

Abstract
Background:
The intimal endothelium is known to condition the underlying medial smooth muscle cell (SMC) layer of the vessel wall, and is highly responsive to receptor-activator of nuclear factor-kB ligand (RANKL) and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), pro-calcific and anti-calcific agents, respectively. In this paper, we tested the hypothesis that RANKL-induced activation of endothelial NF-κB signalling is essential for pro-calcific activation of the underlying SMCs.
Methods:
For these studies, human aortic endothelial and smooth muscle cell mono-cultures (HAECs, HASMCs) were treated with RANKL (0–25 ng/ml ± 5 ng/ml TRAIL) for 72h. Non-contact transwell HAEC:HASMC cocultures were also employed in which the luminal HAECs were treated with RANKL (± 5 ng/ml TRAIL), followed by analysis of pro-calcific markers in the underlying subluminal HASMCs.
Results:
Treatment of either HAECs or HASMCs with RANKL activated the non-canonical NF-κB/p52 and canonical NF-κB/p65 pathways in both cell types. In RANKL ± TRAIL-treated HAECs, recombinant TRAIL, previously demonstrated by our group to strongly attenuate the pro-calcific signalling effects of RANKL, was shown to specifically block the RANKL-mediated activation of non-canonical NF-κB/p52, clearly pointing to the mechanistic relevance of this specific pathway to RANKL function within endothelial cells. In a final series of HAEC:HASMC transwell co-culture experiments, RANKL treatment of HAECs that had been genetically silenced (via siRNA) for the NF-κB2 gene (the molecular forerunner to NF-κB/p52 generation) exhibited strongly attenuated pro-calcific activation of underlying HASMCs relative to scrambled siRNA controls.
Summary:
These in vitro observations provide valuable mechanistic insights into how RANKL may potentially act upon endothelial cells through activation of the alternative NF-κB pathway to alter endothelial paracrine signalling and elicit pro-calcific responses within underlying vascular smooth muscle cells.
Keywords: Endothelial cell, Smooth muscle cell, Calcification, RANKL, TRAIL, NF-κB/p52
Graphical Abstract
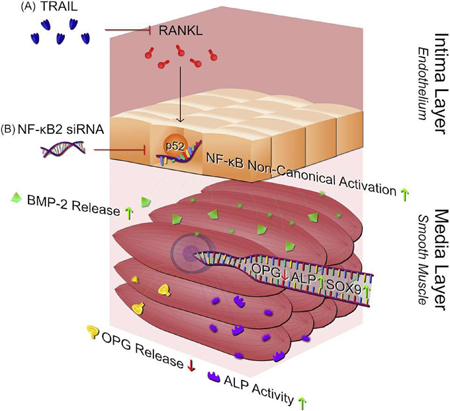
1. Introduction
Elevated level of calcium deposition within the intimal and medial regions of the blood vessel wall give rise to vascular calcification (VC), widely considered a significant risk factor with respect to cardiovascular (CV) morabidity and mortality [1–3]. Much evidence indicates that receptor-activator of nuclear factor-κB ligand (RANKL) display elevated expression in areas of mineralized plaque [4], as well as potent osteoblastic (pro-calcific) actions within blood vessels (for review see [5]).In respect of the latter, RANKL has been shown to promote the osteoblastic activation of vascular smooth muscle cells (VSMCs) via direct VSMC stimulation [6,7], and via stimulation of overlying endothelial cells (ECs) leading to paracrine EC:VSMC signalling [7–9]. Indeed, our own group has recently demonstrated that RANKL can elicit pro-calcific signalling in vascular cells (e.g. increased bone morphogenetic protein-2/BMP-2, alkaline phosphatase/ALP, Runx2, and Sox9 levels, in conjunction with decreased osteoprotegerin/OPG levels) using both mono-culture (direct SMC stimulation) and co-culture (EC:VSMC paracrine stimulation) models [7]. Interestingly, we recently demonstrated that several of the observed pro-calcific actions of RANKL in either model were strongly prevented by RANKL co-incubation with tumour necrosis factor-related apoptosis-inducing ligand (TRAIL), reinforcing the widely held view that TRAIL has vasoprotective potential (for review see [10]).
The signal transduction mechanism(s) regulating the pro-calcific actions of RANKL in the vascular wall, and indeed the RANKL-blocking effects of TRAIL, remain to be properly elucidated, particularly within the context of the EC:VSMC signalling axis. Evidence suggests that the canonical and non-canonical NF-κB pathways may be involved. A study by Panizo et al. for example, demonstrates that direct RANKL treatment of VSMCs leading to calcification may proceed through the non-canonical NF-κB pathway [6]. More recent indirect evidence is presented by Zhan and co-workers, who demonstrate how Exenatide, a glucagon-like peptide-1 (GLP-1) receptor agonist, can inhibit VSMC calcification by disrupting RANKL/NF-κB signalling [11]. The pro-calcific effects of RANKL on VSMCs have also been linked to the induction of proin- flammatory cytokines [12], well established signalling substrates for the NF-κB pathway, and consistent with other studies linking proin- flammatory signalling and NF-κB activation to VSMC calcification [13]. Also worthy of mention (albeit, outside the vascular context), osteoclastic precursor differentiation in response to RANKL/NF-κB signalling is a well characterized phenomenon associated with bone remodelling [14,15].
To our knowledge however, it is not currently known if RANKL elicits NF-kB activation within endothelial cells, and indeed whether or not this may lead to observable pro-calcific responses within underlying VSMCs. To address this important mechanistic question therefore, we have employed human aortic endothelial cells (HAECs) and human aortic smooth muscle cells (HASMCs) to investigate: (i) how RANKL ± TRAIL treatment of either cell type elicits activation of the NF-kB canonical (p65) and non-canonical (p52) pathways and; (ii) how RANKL treatment of HAECs affects pro-calcific activation within underlying co-cultured HASMCs following HAEC/NF-kB silencing.
2. Materials and methods
2.1. Chemicals
Unless otherwise stated, all reagents were purchased from Sigma- Aldrich (Dublin, IRL). Both human aortic endothelial cells (HAECs) and human aortic smooth muscle cells (HASMCs), as well as their respective growth media were purchased from Promocell GmbH (Heidelberg, Germany). Recombinant human RANKL and TRAIL were purchased from R&D Systems (Minneapolis, MN, USA). Primers were sourced from Sigma Aldrich and Eurofins Genomics (Ebersburg, Germany), whilst qPCR reagents were purchased from Roche Diagnostics (West Sussex, UK). Primary antisera (IgG) were purchased from the following sources: Anti-NF-κB/p65 and anti-NF-κB/Phospho-p65 IgG (Cell Signalling Technologies, Danvers, MA, USA); Anti-NF-κB/p52/p100 IgG - this antiserum binds the full-length/p100 and the cleaved/p52 NF-κB fragments (Merck Millipore, Billerica, MA, USA); Anti-GAPDH IgG (R& D Systems, Minneapolis, MN, USA). HRP-conjugated secondary antisera were purchased from Cell Signalling Technologies. ELISA DuoSet kits and alkaline phosphatase (ALP) activity assay kits were purchased from R&D Systems and BioAssay Systems (Hayward, CA, USA), respectively.
