

Abstract
Hydrogels, highly-hydrated crosslinked polymer networks, closely mimic the microenvironment of native extracellular matrix (ECM) and thus present as ideal platforms for three-dimensional cell culture. Hydrogels derived from tissue- and organ-specific decellularized ECM (dECM) may retain bioactive signaling cues from the native tissue or organ that could in turn modulate cell-material interactions and response. In this study, we demonstrate that porcine kidney dECM can be processed to form hydrogels suitable for cell culture and encapsulation studies. Scanning electron micrographs of hydrogels demonstrated a fibrous ultrastructure with interconnected pores, and rheological analysis revealed rapid gelation times with shear moduli dependent upon the protein concentration of the hydrogels. Conditionally-immortalized human glomerular endothelial cells (GEnCs) cultured on top of or encapsulated within hydrogels exhibited high cell viability and proliferation over a one-week culture period. However, gene expression analysis of GEnCs encapsulated within kidney dECM hydrogels revealed significantly lower expression of several relevant genes of interest compared to those encapsulated within hydrogels composed of only purified collagen I.
Keywords: Kidney, Extracellular Matrix (ECM), Decellularized, Hydrogel, Tissue Engineering
I. Introduction
Approximately 15% of the adult population in the United States is affected by chronic kidney disease1, and this percentage is expected to increase both in the United States and worldwide, especially in countries with aging populations and increasing prevalence of hypertension and diabetes mellitus2. Current approaches to modeling kidney development and disease progression include cell culture, animal models, and stem cell-derived organoids3. Traditional cell culture using two-dimensional (2D), monolayer systems are still widely utilized though they lack critical structural signaling cues found in the native three-dimensional (3D) microenvironment4,5. In comparison, animal models, specifically transgenic mouse models, enable superior correlation between genotype and phenotype with the added capacity to directly measure renal function6–8; however, animal models lack utility in efficient, high-throughput drug screening and have inherent limitations in recapitulating human disease9. More recently, advances in stem cell research have led to the development of organoids for a variety of organ systems10–12, including the kidney13–15. However, organoid research is still evolving and additional investigation is required before organoids reach translational potential. Therefore, there is a compelling need for bioengineered kidney tissues as models of development, disease progression, or drug discovery to better understand and prevent the onset of chronic kidney disease and the progression to end-stage renal disease.
Traditional tissue engineering strategies rely on biocompatible materials processed into porous scaffolds for cell seeding and attachment16–18; however, many of the early materials utilized such as poly-lactic-co-glycolic acid or poly-ε-caprolactone only provide mechanical support and structure for cell-seeded constructs19. Within the in vivo physiological environment, cells are surrounded by cell-secreted products collectively referred to as the extracellular matrix (ECM). The ECM is well-known for its role as a support structure and imparting mechanical integrity to tissues and organs; however, the ECM also presents bioactive signals to cells that regulate development and maintenance20–22. By design, the ECM acts as a natural, inductive scaffold that in many ways is analogous to a composite and stimulus-responsive hydrogel23,24.
Hydrogels are crosslinked networks of polymers that have demonstrated considerable potential as a 3D cell culture platform23–27. The polymeric network and highly hydrated environment of hydrogels mimic the microstructure and mechanical properties of ECM and enable diffusion of oxygen, nutrients, and waste through the network23,24. Furthermore, many hydrogels can be formed under mild, cytocompatible conditions amenable to cell encapsulation25,26. Hydrogels derived from isolated components of the ECM such as collagen or gelatin are utilized in drug delivery, cell transplantation, and tissue engineering applications28,29. These single- or even multi-component hydrogels, however, lack the full biochemical complexity of the entire ECM milieu. As research regarding tissue- and organ-derived decellularized ECM (dECM) has gained popularity, investigators have developed methods to process a variety of dECM materials into hydrogels for cell culture applications and minimally-invasive injectable therapies30. While dECM hydrogels derived from some tissue and organ systems, such as the heart31–34, liver35–37, and skeletal muscle33,38–40 have been the focus of several publications, other tissues and organs, for example the kidney, have received little investigation.
The objective of this study was to develop kidney dECM hydrogels as a cell culture platform for bioengineered kidney tissue models and compare cell response to traditional biomaterials such as collagen I hydrogels. Here, we present a method to process kidney dECM into physically crosslinked hydrogels as a substrate for cell culture or a 3D matrix for cell encapsulation. Routine histology (hematoxylin and eosin staining) and DNA quantification demonstrated the efficacy of the kidney decellularization process while immunofluorescence staining revealed retention of key ECM proteins. Rheological characterization of the hydrogels illustrated the time to gelation and measured the plateau shear moduli.
To evaluate the utility of these kidney dECM hydrogels for studies employing kidney-specific cell populations, we investigated the cell response of conditionally-immortalized glomerular endothelial cells (GEnCs) cultured on top of and encapsulated within hydrogels. The conditionally-immortalized GEnCs are a cell line generated from endothelial cells specifically isolated from kidney glomeruli41. These endothelial cells form the capillary loops through which blood flows and is filtered, forming the first layer of the glomerular filtration barrier responsible for the filtering function of the kidney42. Primary GEnCs are difficult to isolate and expand in culture due to the loss of important phenotypic features with increasing passage number41. However, the conditionally-immortalized cell line utilized in this study combines the advantage of continuous cell expansion and passaging when cultured at the permissive temperature while retaining the ability to acquire a mature phenotype when thermoswitched to the non-permissive temperature.
Culture of GEnCs on top of hydrogel substrates confirmed favorable cell viability and proliferation over a twelve-day culture period. Encapsulation of GEnCs within these hydrogel matrices, which has not been previously investigated by others, resulted in favorable cell viability and proliferation during a week in culture, but evaluation of gene expression demonstrated lower fold-change expression for cells encapsulated in kidney dECM hydrogels compared to collagen I hydrogels.
II. Materials & Methods
2.1. Kidney Decellularization
Female Yorkshire pig (3-4 months in age) kidneys were obtained fresh from Northwestern Simulation (Northwestern University Feinberg School of Medicine), following approval by the Northwestern Institutional Animal Care and Use Committee (IACUC), and stored at −80 °C until decellularization. Prior to decellularization, kidneys were thawed for several hours in warm water and then minced into pieces approximately 0.5 cm × 0.5 cm × 0.25 cm in size with a clean razor blade. Kidney pieces were then rinsed with deionized H2O under constant stirring for one day at room temperature with intermittent H2O changes. After rinsing, kidney pieces were decellularized with 0.1% (m/v) sodium dodecyl sulfate (Sigma-Aldrich, #L3771) under constant stirring for two days. After decellularization, kidney dECM was rinsed with deionized H2O under constant stirring for one day with intermittent H2O changes to ensure removal of residual detergent in the tissue. Kidney dECM was frozen at −80 °C until further processing.
2.2. Evaluation and Characterization of Decellularized Kidney Tissues
2.2.1. Histological Analysis
Fresh native and decellularized kidney tissues were fixed in 10% neutral-buffered formalin (Thermo Scientific, #5701) for at least 48 hours before paraffin embedding, sectioning, and routine hematoxylin and eosin staining. After staining, coverslips were mounted with Cytoseal XYL (Thermo Scientific, #8312-4). Slides were imaged on a Zeiss Axioskop upright microscope with a Zeiss AxioCam MRc5 camera.
2.2.2. DNA Quantification
Native and decellularized kidney tissue pieces were lyophilized for at least two days, and the dry weight was recorded. Tissue samples were digested with Proteinase K (Sigma-Aldrich, #P2308) in digestion buffer composed of 0.05 M Tris base, 1 mM CaCl2, pH 8 at 60 °C overnight. DNA was quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, #P7589) following the manufacturer’s protocol and a dsDNA standard included in the kit to generate a standard curve. Fluorescence was measured on a BioTek Cytation 3 Multi-Mode Reader with an excitation wavelength of 480 nm and emission wavelength of 520 nm. Samples were run in triplicate (n = 3) with technical replicates in triplicate. DNA content was normalized to the dry weight of the sample, and data is presented as a percentage of native kidney tissue DNA content.
2.2.3. Immunofluorescence Staining
Paraffin-embedded sections were deparaffinized and rehydrated before antigen retrieval in a Decloaking Chamber™ NxGen (Biocare Medical, model no. DC2012) at 95 °C for 40 min. with antigen retrieval buffer (Abcam, #ab94674). Sections were rinsed with 1× Phosphate-Buffered Saline (PBS, Mediatech, #21-040), permeabilized with 0.05% TWEEN-20 (Sigma-Aldrich, #P9416) for 10 min., and then blocked with Sea Block (Fisher Scientific, #37527) for 30 min. before incubation with primary antibodies. Primary antibodies were diluted in Sea Block as follows: rabbit anti-collagen I at 1:100 (Abcam, #ab34710), rabbit anti-collagen IV at 1:500 (Abcam, #ab6586), and rabbit anti-laminin at 1:200 (Abcam, #ab11575). Sections were incubated with primary antibodies for 1 hr at room temperature, rinsed, and then incubated with secondary antibodies for 1 hr at 37 °C (goat anti-rabbit Alexa Fluor 488, Thermo Fisher, #A11034, diluted 1:300 in Sea Block). After secondary antibody incubation, sections were rinsed and mounted with Mowiol mounting medium containing 4’,6-diamidino-2-phenylindole (DAPI) to stain for cell nuclei. Slides were imaged on a Nikon A1 Confocal Laser Microscope System.
