

Abstract
Triglyceride molecules represent the major form of storage and transport of fatty acids within cells and in the plasma. The liver is the central organ for fatty acid metabolism. Fatty acids accrue in liver by hepatocellular uptake from the plasma and by de novo biosynthesis. Fatty acids are eliminated by oxidation within the cell or by secretion into the plasma within triglyceride-rich very low density lipoproteins. Notwithstanding high fluxes through these pathways, under normal circumstances the liver stores only small amounts of fatty acids as triglycerides. In the setting of overnutrition and obesity, hepatic fatty acid metabolism is altered, commonly leading the accumulation of triglycerides within hepatocytes, and to a clinical condition known as non-alcoholic fatty liver disease (NAFLD). In this review, we describe the current understanding of fatty acid and triglyceride metabolism in the liver and its regulation in health and disease, identifying potential directions for future research. Advances in understanding the molecular mechanisms underlying the hepatic fat accumulation are critical to the development of targeted therapies for NAFLD.
Introduction
The liver is the central organ that controls lipid homeostasis by means of complex, but precisely regulated biochemical, signaling and cellular pathways. Hepatocytes are the main liver parenchymal cells, which control hepatic biochemical and metabolic functions in the liver, including triglyceride metabolism. Additional cell types within the liver include cholangiocytes, Kupffer cells, stellate cells, and endothelial cells, each of which has specialized functions in hepatic pathobiology (261).
This review will focus on the hepatic metabolism of fatty acids (FA) and their neutral storage form, triglycerides (TG), which occurs primarily in hepatocytes. Under normal circumstances on a daily basis, the liver processes large quantities of FA, but stores only small amounts in the form of TG, with steady state TG contents of less than 5% (32). This is because the rates of acquisition of FA by uptake from the plasma and from de novo synthesis within the liver are balanced by rates of FA oxidation and secretion into plasma as TG-enriched very low-density lipoprotein (VLDL-TG). The relatively small quantities of TG stored within the liver are localized in cytoplasmic lipid droplets.
NAFLD is the most common chronic liver disease and is characterized by excess TG accumulation within the liver. It is associated with obesity, type 2 diabetes and dyslipidemia, and commonly occurs in the setting of insulin resistance (22). NAFLD encompasses a spectrum of liver histopathologies from simple hepatic steatosis, often referred to as non-alcoholic fatty liver (NAFL), to non-alcoholic steatohepatitis (NASH) which hepatic steatosis is accompanied by inflammation, cell death and fibrosis (5). Hepatic complications of NASH include cirrhosis and hepatocellular carcinoma. Currently, there are no effective therapies for NAFLD apart from changes in lifestyle that lead to improved physical fitness and weight loss. In order to develop effective pharmacologic therapies, detailed knowledge of the cellular and molecular mechanisms leading to altered hepatic lipid metabolism is essential. In addition to describing the major pathways of hepatic FA and TG metabolism and their regulation, we will discuss recent advances in our understanding of how they are altered in the setting of NAFLD.
Hepatic fatty acid metabolism
FA within the liver originate from either dietary or endogenous sources. Dietary TG are emulsified by bile acids within the intestinal lumen following their hydrolysis primarily by pancreatic lipase (166), which yields sn-2-monoacylglycerols and free FA as products. Following emulsification, these lipid molecules are taken by enterocytes and resynthesized into TG. TG are packaged into chylomicrons, secreted into the lymphatic system and ultimately reach the plasma (112, 122). Much of the chylomicron TG are taken up by muscle and adipose tissue due to the activity of lipoprotein lipase, which is expressed on the luminal surfaces of capillary endothelial cells of these tissues (Fig. 1). TG remaining within the chylomicron remnants are delivered to the liver when these particles are taken up by receptor mediated endocytosis, and FA are released during lysosomal processing of the particles (54).
Figure 1. Major sources for hepatic fatty acids.
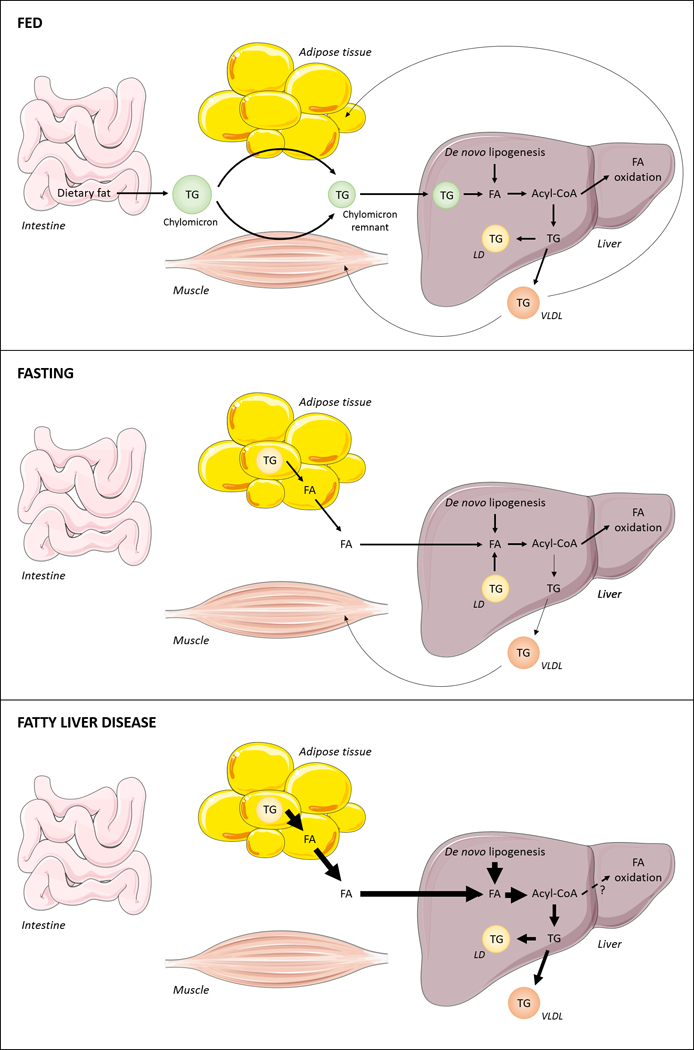
The three major sources for hepatic fatty acids (FA) are dietary lipids, adipose tissue derived-FA and de novo-synthesized FA. After a meal, dietary lipids are hydrolyzed within the intestinal lumen. Upon intestinal uptake, FA are re-esterified to form TG molecules, which are packaged into chylomicrons and delivered primarily to muscle and adipose tissue. The remaining TG present in chylomicron remnants is then transported to the liver and processed intracellularly, leading to FA release within hepatocytes. Carbohydrates, particularly glucose, are utilized in hepatic de novo lipogenesis (DNL) for the production of FA. In order to be metabolized, FAs are activated to form acyl-CoA molecules, which can undergo oxidation or be incorporated into complex lipids. Locally synthesized TG can be stored in intracellular lipid droplets (LDs) or packed into VLDL and secreted into the plasma. Upon fasting, intracellular TG stores are mobilized from adipocytes and hepatocytes to release FA products. Hepatic DNL may also contribute to form an acyl-CoA pool available for energy production, undergoing oxidation by mitochondria, or for VLDL-TG production. In the setting of overnutrition and insulin resistance, hepatic FA levels are increased due to enhanced lipolysis within adipocytes, which leads to increased circulating levels of FA in the bloodstream, and increased hepatic DNL. Excess FA cannot be consumed by oxidative pathways and FA are instead directed towards the synthesis of TG, leading to increased hepatic TG storage and VLDL overproduction. Arrow thickness denotes rates of metabolic activities.
When carbohydrates are abundant, the liver converts glucose into FA, a process referred to as de novo lipogenesis (DNL) (120) (Fig.1). The control of DNL is primarily transcriptional. Plasma insulin activates the endoplasmic reticulum membrane-bound transcription factor sterol regulatory element binding protein 1C (SREBP1c), the N-terminus of which translocates to the nucleus and upregulates all genes in the FA biosynthetic pathway (116). The hepatic uptake of excess plasma glucose promotes the nuclear translocation of carbohydrate response element binding protein (ChREBP), a transcription factor that also upregulates transcription of the majority of FA biosynthetic genes plus pyruvate kinase (89, 205), which increases the availability of citrate for FA synthesis.
Another important source of FA is direct uptake from the plasma. In human subjects, FA from plasma constitute the main source of hepatic triglycerides during fasting (72). Under fasting conditions when plasma insulin concentrations are low, a lipolytic program is initiated in white adipose tissue, which increases the plasma FA pool that is available for uptake by the liver (12, 214) (Fig. 1). FA are largely albumin-bound within the circulation. Several steps are involved in hepatic FA uptake. These include dissociation of FAs from albumin, transport across the hepatocyte plasma membrane, binding to intracellular proteins, and esterification to coenzyme A (CoA). Plasma FA are also the principal source of VLDL-TG in both fasting and fed states (72).
Within the hepatocyte, FA are esterified to glycerol-3-phosphate (G3P) and to cholesterol in order to generate TG or cholesteryl esters, respectively. These neutral lipids can be either stored in cytoplasmic lipid droplets (LDs), or secreted into bloodstream as VLDL particles (135) (Fig. 1). FA within the liver may also be used for the synthesis of other complex lipids, including phospholipids (PL). During fasting, FA are used both as local energy supply as well as substrate for ketone bodies production. Overall, hepatic FA metabolism is tightly regulated by multiple interrelated transcriptional and signaling pathways that remain the subject of intensive investigation and discovery.