2.2. Guidelines and ethical approval
All experimental protocols were carried out in accordance with Dublin City University health and safety regulations. Institutional ethical approval and informed consent were not required for this study (i.e. use of widely commercially available non-transformed human-derived cell lines).
2.3. Cell culture
HAECs obtained from a 23 year old Caucasian male were cultured in endothelial cell growth medium (Promocell GmbH, catalog no. C22020) with the following supplements; fetal calf serum (0.05 ml/ml), endothelial cell growth supplement (0.004 ml/ml), epidermal growth factor (10ng/ml), heparin (90 μg/ml) and hydrocortisone (1 μg/ml). This media was also supplemented with penicillin (100IU/ml) and streptomycin (100 μg/ml). HASMCs obtained from a 19 year old Caucasian male were cultured in smooth muscle cell growth medium (Promocell GmbH, catalog no. C22062) containing the same concentrations of antibiotics, in addition to fetal calf serum (0.05ml/ml), epidermal growth factor (0.5ng/ml), basic fibroblast growth factor (2 ng/ml), and insulin (5 μg/ml). Cells were maintained in a humidified incubator at 37 °C and 5% CO2. Passages 5–10 were used for experimental purposes. Cell number and viability were routinely measured using the advanced detection and accurate measurement (ADAM™) cell counter (Digital Bio, Seoul, KOR) for seeding density purposes, data normalization, and optimization of siRNA transfection conditions (described below). Cell culture experiments were subsequently conducted in two formats: (i) Mono-culture experiments - HAECs and HASMCs were grown to confluency in 6-well dishes and separately treated for 72 h with either RANKL (0–25 ng/ml), TRAIL (0–5 ng/ml), or a combination of both (RANKL: 5 & 25 ng/ml + TRAIL: 5 ng/ml). This treatment period and dose range were previously established by Davenport et al. [8,16] and Harper et al. [7] and yielded robust responses. Post-treatment, cell lysates were harvested as previously described [17] in order to monitor activation of NF-κB/p52 (normalized to NF-κB/p100) and NF-κB/Phospho-p65 (normalized to NF-κB/p65) by Western blotting. All samples were stored at — 80 °C and assayed within three months; (ii) Co-culture experiments - The effect of HAEC paracrine signalling upon HASMC osteoblastic activity was also investigated using a non-contact transwell co-culture model as previously described [7]. Subluminal compartment; HASMCs were seeded at a density of 1.5 × 105 cells per well into standard 6-well culture dishes and grown to confluency. Luminal compartment; HAECs were seeded into permeable (0.4 μm pore) transwell culture inserts (Merck Milli-pore, MA, USA) at a density of 2 × 105 HAECs per insert and grown to confluency. Transwell inserts were then positioned into HASMC plate wells to establish co-culture conditions. A 50:50 mixture of HAE- C:HASMC growth media was employed throughout the subsequent coculture treatment period as previously described [8]. In the first series of co-culture experiments, HAECs were treated for 72 h with either RANKL (0–25 ng/ml), TRAIL (0–5 ng/ml), or a combination of both (RANKL: 5 & 25 ng/ml + TRAIL: 5 ng/ml). Post-treatment, HASMC lysates were harvested as previously described [17] in order to monitor activation of NF-κB/p52 (normalized to NF-κB/p100) and NF-κB/ Phospho-p65 (normalized to NF-κB/p65) by Western blotting. In the second series of co-culture experiments, HAECs were transfected with either 50 nM NF-κB2 siRNA (which codes for the NF-κB/p100 fragment, and so by extension, the NF-κB/p52 fragment) or 50 nM scrambled non-targeting siRNA control and then treated for 72 h with RANKL (0–25 ng/ml). Post-treatment, HASMCs (protein, mRNA) and subluminal conditioned media were harvested for analysis of specific markers of osteoblastic activation (OPG, BMP-2, ALP, Sox9, IL-6, and Runx2). Transfection procedures and optimization are outlined below.
2.4. NF-κB2 siRNA transfection and optimization
All siRNA reagents were purchased from Dharmacon (Lafayette, Colorado, USA). 5 μM siRNA solution was prepared in sterile molecular grade H2O. DharmaFECT™−1 reagent (catalog no. T-2001) and siRNA were individually diluted in antibiotic- and serum-free medium and incubated for 5 min at room temperature. Diluted DharmaFECT™−1 and siRNA were mixed and incubated for a further 20 min at room temperature. The resulting mix was diluted with antibiotic-free complete medium, achieving a final concentration of 50 μM siRNA and a final volume of 10 μl DharmaFECT™−1 per well. Standard media was removed from the cultured HAECs, transfection medium added, and cells were incubated for 48 h (mRNA analysis by qPCR) or 72 h (protein analysis by Western blotting) unless otherwise described. Specific procedures for optimizing siRNA-based knockdown of NF-κB2 in HAECs are outlined as follows: (i) Transfection optimization - HAECs were seeded at a density of 2.5 × 106 cells per well in standard 6-well format for optimisation purposes. Initially, cells were assessed for gene knockdown receptivity using positive control siRNA for endogenous GAPDH (Dharmacon, catalog no. D-001830–01). In this respect, HAECs were exposed to 25 nM GAPDH siRNA across a range of 2.5–10 μl/well DharmaFECT™−1 reagent volumes in order to assess reagent toxicity. Viabilities were maintained above 90% for all DharmaFECT™−1 volumes tested (data not shown). For NF-kB2 gene knockdown optimisation, HAECs were seeded at 2.5 × 106 cells per well and exposed to either 25 nM or 50 nM of NF-κB2 siRNA (Dharmacon, catalog no. L- 003918–00-0005) across a DharmaFECT™−1 volume range of 2.5–10 μl/ well. It was subsequently confirmed that a 50 nM siRNA concentration in 10 μl/well of DharmaFECT™ 1 was the optimal exposure condition for NF-κB2 knockdown in HAECs. Cell viability was maintained above 80% for all conditions tested. Controls routinely included non-targeting scrambled siRNA (Dharmacon, catalog no. D-001206–13-05) and untransfected cells; (ii) NF-κB2 knockdown for co-culture experiments - Prior to co-culture, it was confirmed that siRNA knockdown in HAECs was equally efficient in transwell inserts as compared to standard 6-well culture dishes. HAECs were seeded into semi-permeable transwell inserts at a density of 1.5 × 106 cells per insert, and allowed to grow for 24 h before commencing siRNA knockdown in transfection media over 48 h. HAEC inserts were then removed from the transfection media and transferred to the standard 6-well dishes of confluent HASMCs, thus establishing the co-culture. HAECs were allowed to recover for 6 h in fresh media prior to RANKL treatment for 72 h.