2.2.4. Sulfated Glycosaminoglycan Quantification
Sulfated glycosaminoglycan (sGAG) content was quantified using the 1,9-dimethyl-methylene blue (DMMB) dye assay. Tissue samples were digested with Proteinase K as described previously in section 2.2.2. DMMB dye solution was prepared by dissolving 8.0 mg DMMB dye (Sigma-Aldrich, #341088), 1.52 g glycine (Sigma-Aldrich, #G-8898), and 1.19 g NaCl (Fisher Scientific, #S642) in 500 mL ultrapure water and adjusting the pH to 3.0. Samples were diluted as necessary in 1× Tris-EDTA buffer (Invitrogen, #T1493), and 20 μL of sample was mixed with 200 μL of DMMB dye solution in clear, 96-well plates. Absorbance was measured on a BioTek Cytation 3 Multi-Mode Reader at 530 nm (A530). Samples were run in triplicate (n = 3) with technical replicates in duplicates, compared to a standard curve of chondroitin sulfate (Biocolor, #B1010), and normalized to dry weight of samples.
2.3. Preparation of Hydrogels from Decellularized Extracellular Matrix
Kidney dECM hydrogels were prepared similarly to previously described protocols for other decellularized tissues43. Briefly, frozen kidney dECM was lyophilized for two days, snap frozen, and milled with a Thomas Wiley Mini-Mill Cutting Mill. Milled dECM was enzymatically digested at 10 mg/mL kidney dECM, 1 mg/mL pepsin (Sigma-Aldrich, #P7000), and 0.01 M HCl (Sigma-Aldrich, #H9892) for 48 hours. The resulting pepsin digest was aliquoted and frozen at −80 °C. Aliquots were thawed overnight at 4 °C as needed for experiments. Pre-gel solutions were prepared by neutralizing the pepsin digest with one-tenth the volume of 1.0 M NaOH (Sigma-Aldrich, #S2770), adding one-tenth of the total volume desired of 10× phosphate-buffered saline (PBS, Mediatech, #46-013-CM) to achieve a final concentration of 1×, and diluting the mixture with sterile MilliQ H2O. The final hydrogel was formed after incubating the pre-gel solution at 37 °C for at least one hour. Collagen I hydrogels were prepared similarly from purified porcine collagen I (Advanced BioMatrix, #5169-100ML) as control groups for experimental studies.
2.4. Rheological Characterization
Rheological characterization of hydrogels was performed following a protocol suggested for hydrogels for tissue engineering applications44. Testing was performed using an Anton-Paar MCR 302 rheometer with a 25-mm parallel-plate fixture under strain-controlled conditions. The lower Peltier cell was set to 4 °C for sample loading and then rapidly ramped to 37 °C after lowering the measuring system. Mineral oil was applied to the edges of the fixture and the system was enclosed within a solvent trap to prevent sample dehydration. All samples were mixed fresh, and the pre-gel solution was loaded onto the instrument before testing. For frequency and strain sweeps, samples were incubated on the instrument stage at 37 °C for 30-45 min. to allow hydrogel formation before testing. Final time sweeps were performed for 120 min. at 37 °C, 0.1% strain, and 10 rad/s. All sweeps were run in triplicate (n = 3).
2.5. SEM Analysis
Hydrogels were fixed in 2% (v/v) gluteraldehyde (Sigma-Aldrich, #G5882) and 3% (m/v) sucrose (Sigma-Aldrich, #S7903) in MilliQ H2O for one hour. Samples were dehydrated in a graded ethanol series (30-100%) of 15 min. intervals, critically-point dried with a Tousimis SAMDRI-790 Critical Point Dryer, sputter coated with 5 nm of Au with a Baltec MED-020 Coating System, and imaged on a JEOL Neoscope SEM.
2.6. Cell Culture
Human conditionally-immortalized GEnCs were cultured as described previously41. These are primary cells that have been transfected with a temperature-sensitive SV40-T antigen and the essential catalytic subunit of human telomerase (hTERT) to avoid replicative senescence45. When cultured at the permissive temperature of 33 °C at which expression of these genes is active, the cells maintain an immature state and are able to proliferate. When thermoswitched to the non-permissive temperature of 37 °C, the cells become quiescent and adopt a more mature phenotype that is comparable to freshly isolated GEnCs. Conditionally-immortalized GEnCs were cultured in Endothelial Growth Medium 2 Microvascular (EGM-2 MV BulletKit, Lonza, #CC-3202) containing 5% fetal bovine serum (FBS) and growth factors as supplied with the exception of vascular endothelial growth factor (VEGF). Media supplements included: epidermal growth factor, R3-insulin-like growth factor-1, fibroblast growth factor β, ascorbic acid, hydrocortisone, and gentamicin and amphotericin-B. Media was exchanged every other day for GEnCs, and cells were used at passage 30 or below. GEnCs were cultured for at least five days after thermoswitching to the non-permissive temperature to ensure complete inactivation of the transgenes.
2.7. Cell Studies
2.7.1. Culture on Hydrogel Substrates
Pre-gel solutions were mixed, cast into well-plates, and incubated at 37 °C for one hour. Cells were then prepared for cell seeding: the cells were rinsed once with 1× Dulbecco’s Phosphate-Buffered Saline (DPBS, Mediatech, #21-030), incubated with TrypLE Express (Gibco, #12605-028) for 5 min. at 33 °C, collected and counted, centrifuged, and resuspended at a concentration of 50,000 cells/mL. Cells were then seeded on top of hydrogel substrates and cultured for one week at 33 °C plus an additional five days at 37 °C.
2.7.2. Cell Encapsulation Within Hydrogels
Well-plates were coated with poly(2-hydroxyethylmethacrylate) (poly(2-HEMA); Sigma-Aldrich, #P3932) at least one day prior following established protocols to prevent cell attachment to the well-plate surface46,47. Briefly, 1.2 g of poly(2-HEMA) was added to 95% (v/v) ethanol at 40-60 °C under constant stirring until dissolved and then sterile filtered through a polyethersulfone mesh with a pore size of 0.22 μm (EMD Millipore, #SCGP00525). Sufficient volume of the poly(2-HEMA) solution was added to cover the well surface, and the solution was allowed to evaporate overnight in a biosafety cabinet. To encapsulate cells, cells were rinsed and trypsinized as described previously, then counted. The appropriate volume of cell suspension was aliquoted to obtain the necessary number of cells for encapsulation at a concentration of 1 million cells/mL. The cell suspension was then centrifuged, and pellets were resuspended in a minimal volume of complete media. The pre-gel solutions were then prepared and mixed, and the concentrated cell suspension was added and mixed in with the pre-gel solution to achieve the desired final concentration. The pre-gel solution with encapsulated cells was then cast into well-plates and incubated at 37 °C for at least one hour before complete media was added on top of hydrogels and plates were transferred to 33 °C.
2.8. Cell Viability and Proliferation
2.8.1. Cell Viability Imaging
Cell viability was assessed with the LIVE/DEAD Assay Viability/Cytotoxicity Kit for Mammalian Cells (Molecular Probes, #L3224) following the manufacturer’s protocol. Briefly, samples were rinsed once with 1× DPBS and incubated with 4.0 μM ethidium homodimer and 2.0 μM calcein in 1× DPBS for 30 min. at 33 or 37 °C. Samples were rinsed once more with 1× DPBS and immediately imaged on a Nikon A1R+ Confocal Laser Microscope System.
2.8.2. Quantification of Cell Viability
Live/dead images were separated by channel and images were analyzed in MATLAB R2017a to quantify cell viability. Image pairs were loaded into the software and converted to grayscale, and objects in the images were counted by using a fast two-dimensional peak finder function available on the Mathworks File Exchange website (fastpeakfind.m) that locates local maxima in arrays after applying a threshold. Quantification of cell viability was calculated as follows:
Four independent samples (n = 4) were stained for each experimental group and time point, and three fields-of-view were imaged per sample for a total of twelve images per experimental group per time point.
2.8.3 Cell Proliferation
Samples were digested with Proteinase K digestion buffer as described previously in section 2.2.2. DNA was quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, #P7589) following the manufacturer’s protocol and a dsDNA standard included in the kit to generate a standard curve. Samples for cell proliferation studies were run in quadruplicate (n = 4). Double-stranded DNA content was quantified from cell pellets of 50,000 cells as counted using the Trypan Blue (Gibco, #15250-061) exclusion method to determine the amount of dsDNA per cell (n = 3) to calculate the total cell number from dsDNA content. Proliferation is presented as the total cell number and cell number normalized to the number of cells initially seeded or encapsulated.
2.9. Histology
Samples were fixed in 10% neutral-buffered formalin (Thermo Scientific, #5701) for at least 48 hours before paraffin embedding, sectioning, and routine hematoxylin and eosin staining. After staining, coverslips were mounted with Cytoseal XYL (Thermo Scientific, #8312-4), and slides were imaged on a Zeiss Axioskop upright microscope with a CRI Nuance camera.
2.10. TEM Analysis
Samples were fixed in 2.5% (v/v) glutaraldehyde and 2% (v/v) paraformaldehyde in 0.1 M cacodylate buffer, subsequently post-fixed with 1% osmium tetroxide for one hour then 1% uranyl acetate in maleate buffer for one hour, dehydrated in a graded ethanol and propylene oxide series, and embedded in Epon (polymerized at 60 °C for 48 hours). Ultrathin sections were visualized with a FEI Tecnai Spirit G2 TEM.
2.11. Gene Expression Analysis
RNA was isolated from samples and cell pellets using TRIzol Reagent (Invitrogen, #15596018) following the manufacturer’s protocol, and RNA concentration was measured using a NanoDrop 1000 Spectrophotometer. Reverse transcription and cDNA synthesis was performed using the iScript Reverse Transcription Supermix for RT-qPCR kit (Bio-Rad, #170-8841) with an Applied Biosystems GeneAMP PCR System 9700 following the manufacturer’s protocol. Quantitative PCR was performed with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, #170-5270) and detected using an Applied Biosystems QuantStudio 7 Flex Real-Time PCR System. The thermal profile used was an initial polymerase activation step at 95 °C for 30 sec., followed by 40 amplification cycles of denaturation at 95 °C for 15 sec. and annealing and extension at 60 °C for 60 sec. The mRNA expression of each gene of interest was normalized to cyclophilin A (PPIA) expression, and the relative degree of gene amplification was calculated using the ΔΔCt method: 2[(Ct gene 2 − Ct PPIA 2) − (Ct gene 1 − Ct PPIA 1)]. “Ct gene 1” represents the threshold cycle (Ct) of the target gene of the reference population, cells cultured on tissue culture polystyrene, and “Ct gene 2” the target gene of the sample of interest. Biological replicates were run in quadruplicate (n = 4) with technical replicates in triplicate.
2.12. Immunofluorescence Staining
Fixed and paraffin-embedded sections were processed as described previously in section 2.2.3. for immunofluorescence staining. Primary antibodies were diluted in Sea Block as follows: mouse anti-PECAM-1 at 1:100 (Abcam, #ab187377) and rabbit anti-collagen I at 1:100 (Abcam, #ab24710). Secondary antibodies were diluted in Sea Block as follows: goat anti-mouse Alexa Fluor 488 at 1:500 (Thermo Fisher, #A11029) and goat anti-rabbit Alexa Fluor 555 at 1:500 (Thermo Fisher, #A21429). Slides were mounted with Mowiol mounting medium containing DAPI to stain for cell nuclei and imaged on a Nikon A1 Confocal Laser Microscope System.
2.13. Statistical Analysis
All quantitative data is represented as the mean ± standard error of the mean. Statistical significance was determined using an unpaired two-tailed Student’s t test assuming equal variance with Microsoft Excel (Microsoft). Significance for all statistical analyses was defined as p < 0.05.
III. Results
3.1. Kidney Decellularization
Minced kidney tissue began to lose color during the initial rinse step with deionized H2O as blood was removed. After several hours in detergent, the edges of tissue pieces became white, and after two days, all kidney tissue acquired a blanched appearance (Figure 1). After decellularization, hematoxylin and eosin staining revealed removal of cell nuclei as evidenced by the lack of dark hematoxylin nucleic counterstain present, and glomerular and tubular ECM structures remained identifiable (Figure 2A). Quantification of the amount of double-stranded DNA present within native and decellularized tissues demonstrated that ≥98% of the DNA content (by dry weight) was removed during the decellularization process (Figure 2B). Immunofluorescence staining for key ECM proteins including collagen I, collagen IV, and laminin revealed the presence of these proteins in decellularized tissues, particularly in glomerular structures for collagen IV and laminin (Figure 2C). In addition, results from the DMMB dye assay indicated retention of approximately half of the sGAG content in decellularized tissues in comparison to native tissues when normalized to the lyophilized masses of the samples (Figure 2D).
Figure 1.

Process to form kidney decellularized extracellular matrix hydrogels. The native kidney is minced into pieces approximately 0.5 cm × 0.5 cm × 0.25 cm in size and treated with 0.1% (w/v) sodium dodecyl sulfate to remove all of the cellular material. The remaining decellularized extracellular matrix is lyophilized and milled into a fine powder before being subjected to a pepsin digestion at acidic pH (0.01 M HCl) to disrupt collagen fibril aggregates and solubilize the material. To form a hydrogel, the pepsin digest is kept on ice, neutralized with 1.0 M NaOH, buffered to pH 7.4, and incubated at 37 °C for about an hour.
Figure 2.
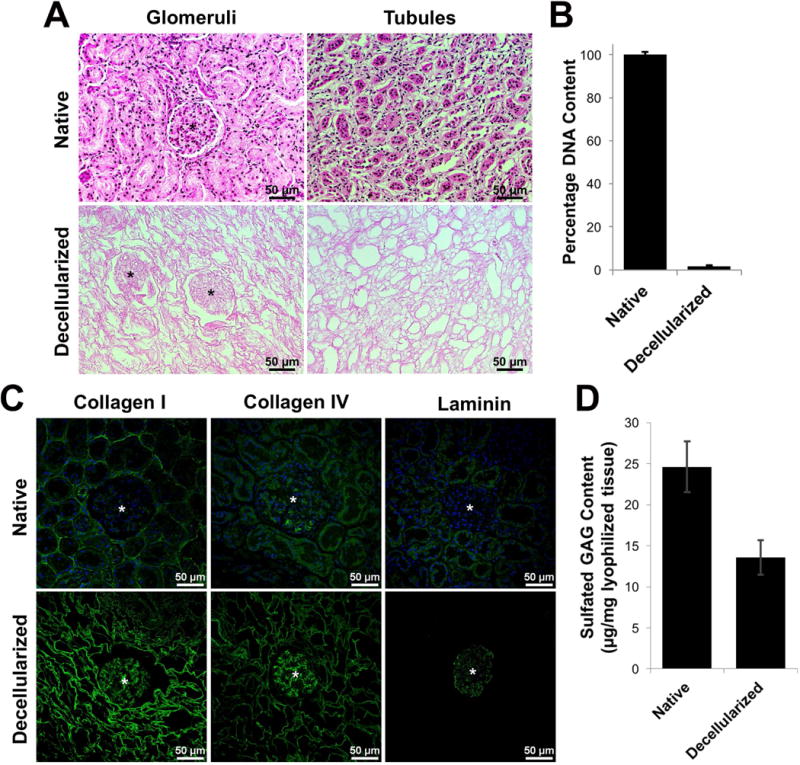
Evaluation and characterization of decellularized kidney tissues. (A) Routine hematoxylin and eosin staining of native and decellularized kidney sections demonstrating removal of cellular nuclei (dark spots) and preservation of the ECM architecture, including glomeruli (*) and tubule structures. (B) Quantification of double-stranded DNA content in native and decellularized kidney tissue. DNA content was normalized to the dry weight of each sample, and data is presented as a percentage of native kidney tissue DNA content (n = 3). (C) Immunofluorescence staining of native and decellularized kidney sections demonstrating retention of collagen I, collagen IV, and laminin in decellularized tissues; glomerular structures indicated by (*). (D) Quantification of sulfated glycosaminoglycan content in native and decellularized kidney tissue. sGAG content was normalized to the dry mass of each sample with samples run in triplicate (n = 3).
3.2 Macroscopic and Ultrastructural Images of Hydrogels
Kidney dECM hydrogels formed at concentrations of 2.5 mg/mL and 5.0 mg/mL retained the cylindrical shape of the mold in which they were cast and were robust enough to be handled with a spatula. Kidney dECM hydrogels at 1.0 mg/mL and collagen I hydrogels 2.5 mg/mL concentrations retained the circular shape but did not meet the height of the original mold. The reduced volume of these hydrogels was likely due to the greater exclusion of water during hydrogel formation which necessitated careful handling of these fragile hydrogels (Figure 3A-D, insets). Ultrastructural imaging via scanning electron microscopy revealed fibrous morphologies of all hydrogels with randomly-oriented fibrils and interconnected pores (Figure 3A-D).
Figure 3.
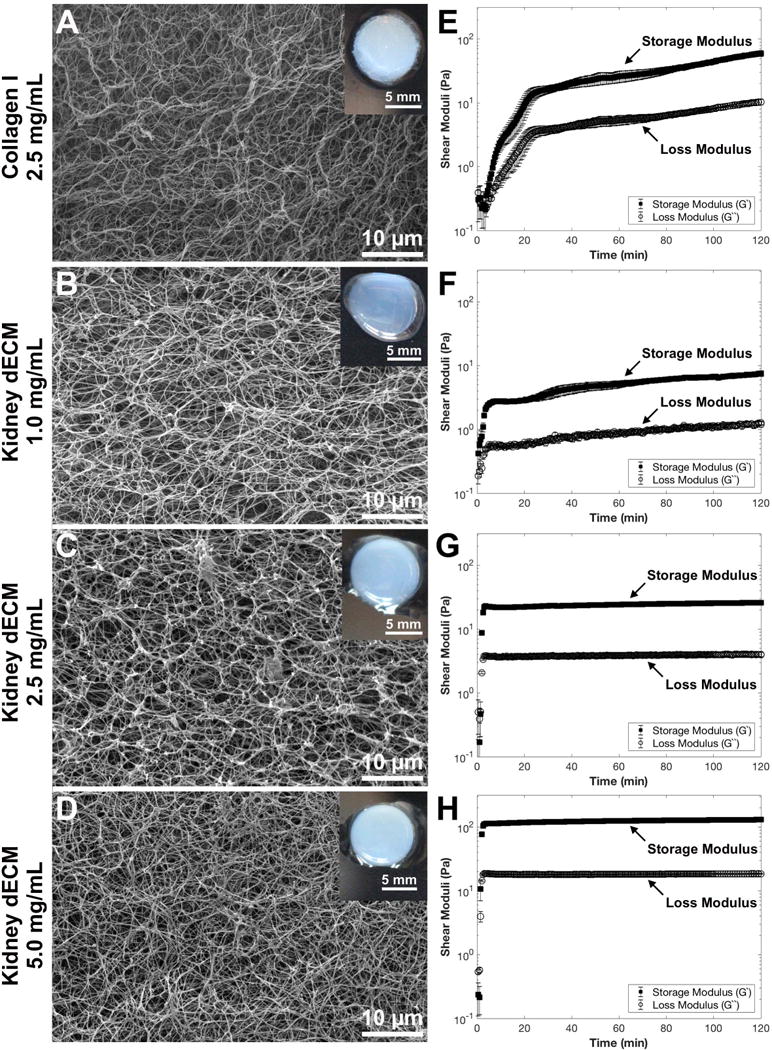
Scanning electron micrographs, macroscopic images (insets), and rheological characterization of collagen I and kidney dECM hydrogels. SEM and macroscopic images of (A) collagen I hydrogel at 2.5 mg/mL, (B) kidney dECM hydrogel at 1.0 mg/mL, (C) kidney dECM hydrogel at 2.5 mg/mL, and (D) kidney dECM hydrogel at 5.0 mg/mL. Rheological characterization of hydrogel formation over time with measured shear moduli plotted on a logarithmic scale of (E) collagen I hydrogels at 2.5 mg/mL, (F) kidney dECM hydrogels at 1.0 mg/mL, (G) kidney dECM hydrogels at 2.5 mg/mL, and (H) kidney dECM hydrogels at 5.0 mg/mL.
3.3 Rheological Characterization of Hydrogels
Rheological testing was performed to measure the change in shear moduli (G’, G’’) over time and rate of gelation, and values are reported in Table 1. The gelation point is reported as the time at which the shear moduli are equal in value44. The time to plateau is reported as the time at which the storage modulus (G’) is approximately an order of magnitude larger than the loss modulus (G’’) and both moduli have reached the plateau phase indicating that a stable hydrogel has formed. The shear moduli of all hydrogels (Figure 3E-H) were characterized by sigmoidal curves with rapid onset of gelation upon ramping the temperature up to 37 °C. Collagen I hydrogels (2.5 mg/mL, Figure 3E) presented the longest time to plateau at 25 min. with an equilibrium G’ value of 15 Pa. The G’ continued to increase to 60 Pa after 120 min. Kidney dECM hydrogels at 1.0 mg/mL (Figure 3F) formed stable hydrogels much more rapidly with the time to plateau being 3.5 min. and an equilibrium G’ value of 2.0-7.5 Pa. Kidney dECM hydrogels at 2.5 mg/mL and 5.0 mg/mL (Figure 3G, 3H) formed stable hydrogels after just 3 min. with equilibrium G’ values of 22-26 Pa and 110-130 Pa, respectively. To confirm formation of a hydrogel, samples were subject to frequency (Supplemental Figure 1A-D) and strain (Supplemental Figure 1E-H) sweeps. The G’ of all samples was independent at frequencies of 25 rad/s and below, and the angular frequency value used for time sweeps was verified to be in the low-frequency plateau region for all samples. Strain sweeps demonstrated the presence of a linear viscoelastic region for all samples, confirming the formation of hydrogels, and the strain value used for time sweeps was within the linear viscoelastic region for all samples. Additionally, all samples exhibited decreasing G’ values after approximately 70% strain with catastrophic failure occurring within the range of 300-500% strain.
Table 1.
Rheological characterization values.
| Collagen I Hydrogels | Kidney dECM Hydrogels | |||
|---|---|---|---|---|
| Concentration (mg/mL) | 2.5 | 1.0 | 2.5 | 5.0 |
| Gelation Point (min) | 5.0 | 1.0 | 2.0 | 1.5 |
| Time to Plateau Phase (min) | 25.0 | 3.5 | 3.0 | 3.0 |
| Equilibrium Storage Modulus, G’ (Pa) | 15.0-60.0 | 2.0-7.5 | 22.0-26.0 | 110.0-130.0 |
| Average Strain Range of Strain Stiffening Behavior | 25.0-70.0% | 12.5-25.0% | 5.0-15.0% | 5.0-10.0% |
3.4 Cell Viability and Proliferation on Hydrogel Substrates
Live/dead staining and image analysis revealed high viability (85-98%) of human conditionally-immortalized GEnCs cultured on collagen I hydrogels (2.5 mg/mL) and kidney dECM hydrogels (2.5 mg/mL) substrates at days 1 and 12 (Figure 4A,B). Quantification of DNA content of samples over time indicated proliferation of GEnCs cultured on both collagen I and kidney dECM hydrogels with cells increasing over six-fold in number during the seven-day culture period at the permissive temperature. Cell proliferation after thermoswitching and culture at the non-permissive temperature between days 7 and 12 decreased, as expected (Figure 4C).
Figure 4.
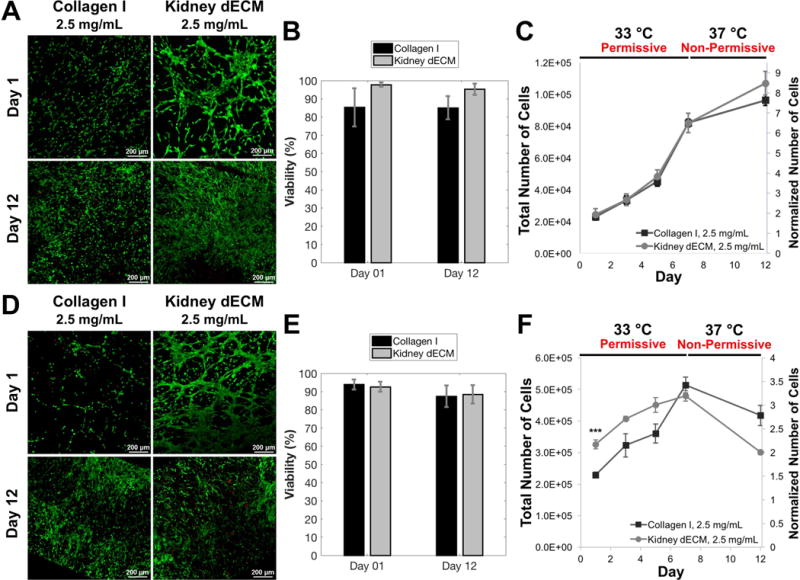
Viability and proliferation of human GEnCs cultured on collagen I hydrogels and kidney dECM hydrogel substrates (A-C) or encapsulated within hydrogels (D-F). (A) Live/dead staining and confocal imaging of GEnCs cultured on collagen I hydrogels or kidney dECM hydrogels at days 1 and 12. (B) Quantification of viability of cells cultured on top of hydrogel substrates presented as the percentage live cells from image analysis. (C) Quantification of cultured GEnC proliferation over twelve days using a Quant-iT PicoGreen dsDNA Assay Kit. Normalized values (right axis) were normalized to the number of cells seeded per sample on day 0. Samples initially seeded with 12,500 cells per sample (50,000 cells/mL). Number of independent measurements, n = 4. (D) Live/dead staining and confocal imaging of GEnCs encapsulated within collagen I hydrogels or kidney dECM hydrogels at days 1 and 12. (E) Quantification of viability of cells encapsulated within hydrogels presented as the percentage live cells from image analysis. (F) Quantification of encapsulated GEnC proliferation over twelve days using a Quant-iT PicoGreen dsDNA Assay Kit. Normalized values (right axis) were normalized to the number of cells encapsulated per sample on day 0. Samples initially seeded with 150,000 cells per sample (1 million cells/mL). Number of independent measurements, n = 4. *** p < 0.005.
3.5 Cellular Encapsulation Within Hydrogels
Live/dead staining and image analysis revealed high cell viability of GEnCs encapsulated within both collagen I hydrogels (2.5 mg/mL) and kidney dECM hydrogels (2.5 mg/mL) at days 1 (92-94%) and 12 (86-88%) (Figure 4D,E). Hydrogels encapsulating cells began to contract after three days in culture and resembled compacted disks after twelve days. Encapsulated GEnCs proliferated during the first seven days when cultured at the permissive temperature, approximately tripling in number for both experimental groups. The cell number was significantly greater (p < 0.005) for cells encapsulated in kidney dECM hydrogels than cells encapsulated in collagen I hydrogels on day 1 with 100,000 more cells present in kidney dECM hydrogels; however, there was no significant difference in cell number between the groups at time points beyond day 1 (Figure 4F). H&E staining of fixed and sectioned samples from days 7 (Supplemental Figure 2) and 12 (Figure 5) showed cell distribution throughout hydrogels and settling of cells during hydrogel formation at the bottom edge of samples. Samples from both groups demonstrated some cell spreading between days 7 and 12 but little presence of cell-cell contacts and no tube formation, which was also confirmed by TEM (Figure 5C,F).
Figure 5.
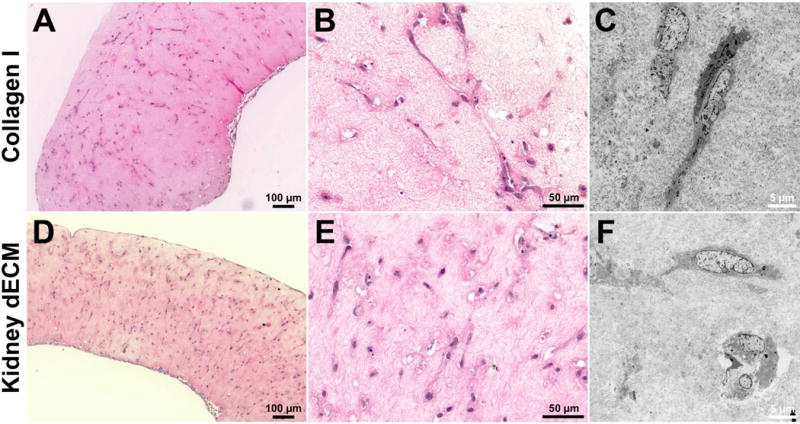
H&E and TEM of GEnCs encapsulated within hydrogels after twelve days in culture. (A, B) H&E stained sections and (C) TEM micrograph of GEnCs encapsulated within collagen I hydrogels. (D, E) H&E stained sections and (F) TEM micrograph of GEnCs encapsulated within kidney dECM hydrogels.
3.6 Gene Expression & Immunofluorescence
Gene expression analysis of encapsulated GEnCs (Figure 6) revealed higher fold-change expression of cells encapsulated in collagen I hydrogels compared to cells encapsulated in kidney dECM hydrogels on days 1 and 7 for all of the genes analyzed except ITGB1. Genes analyzed are involved in cell-cell interactions (Figure 6A-C: PECAM1, CDH5, ICAM2), cell-matrix remodeling (Figure 6D: MMP14), signaling pathways (Figure 6E-H: KDR, TIE1, TEK, PTPRB), endothelial function (Figure 6I-L: VWF, PLVAP, EHD3, EHD4), and cell-matrix binding interactions (Figure 6M,N: ITGA3, ITGB1). Gene expression for both experimental groups was typically highest on day 12, with a few exceptions: PLVAP, TIE1, ITGA3 and ITGB1 for collagen I hydrogels and ITGB1 for kidney dECM hydrogels. Immunofluorescence staining confirmed the expression of platelet endothelial cell adhesion molecule (PECAM-1 or CD31 encoded by PECAM1) by GEnCs encapsulated within both collagen I hydrogels and kidney dECM hydrogels on days 7 and 12 (Figure 7).
Figure 6.
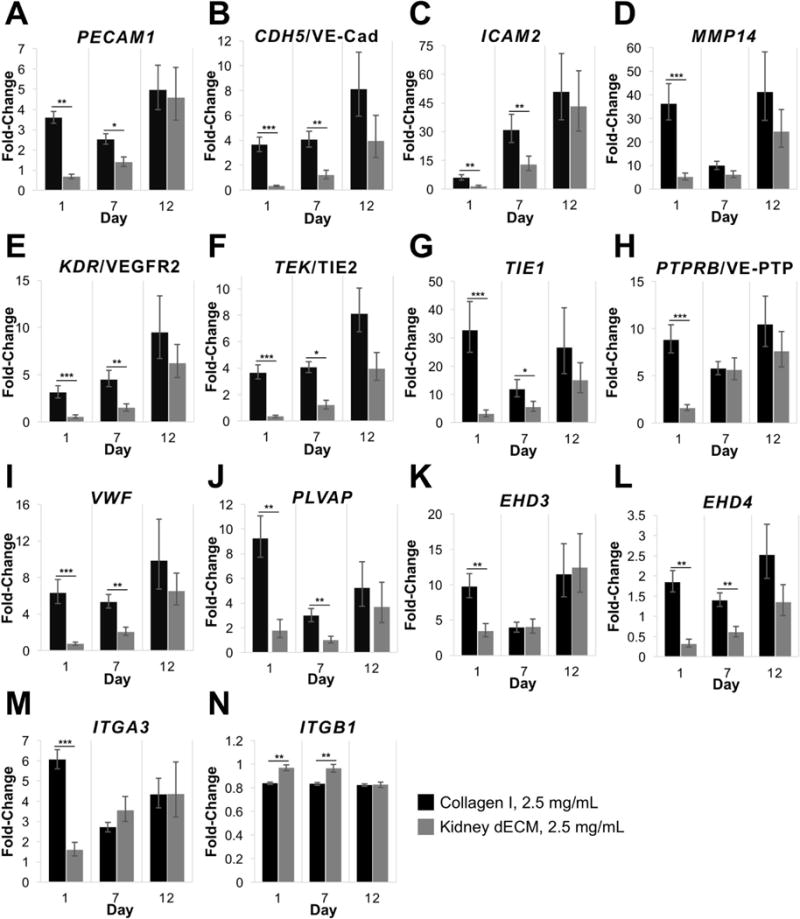
Gene expression of GEnCs encapsulated within hydrogels after 1, 7, and 12 days in culture. (A) PECAM1 encoding for platelet endothelial cell adhesion molecule or CD31. (B) CDH5 encoding for cadherin 5 or vascular endothelial cadherin. (C) ICAM2 encoding for intercellular adhesion molecule 2 or CD102. (D) MMP14 encoding for matrix metalloproteinase 14 or MT1-MMP. (E) KDR encoding for kinase insert domain receptor or vascular endothelial growth factor receptor 2. (F) TEK encoding for tyrosine kinase with immunoglobulin-like and EGF-like domains 2 or TIE2. (G) TIE1 encoding for tyrosine kinase with immunoglobulin-like and EGF-like domains 1 or TIE1. (H) PTPRB encoding for receptor-type tyrosine-protein phosphatase beta or VE-PTP. (I) VWF encoding for von Willebrand Factor. (J) PLVAP encoding for plasmalemma vesicle-associated protein. (K) EHD3 encoding for Eps15 homology domain-containing protein 3. (L) EHD4 encoding for EH domain-containing protein 4. (M) ITGA3 encoding for integrin subunit alpha 3. (N) ITGB1 encoding for integrin subunit beta 1. Values are expressed as fold-change in expression normalized to gene expression of GEnCs cultured on tissue culture polystyrene on day 0. Number of independent measurements, n = 4. Statistical significance denoted by: * p < 0.05, ** p < 0.01, and *** p < 0.001.
Figure 7.
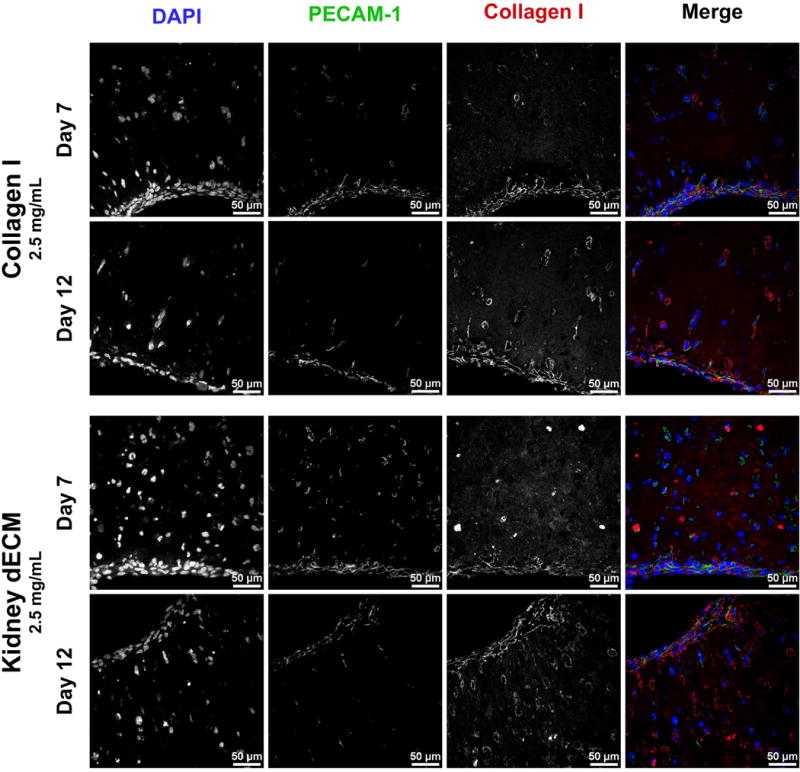
Immunofluorescence staining for PECAM-1 of GEnCs encapsulated in collagen I hydrogels or kidney dECM hydrogels after 7 or 12 days in culture. Merged images: DAPI (blue), PECAM-1 (green), and collagen type I (red).
IV. Discussion
Decellularized ECM is a versatile material that has been utilized in a number of applications over the years ranging from biological wound dressings48 to coatings for cell culture49,50. Although the native architecture of the dECM is lost during processing to form hydrogels, the complex tissue- and organ-specific biochemical signals of the native source are thought to be retained30. As a result, hydrogels derived from dECM, as opposed to those derived from synthetic or purified natural sources, have inherent cell-interactive motifs and bioactive factors. We have demonstrated that porcine kidney dECM can be solubilized and processed to form a mechanically stable hydrogel that supports cell viability and proliferation. Kidney dECM hydrogels were evaluated by rheological characterization and in vitro studies were performed for comparison with purified porcine collagen I hydrogels.
The enzymatic digestion method adopted in this study allows for the solubilization of dECM material without additional purification steps that may result in the removal of desired biochemical components43. The digestion occurs as pepsin, an enzyme that is active only at very acidic pH, cleaves collagen telopeptide bonds and disrupts collagen fibril aggregates51. Once the digest is neutralized, thus inactivating the enzyme, and brought to physiological temperature and salt concentration, the collagen and other matrix peptides self-assemble in an entropy-driven process51,52. The resulting kidney dECM hydrogels possess a loosely organized fibrous organization with an interconnected pore structure as visualized under SEM (Figure 3A-D) that is comparable to collagen I hydrogels and various dECM hydrogels characterized by other groups30.
Gelation kinetics and shear moduli (G’, G’’) of kidney dECM hydrogels were analyzed by parallel-plate rheology. Time sweeps revealed that kidney dECM hydrogels at all the concentrations tested exhibited a decreased lag phase (time of nucleation) and an increased rate of self-assembly in comparison to collagen I hydrogels (Figure 3E-H). This observation agrees with previous time-lapse confocal reflection microscopy studies, which reinforces the idea that additional components present in dECM, such as proteoglycans and glycosaminoglycans, increase the polymerization kinetics of collagen self-assembly53. Evaluation of steady-state G’ values of kidney dECM hydrogels also revealed that increasing dECM concentration led to an increase in G’ value or hydrogel stiffness. This observation agrees with rheological or mechanical characterization of various dECM hydrogels investigated by other groups30, such as liver36 and skeletal muscle39.
Strain sweeps demonstrated that the hydrogels undergo a strain-stiffening behavior (Supplemental Figure 2E-H). This nonlinear elastic property is common among biological materials such as fibrin and is thought to prevent damage to tissues caused by large deformations54. Kidney dECM hydrogels of all concentrations tested exhibited strain-stiffening behavior anywhere between 5-25% strain; however, strain stiffening was much more apparent in lower concentration kidney dECM hydrogels (1.0 and 2.5 mg/mL) as opposed to 5.0 mg/mL hydrogels. Collagen I hydrogels, on the other hand, exhibited strain-stiffening behavior between 25-70% strain. Strain-stiffening behavior is correlated with a material’s network structure55. For physically crosslinked networks, such as the hydrogels examined in this work, strain stiffening is dependent on the flexibility of the polymers or protein filaments. More flexible filaments result in networks that stiffen at higher strains whereas less flexible filaments result in networks that stiffen at lower strains54. This suggests that the collagen fibrils present in the kidney dECM hydrogels are less flexible than those present in the purified collagen I hydrogels. This may be a result of the processing procedure (i.e., pepsin digestion) in combination with the presence of other ECM components that may alter collagen fibril stiffness and overall network structure.
The network structure and overall matrix stiffness act in concert with the matrix components to influence the response of cells exposed to the matrix microenvironment22,56–58. Kidney dECM and collagen I hydrogels of similar concentration (2.5 mg/mL) exhibited similar G’ values based on rheological characterization and were thus employed in cell studies to avoid introducing hydrogel stiffness as a confounding variable during analysis. The concentration for collagen I hydrogels was limited to a maximum of 2.5 mg/mL due to the low concentration of the material supplied by the vendor (a solution of 2.9 mg/mL).
Human conditionally-immortalized GEnCs were employed as a model cell type for investigating the cell response to interactions with kidney dECM hydrogels. This cell line is derived from endothelial cells isolated from kidney glomeruli. Unlike primary cells that are difficult to expand in culture and lose important phenotypic characteristics over time, the conditionally-immortalized GEnCs maintain the capacity to be expanded in culture when cultured at the permissive temperature, but when thermoswitched to the non-permissive temperature become quiescent and adopt a progressively mature phenotype. The in vitro cell studies presented here demonstrate that kidney dECM hydrogels may serve as a biocompatible substrate to support attachment, viability, and proliferation of human GEnCs with results similar to those of collagen I hydrogels (Figures 4A-C). In addition, because these hydrogels are formed under mild conditions and at physiological temperature, they are permissive for cell encapsulation. The cell encapsulation studies presented here demonstrate that human GEnCs exhibit high viability when encapsulated within kidney dECM hydrogels (2.5 mg/mL). However, as the cells began to exert contractile forces on the relatively weak mechanical properties of the collagen I and kidney dECM hydrogels in an effort to spread or migrate59, the hydrogels contracted. Moreover, despite increasing gene expression over the twelve-day culture period of cell-cell interaction proteins PECAM1, CDH5, and ICAM2 (Figure 6A-C) as well as MMP14 (Figure 6D) involved in ECM breakdown60, strong cell-cell interactions were not observed in H&E-stained sections (Figure 5).
In addition to the weak mechanical properties of the hydrogels, it is possible that the biochemical composition of the kidney dECM hydrogels developed in this investigation were not ideal for encapsulation of the GEnCs utilized. Biochemical characterization of decellularized kidney tissues presented in this work was performed, demonstrating retention of key ECM proteins such as collagen I, collagen IV, laminin, and sulfated GAGs. However, decellularized ECM hydrogels are complex and may contain any combination of proteins, cryptic peptides and bioactive motifs, sequestered growth factors, cytokines, chemokines, and even matrix-bound nanovesicles30. Any of these additional bioactive components may present negative or undesired signaling cues to specific cell types. Alternatively, despite increasing gene expression of several growth factor receptors over the culture period, including KDR, TEK, TIE1, and PTPRB (Figures 6E-H), critical bioactive factors may be lost or rendered inactive during decellularization or hydrogel processing61, resulting in poor cell response. For example, VEGF was deliberately excluded from the culture media in these studies following previous investigations utilizing this conditionally-immortalized cell line41,62,63; however, the lack of active and bioavailable VEGF may explain the deficiency of cell-cell contacts formed. Future studies will aim to tune the hydrogel mechanical properties and composition by modifying the processing procedures to include covalent crosslinking methods and strategies to restore critical signaling cues thought to be lost.
The work presented here is the first to explore the use of kidney dECM hydrogels for direct encapsulation of GEnCs; however, this is not the first study to investigate the potential of kidney dECM hydrogels in general. O’Neill, et al. explored the regional specificity of kidney dECM in solubilized, hydrogel, and sheet forms on modulating mouse papilla-derived kidney stem cell growth and metabolism64. The rheological or mechanical properties of the region-specific kidney dECM hydrogels were not investigated in the O’Neill, et al. study; however, the authors demonstrated that kidney stem cells adopted a significantly more proliferative and metabolically active phenotype when cultured in the presence of kidney dECM as opposed to heart or bladder dECM. Furthermore, they also demonstrated that kidney stem cells cultured in the presence of kidney papillae-derived dECM in any form resulted in lower cell proliferation, higher metabolic activity, and altered cell morphology in comparison to cortex- and medulla-derived dECM64. It is thought that the renal papilla represents the native kidney stem cell niche65,66, therefore, it follows that dECM derived from the papillae would maintain a stem cell phenotype as suggested by the results. Furthermore, the authors propose that cortical- and medullary-derived dECM may show potential in directing differentiation of kidney stem cells towards specific renal lineages64.
Nagao, et al. examined the influence of human kidney cortex-derived dECM hydrogels on human kidney peritubular microvascular endothelial cell (HKMEC) phenotype67. Proteomics analysis of the decellularized kidney cortex revealed that collagen IV was present in the highest abundance; however, rheological testing indicated that the complex modulus of the corresponding hydrogel at a concentration of 7.5 mg/mL only reached 15 Pa. This is much weaker than the 110-130 Pa measured for the kidney dECM hydrogels at a concentration of 5.0 mg/mL presented in this study. Nagao, et al. further demonstrated that HKMECs cultured on these hydrogel substrates exhibited a more quiescent and mature phenotype. The addition of collagen I to increase the hydrogel mechanical properties (up to 250 Pa) led to a more activated HKMEC state67. The data presented by the authors coincides with in vivo evidence that an increased collagen I content in the kidney ECM is associated with fibrosclerosis and the development of chronic kidney disease68–70.
Most recently, Magno, et al. investigated the ability to utilize macromolecular crowding as a method for tailoring the fibrillar architecture of kidney dECM hydrogels71. The incorporation of increasing concentrations of a macromolecular crowder, Ficoll400, resulted in faster rates of fibrillogenesis as measured by turbidimetric analysis, generation of thicker fibrils and greater fibril alignment, and reduced hydrogel stiffness. Interestingly, this effect is the opposite of those previously observed for pure collagen I hydrogels where increasing concentrations of macromolecular crowders resulted in a decrease in fibril thickness and reduced fiber alignment72. In addition, the researchers found that the inclusion of Ficoll400 resulted in greater network formation by human umbilical vein endothelial cells (in the presence of VEGF) and morphogenesis of mouse kidney stem cells when cultured on kidney dECM hydrogels71.
Together, this and previous studies demonstrate that kidney-derived dECM hydrogels are able to support cell attachment, viability, and proliferation. Furthermore, region-specific kidney-derived dECM hydrogel substrates have already been shown to modulate cell response and either maintain an immature, stem cell-like phenotype64 or a more mature, quiescent phenotype67. Future studies may thoroughly investigate region-specific kidney-derived dECM composition or the ability to tailor such hydrogels with additives, the corresponding gelation kinetics, and the ultrastructure and rheological characteristics of the resulting hydrogel. Such investigations will continue to delineate some of the factors that influence the response of different cell types or stem cells directly encapsulated within tissue- and organ-specific 3D cell culture microenvironments.
V. Conclusion
Kidney dECM can be digested and processed to form hydrogels that retain biochemical signaling cues of the kidney dECM microenvironment. The kidney dECM hydrogels presented in this study exhibit fibrous ultrastructural networks similar to other dECM hydrogels previously presented by others but unique rheological characteristics from other tissue- and organ-derived dECM hydrogels and from purified collagen I hydrogels. These kidney dECM hydrogels may act as a substrate for cell culture or for encapsulation of cells as a 3D cell culture microenvironment. While only human glomerular endothelial cells were presented in this study, it is anticipated that the hydrogels may serve as a culture environment for any cell type, kidney-specific, stem cell, or otherwise with potential in developing a kidney tissue model for developmental studies and pharmaceutical screening applications.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Alexandra L. Rutz and Phillip E. Lewis for training and advice regarding the development and optimization of kidney decellularized extracellular matrix hydrogels. The authors would also like to thank Dr. Susan E. Quaggin and Dr. Lindsay S. Keir for their guidance regarding glomerular endothelial cell culture and evaluation of cell response. The authors would like to thank Lennell Reynolds, Jr. at the Northwestern University Center for Advanced Microscopy for assistance with TEM sample processing. The authors would like to acknowledge the use of the following research facilities: the Analytical BioNanotechnology Core Facility of the Simpson Querrey Institute at Northwestern University developed by support from the U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University with ongoing support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF NNCI-1542205); the Northwestern University Center for Advanced Microscopy supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center; the Northwestern University Mouse Histology and Phenotyping Laboratory supported by NCI P30-CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center; and the NUSeq Core Facility supported by the Northwestern University Center for Genetic Medicine, Feinberg School of Medicine, and Shared and Core Facilities of the University’s Office for Research.
Author Contributions & Funding Sources
J.S.: Designed experiments, optimized testing parameters, collected and analyzed data, and wrote and edited the manuscript including figures. S.C.S: Provided the conditionally-immortalized glomerular endothelial cell line and assisted with experimental design and manuscript editing. R.N.S.: Co-principal investigator who assisted with experimental design, interpretation of data, and manuscript writing and editing. J.A.W.: Co-principal investigator who assisted with experimental design, interpretation of data, and manuscript writing and editing. This work was supported by a National Institutes of Health Predoctoral Biotechnology Training Program (NIGMS T32 GM008449) and a Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship (NRSA F31 NIDDK 1F31DK108544-01A1) awarded to J.S. This work was supported in part by Merit Review I01BX002660 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Service (J.A.W). The contents do not represent the views of the U.S. Department of Veterans Affairs, the National Institutes of Health, or the United States Government. This research was also supported by a grant from the American Society of Transplantation’s Transplantation and Immunology Research Network (J.A.W), the McCormick Foundation (J.A.W.), and the Google Foundation (R.N.S.). Funding sources had no involvement in study design; collection, analysis, or interpretation of data; in writing of the manuscript; or in the decision to submit the manuscript for publication.
Footnotes
Supplementary Material
Supplementary materials are available online at: [LINK].
Disclosures
The authors declare that they have no conflicts of interest.
References
- 1.2015 Annual Data Report: Epidemiology of Kidney Disease in the United States United States Renal Data System. Bethesda, MD: National Institutes of Diabetes and Digestive and Kidney Disease, National Institutes of Health; 2016. [Google Scholar]
- 2.Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY-M, Yang C-W. Chronic kidney disease: global dimension and perspectives. The Lancet. 2013;382(9888):260–272. doi: 10.1016/S0140-6736(13)60687-X. [DOI] [PubMed] [Google Scholar]
- 3.Oxburgh L, Carroll TJ, Cleaver O, Gossett DR, Hoshizaki DK, Hubbell JA, Humphreys BD, Jain S, Jensen J, Kaplan DL. (Re) Building a Kidney. Journal of the American Society of Nephrology. 2017;28(5):1370–1378. doi: 10.1681/ASN.2016101077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nature reviews Molecular cell biology. 2007;8(10):839–845. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- 5.Antoni D, Burckel H, Josset E, Noel G. Three-dimensional cell culture: a breakthrough in vivo. International journal of molecular sciences. 2015;16(3):5517–5527. doi: 10.3390/ijms16035517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ly JP, Onay T, Quaggin SE. Mouse models to study kidney development, function and disease. Current opinion in nephrology and hypertension. 2011;20(4):382–390. doi: 10.1097/MNH.0b013e328347cd4a. [DOI] [PubMed] [Google Scholar]
- 7.Eddy AA, López-Guisa JM, Okamura DM, Yamaguchi I. Investigating mechanisms of chronic kidney disease in mouse models. Pediatric Nephrology. 2012;27(8):1233–1247. doi: 10.1007/s00467-011-1938-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Sullivan ED, Hughes J, Ferenbach DA. Journal of the American Society of Nephrology 2016. ASN; 2015. Renal Aging Causes and Consequences; p. 121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, Macleod MR. Can animal models of disease reliably inform human studies? PLoS med. 2010;7(3):e1000245. doi: 10.1371/journal.pmed.1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345(6194):1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- 11.Clevers H. Modeling development and disease with organoids. Cell. 2016;165(7):1586–1597. doi: 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- 12.Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nature cell biology. 2016;18(3):246–254. doi: 10.1038/ncb3312. [DOI] [PubMed] [Google Scholar]
- 13.Taguchi A, Kaku Y, Ohmori T, Sharmin S, Ogawa M, Sasaki H, Nishinakamura R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell stem cell. 2014;14(1):53–67. doi: 10.1016/j.stem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 14.Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nature biotechnology. 2015 doi: 10.1038/nbt.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takasato M, Pei XE, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, de Sousa Lopes SMC. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526(7574):564–568. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- 16.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260(5110):920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 17.Vacanti JP, Langer R. Tissue engineering: the design and fabrication of living replacement devices for surgical reconstruction and transplantation. The lancet. 1999;354:S32–S34. doi: 10.1016/s0140-6736(99)90247-7. [DOI] [PubMed] [Google Scholar]
- 18.Lanza R, Langer R, Vacanti JP. Principles of tissue engineering. Academic press; 2011. [Google Scholar]
- 19.Yang S, Leong K-F, Du Z, Chua C-K. The design of scaffolds for use in tissue engineering. Part I. Traditional factors. Tissue engineering. 2001;7(6):679–689. doi: 10.1089/107632701753337645. [DOI] [PubMed] [Google Scholar]
- 20.Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? Journal of theoretical biology. 1982;99(1):31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- 21.Badylak SF, Freytes DO, Gilbert TW. Extracellular matrix as a biological scaffold material: structure and function. Acta biomaterialia. 2009;5(1):1–13. doi: 10.1016/j.actbio.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnology and bioengineering. 2009;103(4):655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geckil H, Xu F, Zhang X, Moon S, Demirci U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine. 2010;5(3):469–484. doi: 10.2217/nnm.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee KY, Mooney DJ. Hydrogels for tissue engineering. Chemical reviews. 2001;101(7):1869–1880. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 26.Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24(24):4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman AS. Hydrogels for biomedical applications. Advanced drug delivery reviews. 2012;64:18–23. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 28.Jonker AM, Löwik DW, van Hest JC. Peptide-and protein-based hydrogels. Chemistry of Materials. 2012;24(5):759–773. [Google Scholar]
- 29.Rutz AL, Shah RN. Protein-Based Hydrogels Polymeric Hydrogels as Smart Biomaterials. Springer; 2016. pp. 73–104. [Google Scholar]
- 30.Saldin LT, Cramer MC, Velankar SS, White LJ, Badylak SF. Extracellular Matrix Hydrogels from Decellularized Tissues: Structure and Function. Acta Biomaterialia. 2016 doi: 10.1016/j.actbio.2016.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ, Christman KL. Naturally derived myocardial matrix as an injectable scaffold for cardiac tissue engineering. Biomaterials. 2009;30(29):5409–5416. doi: 10.1016/j.biomaterials.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singelyn JM, Christman KL. Modulation of material properties of a decellularized myocardial matrix scaffold. Macromolecular bioscience. 2011;11(6):731–738. doi: 10.1002/mabi.201000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ungerleider J, Johnson T, Rao N, Christman K. Fabrication and characterization of injectable hydrogels derived from decellularized skeletal and cardiac muscle. Methods. 2015;84:53–59. doi: 10.1016/j.ymeth.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wassenaar JW, Gaetani R, Garcia JJ, Braden RL, Luo CG, Huang D, DeMaria AN, Omens JH, Christman KL. Evidence for mechanisms underlying the functional benefits of a myocardial matrix hydrogel for post-MI treatment. Journal of the American College of Cardiology. 2016;67(9):1074–1086. doi: 10.1016/j.jacc.2015.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sellaro TL, Ranade A, Faulk DM, McCabe GP, Dorko K, Badylak SF, Strom SC. Maintenance of human hepatocyte function in vitro by liver-derived extracellular matrix gels. Tissue Engineering Part A. 2009;16(3):1075–1082. doi: 10.1089/ten.tea.2008.0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JS, Shin J, Park H-M, Kim Y-G, Kim B-G, Oh J-W, Cho S-W. Liver extracellular matrix providing dual functions of two-dimensional substrate coating and three-dimensional injectable hydrogel platform for liver tissue engineering. Biomacromolecules. 2013;15(1):206–218. doi: 10.1021/bm4015039. [DOI] [PubMed] [Google Scholar]
- 37.Loneker AE, Faulk DM, Hussey GS, D’Amore A, Badylak SF. Solubilized liver extracellular matrix maintains primary rat hepatocyte phenotype in-vitro. Journal of Biomedical Materials Research Part A. 2016;104(4):957–965. doi: 10.1002/jbm.a.35636. [DOI] [PubMed] [Google Scholar]
- 38.DeQuach JA, Lin JE, Cam C, Hu D, Salvatore MA, Sheikh F, Christman KL. Injectable skeletal muscle matrix hydrogel promotes neovascularization and muscle cell infiltration in a hindlimb ischemia model. European cells & materials. 2012;23:400. doi: 10.22203/ecm.v023a31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu Y, Fan X, Tian C, Luo J, Zhang Y, Deng L, Qin T, Lv Q. Decellularization of porcine skeletal muscle extracellular matrix for the formulation of a matrix hydrogel: a preliminary study. Journal of cellular and molecular medicine. 2016 doi: 10.1111/jcmm.12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ungerleider JL, Johnson TD, Hernandez MJ, Elhag DI, Braden RL, Dzieciatkowska M, Osborn KG, Hansen KC, Mahmud E, Christman KL. Extracellular matrix hydrogel promotes tissue remodeling, arteriogenesis, and perfusion in a rat hindlimb ischemia model. JACC: Basic to Translational Science. 2016;1(1–2):32–44. doi: 10.1016/j.jacbts.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, O’Hare MJ, Saleem MA, van den Heuvel LP, Mathieson PW. Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. International Society of Nephrology. 2006;69:1633–1640. doi: 10.1038/sj.ki.5000277. [DOI] [PubMed] [Google Scholar]
- 42.Satchell SC, Braet F. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. American Journal of Physiology-Renal Physiology. 2009;296(5):F947–F956. doi: 10.1152/ajprenal.90601.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freytes DO, Martin J, Velankar SS, Lee AS, Badylak SF. Preparation and rheological characterization of a gel form of the porcine urinary bladder matrix. Biomaterials. 2008;29(11):1630–1637. doi: 10.1016/j.biomaterials.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 44.Zuidema JM, Rivet CJ, Gilbert RJ, Morrison FA. A protocol for rheological characterization of hydrogels for tissue engineering strategies. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2014;102(5):1063–1073. doi: 10.1002/jbm.b.33088. [DOI] [PubMed] [Google Scholar]
- 45.Ali SH, DeCaprio JA. Cellular transformation by SV40 large T antigen: interaction with host proteins. Seminars in Cancer Biology. 2001;11:15–22. doi: 10.1006/scbi.2000.0342. [DOI] [PubMed] [Google Scholar]
- 46.Kuroda Y, Wakao S, Kitada M, Murakami T, Nojima M, Dezawa M. Isolation, culture and evaluation of multilineage-differentiating stress-enduring (Muse) cells. Nature protocols. 2013;8(7):1391–1415. doi: 10.1038/nprot.2013.076. [DOI] [PubMed] [Google Scholar]
- 47.Sun Q, Overholtzer M. Methods for the study of entosis. Necrosis: Methods and Protocols. 2013:59–66. doi: 10.1007/978-1-62703-383-1_5. [DOI] [PubMed] [Google Scholar]
- 48.Elliot RA, Jr, Hoehn JG. Use of commercial porcine skin for wound dressings. Plastic and reconstructive surgery. 1973;52(4):401–405. [PubMed] [Google Scholar]
- 49.Zhang Y, He Y, Bharadwaj S, Hammam N, Carnagey K, Myers R, Atala A, Van Dyke M. Tissue-specific extracellular matrix coatings for the promotion of cell proliferation and maintenance of cell phenotype. Biomaterials. 2009;30(23):4021–4028. doi: 10.1016/j.biomaterials.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeQuach JA, Mezzano V, Miglani A, Lange S, Keller GM, Sheikh F, Christman KL. Simple and high yielding method for preparing tissue specific extracellular matrix coatings for cell culture. PloS one. 2010;5(9):e13039. doi: 10.1371/journal.pone.0013039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hulmes D. Collagen diversity, synthesis and assembly. Springer; 2008. pp. 15–47. Collagen. [Google Scholar]
- 52.Parkinson J, Kadler KE, Brass A. Simple physical model of collagen fibrillogenesis based on diffusion limited aggregation. Journal of molecular biology. 1995;247(4):823–831. doi: 10.1006/jmbi.1994.0182. [DOI] [PubMed] [Google Scholar]
- 53.Brightman AO, Rajwa B, Sturgis J, McCallister M, Robinson J, Voytik-Harbin S. Time-lapse confocal reflection microscopy of collagen fibrillogenesis and extracellular matrix assembly in vitro. Biopolymers. 2000;54(3):222–234. doi: 10.1002/1097-0282(200009)54:3<222::AID-BIP80>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 54.Storm C, Pastore JJ, MacKintosh FC, Lubensky TC, Janmey PA. Nonlinear elasticity in biological gels. Nature. 2005;435(7039):191–194. doi: 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- 55.Erk KA, Henderson KJ, Shull KR. Strain stiffening in synthetic and biopolymer networks. Biomacromolecules. 2010;11(5):1358–1363. doi: 10.1021/bm100136y. [DOI] [PubMed] [Google Scholar]
- 56.Levental I, Georges PC, Janmey PA. Soft biological materials and their impact on cell function. Soft Matter. 2007;3(3):299–306. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 57.Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47(4):1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 58.Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324(5935):1673–1677. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Discher DE, Janmey P, Wang Y-L. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310(5751):1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 60.De Smet F, Segura I, De Bock K, Hohensinner PJ, Carmeliet P. Mechanisms of vessel branching. Arteriosclerosis, thrombosis, and vascular biology. 2009;29(5):639–649. doi: 10.1161/ATVBAHA.109.185165. [DOI] [PubMed] [Google Scholar]
- 61.Gilbert TW, Sellaro TL, Badylak SF. Decellularization of tissues and organs. Biomaterials. 2006;27:3675–3683. doi: 10.1016/j.biomaterials.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 62.Slater SC, Beachley V, Hayes T, Zhang D, Welsh GI, Saleem MA, Mathieson PW, Wen X, Su B, Satchell SC. An in vitro model of the glomerular capillary wall using electrospun collagen nanofibres in a bioartificial composite basement membrane. PloS one. 2011;6(6):e20802. doi: 10.1371/journal.pone.0020802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Slater SC, Ramnath RD, Uttridge K, Saleem MA, Cahill PA, Mathieson PW, Welsh GI, Satchell SC. Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. The international journal of biochemistry & cell biology. 2012;44(9):1482–1490. doi: 10.1016/j.biocel.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 64.O’Neill JD, Freytes DO, Anandappa AJ, Oliver JA, Vunjak-Novakovic GV. The regulation of growth and metabolism of kidney stem cells with regional specificity using extracellular matrix derived from kidney. Biomaterials. 2013;34(38):9830–9841. doi: 10.1016/j.biomaterials.2013.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oliver JA, Maarouf O, Cheema FH, Martens TP, Al-Awqati Q. The renal papilla is a niche for adult kidney stem cells. The Journal of clinical investigation. 2004;114(6):795–804. doi: 10.1172/JCI20921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Al-Awqati Q, Oliver JA. The kidney papilla is a stem cells niche. Stem cell reviews. 2006;2(3):181–184. doi: 10.1007/s12015-006-0046-3. [DOI] [PubMed] [Google Scholar]
- 67.Nagao RJ, Xu J, Luo P, Xue J, Wang Y, Kotha S, Zeng W, Fu X, Himmelfarb J, Zheng Y. Decellularized human kidney cortex hydrogels enhance kidney microvascular endothelial cell maturation and quiescence. Tissue Engineering Part A. 2016;22(19–20):1140–1150. doi: 10.1089/ten.tea.2016.0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mozes MM, Böttinger EP, Jacot TA, Kopp JB. Renal expression of fibrotic matrix proteins and of transforming growth factor-β (TGF-β) isoforms in TGF-β transgenic mice. Journal of the American Society of Nephrology. 1999;10(2):271–280. doi: 10.1681/ASN.V102271. [DOI] [PubMed] [Google Scholar]
- 69.Zeisberg M, Bonner G, Maeshima Y, Colorado P, Müller GA, Strutz F, Kalluri R. Renal fibrosis: collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. The American journal of pathology. 2001;159(4):1313–1321. doi: 10.1016/S0002-9440(10)62518-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruiz-Torres MP, Lopez-Ongil S, Griera M, Diez-Marques ML, Rodriguez-Puyol M, Rodriguez-Puyol D. The accumulation of extracellular matrix in the kidney: consequences on cellular function. Journal of nephrology. 2005;18(3):334. [PubMed] [Google Scholar]
- 71.Magno V, Friedrichs J, Weber HM, Prewitz MC, Tsurkan MV, Werner C. Macromolecular crowding for tailoring tissue-derived fibrillated matrices. Acta Biomaterialia. 2017 doi: 10.1016/j.actbio.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 72.Dewavrin J-Y, Hamzavi N, Shim V, Raghunath M. Tuning the architecture of three-dimensional collagen hydrogels by physiological macromolecular crowding. Acta biomaterialia. 2014;10(10):4351–4359. doi: 10.1016/j.actbio.2014.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.