Fatty acid uptake
Notwithstanding their ability to diffuse across a lipid bilayer (232), FA are taken up by hepatocytes via plasma membrane-associated proteins. A variety of proteins have been associated with long-chain FA transport, including plasma membrane FA-binding protein (FABPpm), FA translocase (FAT)/CD36, caveolin-1, and very long-chain acyl-CoA synthetases (ACSVL/FA transport proteins, also named FATP/solute carrier family 27A1–6, SLC27A1–6) (27, 253) (Fig. 2).
Figure 2. Hepatic fatty acid transport and metabolism.
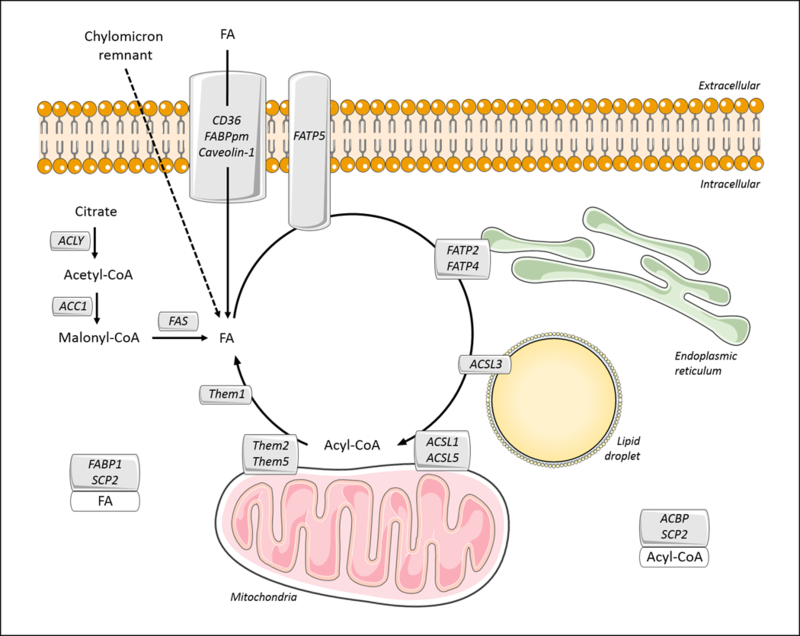
Within the plasma membrane, FA translocase (FAT)/CD36, plasma membrane FA-binding protein (FABPpm), and Caveolin-1 mediate the uptake of fatty acid (FA) that is bound to circulating albumin. Alternatively, hepatic FA can be obtained by the internalization of chylomicron remnant or by de novo lipogenesis. The latter occurs through the activity of three key enzymes: ATP-citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS). In the cytosol, FAs are bound to the fatty acid-binding protein-1 (FABP1) and sterol carrier protein-2 (SCP2), which may control their cellular distribution. FA transport proteins (FATP2, 4 and 5) and long-chain acyl-CoA synthetases (ACSL1, 3 and 5) mediate the activation of long-chain FA to acyl-CoA molecules and their channeling to metabolic pathways. Although associated with mitochondria, ACSL5 may function to promote triglyceride biosynthesis. In the cytosol, acyl-CoAs are bound to acyl-CoA-binding protein (ACBP) or SCP2. Acyl-CoA thioesterases (ACOT)/thioesterase superfamily members (Them1, 2 and 5) appear to counteract ACSL activity by catalyzing the hydrolysis of acyl-CoA molecules into FA and CoA. This may provide additional means of controlling the balance between FA and acyl-CoA within hepatocytes.
The roles of CD36 and FABPpm proteins in FA uptake are established for heart and skeletal muscle, but their function in hepatocytes remains unresolved. Although expression levels are low in the liver, CD36 mRNA levels are positively correlated with hepatic TG contents in a rat model of hepatic steatosis (35) and its protein expression is increased in patients with NAFLD (181). However, the pathophysiological relevance of this protein to the liver is unclear. Whereas CD36 liver-specific deletion in mice leads to reduced hepatic lipid contents and decreased FA uptake in the setting of high fat feeding, suggesting the direct contribution of CD36 to hepatic FA metabolism under these conditions (279), whole body knockout of CD36 in mice significantly impairs FA uptake by heart, adipose tissue, and muscle, but normal uptake is observed in the liver (52). There also seems to be a role for CD36 in hepatic lipogenesis. CD36 is a target gene for the aryl hydrocarbon receptor (AhR), and is required for the TG accumulation observed in the liver in response to its activation (154). In addition, the pregnane X receptor (PXR) induces hepatic CD36 mRNA expression, along with increased FA uptake and TG accumulation in the liver (294). Finally, the CD36 is a transcriptional target of peroxisome proliferator activated receptor (PPAR) γ (259), which is upregulated by PXR (294).
Functional evidence for protein-mediated fatty acid transport in the rat hepatocyte plasma membrane led to the identification of FABPpm as a protein that mediates this activity (247, 249). FABPpm was subsequently identified as mitochondrial aspartate aminotransferase (20, 28). The mRNA encoding this protein is increased in hepatocytes of genetically obese mouse models (180), and the binding of oleate to rat hepatocyte plasma membranes is reduced after treatment with specific antiserum (246). In cultured hepatocytes, ethanol treatment increases mRNA and plasma membrane protein contents, and these changes correlate with increased FA uptake (295). However, the relative contribution of FABPpm to FA uptake by the liver remains unknown.
FATP proteins (1–6) are FA-transporting proteins that are situated in the plasma membrane, as well as intracellular membranes. Because of their capacity to also activate long- or very-long-chain FA (69), these proteins have been re-classified in the very-long-chain acyl-CoA synthetase (ACSVL) family as ACSVL1–6 (106). FATPs couple FA uptake with intracellular FA esterification into acyl-CoAs, a metabolic trapping mechanism that has been referred to as vectorial acylation (77, 258). Although well characterized, neither FATP1 nor FATP4 are normally expressed at substantial levels in the liver (239). Expression of FATP4, an ER membrane-associated isoform (226, 273), is increased in the setting of hepatic steatosis and may play a role in palmitate-mediated lipoapoptosis (226). FATP2 is highly expressed in liver and kidney (85). In HepG2 cells, FATP2 localizes to the ER, but its overexpression increases uptake of long-chain FA in cell culture (148). FATP5 is expressed in the liver, and studies in knockout mice and cell culture systems support a role in FA uptake (70).
Caveolins (types 1–3) are integral membrane proteins that are essential components of caveolae, vesicular membrane microdomains that are rich in cholesterol and sphingolipids (6). Caveolins were initially characterized with respect to cholesterol transport (204). However, their roles in FA transport have been appreciated in experiments using HepG2 hepatoma cells (203), wherein Caveolin-1, CD36, FABPpm, and calcium-independent membrane phospholipase A2 (iPLA2) form a heterotetrameric protein complex within plasma membrane microdomains (248). These complexes promote FA accumulation within caveolae-derived vesicular structures for transport into the cell (247). In support of this mechanism, mice deficient in caveolin-1 show reduced hepatic TG content and impaired LD accumulation (87).
Although multiple factors lead to hepatic steatosis, circulating FA are the main source of hepatic lipids in NAFLD (72). The overflow of FA derived from excessive lipolysis in the adipose tissue contributes to the pathogenesis of insulin resistance in mice and humans (159), leading to both type 2 diabetes (213) and NAFLD (27, 171). Although FA transporters may exert a central role in the control of FA flux into the liver, additional studies are needed to clarify specific functions and physiological contributions of each FA transporter, along with their differential regulation in the context of NAFLD.
De novo lipogenesis
Under normal circumstances in humans, pathways of hepatic DNL appear to be utilized sparingly (114). However, in addition to the enhanced FA uptake, increased DNL can contribute substantially to hepatic steatosis (72). The first step in DNL pathway is catalyzed by ATP-citrate lyase (ACLY), which converts citrate to acetyl-CoA, which is then carboxylated to malonyl-CoA by acetyl-CoA carboxylase (ACC). ACC is followed by a series of reactions that convert malonyl-CoA into palmitate. Fatty acid synthase (FAS) is the key rate-limiting enzyme in palmitate synthesis, and its activity is regulated by multiple mechanisms (125, 143, 218, 228) (Fig. 2). Palmitate can be modified by elongases and desaturases to generate a variety of FA species.
There are two ACC isoforms, cytosolic ACC1 and mitochondrial ACC2, which are encoded by separate genes. The presence of distinct ACC enzymes enables the formation of two distinct pools of malonyl-CoA. ACC1-generated malonyl-CoA is utilized as a substrate by FAS, whereas the malonyl-CoA produced by ACC2 plays a key role in the negative regulation of β-oxidation by inhibiting carnitine palmitoyltransferase 1 (CPT1) (266). ACC1 is highly expressed in lipogenic organs, including the liver, wherein the gene expression and enzyme activity are induced by carbohydrate-rich, low-fat diet, and are suppressed in the setting of starvation or diabetes (139, 149). Whole body disruption of the ACC1 gene results in embryonic lethality, but liver-specific knockout mice are viable and exhibit 70–75% reductions in malonyl-CoA levels and 40–70% reductions in hepatic TG contents (170). Despite similar malonyl-CoA levels as wild type animals, livers of ACC2 knockout mice are characterized by reduced rates of β-oxidation and TG contents. These mice resist to diet-induced obesity and diabetes (2, 3). In keeping with these observations, antisense-mediated knockdown of both ACC1 and ACC2 led to improved hepatic insulin responsiveness in rats with experimental NAFLD (217).
