18BP1‐2) would account for their differential localization to metaphase centromeres and that mutagenesis of these sites might specifically prevent metaphase M18BP1‐1 localization. To test this, we generated chimeric M18BP1 proteins in which regions from one isoform encompassing the CENP‐C binding domain, the CENP‐A nucleosome‐binding domain, or the C terminus were exchanged for the corresponding regions of the other isoform (Fig EV1D). We then assayed localization of these chimeras to metaphase centromeres in M18BP1‐depleted extract. While substitution of M18BP1‐21–570 into M18BP1‐1 partially reduced its localization to metaphase centromeres, substitution of M18BP1‐11–580 into M18BP1‐2 did not confer appreciable metaphase centromeres localization, suggesting that amino acid differences in this region do not strictly control metaphase localization (Fig EV1E). A truncation of M18BP1‐2 comprising amino acids 161–570 was able to localize to metaphase centromeres at levels comparable to M18BP1‐1161–580 and in a CENP‐C‐dependent manner (Fig EV1C). Thus, metaphase localization of M18BP1‐2 may be inhibited by interactions with other regions of the protein outside of 161–570.
We next turned our attention toward conserved features of M18BP1‐1 that might mediate metaphase localization. M18BP1‐1161–580 contains a highly conserved SANTA (SANT‐Associated) domain (Fig 1B), named for its co‐occurrence with DNA‐binding SANT/Myb domains. The SANTA domain contains five highly conserved hydrophobic residues (Fig 2A; Zhang et al, 2006). We previously showed that mutation of these residues to alanine in the SANTA domain of M18BP1‐2 reduced its localization to interphase centromeres by > 90% (French et al, 2017). We tested whether mutation of the SANTA domain in full‐length M18BP1‐1 also affected metaphase localization and found that M18BP1‐1L416A, W419A, F491A, F495A, W499A (M18BP1‐1SANTA) showed a 45 ± 5% reduction in metaphase localization (Fig 2C and D) indicating that the SANTA domain is required for normal levels of metaphase M18BP1 localization.
Figure 2. Mutation of the conserved SANTA domain disrupts M18BP1‐1 binding to CENP‐C and metaphase centromere localization.
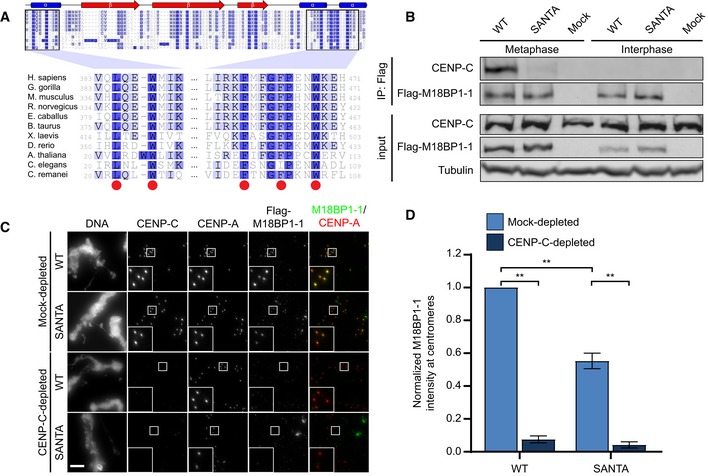
- Alignment of SANTA domain among select eukaryotes. Red circles indicate conserved hydrophobic residues predicted to play a role in protein–protein interactions that were mutated in this study (Zhang et al, 2006). Darker shades of blue represent increased conservation of amino acids in alignment of ˜300 M18BP1 homologues. Secondary structure prediction was reported by Zhang et al (2006).
- Representative Western blot showing disruption of the interaction between M18BP1‐1 and CENP‐C by mutation of the SANTA domain by co‐immunoprecipitation. Extract depleted of endogenous M18BP1 was supplemented with full‐length Flag‐M18BP1‐1WT or Flag‐M18BP1‐1SANTA. The M18BP1‐1 species added to each reaction, and the cell cycle state of the extract is indicated above. The immunoblotted species is indicated at left. Mock precipitations using rabbit reticulocyte lysate without any in vitro translated protein served as a negative control.
- Representative images showing that mutation of the SANTA domain in M18BP1‐1 causes decreased localization to sperm centromeres. Metaphase extracts were depleted of endogenous M18BP1 and either mock‐depleted or depleted of CENP‐C. The extracts were then complemented with WT or SANTA mutant M18BP1‐1. M18BP1‐1 species and depletion conditions are indicated at left; immunolocalized proteins are indicated above. Scale bar, 10 μm. Insets are magnified 3×.
- Quantification of the average Flag‐M18BP1‐1 intensity at centromeres from (C). Values are normalized to centromeric signal of M18BP1‐1WT in mock‐depleted extract. Graph shows the mean ± SEM of three experiments. Significance determined by Welch's unpaired two‐tailed t‐test, **P < 0.005.
Xenopus M18BP1‐1 directly interacts with CENP‐C, and immunodepletion of CENP‐C from egg extract prevents metaphase M18BP1‐1 localization (Moree et al, 2011). We used co‐immunoprecipitation to test whether the reduction in centromere localization of the M18BP1‐1SANTA mutant was due to a disruption of the interaction between M18BP1‐1 and CENP‐C. We added in vitro translated M18BP1‐1WT or M18BP1‐1SANTA to metaphase Xenopus extracts depleted of endogenous M18BP1. While M18BP1‐1WT co‐precipitated CENP‐C from metaphase extract, M18BP1‐1SANTA precipitated little CENP‐C (Fig 2B) suggesting that the SANTA mutant disrupts CENP‐C binding.
We predicted that if metaphase M18BP1 localization were solely mediated by interaction of the SANTA domain with CENP‐C, then M18BP1‐1SANTA localization would not be further reduced following CENP‐C depletion. To test the role of CENP‐C in localizing M18BP1, we depleted CENP‐C from Xenopus egg extracts and measured the localization of M18BP1‐1WT and M18BP1‐1SANTA at centromeres. CENP‐C depletion reduced both M18BP1‐1WT and M18BP1‐1SANTA localization to 8 ± 2 and 4 ± 2% of mock‐depleted levels, respectively (Fig 2C and D). Given that the M18BP1‐1SANTA mutation reduced but did not prevent co‐immunoprecipitation of CENP‐C (Fig 2B), there likely exist additional points of interaction between M18BP1 and CENP‐C or unidentified factors that are depleted with CENP‐C and required for M18BP1 localization.
Amino acids 161–580 comprise the CENP‐C binding domain of M18BP1‐1
To better characterize the regions of M18BP1 required for CENP‐C binding in metaphase, we purified GST‐CENP‐C1191–1400, the previously identified M18BP1‐binding domain (Moree et al, 2011), from Escherichia coli and performed GST pulldowns with a series of M18BP1‐1 truncations expressed by in vitro translation (Figs 3A–C and EV2B). M18BP1‐11–413 and M18BP1‐1414–749, two adjacent but non‐overlapping truncations, bound to CENP‐C, suggesting that portions of each contribute to the interaction between M18BP1 and CENP‐C (Fig 3A–C). M18BP1‐1161–580, which spans regions of M18BP1‐11–413 and M18BP1‐1414–749, bound CENP‐C in vitro (Fig 3A–C). We found that the homologous regions in Xenopus M18BP1‐2 (M18BP1‐2161–570) and in human M18BP1 (HsM18BP1325–480) also bound CENP‐C in vitro (Fig EV2A–D). In contrast with a previous report in mouse (Dambacher et al, 2012), we found that the SANT/Myb domain was not required for CENP‐C binding (Figs 3A–C, and EV2C and D).
Figure 3. M18BP1‐1161–580 recapitulates metaphase‐specific binding to CENP‐C.
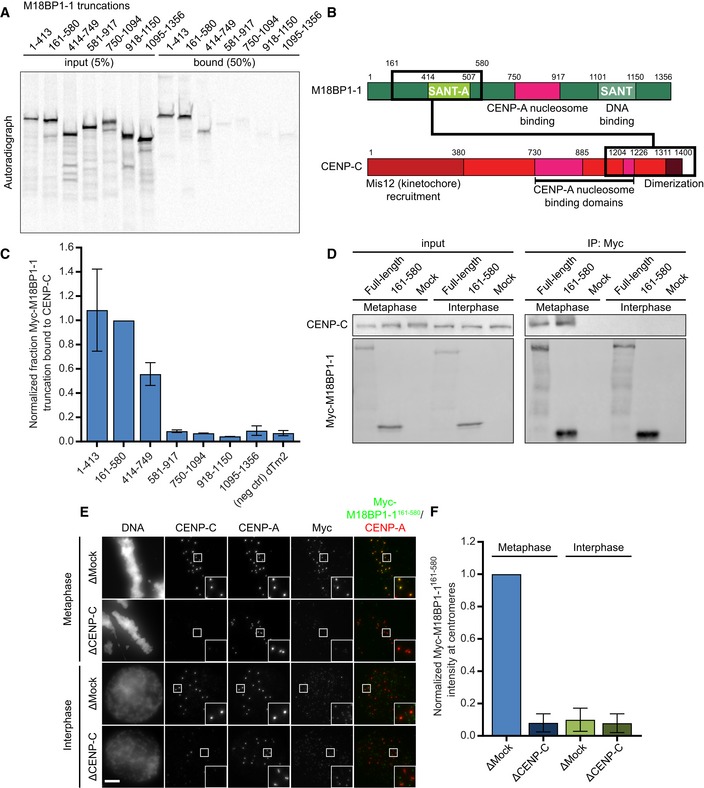
- A representative autoradiograph from a GST‐pulldown of CENP‐C to map the CENP‐C binding domain of M18BP1‐1. Radiolabeled Myc‐M18BP1‐1 truncations (amino acids indicated at the top) were mixed with recombinant GST‐CENP‐C1191–1400. Material bound to glutathione agarose was resolved by SDS–PAGE and visualized by autoradiography (see also Fig EV2).
- Schematic showing the cognate binding domains on M18BP1‐1 (top) and CENP‐C (bottom), indicated by the black boxes, relative to other functional domains.
- Quantification of (A). Bound material as a fraction of the input was calculated from autoradiographs. The graph shows mean fraction bound ± SD of three independent experiments normalized to M18BP1‐1161–580, the CENP‐C binding domain.
- Representative Western blot showing metaphase‐specific binding of M18BP1‐1161–580 to CENP‐C by co‐immunoprecipitation. Extract depleted of endogenous M18BP1 was supplemented with full‐length Myc‐M18BP1‐1 or Myc‐M18BP1‐1161–580. M18BP1‐1 species added to each reaction and cell cycle state of the extract is indicated above. Immunoblotted species indicated at left. Mock precipitations using rabbit reticulocyte lysate without any in vitro translated protein served as a negative control.
- Representative immunofluorescence images showing Myc‐M18BP1‐1161–580 localization at sperm centromeres in extract depleted of endogenous M18BP1. In addition, extract was mock‐depleted or immunodepleted of CENP‐C (indicated at left). Cell cycle state indicated at left, immunolocalized protein indicated above. Scale bar, 10 μm. Insets magnified 3×.
- Quantification of (E). Graph shows mean Myc‐M18BP1‐1161–580 intensity at centromeres ± SEM of two independent experiments normalized to the mock‐depleted, metaphase condition.
Figure EV2. In vitro identification of the CENP‐C binding domain in M18BP1‐1, M18BP1‐2, and human M18BP1.
- Coomassie‐stained gel showing the purity of the GST‐xlCENP‐C1191–1400 and GST‐hsCENP‐C723–943 proteins used for in vitro binding assays.
- Reproduction of the representative GST‐pulldown data from Fig 3 which includes the Coomassie stain showing equivalent recovery of GST‐CENP‐C1191–1400 for all truncations analyzed (left). Bound material as a fraction of the input was calculated from autoradiographs. The graph shows mean fraction bound ± SD of three independent experiments normalized to M18BP1‐1161–580, the CENP‐C binding domain of M18BP1‐1.
- Gel and quantification of GST‐pulldown to map the CENP‐C binding domain of M18BP1‐2. Myc‐M18BP1‐2 truncations (amino acids indicated at the bottom) were translated in reticulocyte lysate in the presence of [35S]‐methionine and mixed with recombinant GST‐CENP‐C1191–1400. Material bound to glutathione agarose was resolved by SDS–PAGE and visualized by autoradiography to assess binding of M18BP1‐2 truncations. Bound material was quantified as in Fig 3B and normalized to M18BP1‐2161–570, the CENP‐C binding domain of M18BP1‐2. Notably, M18BP1‐2161–415 was not sufficient to bind CENP‐C (data not shown). Error bars represent SD of two independent experiments.
- Gel and quantification of GST‐pulldown to map the CENP‐C binding domain of human M18BP1. Pulldowns were performed as in (C), except radiolabeled truncations were mixed with recombinant GST‐hCENP‐C723–943, the M18BP1‐binding domain on human CENP‐C. Data are normalized to M18BP1325–480, the CENP‐C binding domain on human M18BP1. Error bars represent SD of three independent experiments.
M18BP1‐1 binds directly to CENP‐C in metaphase, but not in interphase (Moree et al, 2011). To test whether M18BP1‐1161–580 retained the cell cycle‐dependent interaction with CENP‐C in Xenopus egg extract, we added M18BP1‐1161–580 to metaphase and interphase extract, immunoprecipitated the protein, and assayed CENP‐C co‐precipitation by Western blotting. Similar to full‐length M18BP1‐1, we found that M18BP1‐1161–580 co‐precipitates CENP‐C only from metaphase egg extract (Fig 3D). We next assayed the localization of M18BP1‐1161–580 to sperm centromeres in egg extract and found that M18BP1‐1161–580 associates only with metaphase centromeres. Immunodepletion of CENP‐C from these extracts prevented the localization of M18BP1‐1161–580 to the centromere (Fig 3E and F). Thus, M18BP1‐1161–580 binds directly to CENP‐C, co‐precipitates CENP‐C in metaphase, and localizes to metaphase centromeres dependent on the presence of CENP‐C.
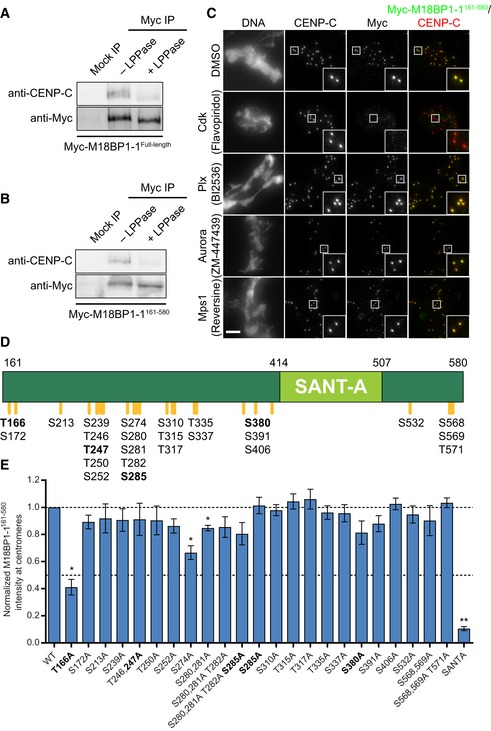
CDK phosphorylation of threonine 166 is required for metaphase M18BP1‐1 localization and CENP‐C binding
During the transition from metaphase to interphase, M18BP1 switches from binding directly to CENP‐C for metaphase centromere localization to binding directly to CENP‐A nucleosomes for interphase centromere localization (Moree et al, 2011; French et al, 2017). We tested whether the interaction between M18BP1 and CENP‐C might be regulated by a shift in mitotic kinase activity as cells exit metaphase. We immunoprecipitated either full‐length M18BP1‐1 or M18BP1‐1161–580 from metaphase extract and treated the immunoprecipitate with λ‐phosphatase. Dephosphorylation by λ‐phosphatase caused dissociation of CENP‐C from both full‐length M18BP1‐1 and M18BP1‐1161–580 precipitates (Fig 4A and B). This suggests that phosphorylation is required to maintain the interaction between M18BP1 and CENP‐C and that regulation of this interaction is preserved in M18BP1‐1161–580.
Figure 4. Identification of metaphase phosphorylation sites in M18BP1‐1 by mass spectrometry.
-
A, BMetaphase binding of M18BP1‐1 to CENP‐C requires phosphorylation. Either full‐length Myc‐M18BP1‐1 (A) or Myc‐M18BP1‐1161–580 (B) was immunoprecipitated from metaphase extract. Half of the immunoprecipitate was treated with λ‐protein phosphatase (+ LPPase) to assess whether binding to CENP‐C required phosphorylation. Immunoprecipitation with an equivalent amount of mouse IgG served as a negative control. Immunoblotted species is indicated at left.
-
CCdk inhibition with flavopiridol prevents M18BP1‐1161–580 localization at metaphase centromeres. Representative images showing Myc‐M18BP1‐1161–580 localization at sperm centromeres in metaphase extract depleted of endogenous M18BP1 following treatment with various mitotic kinase inhibitors. Inhibitor treatment is indicated at left, and immunolocalized protein is indicated above. Scale bar, 10 μm. Insets are magnified 3×. See Fig EV3 for demonstration of inhibitor efficacy.
-
DSchematic of phosphorylation sites in M18BP1‐1161–580 mutated for this study. M18BP1‐1 residue numbers indicated above. Positions of phosphorylation sites are indicated by orange lines and labels below. Bold indicates consensus Cdk phosphorylation sites.
-
EQuantification of immunofluorescence experiments examining localization of M18BP1‐1161–580 phosphorylation site mutants to sperm centromeres in metaphase extract depleted of endogenous M18BP1. Graph shows mean centromere intensity ± SEM of three independent experiments normalized to the WT condition. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005. See also Fig EV4B.
To identify kinases that might regulate M18BP1 localization, we analyzed M18BP1‐1161–580 localization in M18BP1‐depleted metaphase extract treated with inhibitors of several mitotic kinases: cyclin‐dependent kinase (Cdk; flavopiridol), Polo‐like kinase (Plx; BI2536), Aurora kinase (ZM‐447439), and Mps1 (reversine; Fig EV3C–F). Of these, only Cdk inhibition caused loss of M18BP1‐1161–580 localization (Fig 4C). Although Cdk inhibition also drives the extract into interphase, inhibitors of other mitotic kinases had no effect on M18BP1 localization suggesting that Cdk phosphorylation may directly regulate M18BP1.
Figure EV3. Identification of phosphosites in M18BP1‐1161–580 by mass spectrometry.
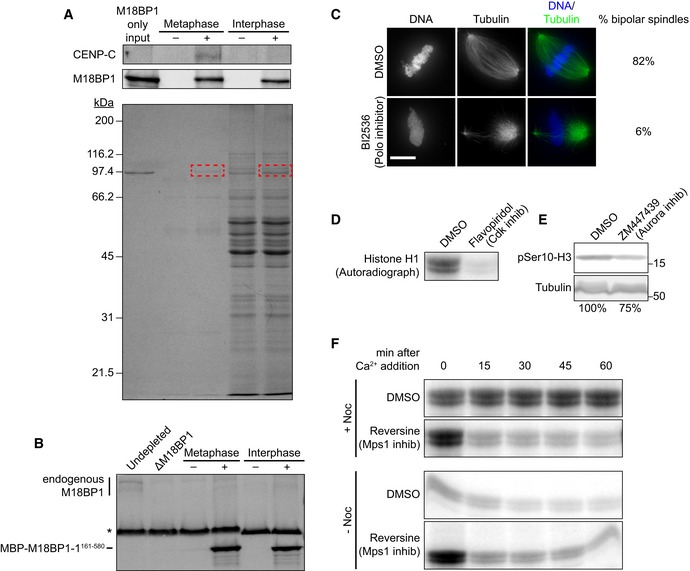
- Pulldowns from metaphase or interphase extract depleted of M18BP1 with (+) or without (−) MBP‐M18BP1‐1161–580. (Top) Immunoblot showing that MBP‐M18BP1‐1161–580 specifically co‐immunoprecipitates CENP‐C from metaphase extract. (Bottom) Coomassie colloidal blue‐stained gel showing material precipitated with α‐MBP antibody‐coated beads. Red boxes indicate bands that were excised and submitted for mass spectrometry.
- Immunoblot showing levels of MBP‐M18BP1‐1161–580 in M18BP1‐depleted extract relative to endogenous M18BP1 levels (undepleted, left lane). Non‐specific band recognized by α‐M18BP1 antibody indicated by asterisk. MBP‐M18BP1‐1161–580 was ˜56‐fold in excess of endogenous M18BP1.
- Representative images showing spindle morphology in cycled metaphase egg extracts treated with DMSO or with the Polo kinase (Plx) inhibitor BI2536. Spindles in BI2536‐treated extract were largely monopolar or asymmetric whereas control extracts showed largely bipolar spindles, similar to the effect of Plx immunodepletion from metaphase egg extract (Budde et al, 2001). Scale bar, 10 μm.
- Autoradiograph showing histone H1 phosphorylation by Cdk in control DMSO‐treated metaphase extracts or extracts treated with the Cdk inhibitor flavopiridol.
- Western blot showing reduction in histone H3 serine 10 phosphorylation by Aurora kinase in extracts treated with ZM447439. See (Gadea & Ruderman, 2005)
- Autoradiographs showing the change in Cdk activity (as determined by histone H1 phosphorylation) in control extracts with and without spindle assembly checkpoint activation brought about by treatment with nocodazole (top, DMSO‐treated). Declining Cdk activity despite nocodazole treatment after Mps1 inhibition (Reversine) indicates bypass of the spindle assembly checkpoint by these inhibitors. See Santaguida et al (2010).
Because M18BP1‐1161–580 is phosphorylated in metaphase extract (Fig 4B), we tested whether M18BP1 phosphorylation might regulate its centromere localization by modulating its interaction with CENP‐C. We purified MBP‐tagged M18BP1‐1161–580 from E. coli and added the purified protein to M18BP1‐depleted egg extract (Fig EV3A and B). We then recovered the protein from the extract with α‐MBP antibody‐coated beads and mapped modification sites on M18BP1‐1161–580 with mass spectrometry (Figs 4D, and EV3A and B). We identified 25 phosphorylated residues, including four minimal consensus Cdk sites (S/T‐P; Fig 4D). We mutated each of these phosphorylation sites to alanine to prevent their modification and assessed the localization of mutant M18BP1‐1161–580 at sperm centromeres in metaphase extract depleted of endogenous M18BP1. Most mutants exhibited a modest (~10%) reduction in metaphase localization (Fig 4E). However, mutation of one conserved Cdk site, T166A, caused a 59 ± 6% decrease in M18BP1‐1161–580 localization (Figs 4E and EV4B). Mutation of the SANTA domain caused an 89 ± 2% decrease in this assay (Fig 4E).
Figure EV4. Metaphase localization of M18BP1‐1 phosphomutants.
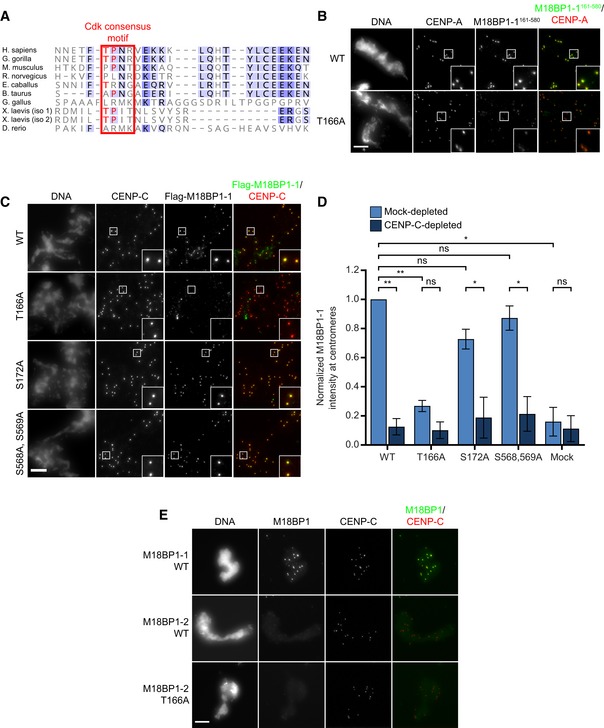
- Conservation of the Xenopus T166 phosphorylation site. Alignment of M18BP1 sequences among select eukaryotes. Red box indicates the position of the Xenopus Cdk site, with conserved T‐P residues indicated in red. Darker shades of blue represent increased conservation of amino acids in alignment of ˜300 M18BP1 homologues.
- Representative immunofluorescence images showing WT and T166A mutant M18BP1‐1161–580 localization at metaphase sperm centromeres (see Fig 4E). Mutant species indicated at left, immunolocalized protein indicated above. Scale bar, 10 μm. Insets are magnified 3×.
- Representative immunofluorescence images showing localization of full‐length M18BP1‐1 phosphorylation site mutants to metaphase centromeres. M18BP1‐1 species indicated at left; immunolocalized species indicated above. Scale bar, 10 μm. Insets are magnified 3×.
- Quantification of (C). Values are normalized to M18BP1‐1WT localization in mock‐depleted extract. Error bars represent SEM of three independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005.
- T166A mutation in M18BP1‐2 does not affect metaphase centromere localization. Metaphase, M18BP1‐depleted extract was supplemented with the M18BP1 species indicated at left and sperm chromatin. Immunolocalized protein indicated above. Scale bar, 10 μm.
When introduced into the full‐length M18BP1 protein, M18BP1‐1T166A caused a substantial 77 ± 3% reduction in metaphase localization whereas two control mutants, M18BP1‐1S172A and M18BP1‐1S568A, S569A, showed only modest localization defects (Figs 5A and C, and EV4C and D). This indicates that Cdk phosphorylation of T166 is primarily responsible for regulating metaphase M18BP1‐1 localization. Mutation of the corresponding residue in M18BP1‐2 showed no effect on metaphase M18BP1‐2 localization (Fig EV4A and E). To test whether the centromere localization defect in M18BP1‐1T166A was due to loss of interaction with CENP‐C, we immunoprecipitated M18BP1‐1 mutants from metaphase extract and found that M18BP1‐1WT, M18BP1‐1S172A, and M18BP1‐1S568A, S569A all co‐precipitated CENP‐C while M18BP1‐1T166A showed dramatically reduced CENP‐C binding (Fig 5B). This indicates that the defect in M18BP1‐1T166A localization to metaphase centromeres likely arises from the disruption of its interaction with CENP‐C.
Figure 5. Phosphorylation of T166 in M18BP1 regulates metaphase localization and binding to CENP‐C.
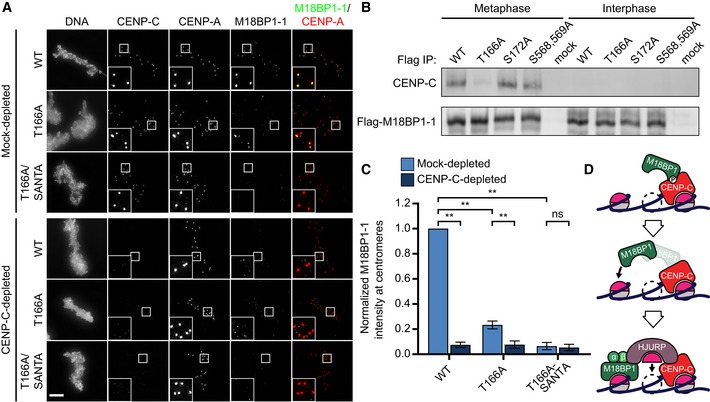
- Representative immunofluorescence images showing M18BP1‐1 mutant localization. M18BP1‐depleted metaphase extract was supplemented with in vitro translated full‐length Flag‐M18BP1‐1 mutants and sperm chromatin. In addition, extract was either mock‐depleted or CENP‐C depleted to assess CENP‐C dependent metaphase localization. M18BP1‐1 species and CENP‐C depletion status are indicated at left; immunolocalized protein is indicated above. Scale bar, 10 μm. Insets are magnified 3×.
- Representative Western blot showing co‐immunoprecipitation of CENP‐C with phosphorylation site mutants of M18BP1‐1. M18BP1‐depleted metaphase or interphase extract was supplemented with in vitro translated Flag‐M18BP1‐1 mutants. M18BP1‐1 mutation and cell cycle state indicated above, and immunoblotted species indicated at right. Mock precipitations using rabbit reticulocyte lysate without any in vitro translated protein served as a negative control.
- Quantification of (A). Flag‐M18BP1‐1 mutant centromere intensity normalized to WT localization in the mock‐depleted condition. Error bars represent SEM from three independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, **P < 0.005.
- Model showing phosphorylation‐dependent binding of Xenopus M18BP1 to CENP‐C during mitosis (top) and the transition to CENP‐A‐dependent binding in interphase (bottom).
We determined the extent to which M18BP1‐1 depends on its interaction with CENP‐C for localization to metaphase centromeres by depleting CENP‐C from egg extract and assaying the localization of the M18BP1‐1T166A mutant. M18BP1‐1T166A localization was further reduced by CENP‐C depletion to 8 ± 3% of mock‐depleted levels (Fig 5A and C), suggesting that T166A does not completely disrupt the interaction between M18BP1 and CENP‐C or that there are additional CENP‐C‐dependent factors required for M18BP1 localization. Our prior in vitro binding experiments indicated that separate elements contained within M18BP1‐11–413 (containing T166) or M18BP1‐1414–749 (containing the SANTA domain) contribute to CENP‐C binding (Fig 3A–C). Furthermore, the SANTA mutation alone did not prevent CENP‐C binding to M18BP1‐1 (Fig 2B). We therefore created an M18BP1‐1T166A, SANTA double mutant. Combining these mutations completely abolished metaphase M18BP1‐1 localization, comparable to that observed after CENP‐C depletion (Fig 5A and C). This indicates not only that both contacts contribute to CENP‐C binding, but also suggests that metaphase M18BP1 localization is mediated exclusively by binding to CENP‐C (Fig 5D).
M18BP1‐1T166A and M18BP1‐1SANTA do not support proper CENP‐A assembly due to defective interphase localization
CENP‐C depletion prevents proper CENP‐A assembly in interphase in part because CENP‐C directly recruits HJURP to centromeres (Moree et al, 2011; Tachiwana et al, 2015; French et al, 2017). Although CENP‐C is not required for interphase M18BP1 localization, it was unclear whether mislocalization of M18BP1 during metaphase might also alter the efficiency of CENP‐A assembly. To uncover a possible requirement for metaphase M18BP1 localization, we asked whether M18BP1 mutants defective in metaphase localization could support interphase CENP‐A assembly.
To assess this, we depleted endogenous M18BP1 from metaphase extract, replaced it with in vitro translated M18BP1‐1 mutants, and assayed the assembly of exogenously added Myc‐CENP‐A at sperm centromeres after release of the extract to interphase (Fig 6A and B). Consistent with previous results, we found that M18BP1 depletion reduced CENP‐A assembly to 39 ± 3% of mock‐depleted levels (Fig 6A and B). Add‐back of both M18BP1 isoforms is necessary to fully rescue CENP‐A assembly on sperm chromatin (Moree et al, 2011; French et al, 2017). However, addition of only wild‐type M18BP1‐1 partially rescued assembly to 62 ± 3% of mock‐depleted levels (Fig 6A and B). Add‐back of M18BP1‐1T166A failed to rescue Myc‐CENP‐A assembly to the same degree, achieving only 53 ± 4% of mock‐depleted levels, and add‐back of M18BP1‐1SANTA rescued Myc‐CENP‐A assembly to 44 ± 3% of mock‐depleted levels (French et al, 2017; Fig 6A and B). Thus, M18BP1 mutants defective in metaphase localization do not fully rescue interphase CENP‐A assembly.
Figure 6. Metaphase targeting mutants of M18BP1‐1 do not support robust CENP‐A assembly.
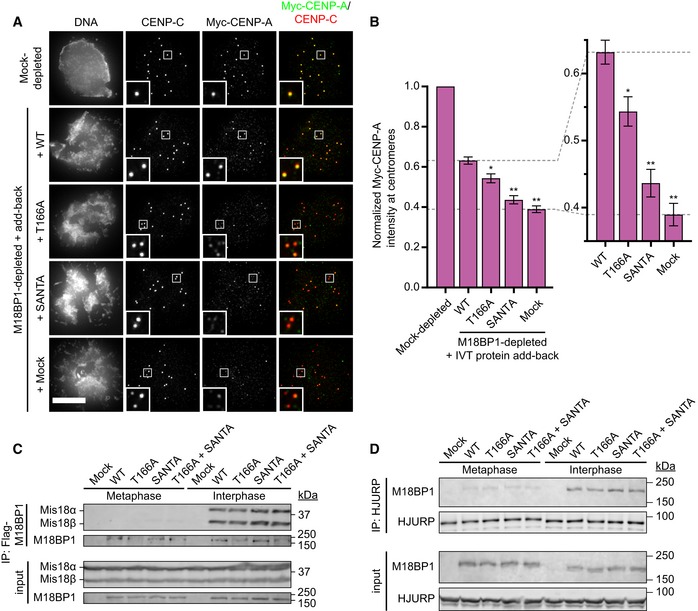
- Representative immunofluorescence images showing incorporation of exogenous Myc‐CENP‐A at centromeres. Sperm nuclei were incubated in M18BP1‐depleted interphase Xenopus egg extracts complemented with the indicated M18BP1‐1 protein. Extracts were supplemented with RNA encoding Myc‐CENP‐A to track new CENP‐A assembly and in vitro translated HJURP. Scale bar, 10 μm. Insets are magnified 3×.
- Quantification of Myc‐CENP‐A loading at Xenopus sperm centromeres in (A). Values are normalized to the centromere signals in mock‐depleted extract. Dashed lines indicate the Myc‐CENP‐A assembly signal observed upon M18BP1 depletion (bottom) and Flag‐M18BP1‐1WT add‐back (top) as points of reference for mutant rescue. Graph shows the mean ± SEM of four independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005.
- Interphase Mis18 complex formation is unaffected by M18BP1‐1 mutations that prevent binding to CENP‐C. Extract depleted of endogenous M18BP1 was supplemented with Myc‐Mis18α, Myc‐Mis18β, and Flag‐M18BP1‐1. M18BP1‐1 species added to each reaction, and cell cycle state of the extract is indicated at the top. Co‐immunoprecipitation of Myc‐Mis18α/β was assessed by α‐Myc immunoblot following Flag precipitation.
- HJURP association with M18BP1‐1 is unaffected by mutations that prevent CENP‐C binding. Extract depleted of endogenous M18BP1 was supplemented with Flag‐M18BP1‐1. M18BP1‐1 species added to each reaction and cell cycle state of the extract are indicated at the top. Co‐immunoprecipitation of Flag‐M18BP1‐1 was assessed by α‐Flag immunoblot following HJURP precipitation.
M18BP1, as part of the Mis18 complex, recruits HJURP to centromeric chromatin for CENP‐A nucleosome assembly (Barnhart et al, 2011; Moree et al, 2011; Nardi et al, 2016; French et al, 2017). To understand why metaphase targeting mutants of M18BP1 were defective in Myc‐CENP‐A assembly, we examined their association with Mis18α, Mis18β, and HJURP by immunoprecipitation followed by Western blotting. We observed interphase‐specific association between Xenopus Mis18α, Mis18β, and M18BP1‐1WT by immunoprecipitation (Fig 6C). M18BP1‐1T166A, M18BP1‐1SANTA, and M18BP1‐1T166A,SANTA bound to Mis18α and Mis18β similar to wild type, suggesting that defects in CENP‐A assembly do not result from defective Mis18 complex assembly. We also assayed binding to HJURP and found that M18BP1‐1WT, M18BP1‐1T166A, M18BP1‐1SANTA, and M18BP1‐1T166A,SANTA all bound to HJURP in interphase (Fig 6D). These data suggest that M18BP1‐1 mutants that are defective in metaphase centromere localization are not compromised in their ability to assemble the Mis18 complex or interact with HJURP.
We had previously found that mutation of the SANTA domain in M18BP1‐2 prevented its localization in interphase (French et al, 2017). Similarly, we found that M18BP1‐1SANTA localization to interphase centromeres was reduced by 76 ± 5% (Fig 7B). Surprisingly, we also found that interphase localization of M18BP1‐1T166A was reduced by 53 ± 3%; M18BP1‐1T166A,SANTA localization was reduced by 81 ± 5% (Fig 7B). Defective interphase localization of these mutants is the likely cause of reduced Myc‐CENP‐A assembly because M18BP1 localization to centromeres in interphase is required for new CENP‐A nucleosome assembly.
Figure 7. Metaphase targeting mutants of M18BP1‐1 inhibit nuclear localization during interphase.
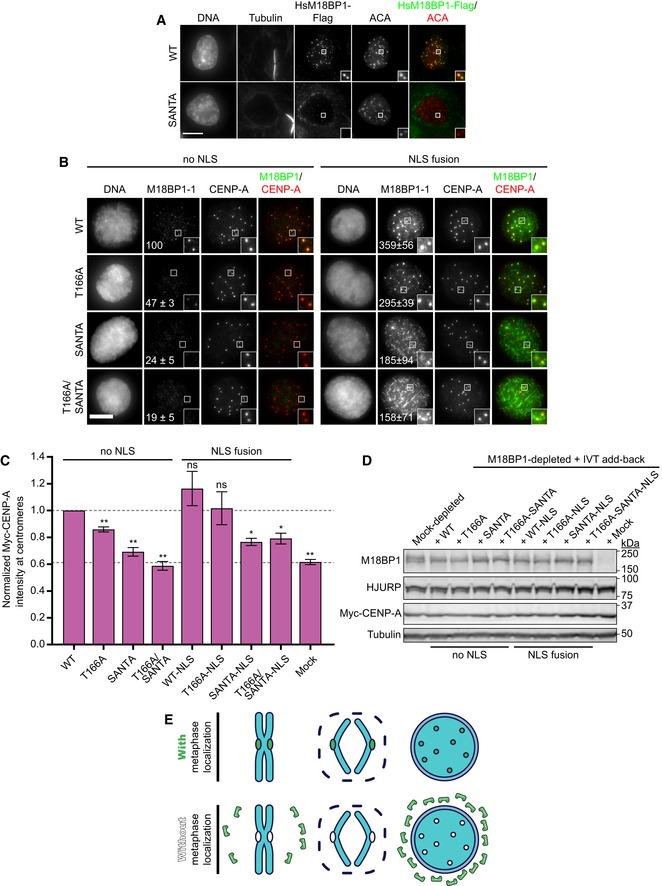
- Representative images showing G1 localization of human M18BP1WT or human M18BP1SANTA in sfGFP‐AID‐M18BP1 DLD1 cells treated with 1 mM IAA for 24 h to remove endogenous M18BP1. Centromeric localization is indicated by localization with ACA (α‐centromere autoantibody serum); early G1 cell cycle state is indicated by midbody staining in the tubulin channel. M18BP1 species indicated at left; immunolocalized protein indicated above. Scale bar, 10 μm. Insets are magnified 3×.
- Representative immunofluorescence images showing localization of full‐length Flag‐M18BP1‐1 (mutant species indicated at left) without (left panels) and with (right panels) fusion to an SV40 NLS to sperm centromeres in interphase extract depleted of endogenous M18BP1. Immunolocalized protein indicated above. Scale bar, 10 μm. Insets are magnified 3×. Quantification of immunofluorescence intensity at centromeres as total integrated fluorescence signal normalized to wild‐type levels. The values shown on each panel are represented as a percentage of wild‐type Flag‐M18BP1‐1 ± SEM from three independent experiments.
- Quantification of Myc‐CENP‐A assembly in M18BP1‐depleted extract complemented with the indicated M18BP1‐1 species. Graph shows mean immunofluorescence intensity at centromeres normalized to reactions complemented with WT M18BP1‐1. Error bars show SEM of at least three independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005.
- Representative Western blot of CENP‐A assembly reactions in (B). Efficient M18BP1 depletion is indicated by comparing lanes 1 and 10. Add‐back of wild‐type or mutant M18BP1‐1 is near endogenous levels.
- Model showing that metaphase localization of M18BP1 (green) promotes M18BP1 retention on chromosomes during nuclear envelope formation to promote centromeric localization during interphase.
Prior work has identified only two mechanisms responsible for interphase M18BP1 localization: binding to Mis18α/β and binding to CENP‐A nucleosomes (Fujita et al, 2007; French et al, 2017; Hori et al, 2017; Pan et al, 2017; Sandmann et al, 2017; Spiller et al, 2017). As described above, immunoprecipitation of Flag‐M18BP1‐1 mutants from extract indicated that interphase‐specific binding to Mis18α/β was intact (Fig 6C). Mutation of T166A or the SANTA domain did not prevent M18BP1 binding to CENP‐A chromatin‐coated beads in vitro, confirming that CENP‐A nucleosome binding was not dramatically impaired (Fig EV5A–C) (French et al, 2017). This suggests that an additional mechanism may act through the CENP‐C binding domain to recruit M18BP1‐1 to interphase centromeres.
Figure EV5. Nuclear exclusion regulates M18BP1 localization at interphase centromeres.
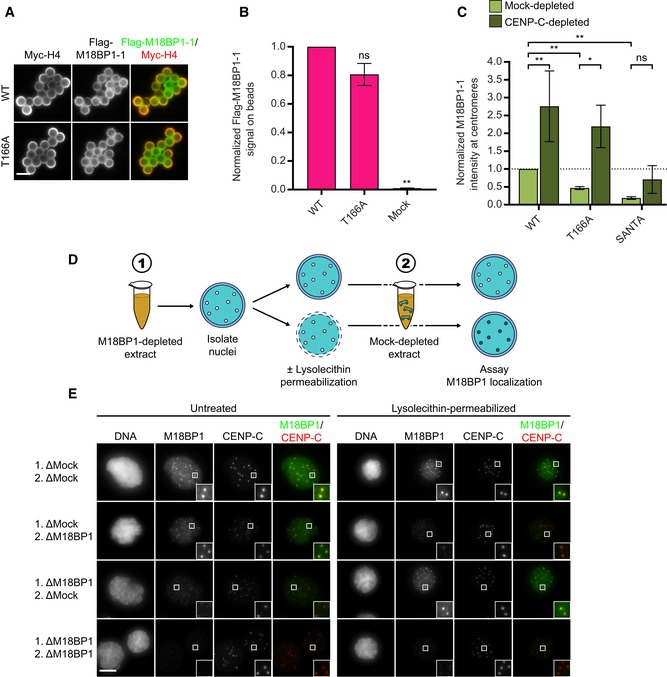
- Representative images of full‐length Flag‐M18BP1‐1WT or Flag‐M18BP1‐1T166A binding to CENP‐A chromatin‐coated beads in reticulocyte lysate. Scale bar, 5 μm.
- Quantification of (A). Flag‐M18BP1‐1 fluorescence on beads was normalized to Myc‐H4 signal to control for the amount of chromatin coating each bead, and then, all conditions were normalized to WT. Error bars show SEM from three independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, ns: not significant, **P < 0.005.
- Metaphase targeting mutants of M18BP1 show increased localization to interphase centromeres following CENP‐C depletion. French et al (2017) showed that this increase depends on the ability of M18BP1 to bind CENP‐A nucleosomes, suggesting that the T166A and SANTA mutations do not prevent CENP‐A nucleosome binding in extract. M18BP1‐1 mutant species indicated below. Values are normalized to M18BP1‐1WT in mock‐depleted interphase extract (dashed line). Error bars represent SEM of three independent experiments. Significance determined by Welch's unpaired two‐tailed t‐test, *P < 0.05, **P < 0.005.
- Schematic showing nuclear transfer and permeabilization scheme to investigate regulation of M18BP1 nuclear localization. Sperm nuclei were prepared in mock‐depleted or M18BP1‐depleted interphase extract (1), recovered by centrifugation, permeabilized, and introduced into fresh mock‐depleted or M18BP1‐depleted interphase extract (2) to assay centromere localization.
- Representative immunofluorescence images of experiment in (D) showing endogenous M18BP1 localization at sperm centromeres in untreated or lysolecithin‐permeabilized nuclei. Incubation scheme indicated at left. Left panels show M18BP1 localization in unpermeabilized nuclei; right panels show localization in lysolecithin‐permeabilized nuclei. Immunolocalized protein indicated above. Scale bar, 10 μm. Insets are magnified 4×.
The SANTA domain plays a conserved role in interphase M18BP1 localization in humans
In contrast to our findings in Xenopus, deletion of the SANTA domain from human, chicken, or Arabidopsis M18BP1 did not prevent M18BP1 localization in these organisms (Lermontova et al, 2013; Stellfox Madison et al, 2016; Hori et al, 2017). To understand the apparent difference in SANTA domain function between Xenopus and human, we generated human cell lines expressing the F‐box protein TIR1 and in which both alleles of M18BP1 were endogenously tagged with a tandem sfGFP‐AID (auxin‐inducible degron) tag, permitting rapid, inducible degradation for complementation studies (Fig EV6A–C; Nishimura et al, 2009; Holland et al, 2012).
Figure EV6. Characterization of M18BP1 localization in human cells.
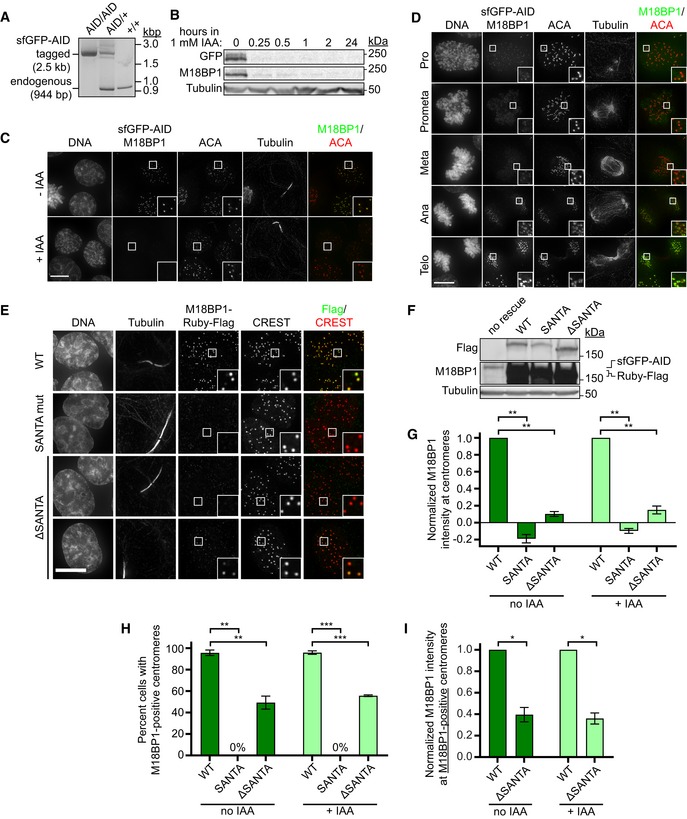
- Agarose gel showing genomic PCR to assess successful integration of the sfGFP‐AID tag at the endogenous M18BP1 locus. Presence of endogenous band only (944 bp) indicates no integration (+/+), presence of the sfGFP‐AID‐M18BP1 band only (2.5 kbp) indicates successful integration at both alleles (AID/AID), and presence of both bands indicates successful integration at only one allele (AID/+).
- Immunoblot showing sfGFP‐AID‐M18BP1 degradation kinetics in the M18BP1AID/AID cell line following supplementation of the medium with 1 mM indole‐3‐acetic acid (IAA). Whole‐cell lysates were harvested at the indicated times following IAA addition and blotted for GFP‐tagged and total M18BP1. Immunoblotted species indicated at left.
- Representative immunofluorescence images showing complete loss of G1 M18BP1 localization in the M18BP1AID/AID cell line within 1 h of IAA addition. Untreated cells are viable and show normal G1 localization of tagged M18BP1, indicating the tagged species remains functional. Midbody staining in the tubulin channel indicates early G1. ACA, α‐centromere autoantibody serum. Scale bar, 10 μm. Insets are magnified 3×.
- Robust centromere localization of endogenous sfGFP‐AID‐M18BP1 was not observed prior to anaphase/telophase in M18BP1AID/AID cells. Cell cycle state indicated at left; immunolocalized protein indicated above. ACA, α‐centromere autoantibody serum. Scale bar 10 μm. Insets are magnified 3×.
- Representative images showing G1 localization of humanM18BP1WT, M18BP1SANTA, or M18BP1 lacking the SANTA domain (M18BP1ΔSANTA) in sfGFP‐AID‐M18BP1 DLD1 cells treated with 1 mM IAA for 24 h to remove endogenous M18BP1. Scale bar, 10 μm. Insets are magnified 3×.
- Immunoblot showing expression of mRuby2‐Flag‐tagged M18BP1 transgenes from (E). Immunoblotted species indicated at left; transgene indicated above. Transgenes are expressed at similar levels to each other (Flag blot), but are in vast excess of endogenous sfGFP‐AID‐M18BP1 levels (M18BP1 blot).
- Quantification of M18BP1 intensity at G1 centromeres in (E). Mutation or deletion of the SANTA domain reduces centromere localization of M18BP1 regardless of the presence of endogenous M18BP1 (i.e., ±IAA). Negative values for M18BP1SANTA arise from background subtraction and reflect the fact that its localization is primarily cytosolic.
- Quantification of percent midbody‐positive G1 cells in (E) showing detectable M18BP1 localization at centromeres. Similar to Stellfox Madison et al (2016), approximately half as many cells are positive for M18BP1ΔSANTA as for M18BP1WT. We observed no cells with centromeric M18BP1SANTA.
- Quantification of M18BP1 intensity at centromeres in cells scored as positive for M18BP1 in (H).
Visualization of endogenous sfGFP‐AID‐M18BP1 signal revealed punctate, centromeric localization in midbody‐positive G1 cells, consistent with previous reports (Fig EV6C; Fujita et al, 2007; Maddox et al, 2007; McKinley & Cheeseman, 2014). We did not observe substantial M18BP1 localization at centromeres during mitosis until anaphase/telophase (Fig EV6D). Upon addition of 1 mM indole‐3‐acetic acid (IAA), M18BP1 protein levels were depleted within 15 min, and centromeric M18BP1 was undetectable at G1 centromeres within 1 h (Fig EV6B and C).
Ectopic expression of wild‐type M18BP1 from a doxycycline‐inducible promoter restored M18BP1 signal at G1 centromeres (Figs 7A and EV6E). Expression of M18BP1SANTA or of M18BP1 lacking the SANTA domain (M18BP1ΔSANTA), however, showed a dramatic reduction in both the amount of M18BP1 localized to centromeres and in the number of G1 cells positive for centromeric M18BP1 (Figs 7A and EV6E–I). Mutation of the SANTA domain caused diffuse cytoplasmic localization and apparent exclusion of M18BP1 from the nucleus (Fig 7A). This demonstrates that the SANTA domain plays a conserved role in M18BP1 localization to interphase centromeres in humans and in Xenopus.
A role for the nuclear envelope in regulating M18BP1 localization
The diffuse, cytoplasmic localization of M18BP1SANTA in human cells suggested a defect in nuclear localization (Fig 7A). Given that the T166A and SANTA mutations in Xenopus M18BP1‐1 did not prevent binding to CENP‐A nucleosomes or to Mis18α/β, we hypothesized that their interphase localization defects resulted from defective nuclear localization.
We first sought to understand how nuclear localization of endogenous M18BP1 is regulated. We isolated nuclei prepared in mock‐depleted or M18BP1‐depleted interphase extract and then reintroduced them into fresh mock‐depleted or M18BP1‐depleted extract to assay M18BP1 centromere localization (Fig EV5D). We found that nuclei assembled in mock‐depleted extract showed persistent localization of M18BP1 after incubation in M18BP1‐depleted extract, suggesting that M18BP1 is efficiently retained in nuclei at centromeres (Fig EV5E). On the other hand, nuclei assembled in M18BP1‐depleted extract and introduced in mock‐depleted extract showed negligible centromeric M18BP1, suggesting that despite the presence of M18BP1 in the cytoplasm little exchange occurred between the nuclear and cytoplasmic pools of M18BP1 (Fig EV5E). To test whether the nuclear envelope limits exchange of nuclear and cytoplasmic M18BP1, we permeabilized nuclei with lysolecithin before their reintroduction into extract (Blow & Laskey, 1988). Upon permeabilization, nuclei assembled in mock‐depleted extract and reintroduced into M18BP1‐depleted extract showed reduced centromeric M18BP1 (Fig EV5E). This suggested that an intact nuclear envelope is required to retain M18BP1 at centromeres and that permeabilization releases this nuclear reservoir. Conversely, permeabilization of nuclei assembled in M18BP1‐depleted extract and reintroduced into mock‐depleted extract showed substantially increased M18BP1 localization (Fig EV5E). Together these results suggest that the nuclear envelope prevents the free exchange of M18BP1 between the cytoplasm and the nucleus.
We hypothesized that if M18BP1 exchange between the cytoplasm and the nucleus is limited, poor interphase localization of metaphase targeting M18BP1‐1 mutants could result from their poor retention at centromeres during nuclear envelope reassembly at mitotic exit and their subsequent exclusion from nuclei. To bypass potential nuclear localization defects of M18BP1‐1 mutants, we fused an SV40 nuclear localization sequence (NLS; PKKKRKV) to M18BP1‐1 and measured localization at interphase centromeres. Flag‐NLS‐M18BP1‐1WT showed 3.6‐fold increase in localization at interphase centromeres relative to Flag‐M18BP1‐1WT lacking an NLS (Fig 7B). Strikingly, Flag‐NLS‐M18BP1‐1T166A localization increased 6.3‐fold relative to Flag‐M18BP1‐1T166A lacking an NLS, achieving localization comparable to Flag‐NLS‐M18BP1WT (Fig 7B). Flag‐NLS‐M18BP1‐1T166A localization was punctate and largely resembled Flag‐NLS‐M18BP1‐1WT localization. Together, this suggests that the T166A mutation may regulate interphase M18BP1‐1 recruitment to centromeres by inhibiting its nuclear localization (Fig 7E).
Flag‐NLS‐M18BP1‐1SANTA and Flag‐NLS‐M18BP1‐1T166A,SANTA showed improved nuclear localization and increased signal at centromeres (Fig 7B). Intriguingly, however, their localization was not centromere‐specific, as was observed for Flag‐NLS‐M18BP1‐1WT or Flag‐NLS‐M18BP1‐1T166A: Flag‐NLS‐M18BP1‐1SANTA signal was observed throughout chromatin with enrichment around centromeres (Fig 7B). This suggests that the SANTA domain plays a dual role, promoting both nuclear retention of M18BP1‐1 at mitotic exit and forming additional interactions that are required for interphase centromere localization.
To assess whether the function of M18BP1‐1 mutants in new CENP‐A nucleosome assembly could be restored by rescuing their nuclear localization, we analyzed exogenous Myc‐CENP‐A assembly in extract depleted of endogenous M18BP1 and complemented with M18BP1‐1 NLS‐fusions (Fig 7C and D). While Flag‐M18BP1‐1T166A showed a 15 ± 2% defect in Myc‐CENP‐A assembly relative to Flag‐M18BP1‐1WT, Flag‐NLS‐M18BP1‐1T166A supported Myc‐CENP‐A levels at levels indistinguishable from wild type (Fig 7C). Thus, rescuing nuclear localization and thereby centromere‐specific localization restored proper CENP‐A assembly. On the other hand, Flag‐NLS‐M18BP1‐1SANTA and Flag‐NLS‐M18BP1‐1T166A,SANTA exhibited only partial restoration of Myc‐CENP‐A assembly relative to extract depleted of M18BP1 (Fig 7C), consistent with these mutants' inability to rescue proper interphase M18BP1‐1 localization.
Discussion
Maintaining one centromere per chromosome across generations is important for faithful genome transmission. This process requires the assembly of CENP‐A nucleosomes every cell cycle, but how CENP‐A assembly factors are recruited to the right locus and how CENP‐A assembly is regulated are incompletely understood. An emerging theme has been that Cdk phosphorylation inhibits premature assembly and that dephosphorylation promotes the localization and function of proteins required for CENP‐A assembly (Silva et al, 2012; McKinley & Cheeseman, 2014; Stankovic et al, 2017). Here, we show that Cdk phosphorylation controls the interaction between M18BP1 and CENP‐C to promote metaphase centromere localization of M18BP1 in Xenopus egg extract. The interaction between CENP‐C and M18BP1 is through the C‐terminal region of CENP‐C containing the conserved CENP‐C motif and the cupin/dimerization domain (Moree et al, 2011), and the N‐terminal region of M18BP1 containing the conserved SANTA domain. Mutations in M18BP1 that break this interaction prevented metaphase M18BP1 localization, suggesting that CENP‐C binding is the primary mechanism for metaphase M18BP1 localization (Fig 5D). It is likely that additional regulatory mechanisms govern M18BP1 localization during mitosis, as M18BP1 binding to CENP‐A nucleosomes is inhibited in metaphase extract (French et al, 2017).
At anaphase, Cdk activity declines. This causes a switch in the mechanism of M18BP1 localization between metaphase and interphase: M18BP1 no longer requires CENP‐C, but rather binds to CENP‐A nucleosomes for interphase centromere localization in Xenopus (Fig 5D; Moree et al, 2011; French et al, 2017). While CENP‐C depletion does not prevent interphase M18BP1 localization, M18BP1 mutants unable to bind CENP‐C showed reduced interphase localization, due in part to defective nuclear localization. This apparent paradox may partly be explained by the fact that CENP‐C occupies potential M18BP1 binding sites on CENP‐A chromatin (French et al, 2017); the liberation of additional M18BP1 binding sites by CENP‐C depletion may allow “extra” M18BP1 to bind centromeres before nuclear envelope assembly. Our data are consistent with a model in which metaphase localization of M18BP1, or at least chromatin association prior to nuclear envelope reformation, promotes proper interphase localization and therefore CENP‐A assembly (Fig 7E).
Metaphase localization of M18BP1 could serve additional functions beyond merely ensuring its retention during nuclear assembly. In interphase, both CENP‐C and M18BP1 are required for HJURP localization and CENP‐A assembly (Moree et al, 2011; French et al, 2017). Metaphase binding of M18BP1 to CENP‐C could ensure their close apposition in the subsequent interphase on adjacent nucleosomes or even opposite faces of the same CENP‐A nucleosome, suggesting a mechanism by which sites for new CENP‐A assembly might be identified prior to G1. Their interaction could also provide an additional layer of regulation to prevent premature HJURP localization. HJURP interacts with CENP‐C1191–1400 during interphase (Tachiwana et al, 2015; French et al, 2017). Binding of M18BP1 to this region in metaphase could competitively inhibit HJURP localization, helping to restrict CENP‐A assembly to G1 (Fig 5D).
While our work demonstrated a role for the SANTA domain in regulating M18BP1 nuclear localization, our inability to rescue centromere‐specific localization of M18BP1‐1SANTA by fusion to an NLS suggests the domain regulates additional aspects of interphase localization. The importance of the SANTA domain is further highlighted by the fact that preventing CENP‐A nucleosome binding by M18BP1 only reduced interphase M18BP1 localization by ~50% (French et al, 2017). Therefore, a major goal moving forward will be to identify the interphase functions of the SANTA domain to understand how the Mis18 complex recognizes CENP‐A chromatin during CENP‐A assembly.
Our work also suggests similarities in how M18BP1 localizes in frog and in human. Previous work in human cells found that removing the SANTA domain caused a ~50% reduction in the number of cells with centromere‐localized M18BP1 (Stellfox Madison et al, 2016). Similar to that work, we found not only that fewer G1 cells were positive for M18BP1ΔSANTA or M18BP1SANTA localization, but that these mutations substantially reduced the total amount of M18BP1 localized to centromeres (Fig EV6E–I). This demonstrates a conserved role for the SANTA domain in G1 localization of human M18BP1. In addition, we found that both human and frog M18BP1 use a homologous domain to directly interact with CENP‐C suggesting conservation of this pathway. CENP‐C‐dependent, metaphase localization of M18BP1 has been previously observed in human (McKinley & Cheeseman, 2014). Metaphase localization may be transient or highly dynamic, however, as centromeric M18BP1 signal remains low prior to anaphase (Fujita et al, 2007; Maddox et al, 2007; Lagana et al, 2010; Silva et al, 2012; Wang et al, 2014; Nardi et al, 2016; Spiller et al, 2017; Stankovic et al, 2017). The cell‐free system we have developed in Xenopus egg extract provides a powerful approach for investigating the conserved cell cycle dynamics of M18BP1 to understand how the Mis18 complex regulates CENP‐A assembly.
Materials and Methods
Experimental model and subject details
Mature X. laevis females (Nasco, LM00535MX) were housed and maintained in the Stanford Aquatic Facility staffed by the Veterinary Service Center. For ovulation, frogs were primed 2–14 days before ovulation by subcutaneous injection of 50 U pregnant mare serum gonadotropin (PMSG; Sigma) at the dorsal lymph sac, then induced 18 h before ovulation with 500 U human chorionic gonadotropin (hCG; Chorulon). During ovulation, frogs were housed individually in 2 l MMR buffer (6 mM Na‐HEPES, pH 7.8, 0.1 mM EDTA, 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 2 mM CaCl2) at 17°C. Animal work was carried out in accordance with the guidelines of the Stanford University Administrative Panel on Laboratory Animal Care (APLAC).
Human DLD1 cell lines (male, pseudo‐diploid, colorectal adenocarcinoma) stably expressing the F‐box protein osTIR1 were a gift from Don Cleveland (Ludwig Cancer Institute, San Diego) (Holland et al, 2012; Fachinetti et al, 2015). Production of sfGFP‐AID‐M18BP1 cells is described below. Cells were cultured in RPMI‐1640 medium supplemented with 10% fetal bovine serum, penicillin/streptomycin, and 2 g/l sodium bicarbonate and maintained in a 37°C/5% CO2 incubator.
Xenopus egg extracts and immunodepletions
CSF‐arrested (metaphase) Xenopus egg extracts were prepared as described previously (Desai et al, 1999; Guse et al, 2012). In brief, eggs were washed with MMR buffer (6 mM Na‐HEPES, pH 7.8, 0.1 mM EDTA, 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 2 mM CaCl2) and then dejellied in MMR + 2% L‐cysteine. Dejellied eggs were washed in CSF‐XB buffer (10 mM K‐HEPES, pH 7.7, 100 mM KCl, 50 mM sucrose, 2 mM MgCl2, 0.1 mM CaCl2, 5 mM EGTA), followed by CSF‐XB containing 10 μg/ml LPC [leupeptin/pepstatin A/chymostatin]. Eggs were packed in a 13 × 51 mm polyallomer tube (Beckman Coulter, 326819) by a low‐speed spin in a tabletop clinical centrifuge for 45 s. After removal of buffer, packed eggs were centrifuged in a SW55Ti rotor (Beckman Coulter) for 15 min at 12,000 g. The soluble cytoplasm was removed by side puncture with a 18‐G needle and supplemented with 50 mM sucrose, 10 μg/ml LPC, 10 μg/ml cytochalasin D, and energy mix (7.5 mM creatine phosphate, 1 mM ATP, and 1 mM MgCl2).
Immunodepletions of CENP‐C and M18BP1 from Xenopus extract were performed essentially as described (Moree et al, 2011; French et al, 2017). Protein A Dynabeads (Invitrogen, 10001D) were washed in TBSTx (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.1% Triton X‐100) and then coupled to affinity purified antibody for 30–60 min at 4°C. For 100 μl extract, 2 μg α‐xCENP‐C antibody [rabbit, raised and purified against xCENP‐C207–296 (Milks et al, 2009)], or 4–5 μg α‐xM18BP1 [rabbit, raised against GST‐xM18BP1‐2161–415 and purified against xM18BP1‐1161–375 (Moree et al, 2011)] was used. An equivalent amount of whole rabbit IgG (Jackson ImmunoResearch Laboratories, Inc.) was used for mock depletions. Antibody‐coupled beads were washed three times in TBSTx and then resuspended in extract. Depletions were rotated at 4°C for 1 h. Beads were removed from extract by 2 × 5‐min magnetizations.
In vitro translation and M18BP1 mutant localization
In vitro translated (IVT) proteins were generated in rabbit reticulocyte lysate supplemented with 20 μM methionine or 4 μCi/μl [35S]‐methionine (Perkin Elmer, NEG709A500UC) using the SP6 TNT Quick‐Coupled Transcription/Translation kit (Promega, L2080) according to the manufacturer's instructions.
To assess localization of specific M18BP1 isoforms, truncations, or mutants, 2 μl of reticulocyte lysate containing in vitro translated protein was added directly to M18BP1‐depleted, CSF‐arrested egg extract (20 μl final reaction volume). For interphase samples, reactions were supplemented with 750 μM CaCl2; paired metaphase reactions were supplemented with an equivalent volume of sperm dilution buffer (50 mM HEPES pH 7.7, 750 mM sucrose, 5 mM MgCl2, 500 mM KCl). Demembranated sperm was added to 3,000/μl, and reactions were incubated at 16–18°C for 75 min. Reactions were then diluted in 1 ml dilution buffer [BRB‐80 (80 mM K‐PIPES, pH 6.8, 1 mM MgCl2, 1 mM EGTA), 0.5% Triton X‐100, 30% glycerol] containing 150 mM KCl and incubated on ice for 5 min. These samples were then fixed by addition of 1 ml dilution buffer containing 4% formaldehyde and spun through cushions of 40% glycerol in BRB‐80 onto poly‐L‐lysine‐coated coverslips to be processed for immunofluorescence.
In Fig 4C to assess the effects of mitotic kinase inhibition on Myc‐M18BP1‐1161–580 localization, extract was treated with the following drugs prior to IVT protein and sperm addition: flavopiridol (10 μM; Sigma, F3055), BI2536 (10 μM; gift from Tarun Kapoor, Rockefeller University), ZM‐447439 (20 μM; gift from James Ferrell, Stanford University), reversine (10 μM; Abcam, ab120921). To minimize the concentration of DMSO added to the extract, 40× drug stocks were prepared in sperm dilution buffer or extract before addition to reactions.
CENP‐A assembly assays
CENP‐A assembly assays on sperm chromatin were performed as previously described (Moree et al, 2011; French et al, 2017). RNAs used to express protein in Xenopus egg extract were prepared with the SP6 mMessage mMachine kit (Life Technologies, AM1340). NotI linearized pCS2+ plasmids were used in the transcription reaction following manufacturer's instructions, using double the amount of DNA. RNA was purified using RNeasy mini columns (QIAGEN). RNA was typically concentrated to at least 1 μg/μl using a SpeedVac (Savant) to avoid excessive dilution of the extract.
CENP‐A assembly reactions (30 μl final volume) contained 1.5 μl xHJURP IVT protein, 0.75 μl Flag‐M18BP1‐1 IVT protein, and 25 ng/μl Myc‐xCENP‐A RNA. CSF‐arrested, M18BP1‐depleted extract (prepared as above) was supplemented with IVT protein and RNA and incubated at 16–18°C for 30 min to permit RNA translation. Cycloheximide was then added to 0.1 mg/ml to terminate translation. Reactions were then released to interphase by addition of 750 μM CaCl2 and supplemented with demembranated sperm chromatin to 3,000/μl. After incubation for 75 min at 16–18°C, assembly reactions were diluted and spun onto coverslips as described above. Quantification of CENP‐A loading (centromeric Myc‐CENP‐A signal) was performed as described below.
Nuclear permeabilization assay
To obtain Xenopus nuclei containing or lacking M18BP1, sperm was incubated in mock‐depleted or M18BP1‐depleted, calcium‐released interphase extracts as described above for 75 min at 16–18°C. Nuclei‐containing extract reactions were then diluted 1:30 with Buffer A (Blow & Laskey, 1988; 15 mM Tris–HCl pH 7.4, 15 mM NaCl, 60 mM KCl, 1 mM beta‐mercaptoethanol, 0.5 mM spermidine, 0.15 mM spermine), and nuclei were pelleted at 2,000 g for 5 min at 4°C. Nuclei were resuspended in 200 μl Buffer A, split into two tubes, and half was treated with 100 μg/ml lysolecithin for 10 min on ice. Nuclei were then diluted with an additional 400 μl Buffer A and pelleted at 2,000 g for 5 min. Nuclei were then resuspended at 55,000/μl in Buffer A and then were added to fresh, calcium‐released extract (released for 60–75 min before addition) at a final concentration of 3,000/μl to assess endogenous or IVT M18BP1 localization as above.
Nuclear permeabilization was verified by incubating an aliquot of nuclei with AlexaFluor 488‐conjugated goat IgG, which is normally excluded by an intact nuclear envelope. Fluorescence signal inside the nucleus indicated successful permeabilization (data not shown).
Immunoprecipitation and λ‐phosphatase treatment
To assay association of full‐length M18BP1‐1 mutants with endogenous CENP‐C, 50 μl reactions containing 5 μl in vitro translated Flag‐M18BP1‐1 in CSF‐arrested or interphase M18BP1‐depleted extract were added to 10 μl Protein A Dynabeads (Invitrogen) coupled to 2.5 μg mouse α‐Flag antibody (Sigma).
To assay association of M18BP1‐1 with Mis18α and Mis18β, 50 μl reactions containing 5 μl Flag‐M18BP1‐1, 2.5 μl Myc‐Mis18α, and 2.5 μl Mis18β IVT proteins in CSF‐arrested or interphase M18BP1‐depleted extract were added to 10 μl Protein A Dynabeads (Invitrogen) coupled to 2.5 μg mouse α‐Flag antibody (Sigma).
To assay association of M18BP1‐1 with HJURP, 50 μl reactions containing 5 μl Flag‐M18BP1‐1 in CSF‐arrested or interphase M18BP1‐depleted extract were added to 10 μl Protein A Dynabeads (Invitrogen) coupled to 2.5 μg rabbit α‐xHJURP antibody.
Following rotation for 1 h at 4°C, beads were collected by magnetization and washed four times in TBSTx. Immunoprecipitates were eluted in sample buffer (50 mM Tris, pH 6.8, 15 mM EDTA, 1 M β‐mercaptoethanol, 3.3% SDS, 10% glycerol, and 1 μg/ml bromophenol blue), resolved by SDS–PAGE, and immunoblotted as described below.
To assess whether phosphorylation was required for the interaction between M18BP1‐1 and CENP‐C, precipitation reactions were prepared as above but scaled to 150 μl. 80 μl of the reaction was added to 20 μl Protein A Dynabeads (Invitrogen) coupled to 5 μg mouse α‐Myc antibody (4A6; Millipore), and 40 μl was added to 10 μl Protein A Dynabeads coupled to 2.5 μg mouse whole IgG (Jackson ImmunoResearch) as a control. Immunoprecipitates were washed three times with TBSTx, then three times with LPPase buffer (50 mM Tris–HCl pH 8.0, 2 mM MnCl2, 0.01% Triton X‐100, 10 mM NaCl). α‐Myc immunoprecipitates were split into two samples, and all samples were resuspended in 50 μl LPPase buffer. Samples were then treated with 200 U lambda phosphatase (P0753S; NEB) or buffer for 30 min at 30°C. Beads were then washed, eluted, and processed for immunoblotting as above.
Histone H1 kinase assays
At the indicated time points, 2–3 μl aliquots of extract were taken from reactions and frozen on dry ice. To measure cyclin‐dependent kinase activity, extract aliquots were thawed and diluted 50‐fold with EB buffer (80 mM β‐glycerol phosphate, 20 mM EGTA, 15 mM MgCl2, pH 7.7). 10 μl of diluted extract was mixed with 8 μl of reaction buffer (32 mM HEPES pH 7.7, 8 mM EGTA, 16 mM MgCl2, 0.5 mM ATP, 0.15 μCi/μl γ‐[32P]‐ATP). For extract samples treated with mitotic kinase inhibitors, H1 kinase reactions included the corresponding inhibitor at the concentration added to extract. 2 μl of 5 mg/ml (10 μg) purified histone H1 (Sigma, 14‐155) was added to each kinase reaction and incubated for 10 min at 30°C. The reaction was stopped by addition of 20 μl of 4× sample buffer (200 mM Tris, pH 6.8, 60 mM EDTA, 4 M β‐mercaptoethanol, 13.2% SDS, 40% glycerol, and 4 μg/ml bromophenol blue), and H1 phosphorylation was detected by autoradiography following SDS–PAGE.
Spindle assembly checkpoint activation upon addition of 10 μg/ml nocodazole and 10,000 sperm/μl was measured by H1 kinase assay as previously described (Chen & Murray, 1997).
GST‐xlCENP‐C1911–1400 and GST‐hsCENP‐C723–943 purification
GST‐Prescission‐xlCENP‐C1191–1400 and GST‐Prescission‐hsCENP‐C723–943 were expressed from modified pGEX plasmid (NEB) in BL21 (DE3) Codon Plus RIPL E. coli. One 2 l culture was grown in 2xYT medium to OD600 of 0.6 at 37°C and then induced with 0.2 mM IPTG for 4 h at 21°C. Bacteria were lysed in GST lysis buffer [10 mM sodium phosphate pH 7.4, 500 mM NaCl, 2.7 mM KCl, 1 mM EDTA, 0.5% Tween‐20, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine hydrochloride, 10 μg/ml LPC (leupeptin, pepstatin A, chymostatin), 0.2 mg/ml lysozyme] by sonication and then clarified by centrifugation at 186,000 g in a Type 45ti rotor (Beckman) for 1 h at 4°C. Clarified lysates were flowed over 2 ml glutathione agarose column (Sigma) equilibrated with GST lysis buffer to capture GST‐tagged protein. Resin was subsequently washed with 40 column volumes lysis buffer containing 0.05% Tween‐20. GST‐CENP‐C1191–1400 was then washed with four column volumes GST wash buffer (50 mM Tris–HCl pH 8.0, 250 mM NaCl, 1 mM DTT) and eluted with GST wash buffer containing 5 mM reduced glutathione. This protein was dialyzed against protein storage buffer (50 mM Tris–HCl pH 8.0, 250 mM NaCl, 1 mM DTT, 10% glycerol), aliquoted, and stored at −80°C.
IVT binding assays
Ten microliter of [35S]‐methionine‐labeled IVT protein was incubated with 1.7 μM GST‐xlCENP‐C1191–1400 or GST‐hsCENP‐C723–943 in 50 mM Tris–HCl pH 8.0, 50 mM NaCl, 0.05% Triton X‐100, and 0.25 mM EDTA. The binding reaction was incubated for 1 h at 4°C. Bound material was recovered using 25 μl glutathione agarose beads, which were then washed three times in 50 mM Tris–HCl pH 8.0, 200 mM NaCl, 0.05% Triton X‐100, and 0.25 mM EDTA. Bound material was separated by SDS–PAGE and visualized by autoradiography. Gels were stained with Coomassie to visualize GST‐CENP‐C recovered.
Fraction bound was quantified using ImageJ (National Institutes of Health). Briefly, a rectangular region of fixed size was drawn around each band in the “input” and “bound” lanes and the mean integrated density measured. To estimate the local background signal, the mean integrated density was also measured for a region of the same size above the bands, and this value was subtracted from the values measured for each band. The “bound” signal was divided by the “input” (adjusted for the amount of the sample loaded per lane) to obtain the fraction bound.
Histone purification
Histones were purified as described previously (Guse et al, 2012; Westhorpe et al, 2015). Briefly, histone H2A and H2B were expressed from a pET3a vector in BL21 (DE3) Codon Plus RIPL E. coli (Agilent Technologies, 230280), grown in 2xYT medium (20 g/l tryptone, 10 g/l yeast extract, and 5 g/l NaCl), and induced for 3 h with 0.25 mM IPTG at an OD600 of 0.6. Bacteria were harvested and resuspended in lysis buffer (20 mM potassium phosphate, pH 6.8, 1 M NaCl, 5 mM 2‐mercaptoethanol, 1 mM PMSF, 1 mM benzamidine, 0.05% NP‐40, and 0.2 mg/ml lysozyme), homogenized using an EmulsiFlex‐C5 (Avestin, Inc.) followed by 1 × 30 s sonication. The lysate was centrifuged (18,000 × g, 20 min, 4°C), and the insoluble pellet was washed in lysis buffer and resuspended in unfolding buffer (7 M guanidine–HCl, 20 mM Tris–HCl, pH 7.5, and 10 mM DTT). After another round of centrifugation, the supernatant from this resuspension was dialyzed into urea buffer (6 M deionized urea, 200 mM NaCl, 10 mM Tris–HCl, pH 8, 1 mM EDTA, 5 mM 2‐mercaptoethanol, and 0.1 mM PMSF). Histones were isolated by chromatography through HiTrap Q and HiTrap S columns in series. Histones were eluted from the HiTrap S column with urea buffer containing 1 M NaCl, dialyzed into water, and lyophilized. To reconstitute H2A/B dimer or H3/H4 tetramer, histones were mixed and resuspended in unfolding buffer followed by dialysis into 2 M NaCl, 10 mM Tris–HCl, pH 7.6, 1 mM EDTA, and 5 mM 2‐mercaptoethanol. H2A/H2B dimers and H3/H4 tetramers were then purified by size‐exclusion chromatography.
Soluble CENP‐A‐Myc‐H4 tetramer was expressed as described (Guse et al, 2012) using the pST39 vector (Tan, 2001). The initial supernatant, isolated as described above, was run through hydroxyapatite (HA; type II 20 μM HA; Bio‐Rad Laboratories) pre‐equilibrated with 20 mM potassium phosphate, pH 6.8. After washing with six column volumes (20 mM potassium phosphate, pH 6.8, 1 M NaCl, and 5 mM 2‐mercaptoethanol), tetramer was eluted with HA buffer containing 3.5 M NaCl. The elution was dialyzed into 10 mM Tris–HCl, pH 7.4, 0.75 M NaCl, 10 mM 2‐mercaptoethanol, and 0.5 mM EDTA. The dialyzed protein was passed through a 1 ml HiTrap SP FastFlow column, washed, and tetramer was eluted over a 20‐column volume gradient into dialysis buffer containing 2 M NaCl. Fractions containing concentrated tetramer were pooled, aliquoted, frozen in liquid nitrogen, and stored at −80°C.
Chromatin bead reconstitution
Biotinylated 19 × 601 array DNA was purified as previously described (Guse et al, 2012). In brief, puC18 containing 19 repeats of the “601” nucleosome positioning sequence (Lowary & Widom, 1998) was purified with a Gigaprep kit (QIAGEN) from SURE2 bacteria grown in Luria Broth media. The plasmid was restriction digested with EcoRI, XbaI, DraI, and HindIII, and the positioning array purified by polyethylene glycol (PEG) precipitation using 0.5% incremental increases in PEG concentration, each with a 10 min, 5,000 × g centrifugation. PEG precipitations with 19 × 601 DNA were dialyzed into TE buffer (10 mM Tris, pH 8 and 0.5 mM EDTA) and concentrated by ethanol precipitation. The EcoRI overhangs were filled with biotin‐14‐dATP (Invitrogen), dCTP, α‐thiο‐dGTP, and α‐thio‐dTTP (ChemCyte) using Klenow fragment 3′‐5′ exo‐ (New England Biolabs).
Reconstituted chromatin containing recombinant nucleosomes was prepared at 2 μM nucleosome concentration by salt dialysis as previously described (Guse et al, 2012). DNA and histones were pooled in high salt buffer (2 M NaCl, 10 mM Tris pH 7.5, 0.25 mM EDTA) and dialyzed over 36 h into low salt buffer (2.5 mM NaCl, 10 mM Tris pH 7.5, 0.25 mM EDTA). H2A/H2B dimers were added at 2.2 × tetramer ratio, and 1.6 tetramers per nucleosome positioning sequence. Reconstituted chromatin was assessed by SyBr Gold (Life Technologies) staining of a 5% acrylamide native PAGE gel after chromatin digestion with AvaI.
Chromatin was bound to streptavidin‐coated M‐280 Dynabeads (Invitrogen, 11205D) prewashed in bead buffer (50 mM Tris pH 7.4, 75 mM NaCl, 0.25 mM EDTA, 0.05% Triton X‐100 and 2.5% polyvinyl alcohol). Chromatin was bound at 2.6 fmol arrays per microgram of bead for 30–60 min at room temperature with constant shaking. After washing in bead buffer, beads were added to rabbit reticulocyte lysate containing in vitro translated protein.
In vitro chromatin binding assays
Reticulocyte lysates containing in vitro translated protein were diluted with 5× CSF‐XBTx (50 mM K‐HEPES, pH 7.7, 500 mM KCl, 250 mM sucrose, 10 mM MgCl2, 0.5 mM CaCl2, 25 mM EGTA, 0.25% Triton X‐100) to a final concentration of 1× CSF‐XBTx. Protein was bound to reconstituted CENP‐A or H3 chromatin by incubating 10 μl diluted IVT protein with 0.25 μl chromatin‐coated beads for 60 min at 21°C. Beads were isolated by magnetization and washed three times with CSF‐XBTx. Beads were fixed for 5 min at 21°C in CSF‐XBTx containing 2% formaldehyde, washed into antibody dilution buffer (20 mM Tris–HCl, pH 7.4, 150 mM NaCl with 0.1% Triton X‐100, and 2% bovine serum albumin), and settled on poly‐L‐lysine‐coated coverslips for 30–60 min before processing for immunofluorescence microscopy.
Human cell line generation
Transfections were carried out with FugeneHD (Promega, E2311) at a ratio of 6 μl FugeneHD:1 μg DNA according to manufacturer's instructions.
CRISPR‐Cas9 engineering was used to generate sfGFP‐AID‐M18BP1 DLD1s. osTIR1/TRex DLD1 cells (gift from Don Cleveland, Ludwig Cancer Institute) were transfected with px458 (Addgene 48138; Ran et al, 2013) modified to replace GFP with mCherry (a gift from Raj Rohatgi, Stanford University) and containing the M18BP1 sgRNA (sgRNA sequence: 5′‐AAGAATTTACTTACCTCCAG‐3′). These cells were co‐transfected at a 1:1 mass ratio with pUC19 containing the coding sequence for sfGFP‐AID flanked by sequences homologous to the 400 bp upstream and downstream of the M18BP1 start codon at the endogenous locus. A silent mutation was introduced at the PAM site to prevent re‐editing. At 48 h after transfection, 25,000 mCherry‐positive cells were sorted to one well of a 6‐well plate by flow cytometry using a Sony SH800Z Cell Sorter to select cells expressing Cas9. Cells were expanded to a 10‐cm dish, and 8 days after the initial sort, single mCherry‐negative and sfGFP‐positive cells were re‐sorted to each well of a 96‐well plate. Successful sfGFP‐AID integrants were identified by immunoblotting with a rabbit α‐GFP antibody and by PCR from genomic DNA (ASON 2497: AGAATAGATCTTGTAGTTGCTGCCT; ASON 2498: GGCTGTTCTTGCTTGTCACG).
To generate complementation lines, AID‐M18BP1 cells were co‐transfected with pcDNA5/FRT/TO containing the full‐length human M18BP1 cDNA with mRuby2 and 3xFlag fused to the C terminus and with pOG44 encoding Flp recombinase (1:1 mass ratio). At 48 h after transfection, culture medium was replaced and supplemented with 50 μg/ml hygromycin to select for stable M18BP1‐mRuby‐Flag integrants. Hygromycin‐resistant colonies were obtained after 1 month of selection and pooled for localization experiments.
Degradation of endogenous CENP‐C or M18BP1 was induced by supplementation of the culture medium with 1 mM indole‐3‐acetic acid (Fisher, 50‐213‐368) for 24 h. Transgene expression was induced by addition of 10 ng/ml doxycycline to the culture medium for 24 h.
To visualize M18BP1 degradation by immunoblotting, cells were collected by trypsinization, washed once in PBS, and then lysed by sonication in lysis buffer (25 mM Tris–HCl pH 7.4, 100 mM NaCl, 5 mM MgCl2, 0.2% Igepal, 10% glycerol, 10 μg/ml LPC, 1 mM PMSF, 1 mM beta‐glycerol‐phosphate, 1 μM microcystin LR). Lysate concentrations were measured by Bradford assay (Bio‐Rad) and normalized across samples by dilution with additional lysis buffer. After addition of sample buffer, immunoblotting was performed as described below.
For localization experiments, cells were grown on acid washed coverglass. Cells were fixed with 3.4% formaldehyde in PBS at 37°C for 5 min and then permeabilized with 0.5% Triton X‐100 in PBS for 5 min at room temperature. Coverslips were then processed for immunofluorescence.
Immunofluorescence
Coverslips were first blocked in antibody dilution buffer (AbDil; 20 mM Tris–HCl, pH 7.4, 150 mM NaCl with 0.1% Triton X‐100, and 2% bovine serum albumin) for 30 min. Coverslips were exposed to primary antibody diluted in AbDil for 15–30 min, washed three times with AbDil, and then exposed to AlexaFluor‐conjugated secondary antibodies (Life Technologies; Jackson ImmunoResearch Laboratories, Inc.). When required, coverslips were blocked with 1 mg/ml whole rabbit or whole mouse IgG in AbDil followed by staining with directly conjugated primary antibodies. Sperm nuclei and human cell coverslips were stained with 10 μg/ml Hoechst 33258 in AbDil for DNA. Coverslips were mounted in 90% glycerol, 10 mM Tris–HCl, pH 8.8, 0.5% p‐phenylenediamine, sealed to a slide with clear nail polish and stored at −20°C.
Primary antibodies used were 2 μg/ml mouse α‐Flag (Sigma, F1804), 0.25–0.5 μg/ml mouse α‐Myc (EMD Millipore, 4A6), 1 μg/ml rabbit‐α‐xCENP‐A, 1 μg/ml rabbit‐α‐xCENP‐C (Milks et al, 2009), 1.5 μg/ml rabbit‐α‐xM18BP1, 0.2 μg/ml mouse α‐tubulin (Sigma, clone DM1α, T6199), and 1:100 human α‐centromere serum (Antibodies, Inc., 15‐234‐0001). Secondary antibodies used were AlexaFluor 488‐, 568‐, and 647‐conjugated goat α‐mouse or α‐rabbit (Life Technologies) and AlexaFluor 647‐conjugated donkey α‐rabbit (Jackson ImmunoResearch Laboratories, Inc.). All secondaries were used at 1–2 μg/ml.
Image acquisition and processing
Imaging was performed on an IX70 Olympus microscope with a DeltaVision system (Applied Precision) a Sedat quad‐pass filter set (Semrock) and monochromatic solid‐state illuminators, controlled via softWoRx 4.1.0 software (Applied Precision). Images of sperm nuclei were acquired using a 60 × 1.4 NA Plan Apochromat oil immersion lens (Olympus). Images of chromatin‐coated beads and human cells were acquired using a 100 × 1.4 NA oil immersion objective (Olympus). Images were acquired with a charge‐coupled device camera (CoolSNAP HQ; Photometrics) and digitized to 12 bits. Z sections were taken at 0.2‐μm intervals (for sperm nuclei and human cells) or 0.4‐μm intervals (for beads). Displayed images of sperm nuclei and human cells are maximum intensity projections of deconvolved z‐stacks. Displayed images of chromatin‐coated beads are a single plane from a z‐stack.
Image analysis of Xenopus sperm (Moree et al, 2011) or chromatin‐coated beads (Westhorpe et al, 2015) was performed using custom software as previously described. For sperm, at least 200 centromeres among at least 15 nuclei were counted in each condition in each experiment. For reconstituted chromatin, ~50–100 beads were quantified per condition per experiment. For human cells, at least 15 pairs of midbody‐positive G1 cells were quantified per condition per experiment.
Immunoblotting
Immunoblots were performed as previously described (Moree et al, 2011; French et al, 2017). Briefly, samples were resolved by SDS–PAGE and transferred onto polyvinylidene fluoride membrane (Bio‐Rad Laboratories). For immunoblotting CENP‐C, samples were transferred in CAPS transfer buffer (10 mM 3‐(cyclohexylamino)‐1‐propanesulfonic acid, pH 11.3, 0.1% SDS, 20% methanol). All other samples were transferred in Tris–glycine transfer buffer (20 mM Tris–HCl, 200 mM glycine). Samples containing 0.75–1.5 μl IVT protein, 1.5 μl Xenopus egg extract, or 50 μg sonicated whole‐cell lysate were loaded per lane.
Membranes were blocked in 4% milk in PBSTx (10 mM sodium phosphate pH 7.4, 150 mM NaCl, 2.7 mM KCl, 0.2% Tween 20), probed with primary antibody, and detected using AlexaFluor 647‐conjugated goat‐α‐rabbit or goat‐α‐mouse antibodies (Jackson ImmunoResearch Laboratories, Inc.). Detection was performed with a fluorescence imager (VersaDoc; Bio‐Rad Laboratories, Inc.). In Figs 2B and 3D, and EV6B and F, M18BP1 was detected using horseradish peroxidase‐conjugated goat‐α‐mouse secondary antibodies followed by chemiluminescence (Thermo Scientific). Chemiluminescent membranes were exposed to X‐ray film for 10–120 s and developed with a Konica SRX‐101A film processor.
Primary antibodies used were 1 μg/ml mouse‐α‐Flag (Sigma, F1804), 1 μg/ml mouse‐α‐Myc (EMD Millipore, 4A6), 5 μg/ml rabbit‐α‐xM18BP1, 1 μg/ml rabbit‐α‐xCENP‐C, 2 μg/ml rabbit‐α‐xHJURP (raised against GST‐xHJURP42–194 and purified against His‐xHJURP42–194), 2 μg/ml rabbit α‐GFP (raised and purified against purified GFP), and 0.2 μg/ml mouse‐α‐tubulin (Dm1α; Sigma). All secondary antibodies were used at 0.4 μg/ml.
Mass spectrometry
For identification of post‐translational modifications of MBP‐Prescission‐xM18BP1‐1161–580, protein was added to 100 μl M18BP1‐depleted metaphase or interphase Xenopus egg extract at 500 nM and incubated for 75 min at 16–18°C. Reactions were then supplemented with 10 mM beta‐glycerol‐phosphate, 1 μM microcystin LR, and 1 mM benzamidine–HCl, and 80 μl of each reaction was then incubated for 2 h with 68 μg rabbit α‐MBP antibody coupled to 3.4 mg Dynabeads (Thermo Fisher, 14301) at 4°C. Beads were re‐isolated by magnetization and washed three times with TBSTx. Proteins were eluted in sample buffer.
Isolated MBP‐Prescission‐xM18BP1‐1161–580 protein separated by SDS–PAGE in 10% acrylamide, stained with Coomassie R‐250, and the protein bands excised. Mass spectrometry was performed by the Taplin Mass Spectrometry Facility. Excised gel bands were cut into approximately 1‐mm3 pieces, reduced with 1 mM dithiothreitol (DTT) for 30 min at 60°C and then alkylated with 5 mM iodoacetamide for 15 min in the dark at room temperature. Gel pieces were trypsin‐digested in the gel, washed, and dehydrated with acetonitrile for 10 min followed by removal of acetonitrile. Gel pieces were dried in a SpeedVac and rehydrated with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing‐grade trypsin (Promega, Madison, WI) at 4°C. Samples were then incubated at 37°C overnight. Peptides were later extracted by removing the ammonium bicarbonate solution, washing once with a solution containing 50% acetonitrile and 1% formic acid, and the extracts were dried in a SpeedVac (~1 h). The samples were then stored at 4°C until analysis. For analysis, samples were reconstituted in 5–10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano‐scale reverse‐phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter × ~30 cm length) with a flame‐drawn tip. After equilibrating the column, each sample was loaded via a Famos autosampler (LC Packings, San Francisco CA). A gradient was formed, and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As each peptide was eluted, they were subjected to electrospray ionization before entering into an LTQ Orbitrap Velos Pro ion‐trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Eluting peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein or translated nucleotide databases with the acquired fragmentation pattern by the software program, Sequest (ThermoFinnigan, San Jose, CA). The modification of 79.9663 mass units to serine, threonine, and tyrosine was included in the database searches to determine phosphopeptides. Phosphorylation assignments were determined by the Ascore algorithm. All databases include a reversed version of all the sequences, and the data were filtered to between a 1 and 2% peptide false discovery rate.
Protein alignment
Protein sequences homologous to the CENP‐C binding domain of Xenopus M18BP1‐1 were identified using NCBI PSI‐BLAST with an input query of the full‐length protein. Multiple sequence alignments were performed with MAFFT (version 7.305b; Katoh et al, 2002) using default parameters.
Author contributions
BTF performed the experiments. BTF and AFS designed the experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank members of the Straight lab for their critical feedback on the project and their comments on this manuscript. B.T.F. was supported by NIH T32 GM007276 and NSF DGE‐1147470. This work was supported by NIH R01 GM074728 to A.F.S.
The EMBO Journal (2019) 38: e100093
References
- Barnhart MC, Kuich PH, Stellfox ME, Ward JA, Bassett EA, Black BE, Foltz DR (2011) HJURP is a CENP‐A chromatin assembly factor sufficient to form a functional de novo kinetochore. J Cell Biol 194: 229–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernad R, Sanchez P, Rivera T, Rodriguez‐Corsino M, Boyarchuk E, Vassias I, Ray‐Gallet D, Arnaoutov A, Dasso M, Almouzni G, Losada A (2011) Xenopus HJURP and condensin II are required for CENP‐A assembly. J Cell Biol 192: 569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow JJ, Laskey RA (1988) A role for the nuclear envelope in controlling DNA replication within the cell cycle. Nature 332: 546–548 [DOI] [PubMed] [Google Scholar]
- Budde PP, Kumagai A, Dunphy WG, Heald R (2001) Regulation of Op18 during spindle assembly in Xenopus egg extracts. J Cell Biol 153: 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF (2010) Dual recognition of CENP‐A nucleosomes is required for centromere assembly. J Cell Biol 189: 1143–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RH, Murray A (1997) Characterization of spindle assembly checkpoint in Xenopus egg extracts. Methods Enzymol 283: 572–584 [DOI] [PubMed] [Google Scholar]
- Dambacher S, Deng W, Hahn M, Sadic D, Frohlich J, Nuber A, Hoischen C, Diekmann S, Leonhardt H, Schotta G (2012) CENP‐C facilitates the recruitment of M18BP1 to centromeric chromatin. Nucleus 3: 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Murray A, Mitchison TJ, Walczak CE (1999) The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro . Methods Cell Biol 61: 385–412 [DOI] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray‐Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni‐Pettinotti G (2009) HJURP is a cell‐cycle‐dependent maintenance and deposition factor of CENP‐A at centromeres. Cell 137: 485–497 [DOI] [PubMed] [Google Scholar]
- Fachinetti D, Diego Folco H, Nechemia‐Arbely Y, Valente LP, Nguyen K, Wong AJ, Zhu Q, Holland AJ, Desai A, Jansen LE, Cleveland DW (2013) A two‐step mechanism for epigenetic specification of centromere identity and function. Nat Cell Biol 15: 1056–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Han JS, McMahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW (2015) DNA sequence‐specific binding of CENP‐B enhances the fidelity of human centromere function. Dev Cell 33: 314–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Logsdon GA, Abdullah A, Selzer EB, Cleveland DW, Black BE (2017) CENP‐A modifications on Ser68 and Lys124 are dispensable for establishment, maintenance, and long‐term function of human centromeres. Dev Cell 40: 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR III, Bassett EA, Wood S, Black BE, Cleveland DW (2009) Centromere‐specific assembly of CENP‐a nucleosomes is mediated by HJURP. Cell 137: 472–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- French BT, Westhorpe FG, Limouse C, Straight AF (2017) Xenopus laevis M18BP1 directly binds existing CENP‐A nucleosomes to promote centromeric chromatin assembly. Dev Cell 42: 190–199 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Hayashi T, Kiyomitsu T, Toyoda Y, Kokubu A, Obuse C, Yanagida M (2007) Priming of centromere for CENP‐A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev Cell 12: 17–30 [DOI] [PubMed] [Google Scholar]
- Gadea BB, Ruderman JV (2005) Aurora kinase inhibitor ZM447439 blocks chromosome‐induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol Biol Cell 16: 1305–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, Straight AF (2011) In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477: 354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Fuller CJ, Straight AF (2012) A cell‐free system for functional centromere and kinetochore assembly. Nat Protoc 7: 1847–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Fujita Y, Iwasaki O, Adachi Y, Takahashi K, Yanagida M (2004) Mis16 and Mis18 are required for CENP‐A loading and histone deacetylation at centromeres. Cell 118: 715–729 [DOI] [PubMed] [Google Scholar]
- Holland AJ, Fachinetti D, Han JS, Cleveland DW (2012) Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci USA 109: E3350–E3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Shang WH, Hara M, Ariyoshi M, Arimura Y, Fujita R, Kurumizaka H, Fukagawa T (2017) Association of M18BP1/KNL2 with CENP‐A nucleosome is essential for centromere formation in non‐mammalian vertebrates. Dev Cell 42: 181–189 [DOI] [PubMed] [Google Scholar]
- Hu H, Liu Y, Wang M, Fang J, Huang H, Yang N, Li Y, Wang J, Yao X, Shi Y, Li G, Xu RM (2011) Structure of a CENP‐A‐histone H4 heterodimer in complex with chaperone HJURP. Genes Dev 25: 901–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen LE, Black BE, Foltz DR, Cleveland DW (2007) Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol 176: 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpen GH, Allshire RC (1997) The case for epigenetic effects on centromere identity and function. Trends Genet 13: 489–496 [DOI] [PubMed] [Google Scholar]
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y (2013) A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP‐C. Science 340: 1110–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IS, Lee M, Park KC, Jeon Y, Park JH, Hwang EJ, Jeon TI, Ko S, Lee H, Baek SH, Kim KI (2012) Roles of Mis18alpha in epigenetic regulation of centromeric chromatin and CENP‐A loading. Mol Cell 46: 260–273 [DOI] [PubMed] [Google Scholar]
- Kral L (2015) Possible identification of CENP‐C in fish and the presence of the CENP‐C motif in M18BP1 of vertebrates. F1000Res 4: 474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagana A, Dorn JF, De Rop V, Ladouceur AM, Maddox AS, Maddox PS (2010) A small GTPase molecular switch regulates epigenetic centromere maintenance by stabilizing newly incorporated CENP‐A. Nat Cell Biol 12: 1186–1193 [DOI] [PubMed] [Google Scholar]
- Lermontova I, Kuhlmann M, Friedel S, Rutten T, Heckmann S, Sandmann M, Demidov D, Schubert V, Schubert I (2013) Arabidopsis kinetochore null2 is an upstream component for centromeric histone H3 variant cenH3 deposition at centromeres. Plant Cell 25: 3389–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence‐directed nucleosome positioning. J Mol Biol 276: 19–42 [DOI] [PubMed] [Google Scholar]
- Maddox PS, Hyndman F, Monen J, Oegema K, Desai A (2007) Functional genomics identifies a Myb domain‐containing protein family required for assembly of CENP‐A chromatin. J Cell Biol 176: 757–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM (2014) Polo‐like kinase 1 licenses CENP‐A deposition at centromeres. Cell 158: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley KL, Cheeseman IM (2016) The molecular basis for centromere identity and function. Nat Rev Mol Cell Biol 17: 16–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiburo MJ, Padeken J, Fulop S, Schepers A, Heun P (2011) Drosophila CENH3 is sufficient for centromere formation. Science 334: 686–690 [DOI] [PubMed] [Google Scholar]
- Milks KJ, Moree B, Straight AF (2009) Dissection of CENP‐C‐directed centromere and kinetochore assembly. Mol Biol Cell 20: 4246–4255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moree B, Meyer CB, Fuller CJ, Straight AF (2011) CENP‐C recruits M18BP1 to centromeres to promote CENP‐A chromatin assembly. J Cell Biol 194: 855–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Montes de Oca R, Lacoste N, Dingli F, Loew D, Almouzni G (2014) Phosphorylation and DNA binding of HJURP determine its centromeric recruitment and function in CenH3(CENP‐A) loading. Cell Rep 8: 190–203 [DOI] [PubMed] [Google Scholar]
- Musacchio A, Desai A (2017) A molecular view of kinetochore assembly and function. Biology (Basel) 6: E5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardi IK, Zasadzińska E, Stellfox ME, Knippler CM, Foltz DR (2016) Licensing of centromeric chromatin assembly through the Mis18α‐Mis18β heterotetramer. Mol Cell 61: 774–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M (2009) An auxin‐based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6: 917–922 [DOI] [PubMed] [Google Scholar]
- Ohzeki J, Bergmann JH, Kouprina N, Noskov VN, Nakano M, Kimura H, Earnshaw WC, Larionov V, Masumoto H (2012) Breaking the HAC Barrier: histone H3K9 acetyl/methyl balance regulates CENP‐A assembly. EMBO J 31: 2391–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J, Shono N, Otake K, Martins NM, Kugou K, Kimura H, Nagase T, Larionov V, Earnshaw WC, Masumoto H (2016) KAT7/HBO1/MYST2 regulates CENP‐A chromatin assembly by antagonizing Suv39 h1‐mediated centromere inactivation. Dev Cell 37: 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer DK, O'Day K, Wener MH, Andrews BS, Margolis RL (1987) A 17‐kD centromere protein (CENP‐A) copurifies with nucleosome core particles and with histones. J Cell Biol 104: 805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Klare K, Petrovic A, Take A, Walstein K, Singh P, Rondelet A, Bird AW, Musacchio A (2017) CDK‐regulated dimerization of M18BP1 on a Mis18 hexamer is necessary for CENP‐A loading. Elife 6: e23352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perpelescu M, Hori T, Toyoda A, Misu S, Monma N, Ikeo K, Obuse C, Fujiyama A, Fukagawa T (2015) HJURP is involved in the expansion of centromeric chromatin. Mol Biol Cell 26: 2742–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann M, Talbert P, Demidov D, Kuhlmann M, Rutten T, Conrad U, Lermontova I (2017) Targeting of Arabidopsis KNL2 to centromeres depends on the conserved CENPC‐k motif in its C terminus. Plant Cell 29: 144–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A (2010) Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol 190: 73–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S, Fukui A, Hikosaka A, Suzuki A, Kondo M, van Heeringen SJ, Quigley I, Heinz S, Ogino H, Ochi H, Hellsten U, Lyons JB, Simakov O, Putnam N, Stites J et al (2016) Genome evolution in the allotetraploid frog Xenopus laevis . Nature 538: 336–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang WH, Hori T, Westhorpe FG, Godek KM, Toyoda A, Misu S, Monma N, Ikeo K, Carroll CW, Takami Y, Fujiyama A, Kimura H, Straight AF, Fukagawa T (2016) Acetylation of histone H4 lysine 5 and 12 is required for CENP‐A deposition into centromeres. Nat Commun 7: 13465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shono N, Ohzeki J, Otake K, Martins NM, Nagase T, Kimura H, Larionov V, Earnshaw WC, Masumoto H (2015) CENP‐C and CENP‐I are key connecting factors for kinetochore and CENP‐A assembly. J Cell Sci 128: 4572–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Bodor DL, Stellfox ME, Martins NM, Hochegger H, Foltz DR, Jansen LE (2012) Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev Cell 22: 52–63 [DOI] [PubMed] [Google Scholar]
- Spiller F, Medina‐Pritchard B, Abad MA, Wear MA, Molina O, Earnshaw WC, Jeyaprakash AA (2017) Molecular basis for Cdk1‐regulated timing of Mis18 complex assembly and CENP‐A deposition. EMBO Rep 18: 894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankovic A, Guo LY, Mata JF, Bodor DL, Cao XJ, Bailey AO, Shabanowitz J, Hunt DF, Garcia BA, Black BE, Jansen LE (2017) A dual inhibitory mechanism sufficient to maintain cell‐cycle‐restricted CENP‐A assembly. Mol Cell 65: 231–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellfox Madison E, Nardi Isaac K, Knippler Christina M, Foltz Daniel R (2016) Differential binding partners of the Mis18α/β YIPPEE domains regulate Mis18 complex recruitment to centromeres. Cell Rep 15: 2127–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler S, Keith KC, Curnick KE, Fitzgerald‐Hayes M (1995) A mutation in CSE4, an essential gene encoding a novel chromatin‐associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev 9: 573–586 [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Muller S, Blumer J, Klare K, Musacchio A, Almouzni G (2015) HJURP involvement in de novo CenH3(CENP‐A) and CENP‐C recruitment. Cell Rep 11: 22–32 [DOI] [PubMed] [Google Scholar]
- Tan S (2001) A modular polycistronic expression system for overexpressing protein complexes in Escherichia coli . Protein Expr Purif 21: 224–234 [DOI] [PubMed] [Google Scholar]
- Van Hooser AA, Ouspenski II, Gregson HC, Starr DA, Yen TJ, Goldberg ML, Yokomori K, Earnshaw WC, Sullivan KF, Brinkley BR (2001) Specification of kinetochore‐forming chromatin by the histone H3 variant CENP‐A. J Cell Sci 114: 3529–3542 [DOI] [PubMed] [Google Scholar]
- Wang J, Liu X, Dou Z, Chen L, Jiang H, Fu C, Fu G, Liu D, Zhang J, Zhu T, Fang J, Zang J, Cheng J, Teng M, Ding X, Yao X (2014) Mitotic regulator Mis18beta interacts with and specifies the centromeric assembly of molecular chaperone holliday junction recognition protein (HJURP). J Biol Chem 289: 8326–8336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhorpe FG, Fuller CJ, Straight AF (2015) A cell‐free CENP‐A assembly system defines the chromatin requirements for centromere maintenance. J Cell Biol 209: 789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhorpe FG, Straight AF (2015) The centromere: epigenetic control of chromosome segregation during mitosis. Cold Spring Harb Perspect Biol 7: a015818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda K, Ando S, Morishita S, Houmura K, Hashimoto K, Takeyasu K, Okazaki T (2000) Human centromere protein A (CENP‐A) can replace histone H3 in nucleosome reconstitution in vitro . Proc Natl Acad Sci USA 97: 7266–7271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou X, Wang W, Deng W, Fang J, Hu H, Wang Z, Li S, Cui L, Shen J, Zhai L, Peng S, Wong J, Dong S, Yuan Z, Ou G, Zhang X, Xu P, Lou J, Yang N et al (2015) Dynamic phosphorylation of CENP‐A at Ser68 orchestrates its cell‐cycle‐dependent deposition at centromeres. Dev Cell 32: 68–81 [DOI] [PubMed] [Google Scholar]
- Zhang D, Martyniuk CJ, Trudeau VL (2006) SANTA domain: a novel conserved protein module in Eukaryota with potential involvement in chromatin regulation. Bioinformatics 22: 2459–2462 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File