2.5. Quantitative real-time PCR (qPCR)
Extraction of total RNA and preparation of cDNA was achieved using the TRIzol™ RNA extraction protocol (ThermoFisher Scientific) and the Applied Biosystems™ high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific), respectively. Prior to cDNA preparation, all RNA samples were routinely pre-treated with DNasel (Sigma- Aldrich). Amplification of target cDNA sequences using gene-specific primers was achieved using the LightCycler®96 real-time PCR system (Roche Diagnostics, West Sussex, UK). PCR reaction mixtures (10 μl) were as follows: 5 μl of FastStart Universal SYBR Green Mastermix (Roche Diagnostics), 1.5 μl of RNase-free water, 2.5 μl of cDNA, 0.5 μl each of 10 μM forward and reverse primers. PCR reaction conditions were as follows: denaturation at 95 °C for 10 min followed by 45 cycles of: (i) denaturation at 95 °C for 10 s; (ii) annealing at 59 °C for 10 s; and (iii) elongation at 72 °C for 10 s. Each cDNA sample was assayed in triplicate and results analysed by the comparative CT method. GADPH was routinely used for normalization purposes. All primer pairs were designed for optimal efficiency according to MIQE guidelines and were pre-screened for correct product size (1% agarose gel electrophoresis). Primer pairs also underwent melt-curve analysis for confirmation of PCR product purity and detection of primer-dimers. GAPDH (238 bp): Forward 5’-gagtcaacggatttggtcgt-3’; Reverse 5’-ttgattttggagggatctcg-3’; ALP (293 bp): Forward 5’-gcctggctacaaggtggtg-3’; Reverse 5’-ggcca- gagcgagcagc-3’; Runx2 (315 bp): Forward 5’-ggtaccagatgggactgtgg-3’; Reverse 5’-gaggcggtcagagaacaaac-3’; Sox9 (85 bp): Forward 5’-agc- gaacgcacatcaagac-3’; Reverse 5’-ctgtaggcgatctgttgggg-3’; OPG (241 bp): Forward 5’-ggcaacacagctcacaagaa-3’; Reverse 5’-ctgggtttgcatgcctttat-3’; BMP-2 (199 bp): Forward 5’-caagccaaacacaaacagcg-3’; Reverse 5’- ccaacgtctgaacaatggca-3’; IL-6 (183 bp): Forward 5’-aaagaggcactggca- gaaaa-3’; Reverse 5’-agctctggcttgttcctcac-3’; NF-κB2 (89 bp): Forward 5’-agaggcttccgatttcgatatgg-3’; Reverse 5’-ggataggtctttcggcccttc-3’.
2.6. Western blotting
Following experimental treatments, HAEC and HASMC lysates were harvested and monitored for levels of NF-κB/p52 (normalized to NF- κB/p100) and NF-κB/Phospho-p65 (normalized to NF-κB/p65) by Western immunoblotting as described by Rochfort et al. with minor modifications [17]. Primary antisera were prepared in TBS-T (tris- buffered saline +0.1% Tween-20) containing 1% BSA at the following dilutions: 1/1000 NF-κB/p52/p100 (rabbit monoclonal IgG), 1/1000 NF-κB/Phospho-p65 and NF-κB/p65 (rabbit polyclonal IgG), and 1/ 5000 GAPDH (mouse monoclonal IgG). Secondary antisera were also prepared in TBST ( + 1% BSA): HRP-conjugated anti-rabbit IgG (1/ 1000) for all NF-κB targets and anti-mouse IgG (1/5000) for GAPDH.
2.7. Enzyme-linked ImmunoSorbent assay (ELISA)
DuoSet™ ELISA Kits (R&D Systems), used in conjunction with F96 Maxisorp™ Nunc-Immuno™ 96-well plates (Bio-Sciences Ltd., Dun Laoghaire, IRL), were employed to accurately measure absolute levels of OPG, BMP-2 and IL-6 within cell lysates and subluminal conditioned media according to the method of Harper et al. [7]. For normalization purposes, OPG and BMP-2 levels in protein lysates were routinely presented as pg/mg of total cellular protein, whilst conditioned media levels were presented as pg/105 cells.
2.8. Alkaline phosphatase activity assay
Previous researchers have successfully employed the Quantichrom™ Kit (BioAssay Systems) to monitor ALP activity, a critical marker of osteoblastic activation [18]. ALP activity was measured in vascular cell conditioned media and total HASMC protein lysate samples according to the method of Harper et al. [7].
2.9. Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Experimental points were performed in duplicate or triplicate with a minimum of three independent experiments (N = 3). Statistical comparisons between control and experimental groups was by ANOVA in conjunction with a Dunnett’s post-hoc test for multiple comparisons. A value of *P <.05 versus control was considered significant. A Student’s t-test was also routinely employed for pairwise comparisons (δP ≤ 0.05).
3. Results
3.1. Effects of RANKL ± TRAIL on NF-kB activation in HAEC mono-cultures
The impact of RANKL ± TRAIL on the activation in HAECs of both the non-canonical (p52) and canonical (p65) NF-κB signalling pathways was investigated. Treatment of HAECs with RANKL (0–25 ng/ml, 72 h) increased NF-κB/p52 levels in a dose-dependent manner. By contrast, treatment with TRAIL (0–25 ng/ml, 72 h) had no effect. Co-incubation of HAECs with RANKL (up to 25 ng/ml) and 5 ng/ml TRAIL however, was subsequently found to completely block the effects of RANKL on NF-κB/p52 induction (Fig. 1A). In parallel studies, treatment of HAECs with RANKL (0–25 ng/ml, 72 h) significantly increased NF-κB/ Phospho-p65 levels, whilst treatment with TRAIL (0–25 ng/ml, 72 h) had no effect. Co-incubation of cells with RANKL (up to 25 ng/ml) and 5 ng/ml TRAIL did not block the effects of RANKL on NF-κB/Phospho- p65 induction (Fig. 1B).
Fig. 1.