FAS expression is ubiquitous, but evidence of elevated transcriptional and enzymatic activities are observed in the liver of murine models of obesity, such as Zucker (fa/fa) rats (227). The transcription of both ACC and FAS genes is enhanced in livers of NAFLD subjects (146). Similar to ACC knockout mice, whole body disruption of FAS leads to embryonic lethality (51). Surprisingly, liver-specific knockout of FAS results in hypoglycemia and hepatic steatosis in mice fed a zero-fat diet (45). Because this phenotype is reversed by treatment with a PPARα agonist, it has been suggested that newly synthesized FA and their derivatives, including phospholipids (44), constitute a distinct pool that provides endogenous ligands for PPARα. This in turn stimulates hepatic gluconeogenesis and FA oxidation (45).
In leptin-deficient obese (ob/ob) mice, increased transcription of Acc1 and Fas is linked to the increased activation of ChREBP (64). Additionally, SREBP-1c and ChREBP can synergistically induce the mRNA expression of Acc and Fas in the liver (65). Expression of both transcriptional factors are upregulated in livers of mouse models of NAFLD (19), however only SREBP-1c is upregulated in patients with NAFLD (115).
Among TG found in VLDL in normal subjects, less than 5% contain FA derived from DNL (68). However, inhibition of DNL impairs production and secretion of VLDL-TG in rats (95). The rate of incorporation of DNL-synthesized FA into VLDL-TG is increased in normal subjects in the setting of high-carbohydrate diet (223, 224), alcohol consumption and infectious states (88), and is positively correlated with increased plasma VLDL-TG content (223). DNL is also increased in obese (67, 244) and NAFLD subjects (68, 88). These observations suggest that hypertriglyceridemia is at least in part attributable to increased availability of DNL-synthesized FA for TG synthesis and VLDL production (223). Ongoing research is needed to clarify mechanisms of altered DNL in a variety of pathogenic conditions, including NAFLD.
Fatty acid activation
Activation by thioesterification to CoA to form fatty acyl-CoA molecules is an obligatory step in the metabolism of long-chain FA. This reaction is catalyzed by members of the long-chain acyl-CoA synthetase (ACSL) family of enzymes (Fig. 2). The five ACSL isoforms found in mammalians have specific tissue distributions, subcellular locations and substrate preferences (106). Additionally, distinct ACSL isoforms play key roles in partitioning fatty acyl-CoAs into different metabolic pathways (57, 106).
ACSL1, the best characterized isoform, is abundantly expressed in a variety of tissues. Because Acsl1 is a target gene of PPARα in rat liver, this enzyme has been associated with FA catabolism (220). ACSL1 interacts physically with CPT1 in the outer mitochondrial membrane of rat hepatocytes, possibly functioning to channel fatty acyl-CoA products into mitochondria for β-oxidation (155). However, this process is not fully understood because liver-specific ACSL1 knockout mice exhibit only modestly decreased rates of β-oxidation and reduced fatty acyl-chain incorporation into newly-synthesized TG, although the total hepatic TG contents remain unchanged (161).
ACSL3 is expressed on lipid droplets in HuH7 hepatoma cells (94), and its expression is up-regulated in livers of ob/ob mice and in mice fed a high-carbohydrate diet (34). Knockdown of ACSL3 in rat hepatocytes reduces DNL through diminished activation of lipogenic transcriptional factors (i.e. PPARγ, ChREBP, SREBP-1c and LXRα) possibly due to the reduced availability of FA species, acyl-CoA or their lipid intermediates (34).
Suggestive of a role for ACSL5 in TG synthesis, mRNA levels are increased by insulin and by SREBP-1c activation (4). In support of this lipogenic role, overexpression of ACSL5 in rat hepatoma cells increases uptake of exogenous FA, as well as its partitioning into TG, but not into PL (174). Knockdown of ACSL5 in rat primary hepatocytes strongly influences the partitioning of fatty acyl-CoAs between lipogenic and lipolytic pathways. In the absence of this enzyme, the incorporation of exogenous and endogenous FA into complex lipids is reduced, the formation of LD is decreased and the oxidation of FA is stimulated (33).
The reaction catalyzed by ACSL isoforms is reversed by the activities of acyl-CoA thioesterases (ACOTs) (Fig. 2). These enzymes catalyze the hydrolysis of acyl-CoA molecules into FA and CoA. Despite their relatively well-investigated enzymatic activities, the biological functions of the fifteen ACOT family members remain less well understood (53, 142, 256). Acots 1–6, which constitute the type I enzymes, are upregulated in the livers of fasted and high fat-fed mice under the transcriptional control of PPARα (79). These observations suggest a role of type I ACOTs in lipid catabolism. Indeed, overexpression of ACOT2 in the liver leads to increased fatty acid oxidation and ketogenesis, although the mechanism underlying this effect remains unclear (182).
ACOTS 7–15 comprise the type II enzymes, and some of these contribute to hepatic FA metabolism. ACOT11 (synonym, Thioesterase superfamily member 1, Them1) is highly expressed in brown adipose tissue, where it functions to suppress energy expenditure (292) by decreasing rates of fat oxidation (196). Acot11 gene expression is also strongly induced in the liver of obese mice (79). Whereas deficiency of this enzyme results in decreased hepatic steatosis, improved glucose homeostasis, and resistance to diet-induced diabetes in high fat fed mice (292), it is unclear whether these phenotypes are indirect effects of increased energy expenditure.
ACOT13 (a.k.a. Them2) is a mitochondria-associated enzyme that is highly expressed in the liver and other oxidative tissues. Them2 knockout mice are resistant to diet-induced hepatic steatosis and exhibit improved glucose metabolism (130). Moreover, Them2-deficiency results in diminished expression of PPARα and HNF4α, possibly due to the role of this enzyme in the control of FA ligands of these nuclear receptors (53) and the reduced FA oxidation (135). Them2 also controls lipid and glucose metabolism by interacting with phosphatidylcholine transfer protein (PC-TP) to suppress hepatic insulin signaling by reducing the activation of both the insulin receptor substrate 2 (IRS2) and the mammalian target of rapamycin (mTOR) (80).
ACOT15 (a.k.a. Them5) appears to play a key role in maintaining the integrity of mitochondrial membranes. Mice lacking ACOT15 experience deficiencies in cardiolipin remodeling in the inner mitochondrial membrane of hepatocytes. This phenotype leads to mitochondrial dysfunction, including altered morphology, decreased mitochondrial FA contents, reduced FA oxidation, and the development of liver steatosis (296).
It is plausible that ACSL and ACOT enzymes coordinate the control of the intracellular balance of fatty acyl-CoAs and FA, intracellular and intra-organelle concentrations of CoA, and the availability of lipid substrates for a variety of metabolic pathways. Both types of enzymes exhibit specific transcriptional patterns of expression that vary depending on cell type and physiological state, and the coordinated regulation of ACSL and ACOT enzymes has been recently described (79). Although elevated hepatic concentrations of long-chain acyl-CoAs have been associated with increased plasma insulin levels and hepatic insulin resistance (48), decreased hepatic fatty acyl-CoA concentrations in the setting of liver-specific deletion of ACSL1 do not prevent the development of insulin resistance in high-fat fed mice (161). As illustrated by these apparently contradictory observations, mechanisms whereby ACSL and ACOT enzymes control FA metabolism within hepatocytes are incompletely understood, and it remains uncertain whether their altered regulation contributes to insulin resistance and related metabolic abnormalities.
Intracellular fatty acid transport
Long-chain FAs and their acyl-CoA derivatives act not only as substrates for lipid synthesis and oxidation, but are also involved in a range of diverse intracellular processes, such as protein palmitoylation, intracellular signaling, and activation of transcription factors (83, 106, 222). Considering this multiplicity of cellular functions, their relative insolubility and potential cytotoxicity (102, 103), it is logical that the intracellular concentrations and localization of free FA and fatty acyl-CoAs are tightly regulated. This is accomplished at least in part due to the activities of lipid binding proteins, several of which have been implicated in the control of the intracellular concentration and the partitioning of long-chain FA and acyl-CoA within hepatocytes. These include the liver fatty acid binding protein (L-FABP, synonym FABP1), acyl-CoA binding protein (ACBP; also named as acyl-CoA-binding domain containing protein-1, ACBD1, or diazepam binding inhibitor, DBI), and sterol carrier protein-2 (SCP2) (8, 103) (Fig. 2).
The FABP protein family comprises 10 members (237). FABP1 is abundant in the cytosol of hepatocytes, comprising 2–5% of the cytosolic protein content (177) and reversibly binds long-chain FA, fatty acyl-CoAs, lysophospholipids, cholesterol and other lipids (14, 36, 254, 255). Fabp1−/− male mice fed standard chow diet exhibit normal gain weight and liver weights (172, 192). However, these animals have reduced total hepatic TG contents as well as decreased rates of FA esterification into TG, with preserved or increased esterification into PL (172, 192). Because the overexpression of FABP1 in L-cell fibroblasts enhances the intracellular traffic of short-, medium-, and long-chain FA from the cytosol to the nucleus (119), it has been conjectured that FABP1 must enter nuclei to promote the entry of FA (222). Indeed, FABP1 directly interacts with PPARα within nuclei of primary hepatocytes (117). This interaction may represent a mechanism for the delivery of FA ligands that activate this key transcription factor (222). FABP1 ablation is protective against diet-induced obesity and hepatic steatosis and improves glucose metabolism in mice fed a Western (high saturated fat, high cholesterol) diet (193). Taken together, these findings suggest a role for FABP1 in FA partitioning into specific metabolic pathways.
ACBP binds medium- and long-chain acyl-CoA with high affinity (84, 207, 211). Levels of ACBP mRNA and protein are highest in the liver (26). In the mouse, ACBP expression is modulated by nutritional status. Fasting results in decreased hepatic ACBP expression levels, whereas high fat feeding increases it (21). Overexpression of ACBP in rat hepatoma cells increases TG content (283). In addition, hepatic TG and PL concentrations are increased in transgenic mice that overexpress ACBP in the liver (118). Under these conditions, microsome-associated glycerol-3-phosphate acyltransferase (GPAT) activity is induced, suggesting that ACBP has a role in the formation and partitioning of an acyl-CoA pool towards glycerolipid synthesis (118).
Expression of SCP-2 is high in liver and intestine (15). This protein has high affinity for binding both long-chain FA and acyl-CoAs, with Kd values in the same range as reported for FABP and ACBP (103, 144, 177). Knockout mice exhibit reduced plasma FA concentrations, decreased hepatic cholesteryl ester and TG contents, as well as increased rates of β-oxidation (225). SCP-2 ablation also results in alterations in the composition and physical properties of lipid rafts from mice primary hepatocytes, including the enrichment in PL contents (9). Moreover, hepatic SCP-2 and FABP1 act to facilitate hydrolysis of cholesteryl ester molecules associated with high-density lipoprotein (HDL) particles and increasing the biliary secretion of bile acids (270) and cholesterol (92).
Because the metabolic fates of FA and acyl-CoA molecules are linked to these intracellular lipid biding proteins, it is tempting to speculate that they could be leveraged to manipulate the flux of lipids between anabolic and catabolic pathways in order to defend against the FA-mediated lipotoxicity, that contributes to the pathogenesis of NAFLD.
Triglyceride metabolism
The assembly of TG molecules constitutes the principal means by which the liver stores and exports FA. Under normal conditions, the liver stores little TG, but exports considerable amounts in the form of VLDL particles that deliver FA to muscle and fat tissue, depending on nutritional status. Whereas previously it was considered that excess TG stores in the setting of NAFLD contributed to lipotoxicity (61), emerging concepts suggest that increased TG storage and VLDL secretion are instead protective against FA-mediated hepatotoxicity.
Triglyceride synthesis
In most mammalian cell types, the G3P pathway is the principal route for the synthesis of TG, contributing over 90% of total TG synthesis (56, 62). The first and rate-limiting step of this pathway is the esterification of long-chain acyl-CoA to G3P, which is catalyzed by mitochondrial and microsomal G3P acyltransferase (GPAT) enzymes. Lysophosphatidic acid (LPA) molecules produced in this reaction are then acylated to form phosphatidic acid (PA) by the acylglycerol-3-phosphate acyltransferases (AGPAT) present in the ER membrane. PA can be converted into cytidine diphosphate diacylglycerol (CDP-DG), which is a substrate for the synthesis of certain glycerolphospholipids and cardiolipins (113, 231) or can be dephosphorylated by phosphatidate phosphohydrolase (PAP, synonym Lipin) to form DG, which serve as precursor molecules for the synthesis of TG, as well as phosphatidylcholine (PC) and phosphatidylethanolamine (PE) (56, 73). DG acyltransferase (DGAT) catalyzes the acylation of DG, constituting the final step of TG synthesis (55). Newly synthesized TG molecules are then directed from ER lipid bilayer to form cytosolic LDs (99, 278) (Fig. 3).
Figure 3. Hepatic triglyceride metabolism.
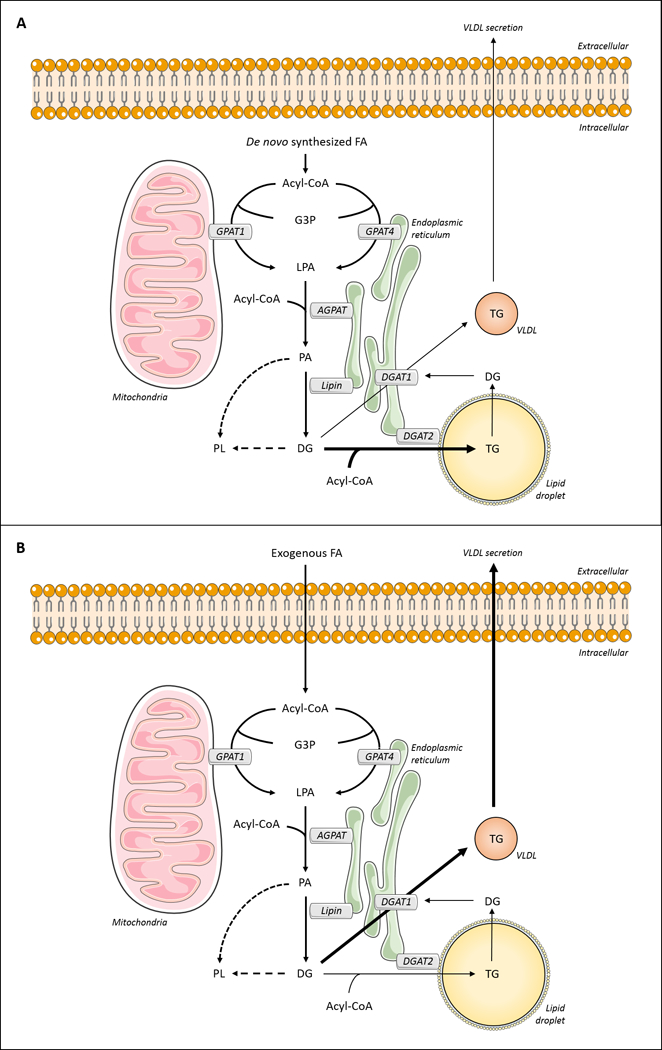
Acyl-CoA molecules can be esterified to glycerol-3-phosphate (G3P) by isoforms of glycerol-3-phosphate acyltransferase (GPAT). In hepatocytes, the isoforms are predominantly GPATs 1 and 4. The produced lysophosphatidic acid (LPA) is acylated by acylglycerol-3-phosphate acyltransferases (AGPATs) to form phosphatidic acid (PA), which can be dephosphorylated by phosphatidic acid phosphatase (PAP) to form diacylglycerol (DG). Both PA and DG can be directed towards phospholipid (PL) synthesis. Additionally, diacylglycerol acyltransferases (DGATs 1 and 2) synthesize triglyceride (TG) by acylation of DG. (A) Fas synthesized de novo are likely to be channeled to VLDL-TG production through DGAT1 activity. (B) Exogenous FA appear to be directed towards TG synthesis for storage in lipid droplets by the activity of DGAT2. In addition, lipid droplet-TG can undergo hydrolysis, with FA re-routed into VLDL by DGAT1.
Four isoforms of GPAT are encoded by distinct genes (276). In the liver, the mitochondria-associated GPAT isoforms (GPAT1 and 2) contribute 30–50% of total GPAT activity (56). GPAT1 is highly expressed in the liver (157). It is induced after fasting/refeeding and in response to insulin, and downregulated via cAMP-dependent signaling pathway during nutrient deprivation (76, 186, 230). GPAT1-deficient mice exhibit increased rates of FA oxidation and concentrations of plasma β-hydroxybutyrate. Moreover, hepatic TG contents are decreased by 60%, suggesting that GPAT1 functions in the partitioning of FA towards TG synthesis and away from oxidation in the liver (110). GPAT1 deficiency also leads to reduced plasma TG levels, reduced VLDL secretion rates and decreased total body weights (110, 191). Accordingly, the overexpression of GPAT1 in rat hepatocytes leads to increased esterification of oleate into LPA, DG and TG, and to increased intracellular TG contents (158, 165, 188). Although highly expressed in the liver, the metabolic role(s) of GPAT2 in the lipid metabolism remain unclear. This isoform is not regulated by either insulin or fasting/refeeding (271), and mouse models with GPAT2 loss or gain of function have yet to be reported.
GPAT3 and GPAT4 account for microsomal GPAT activity. These isoforms are modulated by nutritional status and hormonal variations. Although initially considered to mainly mediate PL synthesis (55), more recent studies have revealed their contributions to TG metabolism in adipose tissue. Specific roles for GPAT3 and GPAT4 in hepatic lipid metabolism remain largely undefined. The overexpression of GPAT4 in HepG2 cells leads to 20% increase in intracellular TG content (189), and its knockout in mice results in 40–50% reduction in hepatic TG (37, 262). It is noteworthy that hepatocytes from mice lacking GPAT4 are able to incorporate de novo synthesized FA into TG (275), suggesting that this isoform could be relevant for the metabolism of exogenous FA.
The second step in TG synthesis is mediated by AGPAT enzymes. Although ten AGPAT enzymes have been identified on the basis of sequence homology, the enzymatic activity for LPA acylation has been confirmed for only a few (252). Certain isoforms initially described as AGPAT were later reclassified into different acyltransferase groups. For example, AGPAT6 and AGPAT8 are currently designated as GPAT4 and GPAT3, respectively (38, 49, 189). AGPAT1 and AGPAT2 are localized in the ER (74, 97, 150, 277) and are highly expressed in the liver (75, 240, 252, 277). However, the contribution of these enzymes to the hepatic metabolism remains unknown.
Lipins-1–3 constitute the PAP enzyme family. Under lipogenic stimuli, these proteins translocate from the cytosol to ER membrane in order to access their substrate PA and contribute the TG biosynthetic pathway (31, 173). Additionally, lipin-1 can localize in the nucleus of hepatocytes, where it acts as a transcriptional co-activator (234). Fasting and glucocorticoid administration result in increased lipin-1 expression in mouse livers (90, 169, 195). This effect is mediated by PPARγ coactivator 1α (PGC-1α) and is required to sustain the fasting-induced expression of PPARα and its downstream targets. Lipin-1 physically interacts with PGC-1α and PPARα in the nucleus, leading to upregulation of oxidative fatty acid metabolism by mitochondria (90). Accordingly, mice lacking lipin-1 exhibit hepatic steatosis (90), enhanced stearoyl-CoA desaturase-1 (Scd1) gene expression, increased rates of VLDL secretion, and increased plasma concentrations of TG without changes in TG synthetic rates (50). Reduced lipin-1 expression is observed in livers of obese diabetic mice, and the overexpression of lipin-1 in this animal inhibits VLDL secretion and improves hepatic insulin responsiveness (50). The effects of lipin-1 on VLDL secretion are independent of PAP enzymatic activity and instead linked to its nuclear receptor interactions (50). In addition to lipin-1, high levels of lipin-2 expression are also observed in the liver (71) and hepatic lipin-3 transcription is increased in lipin-1-deficient mice (71). Moreover, lipin-2 mRNA levels are induced in lipin-1-deficient mice, supporting the hypothesis that lipin isoforms could compensate in order to preserve PAP activity and restore the levels of TG synthesis (107).
In mouse liver, DGAT activity is detected in two microsomal membrane locations: luminal activity (DGAT1, measured as latent – luminal – DGAT activity) and cytosol-accessible activity (predominantly DGAT2, measured as overt DGAT activity) (200). The former contributes to the synthesis of TG that are packaged into VLDL. The latter contributes to the synthesis of a TG pool to be stored in the cytoplasmic LDs (1, 200, 272). Interestingly, the DGAT proteins are genetically unrelated. DGAT1 is part of the membrane bound O-acyl transferase (MBOAT) protein family, which comprises a diverse group of acyltransferases, including acyl-CoA:cholesterol acyltransferase (ACAT) and protein-cysteine N-palmitoyltransferase (286). DGAT2 is a member of diacylglycerol acyltransferase (DAGAT) family that also includes the monoacylglycerol acyltransferase (MGAT) and acyl-CoA wax alcohol acyltransferases (AWAT) (286).
Although DGAT1 and DGAT2 are able to catalyze the same reaction, these enzymes do not appear to be redundant in their respective functions. DGAT1 mRNA is modestly expressed in the liver in comparison to other organs, such as skeletal muscle and small intestine (41). This enzyme is able to catalyze acyltransferase reactions using substrates other than DG. For example, DGAT1 acylates MG, wax esters and retinol in vitro (285). DGAT1 predicted topology combined with results obtained from loss of function studies supports the involvement of this enzyme in both the overt and latent DGAT activities (286, 289). Accordingly, overexpression of DGAT1 in rat hepatoma cells leads to increased intracellular TG contents and also increased rates of VLDL-TG secretion (164). Dgat1 knockout mice have slightly reductions in hepatic TG concentrations when fed a low fat diet, with more prominent decreases being observed in response to high fat feeding (47, 238). High fat fed Dgat1 knockout high fat fed mice are also resistant to obesity and have improved insulin responsiveness (238). Moreover, DGAT1 expression is increased in NAFLD subjects and may contribute to hypertriglyceridemia (146).
By contrast, Dgat2 transcripts are abundant in the liver, and are decreased upon fasting and increased after refeeding (179). DGAT2 exhibits in vitro specificity for the use of DG molecules as substrates (42). The predicted topology of this protein suggests its contribution to overt activity, and this was confirmed in HepG2 cells by studies using DGAT2 inhibitors (98). Accordingly, DGAT2 is found in close association to cytosolic LDs and mitochondria-associated membranes, where enzymes involved in TG synthesis and storage are also localized (178, 242). Dgat2 knockout mice exhibit severe lipopenia and die in the first day after birth (243). The liver-specific overexpression of DGAT2 in chow-fed mouse leads to the development of hepatic steatosis but no abnormalities in plasma glucose and insulin signaling were observed, suggesting that hepatic TG accumulation is not a cause of insulin resistance (183). In agreement with this observation, human DGAT2 genetic variants reduce hepatic steatosis. However, the decrease in fat content does not lead to improved insulin sensitivity (131).
In keeping with specialized cellular functions of the enzymes, gain and loss of function studies in mice have revealed different phenotypes for DGAT1 and DGAT2, without evidence of compensation (243). In fact, a more recent study revealed a unique role of hepatic DGAT2 in its use of de novo-synthesized acyl-CoA and DG as substrates (281). The TG formed by this reaction are stored in cytosolic LDs and ultimately undergoes lipase-mediated hydrolysis. The resulting lipid intermediates (i.e., MG and DG) are available for re-esterification by overt DGAT1 activity present in the cytosolic compartment, followed by TG transport into the ER. These intermediates can also undergo translocation to the ER lumen followed by acylation catalyzed by latent DGAT1 activity (Fig. 3A). Overt and latent activity of DGAT1 contribute to lipoprotein lipidation during VLDL assembly and utilize pre-formed, but not de novo-synthesized acyl-CoA as preferred substrate (Fig. 3B). Therefore, DGAT2 and DGAT1 use independent pools of acyl-CoA for TG synthesis, and act in a sequential and integrated way for TG packaging into nascent VLDL particles (281).
Although hepatic TG accumulation is the major determinant of NAFLD (61), lipid and lipidomic analyses have revealed changes in the compositions of a variety of lipids, including FA, DG, LPA, PA, PL and ceramides (78, 127, 168, 216, 260). The specific contributions of hepatic lipid species to NAFLD remain the subject of active investigation (136).
VLDL assembly and secretion
TG-rich VLDL particles represent the mechanism by which FA are exported from the liver and delivered to muscle for oxidation and adipose tissue for storage, respectively. VLDL assembly is a two-step process that begins in the ER lumen. In the first step, microsomal triglyceride transfer protein (MTP) acts to incorporate a small amount of TG in apoB100 as it is being translated by ribosomes and translocated across the ER membrane (101). In the second step, additional TG is packaged into the nascent apoB100-containing particles as they are traverse from the ER to the Golgi apparatus to form VLDL particles (Fig. 4) (54).
Figure 4. Triglyceride storage and secretion in hepatocytes.
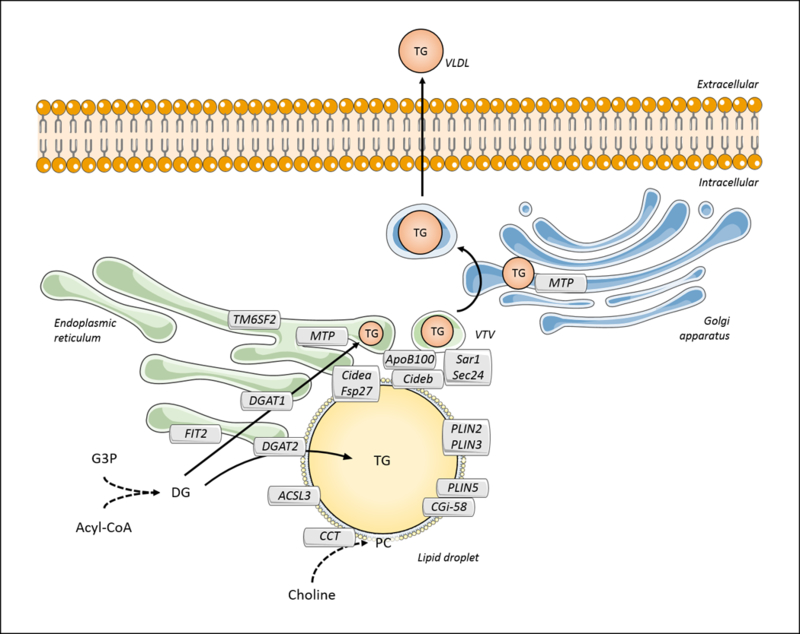
Triglyceride (TG) can be synthesized from diacylglycerol (DG) by diacylglycerol acyltransferases (DGAT1 and 2). DGAT1 preferably provides TG to VLDL during lipidation in the lumen of the endoplasmic reticulum. This process is mediated by microsomal triglyceride transfer protein (MTP), which facilitates the association between TG and apoB100. Transmembrane 6 superfamily member 2 (TM6SF2) may also contribute to VLDL lipidation via yet unknown mechanisms. The nascent VLDL particle is then transferred to the Golgi apparatus through the VLDL transfer vesicle (VTV), followed by a second MTP-mediated lipidation step. VLDL particles are secreted via a vesicle-mediated mechanism. TG can also be formed by the activity of DGAT2, which mainly contributes to storage in lipid droplets. Lipid droplets are delimitated by proteins and a phosphatidylcholine (PC)-enriched surfactant monolayer. Among the lipid-droplet associated proteins, perilipins (PLIN2, 3 and 5) and comparative gene identification-58 (CGI-58) contribute to lipid droplet structure and/or the regulation of lipid droplet-associated enzymes; CTP-phosphocholine cytidylyltransferase (CCT) and acyl-CoA synthetase 3 (ACSL3) may be required for the biosynthesis of lipids; DFF45-like effector (CIDE) proteins, Cidea, Cideb, and Fsp27, and the microsomal fat-inducing transmembrane protein 2 (FIT2) are required for lipid droplet formation, however their specific cellular roles are incompletely understood. Cideb also contributes to VLDL production and secretion via its interaction with Sar1 and Sec24, which are present in VTV.
The link between increased VLDL secretion and metabolic diseases, such as insulin resistance and diabetes, is well stablished (54, 251, 297). The development of insulin resistance leads to hepatic TG accumulation owing to both enhanced FA uptake into the liver and increased DNL. The greater availability of TG together with higher MTP activity promote the overproduction of VLDL particles (10) and greater plasma TG concentrations (54). A positive correlation between plasma TG concentrations and the hepatic steatosis is not always observed (63). Obese leptin-deficient ob/ob mice exhibit decreased VLDL production rates despite marked hepatic steatosis (163). In nondiabetic obese NAFLD subjects, VLDL-TG secretion rates are elevated up to 2-times in comparison to normal subjects (82). In this setting, higher rates of TG accumulation fail to increase VLDL-TG proportionately, suggesting a limited capacity for increasing VLDL production and secretion (81).
Reductions in VLDL secretion can also lead to hepatic steatosis. This occurs in the setting of genetic defects in apoB100 and MTP, which lead to hypobetalipoproteinemia and abetalipoproteinemia, respectively (274). Mipomersen and lomitapide are drugs that respectively target apoB100 and MTP in the interest of reducing plasma LDL cholesterol concentrations by blocking VLDL secretion (58, 263). A mechanism-based side effect of these medications is hepatic steatosis. Finally, a polymorphism in Transmembrane 6 superfamily member 2 (TM6SF2) has been identified by human genome-wide association studies to be associated with hepatic steatosis (147, 236). The polymorphism results in reduced VLDL secretion rates, because of impaired lipidation of nascent VLDL particles (236). Whether the control of VLDL secretion can be leveraged in the management of NAFLD remains unclear.
Lipid droplet biology
LDs are dynamic cellular structures that transiently store lipids. Second to white adipose tissue, the liver has the next greatest capacity to store TG in LDs, such that overnight fasting leads to hepatic LD formation that accommodates FA derived from adipose tissue lipolysis (267). The neutral lipid composition of LDs differs among different hepatic cell types. Whereas hepatocytes harbor TG-enriched LD cores, stellate cell LDs store predominantly vitamin A as retinyl esters (23). The TG-rich cores of LDs in hepatocytes are surrounded by a coat comprised of PL and proteins. The PL are primarily PC and form a monolayer around the neutral lipid core (13). CTP-phosphocholine cytidylyltransferase (CCT) is the rate-limiting enzyme in the Kennedy pathway for PC biosynthesis. Indicative of its role in LD biogenesis, CCT is recruited from the cytosol to expanding LDs in a variety of cell types in order to provide PC for LD surface expansion (278). However, the precise role of CCT on LD formation within hepatocytes is uncertain.
In addition to preserving the LD structure, LD-associated proteins regulate the formation, expansion and contraction of LDs. These proteins include selected enzymes of lipid synthesis, as ACSL3 and DGAT2 (Fig. 4). Changes in the composition and activity of the protein components of the LD have been implicated as contributing to metabolic disease (40, 99, 100, 105). The predominant hepatocellular LD proteins are the members of perilipin (PLIN; former perilipin/ADRP/TIP47, PAT) protein superfamily (PLIN1–5). These proteins in adipocytes are involved both in the stabilization of LD structure as well as in the control of availability of substrates for several LD-associated enzymes (141). Perilipin 1 (PLIN1; synonym Perilipin A) is normally only expressed in LDs from adipocytes, whereas PLIN2 (synonym ADRP) and PLIN3 (synonym TIP47) are associated with LDs in hepatocytes (245). Plin1 mRNA is expressed in livers of subjects with NAFLD (104), and PLIN1 protein is found in LDs of steatotic hepatocytes (245). A key role for PLIN2 in hepatocytes is supported by the observation that it is the most abundant LD protein in the HuH7 hepatocyte cell line (94), and its levels are also increased in steatotic livers of both humans and mice (185) in response to PPARγ activation (197). Overexpression of PLIN2 in rat hepatic stellate cells results in lipid accumulation within LDs (96), while PLIN2-deficient mice exhibit a 60% reduction in hepatic TG content and are resistant to diet-induced hepatic steatosis (46). Liver-specific ablation of PLIN2 in mice results in reduced LD size and is protective against diet-induced NASH (190). PLIN1 is frequently associated with larger LDs in the liver of NAFLD subjects while PLIN2 is predominantly found in smaller LDs (93). The specific targeting of these proteins raises the suggestion of distinct PLIN1 and PLIN2 functions.
Plin3 expression is also induced in livers of high fat-fed mice and its knockdown reduces hepatic TG content, attenuates steatosis, and improves insulin sensitivity and glucose tolerance (39). In hepatocytes, the degradation of PLIN2 and PLIN3 are steps required for lipolysis during nutrient deprivation. The removal of PLIN proteins from LD occurs via chaperone-mediated autophagy (CMA) and facilitates the LD association of components of the lipolytic machinery, including ATGL (134). Moreover, the combined knockdown of Plin2 and Plin3 results in increased insulin resistance in mouse AML12 hepatocytes stimulated with oleate (17). PLIN5 is highly expressed in oxidative tissues, including liver. Hepatic Plin5 mRNA levels are increased after fasting in response to activation of PPARα and/or PPARβ/δ (140). PLIN5 expression is also enhanced in steatotic livers of humans and obese mice, and the knockout of this protein results in decreased hepatic TG content and smaller-sized LDs (268).
In hepatocytes, LDs are tightly associated with ER membrane cisternae, allowing the physical interaction between LD components and microsomal proteins, such as apoB100 (194) (Fig. 4). The fat-inducing transmembrane proteins (FIT1 and 2) are localized in the ER and have the ability to bind DG and TG, with FIT2 showing higher affinity (108). The overexpression on FIT2, the most abundant hepatic FIT isoform, in mouse liver results in increased TG content and LD accumulation (128). These data suggest that FIT2 functions in the binding of neutral lipids synthesized within ER and their channeling to nascent LDs (99).
The DFF45-like effector (CIDE) proteins, Cidea, Cideb, and Fsp27, are associated both to the ER and LDs and have been linked to LD metabolism (100). Recent studies have demonstrated that these proteins act to promote lipid exchange and fusion among LDs (282). However, they are differentially distributed among hepatocytes based on sizes of LD that they harbor. Cideb localizes both to hepatocytes with small and with large LDs, whereas Cidea and Fsp27 specifically localize to hepatocytes containing large LDs (282).
Cidea expression is increased in livers of diabetic mice (137) and a coding region polymorphism is associated with obesity in human subjects (60, 290). Moreover, expression of both Cidea and Fsp27 are upregulated in mice with hepatic steatosis induced by PPARγ activation (176, 264, 288). Fsp27 is a target gene of PPARγ and its transcription is decreased in ob/ob mice with liver-specific knockdown of PPARγ, accompanied by decreased TG content in the liver (175, 176). Cideb is highly expressed in the liver and Cideb knockout mice are resistant to obesity and liver steatosis induced by high-fat diet (160). Moreover, these animals have decreased levels of circulating TG and FA, and exhibit improved hepatic insulin sensitivity. These effects are mediated by the hepatic downregulation of SREBP-1c, resulting in decreased lipogenesis and increased FA oxidation (160). Cideb also interacts with apoB100, and mice lacking Cideb exhibit reduced VLDL secretion and increased hepatic steatosis (284). In addition to its role in VLDL assembly, Cideb has been associated in VLDL export from ER to the Golgi. This occurs through the interaction with components of coat complex II (COPII), namely Sar1 and Sec24, which are present in VLDL transport vesicles (257).
Because NAFLD is characterized by excessive deposition of TG within cytosolic LDs and because proteomic studies have revealed that numerous LD-associated proteins are modulated in human and mice steatotic livers (138, 250), detailed knowledge of the biology hepatocyte LDs could reveal novel targets for therapeutic intervention.
Lipolysis
The LD-associated adipose triglyceride lipase (ATGL, also known as PNPLA2) is the rate-limiting step in TG lipolysis within adipocytes. The resulting DG molecules are then hydrolyzed by the hormone sensitive lipase (HSL) to release monoglycerides (MG). In the final step, the monoacylglycerol lipase (MGL) cleaves MG into glycerol and FA (151) (Fig. 5). Gain and loss of function studies have revealed that hepatic ATGL is required for the lipolysis of TG stored in LDs, controls substrate availability for FA oxidation, and modulates the progression of hepatosteatosis (198, 208, 280). Full activation of ATGL depends on co-activation by the comparative gene identification-58 (CGI-58) (152). Liver-specific ablation of CGI-58 results in NAFLD phenotypes in mice that include hepatic steatosis and fibrosis (109). In the setting of hepatic steatosis, lipolysis mediated by hepatic ATGL is decreased when PLIN5 binds competitively to CGI-58, displacing ATGL (268, 269). Insulin-resistant NAFLD patients who exhibit higher degrees of liver steatosis when compared to NAFLD subjects without insulin resistance, also exhibit decreased CGI-58 mRNA levels. These observations suggest that insulin-resistance could induce hepatic TG accumulation through CGI-58-mediated reductions in ATGL-dependent lipolysis (132). Less is known concerning MGL and HSL activities in hepatocytes, although Hsl expression is down-regulated in livers of NAFLD patients (146).
Figure 5. Lipolysis and fatty acid oxidation in hepatocytes.
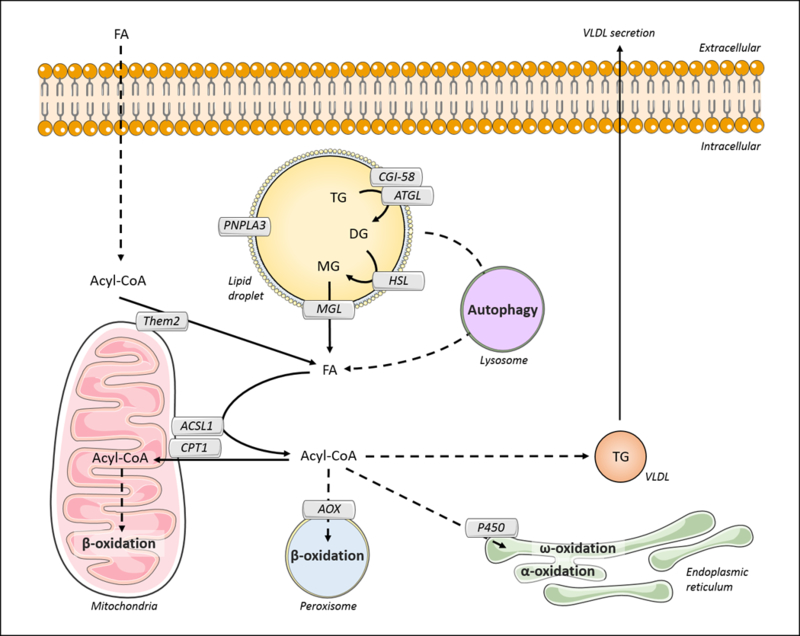
The initiation of hepatic lipolysis depends on the activation of adipose triglyceride lipase (ATGL) through the binding to the comparative gene identification-58 (CGI-58). ATGL catalyzes the hydrolysis of triglyceride (TG), releasing diacylglycerol (DG). This lipid is further hydrolyzed by hormone sensitive lipase (HSL), releasing monoacylglycerol (MG). Monoacylglycerol lipase (MGL) mediates the breakdown of MG into fatty acid (FA) and glycerol. Alternatively, autophagic pathways, such as macroautophagy and chaperone-mediated autophagy (CMA), promote the hydrolysis of LD. FA can also be generated from acyl-CoA by the activity of acyl-CoA thioesterase 13 (ACOT 13; synonym: thioesterase superfamily member 2, Them2). In the mitochondria outer membrane, long-chain acyl-CoA synthetase 1 (ACSL1) converts long-chain FAs to acyl-COAs. ACSL1 interacts physically with carnitine palmitoyltransferase 1 (CPT1). It is likely that ACSL1 channels acyl-CoA to CPT1 to control the availability of substrates for mitochondrial β-oxidation. Additionally, acyl-CoAs can be directed to β-oxidation in the peroxisome, where fatty acyl-CoA oxidase (AOX) represents a rate-limiting step, or ω-oxidation and α-oxidation in the endoplasmic reticulum, mediated by P450 4A family members. Also, part of the acyl-CoA pool can be directed for TG synthesis and VLDL assembly, sustaining the transfer of hepatic lipids to other organs.
In the setting of insulin resistance, enhanced rates of lipolysis within white adipose tissue are a major contributor to the increased FFA plasma levels and hepatic steatosis (159). However, the contribution of TG hydrolysis within hepatocytes to aberrant lipid accumulation in the liver is less clear. Several lipases are present in close association with LDs in adipocytes (30, 66) (Fig. 5), but their contributions to LD TG hydrolysis remain topics of discovery.
The patatin-like phospholipase domain-containing 3 (PNPLA3/adiponutrin) is a TG hydrolase/transacylase that localizes to LD, ER and cytoplasm (124, 212). A single-nucleotide variant of the Pnpla3 gene (I148M) confers increased risk of NAFLD in humans (210). In this connection, livers from transgenic mice expressing this allele show accumulation of inactive PNPLA3 in lipid droplets and increased steatosis when fed a high-sucrose diet (235). Although the genetic association of PNPLA3 with NALFD is robust, key details are still lacking regarding the metabolic activity of this protein.
Autophagy
Autophagy is a lysosome-dependent process that selectively targets, transports and regulates the storage of essential components, including lipids, proteins and carbohydrates. Three types of autophagy have been described in hepatocytes (59). In macroautophagy, LDs or other organelles are engulfed to form autophagosomes, which fuses to lysosome to generate an autolysosome. After hydrolysis by lysosomal enzymes, lipid products are released to the cytosol and recycled by other cellular processes (167). The autophagosome formation relies on several autophagy receptors in addition to the autophagy-related proteins (ATGs) (241). In microautophagy, small vesicles originated from lysosomal membrane invagination mediate the engulfment of cytosolic components (167). In chaperone-mediated autophagy (CMA), lysosomal degradation of specific proteins is regulated upon recognition by CMA receptor (133).
Macroautophagy of LDs (also named lipophagy) is stimulated in hepatocytes during starvation, leading to the release of FA in the cytosol after TG breakdown (233). Additionally, products of lipid anabolism have inhibitory effects in lipophagy (167). Although the mechanisms controlling lipophagy are not well understood, this process is driven by the small GTPase Rab7 protein, which recognizes LDs and mediates their engulfment by autophagosomes (221).
Decreased autophagy has been reported in obesity-related disorders, including hepatosteatosis (219). Altered membrane lipid composition due to high-fat feeding reduces the activity of lysosomal proteins and decreases autophagic activity in mouse liver (145, 209). Impaired CMA leads to hepatosteatosis through at least three mechanisms. First, reduced CMA activity decreases mitochondrial function and β-oxidation. Second, CMA-deficient mice exhibit increased expression of lipogenic enzymes, including DGAT2 (219). Finally, CMA is required for the lipolysis of hepatic TG through hsp70-mediated degradation of PLIN2 and PLIN3 (134). Hence, impaired autophagy in the setting of steatosis exacerbates lipid accumulation within the liver.
Fatty acid oxidation
FA derived from hydrolysis of hepatic TG stores, circulating lipids or DNL, can be oxidized by multiple pathways. Mitochondrial β-oxidation is the primary route for the oxidation of the majority of FA found in hepatocytes, including short- (<C4), medium- (C4-C12), and long-chain (C12-C20) FA. β-oxidation of very long- (C20-C26) and branched-chain FA begins in the peroxisomes. Additional pathways for FA oxidation include α-oxidation and ω-oxidation within the ER and are mediated by cytochrome P450 4A family members (153, 187) (Fig. 5). The expression of genes involved in mitochondrial and extramitochondrial FA oxidation is regulated largely by PPARα activity. PPARα is expressed at high levels in liver, and both whole-body and hepatocyte-specific ablation of PPARα in mice lead to reduced transcription of hepatic genes related to mitochondrial β-oxidation, such as very long chain acyl-CoA dehydrogenase (VLCAD), long chain acyl-CoA dehydrogenase (LCAD), and ACSL1 (7), and genes involved in peroxisomal β-oxidation, including peroxisomal fatty acyl-CoA oxidase (AOX) and cytochrome P450 4A (156, 184). These animals also experience hypoketonemia and increased hepatic steatosis (111, 184).
It is noteworthy that AOX ablation leads to sustained activation of PPARα, likely due to accumulation of PPARα ligands and induction of cytochrome P450 4A (86). Impaired peroxisomal FA oxidation results in the accumulation of dicarboxylic acids within the liver and leads to mitochondrial damage and microvesicular steatosis (153). Alterations in dicarboxylic acid levels are also observed in conditions of FA overload and have been correlated with increased risk of NAFLD development, although the cellular mechanism remains unknown (215, 287). PGC1α and BAF60a form a complex that regulates PPARα transcriptional activity in the liver. Overexpression of BAF60a increases fatty acid oxidation and reduces hepatic steatosis in obese mice (162). Similar phenotypes have been observed in livers of mice overexpressing the PPARα coactivator PGC1β (18).
Notwithstanding the relationship between PPARα activity and hepatic lipid metabolism, the correlation between PPARα expression levels and hepatic steatosis is inconsistent, with reports demonstrating unchanged, increased and decreased expression (16). This is also the case for rates of hepatic mitochondrial β-oxidation. The transcription of CPT1a, is down-regulated in NAFLD (146). Although previous studies revealed decreased hepatic mitochondrial FA oxidation in the setting of hepatic steatosis (123, 202, 206), recent work has demonstrated increased oxidation rates in livers of subjects with hepatic steatosis, metabolic syndrome and NAFLD (16). In livers of ob/ob mice, mitochondrial and peroxisomal oxidative capacities are elevated (29). Mechanisms underlying increased FA oxidation in NAFLD have been reviewed (16). Hypotheses include enhanced activation of PPARα due to increased hepatic FA uptake or biosynthesis.
ER-mediated control of lipid homeostasis
Among its many functions, the ER regulates the synthesis of lipids and proteins, as well as intracellular calcium storage (24). Under physiological conditions, unfolded and misfolded proteins accumulate in the ER lumen. In response to this ER stress, the cell initiates multiple lines of defense. One is the activation of ER-associated degradation (ERAD), a ubiquitin/proteasome protein degradation pathway that relies on E3 ligase complex activation (11). Second, the unfolded protein response (UPR) is initiated (229). This comprises three main signaling pathways mediated by microsomal transmembrane proteins: PKR-like ER kinase (PERK; also known as eukaryotic translation initiation factor 2-α kinase 3, eIF2αk3), inositol requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6) (229). UPR activation alleviates the accumulation of misfolded proteins into the ER through the inhibition of protein translation and the induction of chaperones to increase protein folding capacity, as well as expanding the ER membrane (11).
Lipid accumulation is also associated with chronic ER stress in hepatocytes (293). Lipid-mediated induction of ER stress can be triggered by multiple mechanisms. Obese mice exhibit altered microsomal membrane composition in the liver, with increased PC:PE ratios and variations in PL acyl-chain saturation degree (91, 121). Hence, changes in membrane fluidity leads to altered activity of membrane-associated proteins. For instance, sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) activity is reduced in steatotic mouse livers. In this model, decreased ER Ca2+ content impairs intraluminal Ca2+-dependent chaperones function and is a cause for ER stress (91).
UPR-related proteins directly sense membrane lipid composition in non-hepatic cells. In mouse fibroblasts, PERK and IRE1 are responsive to increased lipid saturation even in the absence of their ER-spanning transmembrane domain (265). However, it is uncertain whether direct lipid sensing mechanisms modulate hepatic ER stress. Because ER stress in turn promotes lipid accumulation in hepatocytes, chronic ER stress may directly contribute to the pathogenesis of NAFLD (291). In cell culture systems, prolonged exposure to oleate or palmitate induces ER stress when these saturated FA are incorporated to ER membrane PL (25). A concomitant reduction in VLDL-TG secretion is attributable to enhanced apoB100 degradation by proteasomal and non-proteasomal mechanisms (43, 199). In this connection, treatment with 4-phenyl butyric acid (PBA), a chemical chaperone, improves ER homeostasis and VLDL secretion after FA exposure (43). Although reduced VLDL secretion due to chronic ER stress results in TG accumulation in McA-RH7777, its potential contribution for the progression of hepatic steatosis was not demonstrated in animal models (199).
ER stress also promotes lipid accumulation in hepatocytes that is attributable to increased lipogenesis (11). In obese mice, hepatic induction of UPR pathway reduces Insig-1 levels and leads to the activation of SREBP, increased expression of lipogenic enzymes and steatosis (129). UPR-mediated activation of PERK may also contribute to hepatic steatosis by increasing expression of VLDL receptors in hepatocytes (126). Moreover, ER stress reduces autophagy through the cross-talk with components of ERAD and UPR in different cell types, exacerbating the hepatic steatosis (11, 201)
In summary, excess lipid load potentially initiates a vicious cycle of hepatic lipid accumulation due to abnormal activity of ER stress-activated pathways, leading to altered lipid homeostasis and TG deposition in the liver (11, 291, 293).
Conclusion
The liver is a main organ for the metabolism of fatty acids and triglycerides and altered activity of metabolic pathways in the setting of overnutrition leads to a common chronic liver disease known as NAFLD. Despite this biomedical relevance, many important questions regarding basic aspects of hepatic lipid metabolism remain unanswered. These include: (a) Which FA transporters are responsible for the increased intracellular concentration of FA in the setting of overnutrition? (b) What is the precise mechanism by which DNL is increased in obese and NAFLD subjects? (c) What is the precise molecular mechanism underlying the partitioning of FA and acyl-CoA among metabolic pathways, which presumably involve ACOT, ACSL and lipid-binding proteins? (d) How are rates of FA oxidation regulated in the setting of overnutrition? In summary, a deeper understanding is needed regarding the precise mechanisms whereby fatty acid uptake and synthesis are tightly balance against oxidation and secretion within the liver. This would be expected to lead to a better understanding of how triglycerides accumulate in many, but not all, individuals who consume excess calories and to new therapeutic strategies for the management of NAFLD.
Didactic Synopsis.
Major teaching points:
A variety of endogenous and exogenous sources provide fatty acids that can be assembled into to triglycerides within the liver in health and disease;
Multiple hepatocellular mechanisms regulate fatty acid uptake, synthesis, transport, and oxidation;
Triglycerides can be stored within hepatocytes, undergo lipolysis, or be exported into the bloodstream depending upon physiological and pathological conditions;
Mitochondria, endoplasmic reticulum, Golgi apparatus, peroxisomes, and lipid droplets are examples of organelles that participate in fatty acid and triglyceride metabolism;
The metabolism of fatty acids and triglyceride is regulated by transcriptional and post-transcriptional mechanisms;
Alterations in fatty acid and triglyceride metabolism lead to non-alcoholic fatty liver disease (NAFLD), which is a common consequence of overnutrition.
Acknowledgements
This work was supported by NIH grants R37DK048873, R01DK056626 and R01DK103046 to D.E.C. M.A.B. is the recipient of NASH Fatty Liver Disease Postdoctoral Research Fellowship from the American Liver Foundation.
Footnotes
Cross-References
Energy metabolism in the liver
Metabolism of lipids in chylomicrons and very low density lipoproteins (legacy)
Obesity
Steatosis in the liver
Triglyceride metabolism (legacy)
References
- 1.Abo-Hashema KAH, Cake MH, Power GW, and Clarke D. Evidence for triacylglycerol synthesis in the lumen of microsomes via a lipolysis-esterification pathway involving carnitine acyltransferases. J Biol Chem 274: 35577–35582, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, and Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291: 2613–2616, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Abu-Elheiga L, Oh WK, Kordari P, and Wakil SJ. Acetyl-CoA carboxylase 2 mutant mice are protected against obesity and diabetes induced by high-fat/high-carbohydrate diets. Proc Natl Acad Sci U S A 100: 10207–10212, 2003. 10.1073/pnas.1733877100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achouri Y, Hegarty BD, Allanic D, Becard D, Hainault I, Ferre P, and Foufelle F. Long chain fatty acyl-CoA synthetase 5 expression is induced by insulin and glucose: Involvement of sterol regulatory element-binding protein-1c. Biochimie 87: 1149–1155, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Ahmed A, Wong RJ, and Harrison SA. Nonalcoholic Fatty Liver Disease Review: Diagnosis, Treatment, and Outcomes. Clin Gastroenterol Hepatol 13: 2062–2070, 2015. 10.1016/j.cgh.2015.07.029 [DOI] [PubMed] [Google Scholar]
- 6.Anderson RGW. The caveolae membrane system. Annu Rev Biochem 67: 1998. [DOI] [PubMed] [Google Scholar]
- 7.Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, and Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPAR alpha). J Biol Chem 273: 5678–5684, 1998. [DOI] [PubMed] [Google Scholar]
- 8.Atshaves BP, Martin GG, Hostetler HA, McIntosh AL, Kier AB, and Schroeder F. Liver fatty acid-binding protein and obesity. J Nutr Biochem 21: 1015–1032, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atshaves BP, McIntosh AL, Payne HR, Gallegos AM, Landrock K, Maeda N, Kier AB, and Schroeder F. SCP-2/SCP-x gene ablation alters lipid raft domains in primary cultured mouse hepatocytes. J Lipid Res 48: 2193–21211, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Avramoglu RK, Basciano H, and Adeli K. Lipid and lipoprotein dysregulation in insulin resistant states. Clin Chim Acta 368: 1–19, 2006. [DOI] [PubMed] [Google Scholar]
- 11.Baiceanu A, Mesdom P, Lagouge M, and Foufelle F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol 12: 710–722, 2016. 10.1038/nrendo.2016.124 [DOI] [PubMed] [Google Scholar]
- 12.Barrows BR, and Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab 91: 1446–1452, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RGW, Liu PS, and Chapman KD. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res 48: 837–847, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Bass NM. Function and regulation of hepatic and intestinal fatty acid binding proteins. Chem Phys Lipids 38: 95–114, 1985. [DOI] [PubMed] [Google Scholar]
- 15.Baum CL, Kansal S, and Davidson NO. Regulation of sterol carrier protein-2 gene expression in rat liver and small intestine. J Lipid Res 34: 729–739, 1993. [PubMed] [Google Scholar]
- 16.Begriche K, Massart J, Robin MA, Bonnet F, and Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58: 1497–1507, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Bell M, Wang H, Chen H, McLenithan JC, Gong DW, Yang RZ, Yu D, Fried SK, Quon MJ, Londos C, and Sztalryd C. Consequences of lipid droplet coat protein downregulation in liver cells: abnormal lipid droplet metabolism and induction of insulin resistance. Diabetes 57: 2037–2045, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bellafante E, Murzilli S, Salvatore L, Latorre D, Villani G, and Moschetta A. Hepatic-specific activation of peroxisome proliferator-activated receptor γ coactivator-1β protects against steatohepatitis. Hepatology 57: 1343–1356, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, