

Short abstract
With the worldwide number of human immunodeficiency virus positive patients stagnant and the increasing emergence of viral strains resistant to current treatment, the development of novel anti-human immunodeficiency virus drug candidates is a perpetual quest of medicinal chemists. Herein, we report a novel group of diarylpyrimidines, non-nucleoside reverse transcriptase inhibitors, which represents an important class of current anti-human immunodeficiency virus therapy. Series of diarylpyrimidines containing o,o-difluorophenyl (A-arm), 4-cyanophenylamino (B-arm), and a small substituent (e.g. NH2, OMe) at positions 2, 4, and 6 of the pyrimidine ring were prepared. The A-arm was modified in the para position (F or OMe) and linked to the central pyrimidine core with a variable spacer (CO, O, NH). Antiviral activities of 20 compounds were measured against wild type human immunodeficiency virus-1 and mutant reverse transcriptase strains (K103N, Y181C) using a cytoprotection assay. To the most promising structural motives belong the o,o-difluoro-p-methoxy A-arm in position 4, and the amino group in position 6 of pyrimidine. Single digit nanomolar activities with no significant toxicity (CC50 > 17,000 nM) were found for compounds 35 (EC50 = 2 nM), 37 (EC50 = 3 nM), and 13 (EC50 = 4 nM) having O, NH, and CO linkers, respectively.
Keywords: Diarylpyrimidine, human immunodeficiency virus, non-nucleoside reverse transcriptase inhibitor, etravirine, rilpivirine
Introduction
Human immunodeficiency virus (HIV), which causes acquired immune deficiency syndrome (AIDS), is one of the greatest threats of the modern age, annually killing one million people worldwide according to the WHO 2016 report.1 Numbers of AIDS patients are not decreasing in spite of billions of dollars being spent on public education, prevention, and treatment. In economically developed countries, current therapy allows the majority of treated patients to live with AIDS for >10 years, while most untreated patients die within two years of AIDS onset.2 Additionally, AIDS treatment is particularly challenging due to mutations that yield new viral strains as well as a growing resistance to marketed drugs.3 Continuing development of new treatments is therefore critical.
Etravirine (ETR)4–6 and rilpivirine (RPV)7,8 are highly potent, FDA approved non-nucleoside reverse transcriptase (RT) inhibitors (NNRTIs) belonging to the diarylpyrimidine (DAPY) class of drugs. (Figure 1).3,9–12 RT is an essential enzyme in the HIV life cycle and DAPY compounds bind to its allosteric hydrophobic site. DAPY analogues, containing a characteristic “horseshoe” or “U-shape” structure,13 have been studied extensively during the last two decades for their high potency and relatively low cytotoxicity. Due to the rapid emergence of multi-drug resistant HIV strains (e.g. K103N and Y181C for NNRTIs), continuous effort has identified a number of new NNRTI drug candidates.13–15
Figure 1.

Structures of FDA approved NNRTIs etravirine (ETR) and rilpivirine (RPV), and a general structure of newly synthesized derivatives.
Various structure–activity relationship studies were performed focusing on characterization and optimization of DAPY structural motives important for high anti-HIV potency.9 The general structure of DAPY analogues consists of a central heteroaryl core (usually pyrimidine) bearing two aryl rings connected using various linkers. Currently, several type of linkers (Y = CO, NH, O) between the pyrimidine core and A-arm are being part of highly potent NNRTIs connecting distinct substitutions on the A and B aromatic rings (Figure 1). Since different ortho substituents lead to the formation of atropoisomers, the A-arm typically contains one para and two identical ortho substituents. The B arm of choice in anti-HIV DAPY research is a 4-cyanophenylamino moiety, attached through position 2 of the pyrimidine ring, as in RPV and ETR.
Well-studied linkers connecting the A-arm to the pyrimidine core include oxygen (ETR) and nitrogen (RPV), which have no or limited capacity for further modifications. Introducing a carbonyl linker has opened new possibilities for expanding this key region of the DAPY structure.
The first published compounds contained an unmodified carbonyl linker,16 which was later expanded by reacting the linker with hydrazine17 or hydroxylamine18 forming Schiff bases. Schiff bases with amines provided, after reduction of the imino double bond, (cyclopropylamino)methylenes19 or (alkylamino)methylenes.20 Further types of carbon-based linkers include halomethylene,21 cyanomethylene,22 and hydroxymethylene23 linkers. Additionally, hydroxy(alkyl)methylene analogues were prepared by reacting alkylmagnesium compounds with the carbonyl group.24 Recently, diatomic linkers for increased conformational flexibility have been described.25
Our previous work,26 as well as other published reports,27 demonstrates that presence of ortho substituents on the A-arm is critical for anti-HIV activity. While RPV and ETR bear two methyl groups, a similar effect on antiviral activity was observed for o,o-difluoro28 or o,o-dichloro29 substitution.
Based on literature data,29 an effective NNRTI DAPY inhibitor consists of a pyrimidine ring bearing a 4-cyanophenylamino arm in position 2 and an o,o-disubstituted phenyl arm connected to position 4 through a short linker.
The goal of our work was to develop novel compounds with increased potency, better resistance profiles, and enhanced drug-like properties to improve the efficacy of current anti-HIV therapy. Our current research focuses on a p-substituted o,o-difluoro A-arm, a standard 4-cyanophenylamino B-arm, preferentially a carbonyl linker, and a substitution at position 6 of the pyrimidine core.
Developing a new synthetic strategy for DAPY methanones in our group26 gave promising compounds that led us to explore advanced analogues modified in positions 2, 4, and 6 of the pyrimidine ring. Such compounds, which combine an o,o-difluorophenyl A-arm with a carbonyl linker, have not been systematically studied through varying the substituents on the pyrimidine ring. Furthermore, compounds bearing nitrogen and oxygen linkers were also prepared.
Results and discussion
Chemistry
The synthetic procedure for preparation of carbonyl linked DAPYs developed in our group26 was used to synthesize a 2,4,6-trisubstituted pyrimidine series. Positions 2, 4, and 6 were substituted with 4-cyanophenylamino, o,o-difluorophenylmethanone, and an amino group, respectively. Initially 2,4,6-trichloropyrimidine was reacted with o,o-difluorobenzaldehyde and sodium hydride using N,N′-dimethylbenzimidazolium iodide as a catalyst (Scheme 1).30,31 Two dichloropyrimidine regioisomers were isolated for each aldehyde with the carbonyl group either in the primarily desired position 4 (compounds 1, 2) or in position 2 (3, 4). The next step in the synthetic sequence was nucleophilic substitution of one chlorine atom by ammonia.32 In the case of 2-(arylmethanone)pyrimidines 3 and 4, only single monoamino products 5 and 6 were formed due to the symmetric nature of the substrates. However, for 4-(arylmethanone)pyrimidines 1 and 2, two monoamino isomers were formed and isolated, with the amino group in position 6 (7, 8), as primarily desired, or in position 2 (9, 10).
Scheme 1.

Conditions and reagents: (a) NaH, N,N′-dimethylbenzimidazolium iodide, dioxane, 60°C and (b) ethanolic ammonia (2.5 M), 25°C.
Each monochloropyrimidine derivative was treated with 4-aminobenzonitrile under Buchwald–Hartwig reaction conditions (Scheme 2).33 Trisubstituted pyrimidine derivatives 11–16 were isolated and the exact positions of each substituent on the pyrimidine ring were assigned using nuclear magnetic resonance (NMR), (more details in the next subsection).
Scheme 2.
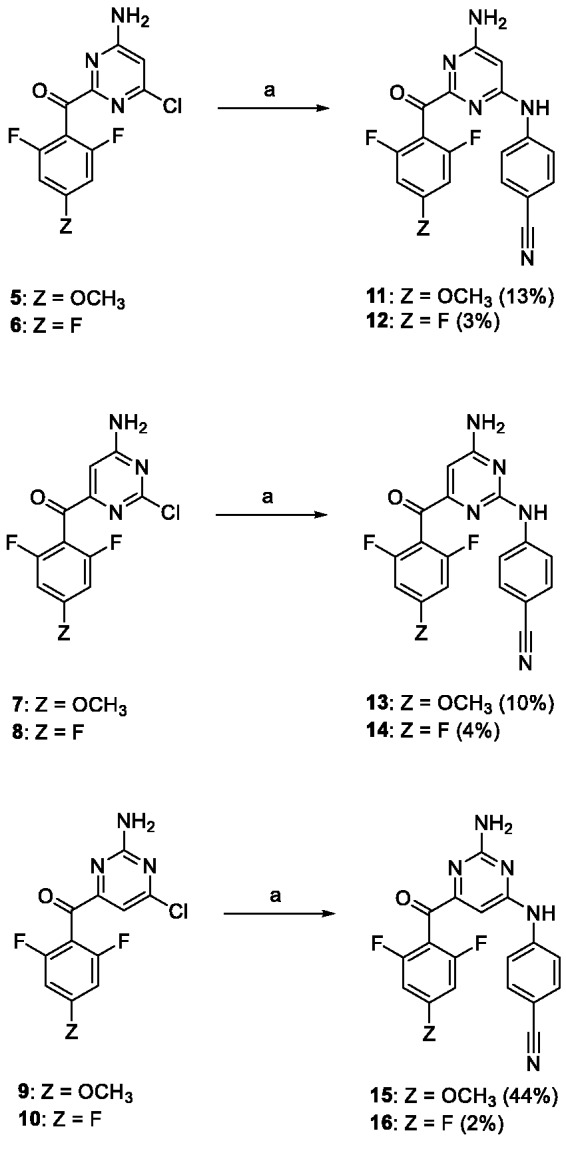
Conditions and reagents: (a) 4-aminobenzonitrile, Pd(OAc)2, XantPhos, Cs2CO3, dioxane, 100°C.
Compounds 13 and 14 were the most potent methanone derivatives, exhibiting low nanomolar activities in an anti-HIV assay (Table 1). These compounds contain the 4-cyanophenylamino moiety in position 2 of the pyrimidine ring, which is in agreement with previously published data.29
Table 1.
Anti-HIV activity (wild type, EC50) and toxicity (CC50) in the MT-4 cell line (n = 3).
| No. | EC50 w.t. (µM) | CC50 (µM) | SI | EC50 K103N (µM) | EC50 Y181C (µM) | X | Y | Z |
|---|---|---|---|---|---|---|---|---|
| 11 | 0.874 | 6.24 | 7.14 | n.d | n.d. | NH2 | 2-CO | OMe |
| 12 | 1.727 | 42.0 | 24.3 | n.d. | n.d. | NH2 | 2-CO | F |
| 13 | 0.004 | 19.6 | 4900 | 0.226 | 0.797 | NH2 | CO | OMe |
| 14 | 0.026 | 24.3 | 935 | 1.703 | 3.899 | NH2 | CO | F |
| 15 | 2.200 | 7.70 | 3.50 | n.d. | n.d. | 2-NH2 | CO | OMe |
| 16 | 40.00 | 0.981 | 0.025 | n.d. | n.d. | 2-NH2 | CO | F |
| 18 | 0.160 | 57.1. | 357 | n.d. | n.d. | Me | CO | F |
| 22 | 0.473 | 9.16 | 19.4 | n.d. | n.d. | OMe | CO | F |
| 27 | 0.019 | 16.1 | 847 | 0.413 | 1.641 | Cl | O | OMe |
| 28 | 0.053 | 14.0 | 264 | 0.657 | 2.533 | Cl | O | F |
| 29 | 0.006 | 16.0 | 2667 | 0.204 | 0.651 | Cl | NH | OMe |
| 30 | 0.027 | 10.2 | 378 | 1.021 | 1.998 | Cl | NH | F |
| 31 | 0.019 | 15.8 | 832 | 0.444 | 4.366 | OMe | O | OMe |
| 32 | 0.054 | 50.0 | 926 | 0.560 | 7.695 | OMe | O | F |
| 33 | 0.007 | 39.0 | 5571 | n.d. | n.d. | OMe | NH | OMe |
| 34 | 0.022 | 9.99 | 454 | 1.207 | 2.015 | OMe | NH | F |
| 35 | 0.002 | 21.7 | 10,850 | 0.036 | 0.637 | NH2 | O | OMe |
| 36 | 0.067 | 19.6 | 293 | 0.067 | 6.662 | NH2 | O | F |
| 37 | 0.003 | 17.2 | 5733 | 0.062 | 0.437 | NH2 | NH | OMe |
| 38 | 0.027 | 34.8 | 1289 | 1.654 | 13.74 | NH2 | NH | F |
| 39a | 0.005 | 57.1 | 11,420 | 0.347 | 0.481 | H | CO | OMe |
| 40a | 0.025 | 57.1 | 2284 | 6.882 | 6.661 | H | CO | F |
| ETR | 0.002 | 5.88 | 2940 | 0.002 | 0.012 | – | – | – |
| EFV | 0.001 | 20.6 | 20,600 | 0.101 | 0.005 | – | – | – |
SI: selectivity index; EFV: efavirenz; ETR: etravirine; n.d.: not determined. CC50/EC50 ratio. Antiviral activities against HIV-1 encoding RT mutations K103N or Y181C were also evaluated.
Synthesis described previously.26
Since derivative 13 (X = NH2, 4.4 nM) demonstrated a similar anti-HIV potency to previously reported26 analogue 39 (Figure 2), which lacks the X = NH2 group (X = H, 4.0 nM), we further investigated position 6 of the pyrimidine ring.
Figure 2.
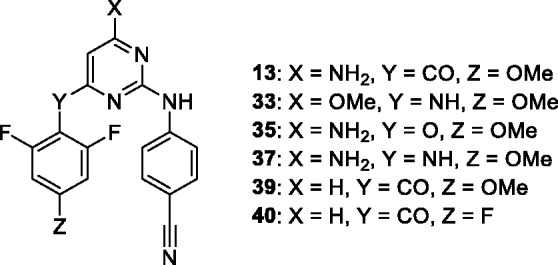
Compounds with low nanomolar anti-HIV activity (wild type). Synthesis of derivatives 39 and 40 was described previously.26
As the first member of this series, 6-methyl (X = Me) derivative 18 was prepared. 1,3,5-Trifluorobenzene was treated with n-butyllithium in tetrahydrofuran (THF) followed by the addition of methyl 2-chloro-6-methylpyrimidine-4-carboxylate to form carbonyl derivative 17 (Scheme 3).34 The next step was Buchwald coupling of crude 17 with 4-aminobenzonitrile giving the desired derivative 18.33
Scheme 3.

Conditions and reagents: (a) butyllithium, THF, –78°C; (b) 4-aminobenzonitrile, Pd(OAc)2, XantPhos, Cs2CO3, dioxane, 100°C.
To introduce a methoxy group to position 6 of pyrimidine, our procedure26 was applied to 4,6-dichloro-2-methylthiopyrimidine (Scheme 4). The synthetic sequence consisted of 6-chloro substitution by sodium methoxide, methylsulfide oxidation followed by ammonolysis, and Buchwald coupling of the intermediate 21 with 4-bromobenzonitrile to give compound 22.
Scheme 4.

Conditions and reagents: (a) NaH, N,N′-dimethylbenzimidazolium iodide, dioxane, 60°C; (b) MeONa, MeOH, reflux; (c) i: mCPBA, CH2Cl2, 0°C, ii: ethanolic ammonia (2.5 M), 25°C; (d) 4-bromobenzonitrile, Pd(OAc)2, XantPhos, Cs2CO3, dioxane, 80°C.
Overall yields in the carbonyl-linked series were moderate to low, due to the formation of isomers, excessive reactivity of the chlorine atom, and other side reactions.
To compare the influence of the carbonyl linker on anti-HIV potency, another series of DAPY compounds with modified linkers was prepared. ETR and RPV, which have an oxygen and nitrogen linker, respectively, served as an inspiration.
Oxygen and nitrogen linked derivatives (31–38) were prepared using a different synthetic route.35 2-Amino-4,6-dichloropyrimidine was used as a starting material for SNAr reactions with an appropriately substituted phenol and aniline (Scheme 5), followed by Buchwald coupling to introduce a 4-cyanophenylamino substitution to position 2 of the pyrimidine ring.33 These monochloro derivatives (27–30) were used as starting materials for subsequent modifications to obtain the desired 6-methoxy and 6-amino compounds 31–38.
Scheme 5.

Conditions and reagents: (a) Y = NH, 2,4,6-trifluoroaniline or 2,6-difluoro-4-methoxyaniline, cat. HCl, dioxane, reflux; Y = O, 2,4,6-trifluorophenol or 2,6-difluoro-4-methoxyphenol, Cs2CO3, DMF, 80°C; (b) 4-bromobenzonitrile, Pd(OAc)2, XantPhos, Cs2CO3, dioxane, 80°C; (c) Y = NH, MeONa, MeOH, heating; Y = O, MeONa, MeOH, reflux; (d) i: 4-methoxybenzylamine, Pd(OAc)2, XantPhos, Cs2CO3, dioxane, 80°C; ii: CF3COOH, 25°C; (e) i: NaN3, DMF, MW, 130°C; ii: triphenylphosphine, THF, 25°C, then water, HCl (cat.).
NMR studies
Isomeric compounds bearing the carbonyl bridge in positions 2 and 4 exhibit very similar proton and carbon shifts in their respective NMR spectra. Due to the overall lack of protons in these molecules, unambiguous assignment of carbon signals and structure confirmation was complicated. Based on standard one-dimensional experiments combined with heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC) spectra, it was possible to assign the signals of aromatic substituents on the pyrimidine ring. However, distinguishing between substituent positions on the pyrimidine moiety in isomeric compounds 13/15 and 14/16 was complicated due to the absence of cross-peaks Py-H5/C = O. In order to determine the correct positions of the substituents, we used a similar approach to Joshi et al.,36 where analogous pyrimidine derivatives substituted in positions 2, 4, and 6 were analyzed using NOESY spectra. For compounds 15 and 16, our conformational study indicated a sterical proximity of the aniline N–H proton with an aromatic proton in position 5 of the pyrimidine ring. In the 2D-NOESY spectra,37 the cross-peak of H-5 with N–H was observed only with the aniline in position 4 of the pyrimidine ring (compounds 15 and 16).
Biological activity
Entire series of compounds was tested for its in vitro anti-HIV activity (Table 1) using a previously described five-day multi-cycle assay that measures protection from virus-induced cytopathic effects in MT-4 cells acutely infected with HIV-1 (IIIB strain).38–40 Another evidence that studied compounds are specific inhibitors of HIV RT is a X-Ray co-crystal structure of compound 39 with the HIV-RT enzyme.26 It was confirmed that a 4-cyanophenylamino B-arm in position 2 of the pyrimidine ring is essential for high antiviral potency (13 and 14; 4 and 26 nM, respectively). All compounds bearing another substituent in this position were less potent (11, 12, 15, and 16 in micromolar range).
In the CO linker series, polarity of the X substituents directly impacts biological activity. The X = NH2 substituent (compound 14, Z = F, 26 nM) had higher biological activity than the less polar X = Me (18, 160 nM) and X = OMe (22, 473 nM) derivatives, showing a negative effect of low polarity groups on biological activity.
This trend was not observed for the O-linked series (Z = F), where compounds with X = Cl (28, 53 nM) and X = OMe (32, 54 nM) groups exhibited marginally higher activity than the X = NH2 (36, 67 nM) analogues. In the NH-linked series (Z = F), all derivatives (30, 34, 38) exhibited activity in the range 22–27 nM.
The influence of the linker (in the Z = F series) is demonstrated for the compounds with X = OMe, where the measured activity for the CO, O, and NH linker is 473 nM (22), 54 nM (32), and 22 nM (34), respectively.
All compounds with single digit nanomolar activities contain the electron donating p-methoxy group (Z = OMe). Compounds 29 (X = Cl, 6 nM) and 33 (X = OMe, 7 nM) with a nitrogen linker (Y = NH) exhibited excellent activities despite the presence of a less polar substituent in position 6. Regardless of the linker, these compounds (X = NH2, Z = OMe) demonstrated comparable single digit activities: 35 (2 nM), 37 (3 nM), and 13 (4 nM) (for Y = O, NH, CO, respectively).
Two compounds with an oxygen linker (Y = O), where Z = OMe, possessed slightly lower activity for the X = Cl (27, 19 nM) or X = OMe (31, 19 nM) groups.
Direct comparison of X = NH2 carbonyl-linked compounds 13 (Z = OMe, 4 nM) and 14 (Z = F, 26 nM) with previously published X = H analogues 39 (Z = OMe, 5 nM) and 40 (Z = F, 25 nM) showed minimal impact of this substitution on anti-HIV activity. However, the X = NH2 and CO-linker introduce a vast chemical space for further structural optimization.
Selected derivatives were also tested against clinically relevant K103N and Y181C HIV RT mutants (Table 1). In all cases, the compounds were less potent against the mutants (36 nM–13.7 µM) than the wild type (2 nM–67 nM) for selected derivatives.
The selectivity index (SI) was calculated for all compounds to evaluate the toxicity–activity ratio (Table 1). The highest SI was found for compounds 39 (SI = 11,420) and 35 (SI = 10,850), which contain CO and O linkers, respectively. These values are higher than for ETR (SI = 2940) but lower than for efavirenz (SI = 20,600).
In summary, three regioisomers of 2,4,6-trisubstituted pyrimidine were prepared with the A-arm connected via a CO linker. Supported by our previous research, as well as data available for marketed drugs, compounds 13 and 14 with a 4-cyanophenylamino B-arm in pyrimidine position 2 demonstrated the highest activities.
Additionally, a series of derivatives with the 2,4,6-trifluorophenyl A-arm (Z = F) was prepared and the influence of the linker (CO, O, NH) and the substitution in position 6 of the pyrimidine core (Cl, OMe, NH2, Me) were investigated. Fluorine analogues (Z = F) had anti-HIV activities ranging from 22 to 55 nM. These data correspond well with the X = H analogue 40 (25 nM).
Compounds bearing the 4-methoxy-2,6-difluorophenyl A-arm showed an increase of approximately an order of magnitude in antiviral activity compared to the 2,4,6-trifluorophenyl series. The best X substituent on the pyrimidine core is the NH2 group with 4.4 nM, 2.9 nM, and 2.3 nM activities for the CO, NH, and O linkers, respectively. Again, demonstrating good agreement with our previously reported X = H analogue 39 (4.0 nM).26
Molecular modeling
Glide XP (Schrodinger 2015) was used to investigate the binding modes of compound 35 with the O-linked o,o-difluorophenyl A-arm. In comparison to the solved ETR X-ray cocrystal structure (PDB: 3M8P),41 docking predicts that the aminopyrimidine core of 35 overlays well with the ETR structure regardless of the absence of bromine in position 5 (Figure 3, left). The rotation angles between the core and the A-arm are also similar, with values of 78° and 74° for ETR and 35, respectively (Figure 3, right). Additionally, o,o-difluoro and p-methoxy substitutions are predicted not to disturb the overall binding mode of these NNRTIs.
Figure 3.

Docking of compound 35 in to the X-ray structure of ETR in RT (PDB: 3M8P). Side (left) and top (right) view into the allosteric binding pocket.
Conclusions
A new series of trisubstituted DAPY analogues was prepared starting from commercially available trisubstituted pyrimidines and evaluated for their anti-HIV potency against wild type and two clinically relevant mutant strains (K103N and Y181C). Several inhibitors exerted single digit nanomolar potency.
Each compound consists of a small substituent in the position 6 of the central core (Cl, OMe, NH2, Me), 4-cyanophenylamino B-arm and p-substituted o,o-difluorophenyl A-arm connected to the central pyrimidine core through a variable linker (Y = CO, NH, O). Influence of the A-arm para substituent (F, OMe) was also investigated.
Our data show that 4-cyanophenylamino B-arm is indispensable for high antiviral activity and OMe is clearly the best para substituent of the A-arm. Influence of the C-6 substitution of the central core appears to vary based on the linker connecting A-arm to the pyrimidine core. In the case of CO linker, the biological activities dramatically decrease with decreasing polarity of the substituent; however, in the NH and O linker series it had only marginal effect.
Evaluation of the most suitable linker showed rather small impact on the resulting anti-HIV potency in the most active series of compounds. This is very interesting as only the CO linker has any space for further derivatization and it will be investigated in our further work, which will be especially aimed at improving activity against mutants.
Experimental
Chemistry
Chemical reagents and analytical grade solvents were used as received from commercial sources. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III NMR spectrometer at 600.1 MHz (for 1H) equipped with a 5-mm TCI cryoprobe head in DMSO-d6 (Aldrich, 99.8% D). Chemical shifts are reported in δ, residual solvent peaks (DMSO-d6 1H 2.5 ppm and 13C 39.5 ppm) were used as references. For details concerning the experimental techniques and methods, see the NMR studies section. The following abbreviations were used to describe peak patterns: Py: pyrimidine part of molecule; An: aromatic system bonded to amino group (B-arm); Ar: ortho-substituted aromatic system (A-arm); b: broad; s: singlet; d: doublet, t: triplet. The high-resolution mass spectra (HRMS) were measured on an LTQ Orbitrap XL spectrometer (Thermofisher Scientific) using ESI ionization or GC/TOF-MS GCT Premier (Waters) using EI ionization. Reaction progress was monitored either with thin-layer chromatography (TLC) on silica gel plates and UV visualization at 254 nm or with a Waters UPLC/MS system using a water–acetonitrile gradient (0.1% formic acid as modifier) on Waters BEH 1.7 μ C18 130 Å, 100 × 2.10 mm, flow 0.5 mL/min. Flash chromatography separations were performed on silica gel or C18 modified silica gel (300–400 mesh) using a Teledyne Isco system.
Microwave-assisted reactions were carried out in a CEM Discover (Explorer) microwave aparatus, with a 24-position system for 10-mL vessels sealed with Teflon septa. It was operated at a frequency of 2.45 GHz with continuous irradiation power from 0 to 300 W and IR monitored temperature. The solutions were steadily stirred during the reaction.
All final derivatives were lyofilized and thus isolated as amorphous solids. Derivatives 39 and 40 were prepared according to the previously described procedure.26
General procedure A: The appropriate benzaldehyde (1 eq.) and N,N′-dimethylbenzimidazolium iodide (0.5 eq.) were dissolved in dry 1,4-dioxane under an argon atmosphere. 2,4,6-Trichloropyrimidine (1 eq.) was added followed by NaH (60% in mineral oil, 3.5 eq.). The reaction mixture was heated at 60°C for 16 h. The solution was added to a mixture of water (100 mL) and ethyl acetate (EtOAc) (100 mL). The layers were separated and the water layer was further extracted with EtOAc (2 × 100 mL). The combined organic phases were dried over sodium sulfate, filtered, and evaporated. The products were isolated by flash silica gel chromatography (gradient from hexane to EtOAc, 0–100%) followed by reverse phase flash chromatography (gradient from water to MeOH, 0–100%).
General procedure B: Appropriate dichloro derivative (1 eq.) was dissolved in ethanolic ammonia (2.5 M, 50 eq.), and the reaction mixture was stirred at room temperature for 16 h. The solution was diluted with water (100 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were collected, dried over sodium sulfate, filtered, and evaporated. The products were isolated by flash silica gel chromatography (gradient from hexane to EtOAc, 0–100%) followed by reverse phase flash chromatography (gradient from water to MeOH, 0–100%).
General procedure C: Appropriate chloro derivative (1 eq.), aniline (1.1 eq.), palladium(II) acetate (0.1 eq.), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (0.2 eq.) and cesium carbonate (2 eq.) were mixed in dry 1,4-dioxane under argon atmosphere. The reaction mixture was heated at 80°C for 3 h. After cooling to room temperature, the mixture was diluted with water (50 mL) and extracted with EtOAc (3 × 40 mL). The combined organic layers were dried over sodium sulfate, filtered, and evaporated. The product was isolated by flash silica gel chromatography (gradient from hexane to EtOAc, 0–100%) followed by reverse phase flash chromatography (gradient from water to MeOH, 0–100%).
General procedure D: Appropriate amino derivative (1 eq.), 4-bromobenzonitrile (1.5 eq.), palladium(II) acetate (0.1 eq.), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (0.2 eq.) and cesium carbonate (2 eq.) were mixed in dry 1,4-dioxane under argon atmosphere. The reaction mixture was heated at 80°C for 5 h. After cooling to room temperature, the mixture was diluted with water (50 mL) and extracted with EtOAc (3 × 40 mL). The combined organic layers were dried over sodium sulfate, filtered, and evaporated. The product was isolated by flash silica gel chromatography (gradient from hexane to EtOAc, 0–100%) followed by reverse phase flash chromatography (gradient from water to MeOH, 0–100%).
Preparation of (2,6-dichloropyrimidin-4-yl)(2,6-difluoro-4-methoxyphenyl)methanone (1) and (4,6-dichloropyrimidin-2-yl)(2,6-difluoro-4-methoxyphenyl)methanone (3)
Treatment of 2,4,6-trichloropyrimidine (5.26 g, 29 mmol) with 2,6-difluoro-4-methoxybenzaldehyde (4.50 g, 26 mmol) according to general procedure A gave 1 (2.95 g, 36%) and 3 (1.40 g, 17%) as white solids.
(1) HRMS (EI): Calculated for C12H6Cl2O2N2F2 [M + H]+: 317.9774, Found: 317.9776; UPLC/MS (m/z) 318.87 [M + H]+, Tr 4.96 min.
(3) HRMS (EI): Calculated for C12H6Cl2O2N2F2 [M + H]+: 317.9774, Found: 317.9773; UPLC/MS (m/z) 318.79 [M + H]+, Tr 4.70 min.
Preparation of (2,6-dichloropyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone (2) and (4,6-dichloropyrimidin-2-yl)(2,4,6-trifluorophenyl)methanone (4)
Treatment of 2,4,6-trichloropyrimidine (4 g, 22 mmol) with 2,4,6-trifluorobenzaldehyde (3.52 g, 22 mmol) according to general procedure A gave 2 (700 mg, 10%) and 4 (270 mg, 4%) as white solids.
(2) HRMS (EI): Calculated for C11H3Cl2N2OF3 [M + H]+: 305.9575, Found: 305.9578; UPLC/MS (m/z) 307.02 [M + H]+, Tr 4.67 min.
(4) HRMS (ESI): Calculated for C11H3Cl2N2OF3 [M + H]+: 305.9575, Found: 305.9572; UPLC/MS (m/z) 307.02 [M + H]+, Tr 5.23 min.
Preparation of (4-amino-6-chloropyrimidin-2-yl)(2,6-difluoro-4-methoxyphenyl)methanone (5)
Treatment of 3 (1.4 g, 4.4 mmol) with ethanolic ammonia (2.5 M, 30 mL) according to general procedure B gave 5 as a white solid (0.7 g, 70%).
(5) 1H NMR (600.1 MHz, DMSO-d6) δ 7.74 (bs, 1H, NH), 7.60 (bs, 1H, NH), 6.93–6.87 (m, 2H, Ar-meta), 6.59 (s, 1H, Py-H5), 3.87 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 185.35 (s, CO), 165.23 (s, Py-C4), 164.06 (t, JCF = 15.0 Hz, Ar-para), 161.93 (s, Py-C2), 161.75 (dd, JCF = 10.2, 251.6 Hz, Ar-ortho), 158.20 (s, Py-C6), 108.12 (t, JCF = 17.8 Hz, Ar-ipso), 104.33 (d, Py-C5), 99.05 (dd, JCF = 4.5, 23.8 Hz, Ar-meta), 56.72 (s, CH3); HRMS (EI): Calculated for C12H8ClN3O2F2 [M + H]+: 299.0273, Found: 299.0276; UPLC/MS (m/z) 299.74 [M + H]+, Tr 4.02 min.
Preparation of (4-amino-6-chloropyrimidin-2-yl)(2,4,6-trifluorophenyl)methanone (6)
Treatment of 4 (400 mg, 1.3 mmol) with ethanolic ammonia (2.5 M, 30 mL) according to general procedure B gave 6 (152 mg, 41%).
(6) 1H NMR (600.1 MHz, DMSO-d6) δ 7.76 (bs, 1H, NH2), 7.65 (bs, 1H, NH2), 7.44–7.36 (m, 2H, Ar-meta), 6.63 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 187.19 (s, CO), 165.27 (s, Py-C2), 163.99 (dt, JCF = 16.1, 252.3 Hz, Ar-para), 160.61 (ddd, JCF = 10.0, 16.0, 252.3 Hz, Ar-ortho), 160.48 (s, Py-C6), 158.23 (s, Py-C4), 112.86 (td, JCF = 4.6, 19.4 Hz, Ar-ipso), 104.89 (d, Py-C5), 101.65 (td, JCF = 5.2, 26.9, Ar-meta); HRMS (EI): Calculated for C11H5ClN3OF3 [M + H]+: 287.0073, Found: 287.0076; UPLC/MS (m/z) 287.74 [M + H]+, Tr 4.02 min.
Preparation of (6-amino-2-chloropyrimidin-4-yl)(2,6-difluoro-4-methoxyphenyl)methanone (7) and (2-amino-6-chloropyrimidin-4-yl)(2,6-difluoro-4-methoxyphenyl)methanone (9)
Treatment of 1 (2.95 g, 9.3 mmol) with ethanolic ammonia (2.5 M, 30 mL) according to general procedure B gave compound 7 (1.35 g, 48%) and compound 9 (0.45 g, 16%) as white solids.
(7) 1H NMR (600.1 MHz, DMSO-d6) δ 7.97 (bs, 1H, NH2), 7.88 (bs, 1H, NH2), 6.94–6.88 (m, 2H, Ar-meta), 6.88 (s, 1H, Py-H5), 3.87 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 187.26 (s, CO), 166.25 (s, Py-C2), 163.83 (t, JCF = 14.7 Hz, Ar-para), 161.24 (dd, JCF = 10.4, 250.7 Hz, Ar-ortho), 160.17 (s, Py-C4), 159.97 (s, Py-C6), 107.58 (t, JCF = 19.1 Hz, Ar-ipso), 102.41 (d, Py-C5), 98.93 (dd, JCF = 4.3, 23.8 Hz, Ar-meta), 56.65 (q, CH3); HRMS (EI): Calculated for C12H8ClN3O2F2 [M + H]+: 299.0273, Found: 299.0275; UPLC/MS (m/z) 299.82 [M + H]+, Tr 4.11 min.
(9) 1H NMR (600.1 MHz, DMSO-d6) δ 7.52 (bs, 1H, NH), 7.03 (s, 1H, Py-H5), 6.94–6.89 (m, 2H, Ar-meta), 3.87 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 187.56 (s, CO), 164.08 (t, JCF = 14.8 Hz, Ar-para), 163.62 (s, Py-C6), 163.48 (s, Py-C4), 161.64 (s, Py-C2), 161.51 (dd, JCF = 10.2, 251.4 Hz, Ar-ortho), 108.67 (t, JCF = 18.5 Hz, Ar-ipso), 105.74 (d, Py-C5), 99.04 (dd, JCF = 4.3, 23.9 Hz, Ar-meta), 56.73 (s, CH3); HRMS (EI): Calculated for C12H8ClN3O2F2 [M + H]+: 299.0273, Found: 299.0270; UPLC/MS (m/z) 299.82 [M + H]+, Tr 4.41 min.
Preparation of (4-amino-2-chloropyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone (8) and (2-amino-6-chloropyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone (10)
Treatment of 2 (400 mg, 1.3 mmol) with ethanolic ammonia (2.5 M, 30 mL) according to general procedure B gave 8 (142 mg, 38%) and 10 (60 mg, 16%) as white solids.
(8) 1H NMR (600.1 MHz, DMSO-d6) δ 8.03 (bs, 1H, NH2), 7.93 (bs, 1H, NH2), 7.45–7.39 (m, 2H, Ar-meta), 6.97 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 187.16 (s, CO), 166.25 (s, Py-C6), 163.98 (dt, JCF = 5.9, 252.3 Hz, Ar-para), 160.30 (ddd, JCF = 10.1, 16.0, 252.2 Hz, Ar-ortho), 160.13 (s, Py-C4), 158.77 (s, Py-C2), 112.04 (td, JCF = 4.5, 20.1 Hz, Ar-ipso), 102.60 (d, Py-C5), 101.63 (td, JCF = 4.9, 24.6 Hz, Ar-meta); HRMS (EI): Calculated for C11H5ClN3OF3 [M + H]+: 287.0073, Found: 287.0074; UPLC/MS (m/z) 287.82 [M + H]+, Tr 4.40 min.
(10) 1H NMR (600.1 MHz, DMSO-d6) δ 7.55 (bs, 2H, NH2), 7.11 (s, 1H, Py-H5), 7.46–7.39 (m, 2H, Ar-meta); 13C NMR (150.9 MHz, DMSO-d6) δ 187.50 (s, CO), 164.11 (dt, JCF = 5.9, 252.3 Hz, Ar-para), 163.58 (s, Py-C6), 162.11 (s, Py-C4), 161.99 (s, Py-C2), 160.50 (ddd, JCF = 10.1, 16.0, 252.8 Hz, Ar-ortho), 112.15 (td, JCF = 4.5, 19.8 Hz, Ar-ipso), 105.77 (s, Py-C5), 101.71 (td, JCF = 4.5, 19.8 Hz, Ar-meta); HRMS (EI): Calculated for C11H5ClN3OF3 [M + H]+: 287.0073, Found: 287.0076; UPLC/MS (m/z) 287.82 [M + H]+, Tr 4.11 min.
Preparation of 4-((6-amino-2–(2,6-difluoro-4-methoxybenzoyl)pyrimidin-4-yl)amino)benzonitrile (11)
Treatment of 5 (250 mg, 0.83 mmol) with 4-aminobenzonitrile (150 mg, 1.27 mmol) according to general procedure C gave 11 (40 mg, 13%) as a yellow solid.
(11) 1H NMR (600.1 MHz, DMSO-d6) δ 9.77 (bs, 1H, NH), 7.65–7.61 (m, 2H, An-ortho), 7.58–7.54 (m, 2H, An-meta), 6.99 (bs, 2H, NH2), 6.94–6.89 (m, 2H, Ar-meta), 5.98 (s, 1H, Py-H5), 3.88 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 187.10 (s, CO), 164.60 (s, Py-C2), 163.18 (t, JCF = 14.6 Hz, Ar-para), 161.12 (dd, JCF = 10.8, 249.2 Hz, Ar-ortho), 160.66 (s, Py-C4), 159.74 (s, Py-C6), 145.11 (s, An-ipso), 132.94 (s, An-meta), 119.52 (s, An-CN), 118.35 (s, An-ortho), 109.40 (t, JCF = 19.6 Hz, Ar-ipso), 102.16 (s, An-para), 98.75 (dd, JCF = 4.6, 23.3 Hz, Ar-meta), 87.70 (d, Py-C5), 56.63 (s, CH3); HRMS (ESI): Calculated for C19H14N5O2F2 [M + H]+: 382.1110, Found: 382.1112; UPLC/MS (m/z) 381.95 [M + H]+, Tr 4.18 min.
Preparation of 4-((6-amino-2–(2,4,6-trifluorobenzoyl)pyrimidin-4-yl)amino)benzonitrile (12)
Treatment of 6 (300 mg, 1 mmol) with 4-aminobenzonitrile (130 mg, 1.1 mmol) according to general procedure C gave 12 (10 mg, 3%) as a white solid.
(12) 1H NMR (600.1 MHz, DMSO-d6) δ 9.80 (bs, 1H, NH), 7.60–7.57 (m, 2H, An-ortho), 7.57–7.54 (m, 2H, An-meta), 7.45–7.40 (m, 2H, Ar-meta), δ 7.04 (bs, 2H, NH2), 6.00 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 186.86 (s, CO), 164.66 (s, Py-C2), 163.37 (dt, JCF = 5.8, 250.7 Hz, Ar-para), 159.94 (ddd, JCF = 10.7, 16.0, 250.2, Ar-ortho), 159.73 (s, Py-C4), 159.37 (s, Py-C6), 144.96 (s, An-ipso), 132.83 (s, An-meta), 119.43 (s, CN), 118.27 (s, An-ortho), 114.20 (td, JCF = 5.3, 10.4 Hz, Ar-ipso), 105.27 (s, An-para), 101.38 (td, JCF = 4.8, 26.1 Hz, Ar-meta), 88.16 (s, Py-C5); HRMS (ESI): Calculated for C18H11N5OF3 [M + H]+: 370.0910, Found: 370.0912; UPLC/MS (m/z) 370.02 [M + H]+, Tr 4.19 min.
Preparation of 4-((4-amino-6–(2,6-difluoro-4-methoxybenzoyl)pyrimidin-2-yl)amino)benzonitrile (13)
Treatment of 7 (250 mg, 0.83 mmol) with 4-aminobenzonitrile (150 mg, 1.27 mmol) according to general procedure C gave 13 (30 mg, 10%) as a yellow solid.
(13) 1H NMR (600.1 MHz, DMSO-d6) δ 9.74 (bs, 1H, NH), 7.85–7.81 (m, 2H, An-ortho), 7.55–7.50 (m, 2H, An-meta), 7.27 (bs, 2H, NH2), 6.97–6.92 (m, 2H, Ar-meta), 6.55 (s, 1H, Py-H5), 3.89 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 189.33 (s, CO), 165.14 (s, Py-C4), 163.23 (t, JCF = 14.5 Hz, Ar-para), 160.83 (dd, JCF = 10.9, 248.7 Hz, Ar-ortho), 159.53 (s, Py-C2), 159.01 (s, Py-C6), 145.33 (s, An-ipso), 132.62 (s, An-meta), 119.68 (s, An-CN), 118.16 (s, An-ortho), 108.73 (t, JCF = 20.5 Hz, Ar-ipso), 101.83 (s, An-para), 98.78 (dd, JCF = 4.6, 23.8 Hz, Ar-meta), 96.39 (s, Py-C5), 56.65 (s, CH3); HRMS (ESI): Calculated for C19H14N5O2F2 [M + H]+: 382.1110, Found: 382.1111; UPLC/MS (m/z) 381.87 [M + H]+, Tr 4.42 min.
Preparation of 4-((4-amino-6–(2,4,6-trifluorobenzoyl)pyrimidin-2-yl)amino)benzonitrile (14)
Treatment of 8 (200 mg, 0.7 mmol) with 4-aminobenzonitrile (91 mg, 0.8 mmol) according to general procedure C gave 14 (10 mg, 4%) as a white solid.
(14) 1H NMR (600.1 MHz, DMSO-d6) δ 9.74 (bs, 1H, NH), 7.78–7.74 (m, 2H, An-ortho), 7.54–7.50 (m, 2H, An-meta), 7.49–7.42 (m, 2H, Ar-meta), 7.31 (bs, 2H, NH2), 6.64 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 189.20 (s, CO), 165.14 (s, Py-C4), 163.55 (dt, JCF = 5.8, 250.8 Hz, Ar-para), 159.84 (ddd, JCF = 10.7, 15.8, 250.5 Hz, Ar-ortho), 159.60 (s, Py-C2), 157.67 (s, Py-C6), 145.17 (s, An-ipso), 132.50 (d, An-meta), 119.60 (s, CN), 118.10 (s, An-ortho), 113.28 (td, JCF = 4.4, 19.7 Hz, Ar-ipso), 102.00 (s, An-para), 101.47 (td, JCF = 4.8, 23.9 Hz, Ar-meta), 96.55 (s, Py-C5); HRMS (ESI): Calculated for C18H10ON5F3Na [M+Na]+: 392.0730, Found: 392.0731; UPLC/MS (m/z) 369.87 [M + H]+, Tr 4.20 min.
Preparation of 4-((4-amino-6–(2,6-difluoro-4-methoxybenzoyl)pyrimidin-2-yl)amino)benzonitrile (15)
Treatment of 9 (250 mg, 0.8 mmol) with 4-aminobenzonitrile (104 mg, 0.9 mmol) according to general procedure C gave 15 (133 mg, 44%) as a red solid.
(15) 1H NMR (600.1 MHz, DMSO-d6) δ 10.03 (bs, 1H, NH), 8.02–7.97 (m, 2H, An-ortho), 7.75–7.71 (m, 2H, An-meta), 6.92–6.85 (m, 2H, Ar-meta), 6.82 (bs, 2H, NH2), 6.57 (s, 1H, Py-H5), 3.86 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 189.41 (s, CO), 163.27 (t, JCF = 14.6 Hz, Ar-para), 163.27 (s, Py-C2), 161.53 (s, Py-C6), 161.05 (dd, JCF = 10.8, 249.6 Hz, Ar-ortho), 160.85 (s, Py-C4), 144.61 (s, An-ipso), 133.11 (s, An-meta), 119.49 (s, An-CN), 119.03 (s, An-ortho), 108.64 (t, JCF = 19.7 Hz, Ar-ipso), 103.02 (s, An-para), 98.75 (dd, JCF = 4.4, 23.5 Hz, Ar-meta), 95.52 (s, Py-C5), 56.59 (s, CH3); HRMS (ESI): Calculated for C19H14N5O2F2 [M + H]+: 382.1110, Found: 382.1112; UPLC/MS (m/z) 383.681 [M + H]+, Tr 3.86 min.
Preparation of 4-((2-amino-6–(2,4,6-trifluorobenzoyl)pyrimidin-4-yl)amino)benzonitrile (16)
Treatment of 10 (150 mg, 0.5 mmol) with 4-aminobenzonitrile (65 mg, 0.6 mmol) according to general procedure C gave 16 (32 mg, 17%) as a white solid.
(16) 1H NMR (600.1 MHz, DMSO-d6) δ 10.05 (bs, 1H, NH), 8.02–7.98 (m, 2H, An-ortho), 7.75–7.71 (m, 2H, An-meta), 7.42–7.36 (m, 2H, Ar-meta), 6.84 (bs, 2H, NH2), 6.65 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 189.26 (s, CO), 163.76 (dt, JCF = 15.4, 250.8 Hz, Ar-para), 163.38 (s, Py-C2), 161.56 (s, Py-C6), 160.10 (ddd, JCF = 10.1, 16.5, 251.8 Hz, Ar-ortho), 159.49 (s, Py-C4), 113.14 (td, JCF = 4.8, 20.7 Hz, Ar-ipso), 101.47 (td, JCF = 4.8, 22.8 Hz, Ar-meta), 95.59 (s, Py-C5); HRMS (ESI): Calculated for C18H11N5OF3 [M + H]+: 370.0910, Found: 370.0911; UPLC/MS (m/z) 369.95 [M + H]+, Tr 3.78 min.
Preparation of 4-((4-methyl-6–(2,4,6-trifluorobenzoyl)pyrimidin-2-yl)amino)benzonitrile (18)
A solution of butyllithium (1.6 M in hexanes, 6.1 mL, 9.8 mmol) was slowly added to a precooled (–78°C) solution of 1,3,5-trifluorobenzene (1 mL, 10 mmol) in dry THF (10 mL). The reaction mixture was stirred at –78°C for 2 h, after which a solution of methyl 2-chloro-6-methylpyrimidine-4-carboxylate (1 g, 5.4 mmol) in dry THF (10 mL) was slowly added and this mixture was slowly allowed to reach room temperature. The resulting solution was poured into EtOAc (100 mL) and water (100 mL) mixture, the organic layer was separated, washed with brine (100 mL), and dried over sodium sulfate. Crude intermediate 17 was isolated by flash chromatography on silica gel (gradient from hexane to EtOAc, 0–100%). UPLC/MS (m/z) 286.84 [M + H]+, Tr 4.67 min.
Treatment of crude 17 (50 mg) with 4-aminobenzonitrile (26 mg, 0.22 mmol) according to general procedure C gave 18 (55 mg, overall yield 3%) as a yellow solid.
(18) 1H NMR (600.1 MHz, DMSO-d6) δ 10.47 (bs, 1H, NH), 7.74–7.71 (m, 2H, An-ortho), 7.61–7.57 (m, 2H, An-meta), 7.53–7.47 (m, 2H, Ar-meta), 7.46 (s, 1H, Py-H5), 2.56 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 188.58 (s, CO), 171.67 (s, Py-C6), 163.94 (dt, JCF = 5.9, 251.7 Hz, Ar-para), 160.08 (ddd, JCF = 10.5, 15.8, 251.4 Hz, Ar-ortho), 159.30 (s, Py-C4), 158.44 (s, Py-C2), 144.33 (s, An-ipso), 132.75 (s, An-meta), 119.34 (s, CN), 118.26 (s, An-ortho), 112.55 (td, JCF = 4.5, 11.0 Hz, Ar-ipso), 109.77 (s, Py-C5), 102.93 (s, An-para), 101.67 (td, JCF = 4.6, 23.6 Hz, Ar-meta), 23.96 (s, CH3); HRMS (EI): Calculated for C19H11N4OF3 [M + H]+: 368.0885, Found: 368.0895; UPLC/MS (m/z) 368.97 [M + H]+, Tr 4.69 min.
Preparation of (6-chloro-2-(methylthio)pyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone (19)
Treatment of 4,6-dichloro-2-(methylthio)pyrimidine (1 g, 5 mmol) with 2,4,6-trifluorobenzaldehyde (0.8 g, 5 mmol) according to general procedure A gave 19 (240 mg, 16%) as a white solid.
(19) 1H NMR (600.1 MHz, DMSO-d6) δ 7.87 (s, 1H, Py-H5), 7.48–7.43 (m, 2H, Ar-meta), 2.43 (s, 3H, SCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 186.07(s, CO), 173.43 (s, Py-C2), 164.45 (dt, JCF = 16.1, 253.1 Hz, Ar-para), 162.37 (s, Py-C4), 160.67 (ddd, JCF = 10.8, 16.1, 253.7 Hz, Ar-ortho), 159.84 (s, Py-C6), 114.19 (d, Py-C5), 111.33 (td, JCF = 4.5, 19.2 Hz, Ar-ipso), 101.79 (td, JCF = 3.1, 26.8 Hz, Ar-meta), 13.70 (s, SCH3); HRMS (ESI): Calculated for C12H7ClN2F3S [M + H]+: 318.9914, Found: 318.9911; UPLC/MS (m/z) 319.121 [M + H]+, Tr 5.23 min.
Preparation of (6-methoxy-2-(methylthio)pyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone (20)
Compound 19 (1 g, 3 mmol) and sodium methoxide (430 mg, 9 mmol) were dissolved in methanol (30 mL), and the reaction mixture was heated to reflux for 16 h. The solution was diluted with water (60 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash silica gel chromatography (gradient from cyclohexane to MeOH, 0–30%) gave 20 (300 g, 32%) as a white solid.
(20) 1H NMR (600.1 MHz, DMSO-d6) δ 7.41–7.35 (m, 2H, Ar-meta), 7.11 (s, 1H, Py-H5), 4.00 (s, 3H, OCH3), 2.39 (s, 3H, SCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 187.13 (s, CO), 172.23 (s, Py-C2), 170.08 (s, Py-C6), 163.96 (dt, JCF = 16.0, 252.2 Hz, Ar-para), 160.29 (ddd, JCF = 10.0, 16.0, 252.3 Hz, Ar-ortho), 159.37 (s, Py-C4), 112.03 (td, JCF = 4.6, 20.1 Hz, Ar-ipso), 101.39 (td, JCF = 5.3, 26.9 Hz, Ar-meta), 101.34 (s, Py-C5), 54.61 (s, OCH3), 13.34 (s, SCH3); HRMS (ESI): Calculated for C13H10O2N2F3S [M + H]+: 315.0410, Found: 315.0408; UPLC/MS (m/z) 315.164 [M + H]+, Tr 5.18 min.
Preparation of (2-amino-6-methoxypyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone (21)
A solution of 20 (300 mg, 1 mmol) in dichloromethane (30 mL) was cooled to 0°C, and a solution of 3-chloroperbenzoic acid (471 mg, 3 mmol) in dichloromethane (30 mL) was added dropwise. The reaction mixture was stirred for 1 h at 0°C, diluted with dichloromethane (10 mL) and extracted with a solution of NaHCO3 (1 × 20 mL), brine (1 × 20 mL) and water (1 × 20 mL). The organic layer was dried over magnesium sulfate, filtered, and evaporated to give 400 mg of the crude intermediate (6-methoxy-2-(methylsulfonyl)pyrimidin-4-yl)(2,4,6-trifluorophenyl)methanone. The intermediate was subsequently dissolved in ethanolic ammonia (2.5 M ammonia in ethanol, 50 eq.) and the solution was stirred at room temperature for 16 h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 21 as a white solid (50 mg, 18%).
(21) 1H NMR (600.1 MHz, DMSO-d6) δ 7.41–7.35 (m, 2H, Ar-meta), 6.49 (s, 1H, Py-H5), 3.88 (s, OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 188.73 (s, Ar-ipso), 171.09 (s, Py-C6), 163.77 (s, Py-C2), 162.16 (dt, JCF = 15.9, 251.4 Hz, Ar-para), 161.30 (s, Py-C4), 160.20 (ddd, JCF = 10.4, 15.9, 251.6 Hz, Ar-ortho), 113.00 (td, JCF = 4.7, 20.5 Hz, Ar-ipso), 101.48 (td, JCF = 4.8, 26.8 Hz, Ar-meta), 93.82 (s, Py-C5), 53.64 (s, OCH3); HRMS (ESI): Calculated for C12H9O2N3F3 [M + H]+: 284.0641, Found: 284.0640; UPLC/MS (m/z) 284.164 [M + H]+, Tr 4.26 min.
Preparation of 4-((4-methoxy-6–(2,4,6-trifluorobenzoyl)pyrimidin-2-yl)amino)benzonitrile (22)
Treatment of 21 (200 mg, 0.7 mmol) with 4-bromobenzonitrile (191 mg, 5 mmol) according to general procedure D gave 22 (90 mg, 34%) as a white solid.
(22) 1H NMR (600.1 MHz, DMSO-d6) δ 7.78–7.74 (m, 2H, An-ortho), 7.63–7.59 (m, 2H, An-meta), 7.52–7.46 (m, 2H, Ar-meta), 6.94 (s, 1H, Py-H5), 4.03 (s, 3H, OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 187.90 (s, CO), 171.25 (s, Py-C6), 163.92 (dt, JCF = 15.7, 251.9 Hz, Ar-para), 160.14 (s, Py-C2), 160.09 (ddd, JCF = 9.8, 16.0, 251.3 Hz, Ar-ortho), 159.18 (s, Py-C4), 144.19 (s, An-ipso), 132.78 (s, An-meta), 119.30 (s, CN), 118.66 (s, An-ortho), 112.66 (td, JCF = 4.5, 20.7 Hz, Ar-ipso), 101.66 (td, JCF = 5.2, 24.9 Hz, Ar-meta), 97.98 (s, Py-C5), 54.63 (s, OCH3); HRMS (ESI): Calculated for C19H11O2N4F3Na [M+Na]+: 407.0726, Found: 407.0723; UPLC/MS (m/z) 385.219 [M + H]+, Tr 5.06 min.
Preparation of 4-chloro-6–(2,6-difluoro-4-methoxyphenoxy)pyrimidin-2-amine (23)
2-Amino-4,6-dichloropyrimidine (1.6 g, 9.8 mmol), 2,6-difluoro-4-methoxyphenol (1.72 g, 10.8 mmol), and cesium carbonate (3.2 g, 9.8 mmol) were dissolved in N,N-dimethylformamide (DMF) (40 mL), and the reaction mixture was heated at 80°C for 1 h. The solution was diluted with water (70 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash silica gel chromatography (gradient from cyclohexane to AcOEt, 0–40%) gave 23 (2 g, 58%) as a white solid.
(23) 1H NMR (600.1 MHz, DMSO-d6) δ 7.25 (bs, 2H, NH2), 6.96–6.90 (m, 2H, Ar-meta), 6.49 (s, 1H, Py-H5), 3.79 (s, 3H, OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 169.31 (s, Py-C2), 162.73 (s, Py-C6), 161.16 (s, Py-C2), 157.42 (t, JCF = 13.0 Hz, Ar-para), 155.12 (dd, JCF = 7.0, 245.8 Hz, Ar-ortho), 121.32 (t, JCF = 16.4 Hz, Ar-ipso), 99.00 (dd, JCF = 4.2, 21.5 Hz, Ar-meta), 93.79 (s, Py-C5), 56.28 (s, OCH3); HRMS (ESI): Calculated for C11H9ClO2N3F2 [M + H]+: 288.0346, Found: 288.0347; UPLC/MS (m/z) 288.21 [M + H]+, Tr 4.26 min.
Preparation of 4-chloro-6–(2,4,6-trifluorophenoxy)pyrimidin-2-amine (24)
2-Amino-4,6-dichloropyrimidine (2 g, 12 mmol), 2,4,6-trifluorophenol (1.95 g, 13 mmol), and cesium carbonate (5.9 g, 18 mmol) were dissolved in DMF (50 mL), and the reaction mixture was heated at 80°C for 4 h. The solution was diluted with water (70 mL) and extracted with EtOAc (3 × 50 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 24 (2.32 g, 65%) as a white solid.
(24) 1H NMR (600.1 MHz, DMSO-d6) δ 7.48–7.40 (m, 2H, Ar-meta), 7.35 and 7.23 (bs, 2H, NH2), 6.55 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 168.82 (s, Py-C4), 162.64 (s, Py-C6), 161.36 (s, Py-C2), 158.78 (dt, JCF = 4.3, 245.0 Hz, Ar-para), 154.87 (ddd, JCF = 6.5, 16.0, 249.2 Hz, Ar-ortho), 125.04 (s, JCF = 5.2, 16.0 Hz, Ar-ipso), 101.70 (ddd, JCF = 4.7, 21.5, 25.5 Hz, Ar-meta), 93.80 (s, Py-C5); HRMS (ESI): Calculated for C10H6ClON3F3 [M + H]+: 276.0146, Found: 276.0149; UPLC/MS (m/z) 275.66 [M + H]+, Tr 4.50 min.
Preparation of 6-chloro-N4-(2,6-difluoro-4-methoxyphenyl)pyrimidin-2,4-diamine (25)
2-Amino-4,6-dichloropyrimidine (500 mg, 3 mmol) and 2,6-difluoro-4-methoxyaniline (525 mg, 3.3 mmol) were dissolved in dioxane (40 mL) and a catalytic amount of HCl was added. The reaction mixture was heated to reflux for 3 h after which it was diluted with water (60 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 25 (600 mg, 70%) as a white solid.
(25) 1H NMR (600.1 MHz, DMSO-d6) δ 8.67 (bs, 1H, NH), 6.86 (s, 1H, Py-H5), 6.85–6.80 (m, 2H, Ar-meta), 5.84 (bs, 2H, 2-NH2); 13C NMR (150.9 MHz, DMSO-d6) δ 162.95 (s, Py-C2), 162.83 (s, Py-C4), 160.97 (s, Py-C6), 159.05 (dd, JCF = 7.9, 245.8 Hz, Ar-ortho), 158.61 (t, JCF = 13.7 Hz, Ar-para), 107.84 (t, JCF = 16.9 Hz, Ar-ipso), 107.53 (s, Py-C5), 98.53 (dd, JCF = 4.7, 23.5 Hz, Ar-meta), 56.15 (s, OCH3); HRMS (ESI): Calculated for C11H10ClON4F2 [M + H]+: 287.0506, Found: 287.0507; UPLC/MS (m/z) 287.22 [M + H]+, Tr 3.72 min.
Preparation of 6-chloro-N4-(2,4,6-trifluorophenyl)pyrimidin-2,4-diamine (26)
2-Amino-4,6-dichloropyrimidine (2 g, 12 mmol) and 2,4,6-trifluoroaniline (1.94 g, 13 mmol) were dissolved in dioxane (50 mL) and a catalytic amount of HCl was added. The reaction mixture was heated to reflux for 16 h, diluted with water (80 mL), and extracted with EtOAc (3 × 50 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash silica gel chromatography (gradient from CHCl3 to MeOH, 0–15%) gave 26 (2.52 g, 71%) as a white solid.
(26) 1H NMR (600.1 MHz, DMSO-d6) δ 8.87 (bs, 1H, NH), 7.33–7.21 (m, 2H, Ar-meta), 6.61 (bs, 2H, NH2), 5.91 (bs, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 163.16 (s, Py-C4), 162.91 (s, Py-C6), 159.77 (dt, JCF = 15.5, 245.0 Hz, Ar-para), 159.49 (ddd, JCF = 7.4, 15.8, 248.7 Hz, Ar-ortho), 158.68 (s, Py-C2), 112.46 (td, JCF = 4.7, 16.8 Hz, Ar-ipso), 101.02 (td, JCF = 5.5, 22.0 Hz, Ar-meta), 92.51 (d, Py-C5); HRMS (ESI): Calculated for C10H7ClN4F3 [M + H]+: 275.0306, Found: 275.0307; UPLC/MS (m/z) 274.56 [M + H]+, Tr 3.87 min.
Preparation of 4-((4-chloro-6–(2,6-difluoro-4-methoxyphenoxy)pyrimidin-2-yl)amino)benzonitrile (27)
Treatment of 23 (600 mg, 2 mmol) with 4-bromobenzonitrile (546 mg, 3 mmol) according to general procedure D gave 27 (450 mg, 58%) as a white solid.
(27) 1H NMR (600.1 MHz, DMSO-d6) δ 10.62 (bs, 1H, NH), 7.61–7.52 (m, 4H, An-ortho and An-meta), 7.08–7.03 (m, 2H, Ar-meta), 7.02 (s, 1H, Py-H5), 3.86 (m, 3H, OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 169.19 (s, Py-C4), 161.41 (s, Py-C6), 158.03 (s, Py-C2), 157.94 (t, JCF = 12.8 Hz, Ar-para), 155.06 (td, JCF = 6.9, 246.2 Hz, Ar-ortho), 143.42 (s, An-ipso), 132.76 (s, An-meta), 121.27 (t, JCF = 16.4 Hz, Ar-ipso), 119.13 (s, CN), 118.85 (s, An-ortho), 103.83 (s, An-para), 99.19 (dd, JCF = 4.0, 21.3 Hz, Ar-meta), 97.91 (s, Py-C5), 56.48 (s, OCH3); HRMS (ESI): Calculated for C18H11O2ClN4F2 [M + H]+: 389.0611, Found: 389.0608; UPLC/MS (m/z) 389.27 [M + H]+, Tr 5.34 min.
Preparation of 4-((4-chloro-6–(2,4,6-trifluorophenoxy)pyrimidin-2-yl)amino)benzonitrile (28)
Treatment of 24 (900 mg, 3.3 mmol) with 4-bromobenzonitrile (910 mg, 5 mmol) according to general procedure D gave 28 (1.07 g, 74%) as a white solid.
(28) 1H NMR (600.1 MHz, DMSO-d6) δ 10.65 (bs, 1H, NH), 7.62–7.58 (m, 2H, An-ortho), 7.59–7.53 (m, 2H, Ar-meta), 7.56–7.51 (m, 2H, An-meta), 7.06 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 168.69 (s, Py-C4), 161.61 (s, Py-C6), 159.16 (dt, JCF = 14.5, 246 Hz, Ar-para), 158.03 (s, Py-C2), 154.80 (ddd, JCF = 6.4, 16.0, 248.5 Hz, Ar-ortho), 143.37 (s, An-ipso), 132.78 (s, An-meta), 125.02 (td, JCF = 5.5, 16.0 Hz, Ar-ipso), 119.10 (s, CN), 118.85 (s, An-ortho), 103.91 (s, An-para), 101.93 (td, JCF = 4.9, 21.2 Hz, Ar-meta), 97.90 (s, Py-C5); HRMS (ESI): Calculated for C17H8ClON4F3Na [M+Na]+: 399.0231, Found: 399.0232; UPLC/MS (m/z) 377.15 [M + H]+, Tr 5.00 min.
Preparation of 4-((4-chloro-6-((2,6-difluoro-4-methoxyphenyl)amino)pyrimidin-2-yl)amino)benzonitrile (29)
Treatment of 25 (600 mg, 2 mmol) with 4-bromobenzonitrile (191 mg, 5 mmol) according to general procedure D gave 29 (240 mg, 31%) as a white solid.
(29) 1H NMR (600.1 MHz, DMSO-d6) δ 10.10 (bs, 1H, An-NH), 9.28 (bs, 1H, Ar-NH), 7.68–7.61 (m, 2H, An-ortho), 7.57–7.48 (m, 2H, An-meta), 6.97–6.92 (m, 2H, Ar-meta), 6.35 (bs, 1H, Py-H5), 3.84 (s, 3H, OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 163.39 (s, Py-C4), 158.99 (t, JCF = 15.2 Hz, Ar-para), 158.81 (dd, JCF = 7.4, 245.1 Hz, Ar-ortho), 158.54 (s, Py-C2, Py-C6), 144.47 (s, An-ipso), 132.66 (d, An-meta), 119.43 (s, CN), 118.40 (s, An-ortho), 107.63 (t, JCF = 14.8 Hz, Ar-ipso), 102.64 (s, An-para), 98.66 (td, JCF = 5.0, 23.3 Hz, Ar-meta), 96.17 (s, Py-C5), 56.32 (s, OCH3); HRMS (ESI): Calculated for C18H12ClON5F2Na [M+Na]+: 410.0591, Found: 410.0587; UPLC/MS (m/z) 388.08 [M + H]+, Tr 4.93 min.
Preparation of 4-((4-chloro-6-((2,4,6-trifluorophenyl)amino)pyrimidin-2-yl)amino)benzonitrile (30)
Treatment of 26 (1 g, 3.6 mmol) with 4-bromobenzonitrile (9.83 g, 5.4 mmol) according to general procedure D gave 30 (1.1 g, 81%) as a white solid.
(30) 1H NMR (600.1 MHz, DMSO-d6) δ 10.13 (bs, 1H, An-NH), 9.47 (bs, 1H, Ar-NH), 7.68–7.61 (m, 2H, An-ortho), 7.59–7.53 (m, 2H, An-meta), 7.45–7.38 (m, 2H, Ar-meta), 6.34 (bs, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 162.94 (s, Py-C4), 160.06 (dt, JCF = 15.4, 246.3 Hz, Ar-para), 158.47 (s, Py-C2), 158.42 (s, Py-C6), 158.27 (ddd, JCF = 7.2, 15.8, 249.5 Hz, Ar-ortho), 144.25 (s, An-ipso), 132.50 (s, An-meta), 119.21 (s, CN), 118.38 (s, An-ortho), 112.08 (td, JCF = 4.0, 17.8 Hz, Ar-ipso), 102.76 (s, An-para), 101.52 (td, JCF = 5.3, 23.5 Hz, Ar-meta), 95.90 (s, Py-C5); HRMS (ESI): Calculated for C17H10ClN5F3 [M + H]+: 376.0571, Found: 376.0568; UPLC/MS (m/z) 375.9 [M + H]+, Tr 4.45 min.
Preparation of 4-((4–(2,6-difluoro-4-methoxyphenoxy)-6-methoxypyrimidin-2-yl)amino)benzonitrile (31)
Compound 27 (150 mg, 0.4 mmol) and sodium methoxide (65 mg, 1.2 mmol) were dissolved in methanol (30 mL), and the reaction mixture was heated to reflux for 16 h. The solution was diluted with water (60 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 31 as a white solid (80 mg, 53%).
(31) 1H NMR (600.1 MHz, DMSO-d6) δ 7.03–6.97 (m, 2H, Ar-meta), 7.69–7.63 (m, 2H, An-ortho), 7.57–7.51 (m, 2H, An-meta), 6.12 (s, 1H, Py-H5), 3.95 (s, 3H, Py-OCH3), 3.84 (s, 3H, Ar-OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 172.23 (s, Py-C4), 169.64 (s, Py-C6), 157.99 (s, Py-C2), 157.50 (t, JCF = 12.9 Hz, Ar-para), 155.35 (dd, JCF = 7.1, 245.7 Hz, Ar-ortho), 144.25 (s, An-ipso), 132.64 (s, An-meta), 121.86 (t, JCF = 16.3 Hz, Ar-ipso), 119.31 (s, CN), 118.61 (s, An-ortho), 102.95 (s, An-para), 98.97 (dd, JCF = 4.3, 21.0 Hz, Ar-meta), 82.10 (s, Py-C5), 56.38 (s, Ar-OCH3), 54.46 (s, Py-OCH3); HRMS (ESI): Calculated for C19H15ClO3N4F2 [M + H]+: 385.1107, Found: 385.1108; UPLC/MS (m/z) 385.31 [M + H]+, Tr 5.26 min.
Preparation of 4-((4-methoxy-6–(2,4,6-trifluorophenoxy)pyrimidin-2-yl)amino)benzonitrile (32)
Compound 28 (250 mg, 0.8 mmol) and sodium methoxide (134 mg, 0.8 mmol) were dissolved in methanol (40 mL), and the reaction mixture was heated to reflux for 24 h. The solution was diluted with water (70 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash silica gel chromatography (gradient from cyclohexane to EtOAc, 0–40%) followed by flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave product 32 (80 mg, 31%) as a white solid.
(32) 1H NMR (600.1 MHz, DMSO-d6) δ 10.12 (bs, 1H, NH), 7.63–7.58 (m, 2H, An-ortho), 7.57–7.53 (m, 2H, An-meta), 7.50–7.43 (m, 2H, Ar-meta), 6.15 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 172.55 (s, Py-C6), 169.38 (s, Py-C4), 159.05 (dt, JCF = 14.9, 245.3 Hz, Ar-para), 158.15 (s, Py-C2), 155.25 (ddd, JCF = 6.7, 15.9, 248.9 Hz, Ar-ortho), 144.32 (s, An-ipso), 132.90 (s, An-meta), 125.80 (td, JCF = 5.1, 15.9 Hz, Ar-ipso), 119.52 (s, CN), 118.85 (s, An-ortho), 103.30 (s, An-para), 101.89 (td, JCF = 4.9, 27.1 Hz, Ar-meta), 82.44 (s, Py-C5); HRMS (ESI): Calculated for C18H12O2N4F3 [M + H]+: 373.0910, Found: 373.0908; UPLC/MS (m/z) 373.200 [M + H]+, Tr 5.21 min.
Preparation of 4-((4-((2,6-difluoro-4-methoxyphenyl)amino)-6-methoxypyrimidin-2-yl)amino)benzonitrile (33)
Compound 29 (100 mg, 0.3 mmol) and sodium methoxide (65 mg, 1.2 mmol) were dissolved in methanol (20 mL). The reaction mixture was heated under microwave (MW) conditions (100°C, 4 h), after which it was diluted with water (50 mL) and extracted with EtOAc (3 × 30 mL). Organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 33 (25 mg, 22%) as a white solid.
(33) 1H NMR (600.1 MHz, DMSO-d6) δ 9.64 (bs, 1H, An-NH), 8.68 (bs, 1H, Ar-NH), 7.85–7.75 (m, 2H, An-ortho), 7.57–7.50 (m, 2H, An-meta), 6.92–6.87 (m, 2H, Ar-meta), 5.50 (bs, 1H, Py-H5), 3.84 (s, 3H, Py-OCH3), 3.83 (s, 3H, Ar-OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 170.27 (s, Py-C6), 164.00 (s, Py-C4), 159.10 (dd, JCF = 8.0, 245.3 Hz, Ar-ortho), 158.53 (t, JCF = 14.2 Hz, Ar-ipso), 152.25 (s, Py-C2), 145.25 (s, An-ipso), 132.55 (s, An-meta), 119.62 (s, CN), 118.21 (s, An-ortho), 108.57 (t, JCF = 15.2 Hz, Ar-ipso), 101.78 (s, An-para), 98.52 (dd, JCF = 4.5, 23.6 Hz, Ar-meta), 79.43 (s, Py-C5), 56.21 (s, Ar-OCH3), 53.42 (s, Py-OCH3); HRMS (ESI): Calculated for C19H16O2N5F2 [M + H]+: 384.1267, Found: 384.1264; UPLC/MS (m/z) 384.33 [M + H]+, Tr 4.88 min.
Preparation of 4-((4-methoxy-6-((2,4,6-trifluorophenyl)amino)pyrimidin-2-yl)amino)benzonitrile (34)
Compound 30 (480 mg, 1.3 mmol) and sodium methoxide (280 mg, 5.2 mmol) were dissolved in methanol (50 mL). The reaction mixture was refluxed for 16 h. The solution was diluted with water (80 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 34 (270 mg, 56%) as a white solid.
(34) 1H NMR (600.1 MHz, DMSO-d6) δ 9.67 (bs, 1H, An-NH), 8.89 (bs, 1H, Ar-NH), 7.78–7.74 (m, 2H, An-ortho), 7.57–7.53 (m, 2H, An-meta), 7.39–7.34 (m, 2H, Ar-meta), 5.58 (bs, 1H, Py-H5), 3.86 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 170.31 (s, Py-C6), 163.45 (s, Py-C4), 159.66 (dt, JCF = 15.4, 245.6 Hz, Ar-para), 158.52 (ddd, JCF = 7.4, 15.8, 248.2 Hz, Ar-ortho), 158.40 (s, Py-C2), 145.13 (s, An-ipso), 132.57 (s, An-meta), 119.58 (s, CN), 118.17 (s, An-ortho), 113.14 (td, JCF = 3.1, 15.5 Hz, Ar-ipso), 101.90 (s, An-para), 101.01 (td, JCF = 4.8, 22.5 Hz, Ar-meta), 79.89 (s, Py-C5), 53.48 (s, CH3); HRMS (ESI): Calculated for C18H13ON5F3 [M + H]+: 372.1066, Found: 372.1067; UPLC/MS (m/z) 372.04 [M + H]+, Tr 4.46 min.
Preparation of 4-((4-amino-6–(2,6-difluoro-4-methoxyphenoxy)pyrimidin-2-yl)amino)benzonitrile (35)
A mixture of 27 (200 mg, 0.5 mmol), 4-methoxybenzylamine (137 mg, 1 mmol), palladium(II) acetate (11 mg, 0.05 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (58 mg, 0.1 mmol), and cesium carbonate (325 mg, 1 mmol) in dry dioxane (6 mL) was heated at 80°C under argon atmosphere for 2 h. The mixture was diluted with water (40 mL) and extracted with EtOAc (3 × 30 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave the intermediate 4-((4–(2,6-difluoro-4-methoxyphenoxy)-6-((4-methoxybenzyl)amino)pyrimidin-2-yl)amino)benzonitrile, which was subsequently dissolved in CF3COOH (3 mL) and stirred for 16 h. Volatiles were evaporated, and flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 35 (20 mg, 11%) as a white solid.
(35) 1H NMR (600.1 MHz, DMSO-d6) δ 9.58 (bs, 1H, NH), 7.73–7.68 (m, 2H, An-ortho), 7.50–7.46 (m, 2H, An-meta), 7.00–6.93 (m, 2H, Ar-meta), 6.83 (bs, 2H, NH2), 5.58 (s, 1H, Py-H5), 3.83 (s, 3H, OCH3); 13C NMR (150.9 MHz, DMSO-d6) δ 168.42 (s, Py-C4), 166.35 (s, Py-C6), 158.61 (Py-C2), 157.16 (t, JCF = 13.0 Hz, Ar-para), 155.57 (dd, JCF = 7.3, 245.6 Hz, Ar-ortho), 145.19 (s, An-ipso), 132.46 (s, An-meta), 119.58 (s, CN), 118.2 (s, An-ortho), 101.77 (s, An-para), 98.91 (dd, JCF = 4.6, 21.1 Hz, Ar-meta), 78.80 (s, py-C5), 56.32 (s, OCH3); HRMS (ESI): Calculated for C18H14O2N5F2 [M + H]+: 370.1110, Found: 370.1105; UPLC/MS (m/z) 370.34 [M + H]+, Tr 4.68 min.
Preparation of 4-((4-amino-6–(2,4,6-trifluorophenoxy)pyrimidin-2-yl)amino)benzonitrile (36)
A mixture of 28 (100 mg, 0.3 mmol), 4-methoxybenzylamine (82 mg, 0.6 mmol), palladium(II) acetate (7 mg, 0.03 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (35 mg, 0.06 mmol), and cesium carbonate (195 mg, 0.6 mmol) in dry dioxane (5 mL) was heated at 80°C under an argon atmosphere for 16 h. The mixture was diluted with water (60 mL) and extracted with EtOAc (3 × 40 mL). The organic layers were collected and combined, dried over magnesium sulfate, filtered and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave the intermediate 4-((4-((4-methoxybenzyl)amino)-6–(2,4,6-trifluorophenoxy)pyrimidin-2-yl)amino)benzonitrile, which was subsequently dissolved in CF3COOH (3 mL) and stirred for 16 h. Volatiles were evaporated under vacuum, and flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 36 (10 mg, 20%) as a white solid.
(36) 1H NMR (600.1 MHz, DMSO-d6) δ 9.59 (bs, 1H, NH), 7.68–7.65 (m, 2H, An-ortho), 7.52–7.48 (m, 2H, An-meta), 7.48–7.43 (m, 2H, Ar-meta), 6.89 (bs, 2H, NH2), 5.63 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 167.91 (s, Py-C4), 166.39 (s, Py-C2), 158.52 (dt, JCF = 14.5, 245.0 Hz, Ar-para), 158.52 (s, Py-C6), 155.29 (ddd, JCF = 6.7, 15.7, 244 Hz, Ar-ortho), 145.07 (s, An-ipso), 132.45 (s, An-meta), 125.92 (td, JCF = 5.1, 16.5 Hz, Ar-ipso), 119.10 (s, CN), 101.89 (s, An-para), 101.50 (td, JCF = 4.2, 20.9 Hz, Ar-meta), 78.95 (s, Py-C5); HRMS (ESI): Calculated for C17H11ON5F3 [M + H]+: 358.0910, Found: 358.0912; UPLC/MS (m/z) 358.181 [M + H]+, Tr 4.55 min.
Preparation of 4-((4-amino-6-((2,6-difluoro-4-methoxyphenyl)amino)pyrimidin-2-yl)amino)benzonitrile (37)
Compound 29 (250 mg, 0.7 mmol) and NaN3 (182 mg, 2.8 mmol) were dissolved in DMF (4 mL), and the reaction mixture was heated under MW conditions at 140°C for 1 h. The solution was diluted with water (40 mL) and extracted with EtOAc (3 ×30 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Evaporation gave 50 mg of the crude intermediate 4-((4-azido-6-((2,6-difluoro-4-methoxyphenyl)amino)pyrimidin-2-yl)amino)benzonitrile, which was subsequently dissolved in THF (10 mL) with triphenylphosphine (26 mg, 0.1 mmol). The reaction mixture was stirred at room temperature for 16 h. Water (4 mL) and a catalytic amount of HCl were then added, and the solution was stirred for 16 h. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 37 (30 mg, 11%) as a white solid.
(37) 1H NMR (600.1 MHz, DMSO-d6) δ 7.75 (bs, 1H, An-NH), 7.62–7.59 (m, 2H, An-ortho), 7.41–7.38 (m, 2H, An-meta), 6.52–6.48 (m, 2H, Ar-meta), 6.27 (bs, 1H, Ar-NH), 5.07 (bs, 1H, Py-H5), 4.69 (bs, 2H, NH2), 3.76 (s, 3H, CH3); 13C NMR (150.9 MHz, DMSO-d6) δ 164.15 (s, Py-C6), 163.05 (s, Py-C2), 159.35 (dt, JCF = 7.4, 248.5 Hz, Ar-para), 159.01 (d, JCF = 13.3 Hz, Ar-ortho), 158.79 (s, Py-C4), 144.45 (s, An-ipso), 132.81 (s, An-meta), 119.75 (s, CN), 118.20 (s, An-ortho), 108.08 (d, JCF = 17.0 Hz, Ar-ipso), 103.24 (s, An-para), 98.29 (td, JCF = 4.3, 22.9 Hz, Ar-meta), 77.77 (s, Py-C5), 55.90 (s, CH3); HRMS (ESI): Calculated for C18H15ON6F2 [M + H]+: 369.1271, Found: 369.1270; UPLC/MS (m/z) 369.236 [M + H]+, Tr 3.64 min.
Preparation of 4-((4-amino-6-((2,4,6-trifluorophenyl)amino)pyrimidin-2-yl)amino)benzonitrile (38)
Compound 30 (200 mg, 0.5 mmol) and NaN3 (98 mg, 1.5 mmol) were dissolved in DMF (7 mL), and the reaction mixture was heated under MW conditions at 130°C for 2 h. The solution was diluted with water (40 mL) and extracted with EtOAc (3 ×30 mL). The organic layers were collected, dried over magnesium sulfate, filtered, and evaporated under vacuum. Flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 50 mg of the intermediate 4-((4-azido-6-((2,4,6-trifluorophenyl)amino)pyrimidin-2-yl)amino)benzonitrile, which was subsequently dissolved in THF (10 mL) with triphenylphosphine (34 mg, 0.13 mmol). The reaction mixture was stirred at room temperature for 16 h. Water (4 mL) and catalytic amount of HCl were then added, and the resulting solution was stirred at room temperature for 16 h. Volatiles were evaporated under vacuum, and flash reverse phase chromatography (gradient from water to MeOH, 0–100%) gave 38 (10 mg, 20%) as a white solid.
(38) 1H NMR (600.1 MHz, DMSO-d6) δ 9.20 (bs, 1H, An-NH), 8.39 (bs, 1H, Ar-NH), 7.82–7.78 (m, 2H, An-ortho), 7.50–7.46 (m, 2H, An-meta), 7.33–7.27 (m, 2H, Ar-meta), 6.22 (bs, 2H, NH2), 5.31 (s, 1H, Py-H5); 13C NMR (150.9 MHz, DMSO-d6) δ 164.63 (s, Py-C6), 162.10 (s, Py-C4), 159.54 (dt, JCF = 15.3, 245.5 Hz, Ar-para), 158.97 (s, Py-C2), 158.88 (ddd, JCF = 7.7, 15.8, 248.2 Hz, Ar-ortho), 146.08 (s, An-ipso), 132.56 (s, An-meta), 120.00 (s, CN), 117.96 (s, An-ortho), 113.97 (dd, JCF = 4.7, 16.9 Hz, Ar-ipso), 100.99 (s, An-para), 100.95 (td, JCF = 5.7, 21.4 Hz, Ar-meta), 78.06 (s, Py-C5); HRMS (ESI): Calculated for C17H12N6F3 [M + H]+: 357.1071, Found: 357.1071; UPLC/MS (m/z) 357.236 [M + H]+, Tr 3.72 min.
MT-4 antiviral and cytotoxicity assay
Compounds were tested in a high-throughput 384-well assay format for their ability to inhibit the virus replication-induced cytopathic effect in MT-4 cell cultures acutely infected with HIV-1 (IIIB strain). Compounds were serially diluted (1:3) in DMSO on 384-well polypropylene plates and further diluted 200-fold into complete RPMI media (10% FBS, 1% P/S) using Biotek Micro Flow and Agilent ECHO acoustic dispenser. Each plate contained up to eight test compounds, with negative (no drug control) and 5 µM AZT positive controls. MT-4 cells were pre-infected with 10 µL of either RPMI (mock-infected) or a fresh 1:250 dilution of an HIV-1 (IIIB) concentrated virus stock. Infected and uninfected MT-4 cells were further diluted in complete RPMI media and added to each plate using a Micro-Flow dispenser. After five days of incubation in a humidified and temperature controlled incubator (37°C), Cell Titer Glo (Promega) was added to the assay plates to quantify the amount of luciferase. EC50 and CC50 values were defined as the compound concentration that causes a 50% decrease in luminescence signal and were calculated using a sigmoidal dose–response model to generate curve fits.26
Acknowledgement
This study is a part of the research project RVO61388963 of the Institute of Organic Chemistry and Biochemistry, Czech Academy of Sciences. We would like to thank Amberlyn Peterson for language editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the subvention for development of research organization (Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, RVO: 61388963).
References
- 1.World Health Organization, Geneva, Switzerland. www.who.int (accessed 4 September 2018).
- 2.Poorolajal J, Hooshmand E, Mahjub H, et al. Survival rate of AIDS disease and mortality in HIV-infected patients: a meta-analysis. Public Health 2016; 139: 3–12. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E. Non‐nucleoside reverse transcriptase inhibitors (NNRTIs). Past Present and Future Chem Biodivers 2004; 1: 44–64. [DOI] [PubMed] [Google Scholar]
- 4.Madruga JV, Cahn P, Grinsztejn B, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-1: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007; 370: 29–38. [DOI] [PubMed] [Google Scholar]
- 5.Lazzarin A, Campbell T, Clotet B, et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 2007; 370: 39–48. [DOI] [PubMed] [Google Scholar]
- 6.Geretti AM. Shifting paradigms: the resistance profile of etravirine. J Antimicrob Chemother 2008; 62: 643–647. [DOI] [PubMed] [Google Scholar]
- 7.Janssen PA, Lewi PJ, Arnold E, et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1 E)-2-cyanoethenyl]-2, 6-dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (R278474, rilpivirine). J Med Chem 2005; 48: 1901–1909. [DOI] [PubMed] [Google Scholar]
- 8.Baert L, van G, Klooster T, et al. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur J Pharm Biopharm 2009; 72: 502–508. [DOI] [PubMed] [Google Scholar]
- 9.de Béthune M-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res 2010; 85: 75–90. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds C, de Koning CB, Pelly SC, et al. In search of a treatment for HIV–current therapies and the role of non-nucleoside reverse transcriptase inhibitors (NNRTIs). Chem Soc Rev 2012; 41: 4657–4670. [DOI] [PubMed] [Google Scholar]
- 11.De Clercq E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res 1998; 38: 153–179. [DOI] [PubMed] [Google Scholar]
- 12.Zhan P, Chen X, Li D.et al. HIV‐1 NNRTIs: structural diversity, pharmacophore similarity, and impliations for drug design. Med Res Rev 2013; 33: E1–72. [DOI] [PubMed] [Google Scholar]
- 13.Lansdon EB, Brendza KM, Hung M, et al. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J Med Chem 2010; 53: 4295–4299. [DOI] [PubMed] [Google Scholar]
- 14.Li D, Zhan P, De Clercq E, et al. Strategies for the design of HIV-1 non-nucleoside reverse transcriptase inhibitors: lessons from the development of seven representative paradigms. J Med Chem 2012; 55: 3595–3613. [DOI] [PubMed] [Google Scholar]
- 15.Zhan P, Pannecouque C, De Clercq E, et al. Anti-HIV drug discovery and development: current innovations and future trends: miniperspective. J Med Chem 2015; 59: 2849–2878. [DOI] [PubMed] [Google Scholar]
- 16.Chen FE, Zeng Z-S, Liang Y-H. Feng, Patent CN 101723903, P.R.C. Fudan University, China, 2010.
- 17.Ma XD, Yang SQ, Gu SX, et al. Synthesis and anti‐HIV activity of Aryl‐2‐[(4‐cyanophenyl) amino]‐4‐pyrimidinone hydrazones as potent non‐nucleoside reverse transcriptase inhibitors. ChemMedChem 2011; 6: 2225–2232. [DOI] [PubMed] [Google Scholar]
- 18.Feng X-Q, Zeng Z-S, Liang Y-H, et al. Synthesis and biological evaluation of 4-(hydroxyimino) arylmethyl diarylpyrimidine analogues as potential non-nucleoside reverse transcriptase inhibitors against HIV. Bioorg Med Chem 2010; 18: 2370–2374. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Meng G, Zheng A, et al. Design and synthesis of a new series of cyclopropylamino-linking diarylpyrimidines as HIV non-nucleoside reverse transcriptase inhibitors. Eur J Pharm Sci 2014; 62: 334–341. [DOI] [PubMed] [Google Scholar]
- 20.Meng G, Liu Y, Zheng A, et al. Design and synthesis of a new series of modified CH-diarylpyrimidines as drug-resistant HIV non-nucleoside reverse transcriptase inhibitors. Eur J Med Chem 2014; 82: 600–611. [DOI] [PubMed] [Google Scholar]
- 21.Yan Z-H, Wu H-Q, Chen W-X, et al. Synthesis and biological evaluation of CHX-DAPYs as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem 2014; 22: 3220–3226. [DOI] [PubMed] [Google Scholar]
- 22.Zeng ZS, Liang YH, Feng XQ, et al. Lead optimization of diarylpyrimidines as non‐nucleoside inhibitors of HIV‐1 reverse transcriptase. ChemMedChem 2010; 5: 837–840. [DOI] [PubMed] [Google Scholar]
- 23.Gu S-X, He Q-Q, Yang S-Q, et al. Synthesis and structure–activity relationship of novel diarylpyrimidines with hydromethyl linker (CH (OH)-DAPYs) as HIV-1 NNRTIs. Bioorg Med Chem 2011; 19: 5117–5124. [DOI] [PubMed] [Google Scholar]
- 24.Yan Z-H, Huang X-Y, Wu H-Q, et al. Structural modifications of CH (OH)-DAPYs as new HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem 2014; 22: 2535–2541. [DOI] [PubMed] [Google Scholar]
- 25.Gu S-X, Qiao H, Zhu Y-Y, et al. A novel family of diarylpyrimidines (DAPYs) featuring a diatomic linker: design, synthesis and anti-HIV activities. Bioorg Med Chem 2015; 23: 6587–6593. [DOI] [PubMed] [Google Scholar]
- 26.Šimon P, Baszczyňski O, Šaman D, et al. Novel (2,6-difluorophenyl)(2-(phenylamino)pyrimidin-4-yl)methanones with restricted conformation as potent non-nucleoside reverse transcriptase inhibitors against HIV-1. Eur J Med Chem 2016; 122: 185–195. [DOI] [PubMed] [Google Scholar]
- 27.Mordant C, Schmitt B, Pasquier E, et al. Synthesis of novel diarylpyrimidine analogues of TMC278 and their antiviral activity against HIV-1 wild-type and mutant strains. Eur J Med Chem 2007; 42: 567–579. [DOI] [PubMed] [Google Scholar]
- 28.Rotili D, Tarantino D, Artico M, et al. Diarylpyrimidine−dihydrobenzyloxopyrimidine hybrids: new, wide-spectrum anti-HIV-1 agents active at (sub)-nanomolar level. J Med Chem 2011; 54: 3091–3096. [DOI] [PubMed] [Google Scholar]
- 29.Ludovici DW, De Corte BL, Kukla MJ, et al. Evolution of anti-HIV drug candidates. Part 3: diarylpyrimidine (DAPY) analogues. Bioorg Med Chem Lett 2001; 11: 2235–2239. [DOI] [PubMed] [Google Scholar]
- 30.Bedford ST, Benwell KR, Brooks T, et al. of potent and selective functional antagonists of the human adenosine A 2B receptor. Bioorg Med Chem Lett 2009; 19: 5945–5949. [DOI] [PubMed] [Google Scholar]
- 31.Miyashita A, Matsuda H, Iijima C, et al. Catalytic action of azolium salts. ii. aroylation of 4-chloroquinazolines with aromatic aldehydes catalyzed by 1, 3-dimethylbenzimidazolium iodide. Chem Pharm Bull 1992; 40: 43–48. [Google Scholar]
- 32.Procházková E, Čechová L, Tarábek J, et al. Tunable push–pull interactions in 5-nitrosopyrimidines. J Org Chem 2016; 81: 3780–3789. [DOI] [PubMed] [Google Scholar]
- 33.Surry DS, Buchwald SL. Dialkylbiaryl phosphines in Pd-catalyzed amination: a user’s guide. Chem Sci 2011; 2: 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantrell AS, Engelhardt P, Högberg M, et al. Phenethylthiazolylthiourea (PETT) compounds as a new class of HIV-1 reverse transcriptase inhibitors. 2. Synthesis and further structure−activity relationship studies of PETT analogs. J Med Chem 1996; 39: 4261–4274. [DOI] [PubMed] [Google Scholar]
- 35.Chen X, Liu X, Meng Q, et al. Novel piperidinylamino-diarylpyrimidine derivatives with dual structural conformations as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem Lett 2013; 23: 6593–6597. [DOI] [PubMed] [Google Scholar]
- 36.Joshi S, Maikap GC, Titirmare S, et al. An improved synthesis of etravirine. Org Process Res Dev 2010; 14: 657–660. [Google Scholar]
- 37.Thrippleton MJ, Keeler J. Elimination of zero‐quantum interference in two‐dimensional NMR spectra. Angew Chem 2003; 115: 4068–4071. [DOI] [PubMed] [Google Scholar]
- 38.Pauwels R, De Clercq E, Desmyter J.et al. Sensitive and rapid assay on MT-4 cells for detection of antiviral compounds against the AIDS virus. J Virol Methods 1987; 16: 171–185. [DOI] [PubMed] [Google Scholar]
- 39.Andries K, Azijn H, Thielemans T.et al. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother 2004; 48: 4680–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen PA, Lewi PJ, Arnold E.et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1 E)-2-cyanoethenyl]-2, 6-dimethylphenyl] amino]-2-pyrimidinyl] amino] benzonitrile (R278474, rilpivirine). J Med Chem 2005; 48: 1901–1909. [DOI] [PubMed] [Google Scholar]
- 41.Kertesz DJ, Brotherton-Pleiss C, Yang M, et al. Discovery of piperidin-4-yl-aminopyrimidines as HIV-1 reverse transcriptase inhibitors. N-benzyl derivatives with broad potency against resistant mutant viruses. Bioorg Med Chem Lett 2010; 20: 4215–4218. [DOI] [PubMed] [Google Scholar]