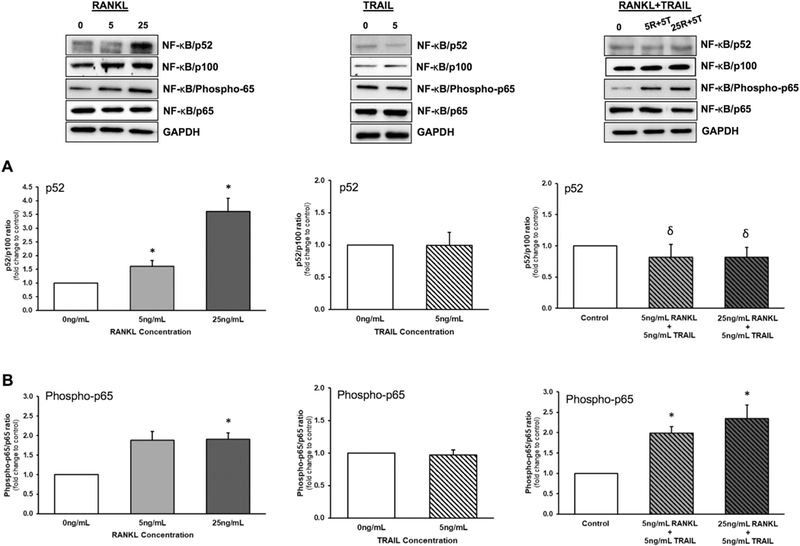
Effects of RANKL and TRAIL on NF-κB activation in HAECs. Endothelial cells were treated for 72 h with RANKL (0–25 ng/ml), TRAIL (0–25 ng/ml), or a combination of both ligands (see key below). Cells were then monitored by Western blotting for activation of NF-κB as follows: (A) p52/p100 ratio; and (B) Phospho- p65/p65 ratio. Histograms represent fold change in band intensity relative to untreated controls. Blots are representative. *P ≤.05 versus 0 ng/ml RANKL (or control); δP ≤.05 versus 5 and 25 ng/ml RANKL alone, respectively. Key: 5R, 5 ng/ml RANKL; 25R, 25 ng/ml RANKL; 5T, 5 ng/ml TRAIL.
3.2. Effects of RANKL ± TRAIL on HASMC mono-cultures
The impact of RANKL ± TRAIL on the activation of both the non-canonical and canonical NF-κB signalling pathways in HASMCs was next investigated. Treatment of HAECs with RANKL (0–25 ng/ml, 72 h) increased both NF-κB/p52 and NF-κB/Phospho-p65 levels, with maximal activation achieved at just 5 ng/ml RANKL (Fig. 2A,B), whilst treatment with TRAIL (5 ng/ml, 72 h) was seen to significantly increase NF-κB/Phospho-p65 levels (Fig. 2B). Finally, co-incubation of HASMCs with RANKL (up to 25 ng/ml) and 5 ng/ml TRAIL did not block the effects of RANKL on either NF-κB/p52 or NF-κB/Phospho-p65 induction (Fig. 2A,B).
Fig. 2.
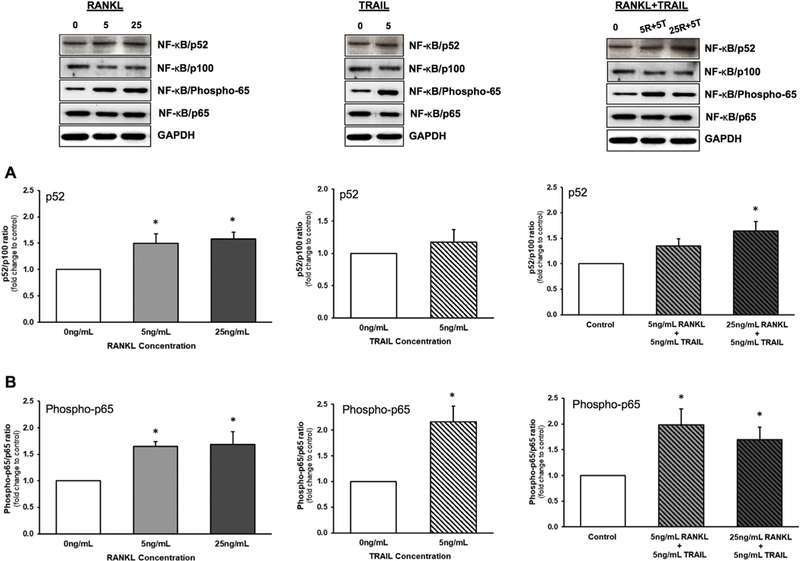
Effects of RANKL and TRAIL on NF-κB activation in HASMCs. Smooth muscle cells were treated for 72 h with RANKL (0–25 ng/ml), TRAIL (0–25 ng/ml), or a combination of both ligands (see key below). Cells were then monitored by Western blotting for activation of NF-κB as follows: (A) p52/p100 ratio; and (B) Phosphop65/ p65 ratio. Histograms represent fold change in band intensity relative to untreated controls. Blots are representative. *P ≤.05 versus 0 ng/ml RANKL (or control). Key: 5R, 5 ng/ml RANKL; 25R, 25 ng/ml RANKL; 5T, 5 ng/ml TRAIL.
3.3. Paracrine effects of RANKL ± TRAIL on HASMCs in a HAEC:HASMC co-culture model
In a final iteration of this experiment using the transwell co-culture model, we investigated how treatment of HAECs with RANKL ± TRAIL might affect activation of both the non-canonical and canonical NF-κB signalling pathways in underlying HASMCs. Treatment of HAECs with either RANKL (0–25 ng/ml, 72 h), TRAIL (5 ng/ml, 72 h), or a combination of both ligands, had no statistically significant effects on levels of NF-KB/p52 (Fig. 3A) and NF-κB/Phospho-p65 (Fig. 3B) in underlying HASMCs. In a further series of control experiments, the transendothelial permeability kinetics of RANKL and TRAIL within the HAEC:HASMC co-culture model was assessed. Confluent luminal HAECs were treated for 72 h with either RANKL (25 ng/ml) or TRAIL (5 ng/ml). Levels of recombinant RANKL (890pg/ml) and TRAIL (538pg/ml) measured within the subluminal HASMC space after 72 h corresponded to just 1.78% and 5.36% of the luminal ligand dose, respectively, clear evidence of a strong endothelial barrier and minimal cross-reactivity of ligands with underlying HASMCs (Supplementary Fig. S1A). Moreover, neither ligand had any effect on HAEC barrier function, monitored according to the method of Rochfort et al. [17] (Supplementary Fig. S1B).
Fig. 3.
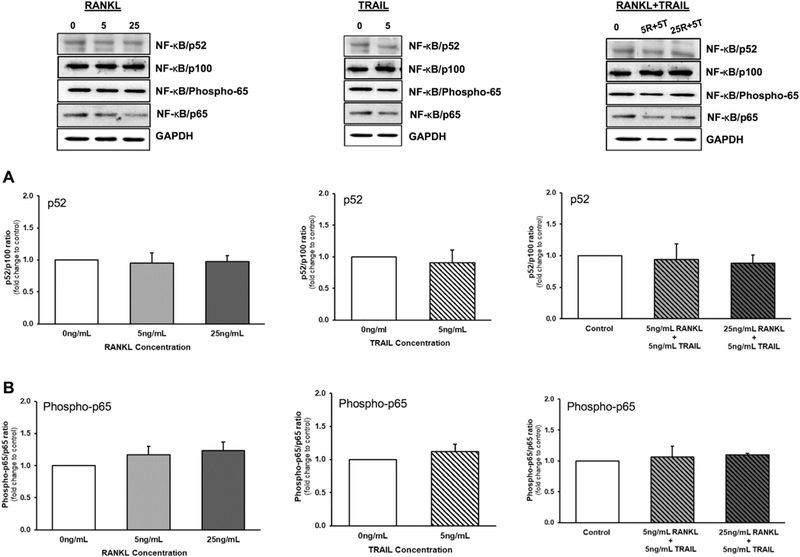
Effects of RANKL and TRAIL on NF-κB activation in a HAEC:HASMC transwell co-culture model. HAECs were treated for 72 h with RANKL (0–25 ng/ml), TRAIL (0–25ng/ml), or a combination of both ligands (see key below). HASMCs were then harvested and monitored by Western blotting for activation of NF-κB as follows: (A) p52/p100 ratio; and (B) Phospho-p65/p65 ratio. Histograms represent fold change in band intensity relative to untreated controls. Blots are representative. Key: 5R, 5 ng/ml RANKL; 25R, 25 ng/ml RANKL; 5T, 5 ng/ml TRAIL.
3.4. Efficacy of the NF-κB2 siRNA transfection in HAECs
To optimize NF-κB2 siRNA transfection conditions in HAECs, numerous parameters were considered: (i) volume of DharmaFECT™−1 transfection reagent; (ii) siRNA concentration; (iii) reduction in target gene expression; and (iv) effects on cell viability. Our optimization studies demonstrated that increasing volume (0–10 μl) of DharmaFECT™−1 reagent had only nominal effects on HAEC viability, with 2.5 μl exhibiting no statistically significant effects on cell viability (Fig. 4A,B - right hand side/RHS). Moreover, 50 nmol/l NF-κB2 siRNA delivered significantly greater knockdown than 25 nM NF-κB2 siRNA at all volumes of reagent tested (Fig. 4A,B - left hand side/LHS). The transfection condition selected for all subsequent experiments was therefore 50 nM NF-κB2 siRNA in 10 μl of DharmaFECT™−1 reagent, routinely yielding ~90% knockdown of mRNA and ~45–48% knockdown of the associated p100 (and p52) protein fragment (Fig. 5A – LHS & RHS). By contrast, transfection of HAECs with 50 nM scrambled nt- siRNA in 10 μl of DharmaFECT™−1 reagent had no significant effect on either NF-κB2 mRNA or protein levels (Fig. 5B - LHS & RHS). In the interests of siRNA quality control and specificity, the potential offtarget effects of NF-κB2 siRNA (Supplementary Fig. S2) and non-targeting scrambled siRNA (Supplementary Fig. S3) on markers of osteoblastic activation in HAECs were also assessed. Markers included ALP, BMP-2, OPG, Runx2 and IL-6. Across all markers tested, off-target effects in HAECs we’re either statistically insignificant or relatively trivial, with the exception of IL-6. Transfection of HAECs with NF-kB2 siRNA was seen to concurrently increase IL-6 mRNA levels, whilst decreasing IL-6 release (Supplementary Fig. S2C,F).
Fig. 4.
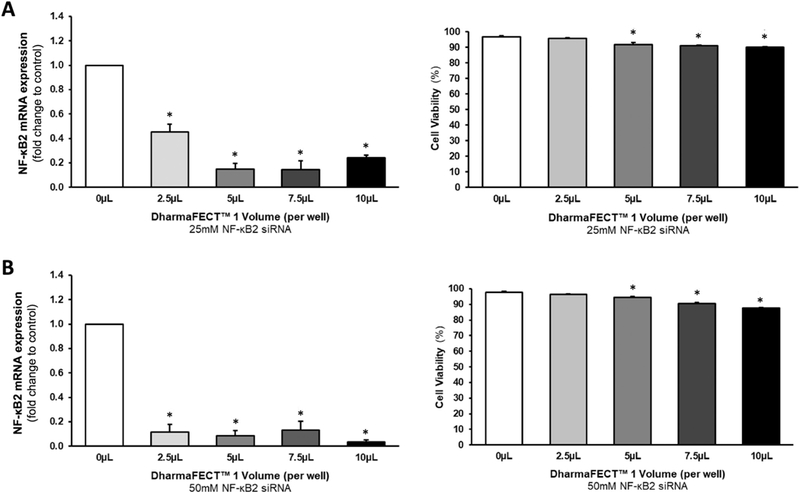
Optimization of NF-κB2 siRNA transfection in HAECs. Cells were transfected with either (A) 25nmol/l NF-κB2 siRNA or (B) 50 nM NF-κB2 siRNA using variable volumes (2.5–10 μl) of DharmaFECT™−1 transfection reagent. Post-transfection, cells were harvested and monitored for reduction in mRNA levels (LHS) and cell viability (RHS) using RT-qPCR and ADAM™ cell counting, respectively. *P ≤.05 versus 0 μl control. Key: LHS, left hand side; RHS, right hand side; nt, non-targeting/scrambled.
Fig. 5.
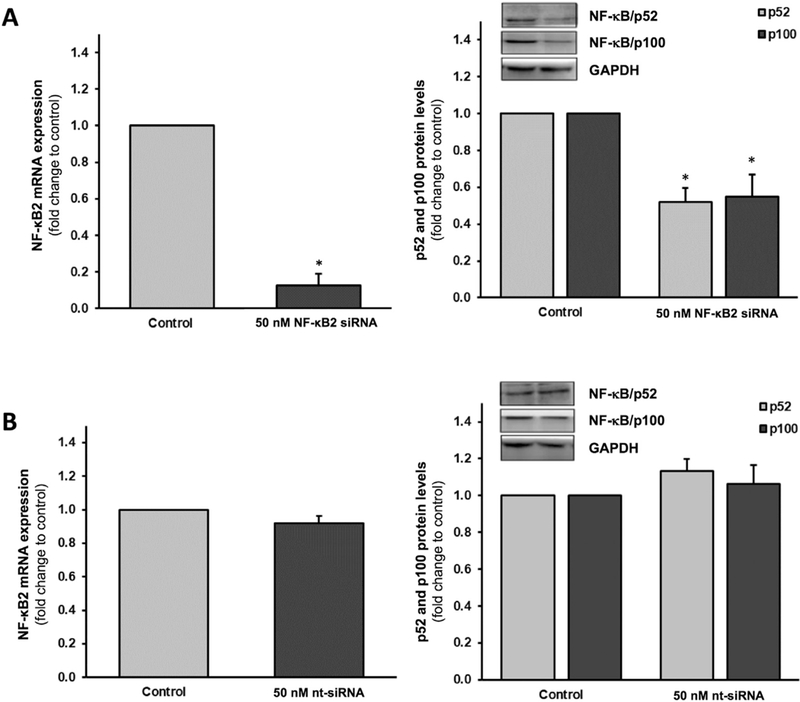
Efficacy of the NF-κB2 siRNA in HAECs. Cells were transfected with either (A) 50 nM NF-κB2 siRNA or (B) 50 nM scrambled nt-siRNA control. Post-transfection, cells were harvested and monitored for reduction in mRNA levels (LHS), as well as p52 and p100 protein levels (RHS), using RT-qPCR and Western blotting, respectively. *P ≤.05 versus corresponding control. Blots (RHS) are representative. Key: LHS, left hand side; RHS, right hand side; nt, non-targeting/ scrambled.
3.5. Effect of NF-κB2 HAEC silencing on osteoblastic activation in co-cultured HASMCs
HAECs and HASMCs were grown in a transwell co-culture format as previously described [7]. HAECs within the luminal transwell compartment were transfected with 50 nM NF-κB2 siRNA (or non-targeting scrambled siRNA controls). Post-transfection, the HAECs were then treated for 72 h with RANKL (0–25ng/ml) to induce osteoblastic signalling within underlying subluminal HASMCs. The following trends were noted within the HASMCs: (i) RANKL caused a decrease in OPG mRNA expression, which was completely blocked by silencing of NF- κB2 (Fig. 6A); (ii) Whilst NF-κB2 silencing caused a significant general increase in OPG release levels relative to scrambled controls, the RANKL-induced decrease in OPG release rates (~39–44%) was unchanged by NF-κB2 silencing (Fig. 6B); (iii) RANKL caused an increase in ALP mRNA expression, which was completely blocked by silencing of NF-κB2 (Fig. 6C); (iv) RANKL caused an increase in ALP activity, which was almost completely blocked by silencing of NF-κB2 (Fig. 6D); (v) RANKL caused a moderate increase in BMP-2 release, which was completely blocked by silencing of NF-κB2 (Fig. 6E); and (vi) Whilst NF-κB2 silencing caused a significant general decrease in Sox9 mRNA levels relative to scrambled controls, the RANKL-induced increase in Sox9 expression was completely blocked by NF-κB2 silencing (Fig. 6F).
Fig. 6.
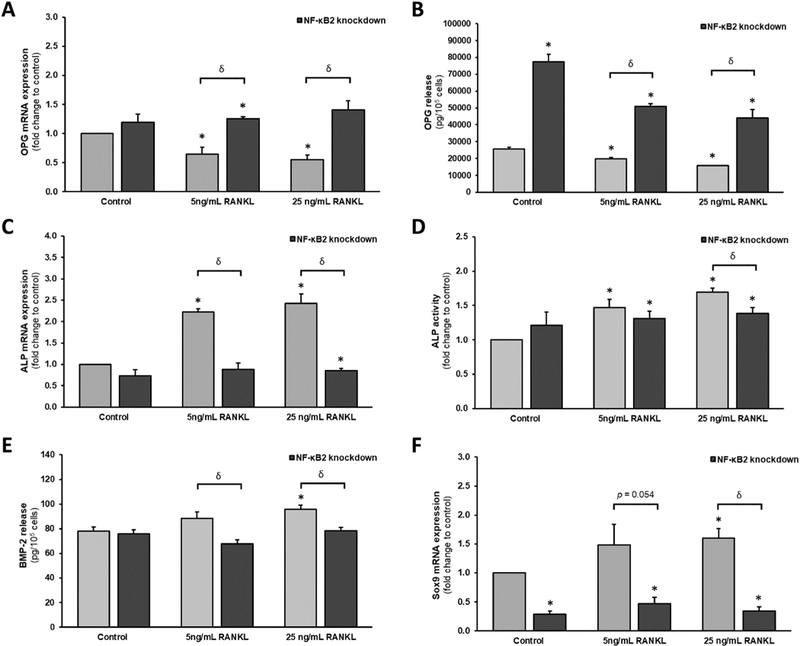
Effects of endothelial NF-κB2 gene silencing on RANKL-induced osteoblastic signalling in a transwell vascular cell co-culture model. HAECs within the luminal transwell compartment were transfected with 50 nM NF-κB2 siRNA (or nt-siRNA controls). Post-transfection, HAECs were treated for 72 h with RANKL (0-25 ng/ml) to induce osteoblastic signalling within underlying subluminal HASMCs. HASMCs were subsequently harvested and monitored for; (A) OPG mRNA; (B) OPG release; (C) ALP mRNA; (D) ALP activity; (E) BMP-2 release; and (F) Sox9 mRNA. *P ≤.05 versus unsilenced 0 ng/ml RANKL control. Key: nt, non-targeting/scrambled.
4. Discussion
The signal transduction mechanism(s) underpinning the pro-calcific actions of RANKL within the vascular wall remain to be properly elucidated. In view of the potential value of NF-κB as a therapeutic target in the management of diabetic microvascular complications (for review see [19]), we tested the hypothesis that NF-κB pathway activation within the intimal endothelium was centrally involved. Within the scientific literature, both direct and indirect evidence demonstrates how RANKL-treatment of VSMCs may elicit calcification via NF-κB pathway activation within these cells [6,11–15]. However, given the relevance of the endothelium to the continual paracrine conditioning of the medial SMC layer within the vascular wall, as well as the responsiveness of endothelial cells to pro-calcific agents such as RANKL [8], it is noteworthy that there have thus far been no studies to date investigating whether or not RANKL elicits NF-κB activation in endothelial cells, or indeed the subsequent consequences of this for underlying VSMCs. To address this important mechanistic question, we have employed HAECs and HASMCs to investigate how RANKL treatment of either cell type elicits activation of the NF-κB/p52 (non-canonical/alternative) and NF-κB/p65 (canonical) pathways and importantly, to test whether or not the TRAIL-dependent blockade of RANKL actions recently demonstrated by our group [Harper et al., 2017] can be mediated through one of these pathways. With specific regard to our hypothesis, we also examine how RANKL treatment of HAECs affects pro-calcific activation within underlying co-cultured HASMCs following NF-κB gene silencing within the HAEC monolayer.
Our group recently demonstrated that RANKL could elicit pro-osteoblastic signalling in HAECs and HASMCs when either cell type was individually treated with RANKL [7]. Moreover, when HAECs and HASMCs were co-cultured within a transwell model, treatment of luminal HAECs with RANKL led to the pro-calcific activation of subluminal HASMCs, clearly testifying to the paracrine nature of this signalling axis [7]. We also presented clear evidence that TRAIL could block several key pro-calcific actions of RANKL within these vascular models. The current paper adds mechanistic depth to these earlier results by investigating the role of the NF-κB pathway in these models. In this respect, we began by demonstrating that RANKL (but not TRAIL) could robustly activate both the NF-κB/p52 and NF-κB/p65 pathways in HAECs. Interestingly, co-incubation of HAECs with TRAIL was found to block RANKL-dependent activation of the non-canonical p52 pathway, but not the canonical p65 pathway in these cells. It can also be noted that the inability of TRAIL to directly activate NF-κB in our HAECs is consistent with an earlier report by Seccherio et al., who reported a similar lack of effect of TRAIL in human umbilical vein endothelial cells (HUVECs) [20]. In follow-up studies with HASMCs, we again show that RANKL can activate both NF-κB pathways [6,11], although co-incubation of cells with TRAIL did not block activation of either pathway (in fact, TRAIL was actually seen to independently activate NF-κB/p65 in HASMCs, consistent with previous studies by Ka-vurma and co-workers [21]). Finally, using a HAEC:HASMC transwell co-culture model, we observed that RANKL ± TRAIL treatment of HAECs within the luminal compartment did not lead to NF-κB pathway activation (either non-canonical or canonical) in the underlying HASMCs of the subluminal compartment, an observation which is also consistent with non-leakage of RANKL and TRAIL from the luminal into the sub-luminal compartment of the co-culture.
These findings essentially demonstrate: (i) the ability of RANKL to activate both the NF-κB/p52 and NF-κB/p65 pathways in endothelial cells and; (ii) the ability of TRAIL to robustly counteract RANKL- mediated activation of the non-canonical NF-κB/p52 pathway in HAECs. When viewed in light of the earlier findings of Harper et al. [7], they also suggest a clear role for the non-canonical NF-κB/p52 pathway in mediating the pro-calcific actions of RANKL acting across the endothelium in a paracrine manner to the underlying smooth muscle cells. In order to properly confirm this, we decided to investigate the effects of attenuating the non-canonical NF-κB/p52 pathway in HAECs. Specifically, we employed siRNA transfection of HAECs to silence the NF- κB2 gene (which codes for the NF-κB/p100 fragment, and thus by extension, the NF-κB/p52 fragment involved in nuclear translocation during alternative pathway activation). In this respect, we achieved close to 90% and 50% NF-κB2 knockdown at the mRNA and protein levels, respectively, with no significant effects on cell viability. Importantly, the NF-κB2 siRNA had no effects on either total or phosphorylated levels of NF-κB/p65, ruling out any off-target effects (Supplementary Fig. S4). It can be noted that several recent studies have successfully employed siRNA strategies to inhibit NF-κB pathways in various cell types including vascular cells, achieving comparable levels of knockdown to those indicated in our study [13,22,23]. Using our transwell co-culture model, we next treated siRNA-transfected HAECs (i.e. both scrambled siRNA controls and NF-κB2 knockdowns) with RANKL (5 or 25 ng/ml) and monitored underlying co-cultured HASMCs for changes in pro-calcific indices, namely, levels of OPG, ALP, BMP-2, and Sox9. RANKL treatment of scrambled HAEC controls led to predictably significant increases in the pro-calcific status of co-cultured HASMCs (elevated ALP, Sox9 and BMP-2 levels, in conjunction with reduced OPG levels) [7]. In NF-κB2-silenced HAECs however, RANKL treatment of HAECs had little or no effect on the underlying HASMCs relative to scrambled controls. These observations confirm our hypothesis that RANKL induction of the non-canonical NF-κB/p52 pathway in HAECs is essential for eliciting pro-calcific activation within co-cultured HASMCs. Also worthy of mention, we noted in separate experiments that β-glycerophosphate (a known inducer of vascular calcification in SMCs) can activate the non-canonical (but not the canonical) NF-κB pathway in HAECs (data not shown). Finally, a recent study by Patel et al. adds some weight to this EC:SMC regulatory concept [22]. These researchers present a model of hypoxia-induced vascular remodelling in which exposure of human pulmonary micro- vascular endothelial cells (HPMvECs) to hypoxia (1%, 24 h) elicited increased expression of endothelial ICAM-1 and endothelin-1 and yielded endothelial-conditioned media that, when incubated with reporter smooth muscle cells, could significantly enhance SMC proliferation. Importantly for this study, siRNA-mediated silencing of canonical NF-κB/p65 (RelA) in HPMvECs could significantly reduce this hypoxia-driven effect in SMCs.
5. Conclusions
In conclusion, treatment of HAECs with RANKL can activate the non-canonical NF-κB/p52 (and canonical NF-κB/p65) pathway in these cells. TRAIL, previously demonstrated by our group to strongly attenuate the pro-calcific signalling effects of RANKL [7], was shown to specifically block the RANKL-mediated activation of non-canonical NF-κB/p52 in HAECs, clearly pointing to the mechanistic relevance of this specific pathway to RANKL function within the endothelium. This was subsequently confirmed in a HAEC:HASMC transwell co-culture experiment, whereby RANKL treatment of HAECs genetically silenced (via siRNA) for the NF-κB2 gene (the molecular forerunner to NF-κB/p52) exhibited strongly attenuated pro-calcific activation of underlying HASMCs relative to scrambled siRNA controls. These in vitro observations provide valuable mechanistic insights into how RANKL may potentially act upon endothelial cells through activation of the alternative NF-κB/p52 pathway to alter endothelial paracrine signalling and elicit pro-calcific responses within underlying vascular smooth muscle cells.
Supplementary Material
Acknowledgements
The authors wish to acknowledge the generous financial support (to EH) from the DCU O’Hare Scholarship and the Government of Ireland/Irish Research Council Postgraduate Scholarship scheme (Grant reference G0IPG/2015/3758). Support (to PMC) was also provided through the Science Foundation Ireland US-Ireland R&D Partnership Programme (Grant reference 14/US/B3116).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cellsig.2018.04.004.
Conflict of interests
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- [1].Abedin M, Tintut Y, Demer LL, Vascular calcification: mechanisms and clinical ramifications, Arterioscler. Thromb. Vasc. Biol. 24 (2004) 1161–1170. [DOI] [PubMed] [Google Scholar]
- [2].Wexler L, Brundage B, Crouse J, Detrano R, Fuster V, Maddahi J, Rumberger J, Stanford W, White R, Taubert K, Coronary artery calcification: pathophysiology, epidemiology, imaging methods, and clinical implications - a statement for health professionals from the American Heart Association writing group, Circulation 94 (1996) 1175–1192. [DOI] [PubMed] [Google Scholar]
- [3].Demer LL, Tintut Y, Vascular calcification: pathobiology of a multifaceted disease, Circulation 117 (2008) 2938–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Higgins CL, Isbilir S, Basto P, Chen IY, Vaduganathan M, Vaduganathan P, Reardon MJ, Lawrie G, Peterson L, Morrisett JD, Distribution of alkaline phosphatase, osteopontin, RANK ligand and osteoprotegerin in calcified human carotid atheroma, Protein J. 34 (2015) 315–328. [DOI] [PubMed] [Google Scholar]
- [5].Harper E, Forde H, Davenport C, Rochfort KD, Smith D, Cummins PM, Vascular calcification in type-2 diabetes and cardiovascular disease: integrative roles for OPG, RANKL and TRAIL, Vasc. Pharmacol. 82 (2016) 30–40. [DOI] [PubMed] [Google Scholar]
- [6].Panizo S, Cardus A, Encinas M, Parisi E, Valcheva P, Lopez-Ongil S, Coll B, Fernandez E, Valdivielso JM, RANKL increases vascular smooth muscle cell calcification through a RANK-BMP4-dependent pathway, Circ. Res. 104 (2009) 1041–1048. [DOI] [PubMed] [Google Scholar]
- [7].Harper E, Rochfort KD, Forde H, Davenport C, Smith D, Cummins PM, TRAIL attenuates RANKL-mediated osteoblastic signalling in vascular cell mono-culture and co-culture models, PLoS One 12 (2017) e0188192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Davenport C, Harper E, Forde H, Rochfort KD, Murphy RP, Smith D, Cummins PM, RANKL promotes osteoblastic activity in vascular smooth muscle cells by upregulating endothelial BMP-2 release, Int. J. Biochem. Cell Biol. 77 (2016) 171–180. [DOI] [PubMed] [Google Scholar]
- [9].Osako MK, Nakagami H, Koibuchi N, Shimizu H, Nakagami F, Koriyama H, Shimamura M, Miyake T, Rakugi H, Morishita R, Estrogen inhibits vascular calcification via vascular RANKL system: common mechanism of osteoporosis and vascular calcification, Circ. Res. 107 (2010) 466–475. [DOI] [PubMed] [Google Scholar]
- [10].Forde H, Harper E, Davenport C, Rochfort KD, Wallace R, Murphy RP, Smith D, Cummins PM, The beneficial pleiotropic effects of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) within the vasculature: a review of the evidence, Atherosclerosis 247 (2016) 87–96. [DOI] [PubMed] [Google Scholar]
- [11].Zhan JK, Tan P, Wang YJ, Wang Y, He JY, Tang ZY, Huang W, Liu YS, Exenatide can inhibit calcification of human VSMCs through the NF-kappaB/ RANKL signaling pathway, Cardiovasc. Diabetol. 13 (2014) 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Deuell KA, Callegari A, Giachelli CM, Rosenfeld ME, Scatena M, RANKL enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: dependence on IL-6 and TNF-a, J. Vasc. Res. 49 (2012) 510–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hénaut L, Sanz AB, Martin-Sanchez D, Carrasco S, Villa-Bellosta R, Aldamiz- Echevarria G, Massy ZA, Sanchez-Nino MD, Ortiz A, TWEAK favours phosphate- induced calcification of vascular smooth muscle cells through canonical and non- canonical activation of NFkB, Cell Death Dis. 7 (2016) e2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, Takeshita S, Wagner EF, Noda M, Matsuo K, Xing L, Boyce BF, NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1, J. Biol. Chem. 282 (2007) 18245–18253. [DOI] [PubMed] [Google Scholar]
- [15].Yu M, Qi X, Moreno JL, Farber DL, Keegan AD, NF-κB signaling participates in both RANKL- and IL-4-induced macrophage fusion: receptor cross-talk leads to alterations in NF-kB pathways, J. Immunol. 187 (2011) 1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Davenport C, Mahmood WA, Forde H, Ashley DT, Agha A, McDermott J, Sreenan S, Thompson CJ, McGrath F, McAdam B, Cummins PM, Smith D, The effects of insulin and liraglutide on osteoprotegerin and vascular calcification in vitro and in patients with type 2 diabetes, Eur. J. Endocrinol. 173 (2015) 53–61. [DOI] [PubMed] [Google Scholar]
- [17].Rochfort KD, Collins LE, Murphy RP, Cummins PM, Downregulation of blood- brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase- dependent ROS generation: consequences for interendothelial adherens and tight junctions, PLoS One 9 (2014) e101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wan Y, Chong LW, Evans RM, PPAR-gamma regulates osteoclastogenesis in mice, Nat. Med. 13 (2007) 1496–1503. [DOI] [PubMed] [Google Scholar]
- [19].Suryavanshi SV, Kulkarni YA, NF-κβ: a potential target in the management of vascular complications of diabetes, Front. Pharmacol. 8 (2017) 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Secchiero P, Gonelli A, Carnevale E, Milani D, Pandolfi A, Zella D, Zauli G, TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways, Circulation 107 (2003) 2250–2256. [DOI] [PubMed] [Google Scholar]
- [21].Kavurma MM, Schoppet M, Bobryshev YV, Khachigian LM, Bennett MR, TRAIL stimulates proliferation of vascular smooth muscle cells via activation of NF- kappaB and induction of insulin-like growth factor-1 receptor, J. Biol. Chem. 283 (2008) 7754–7762. [DOI] [PubMed] [Google Scholar]
- [22].Patel H, Zaghloul N, Lin K, Liu SF, Miller EJ, Ahmed M, Hypoxia-induced activation of specific members of the NF-kB family and its relevance to pulmonary vascular remodelling, Int. J. Biochem. Cell Biol. 92 (2017) 141–147. [DOI] [PubMed] [Google Scholar]
- [23].Wang J, Yi S, Zhou J, Zhang Y, Guo F, The NF-κB subunit RelB regulates the migration and invasion abilities and the radio-sensitivity of prostate cancer cells, Int. J. Oncol. 49 (2016) 381–392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.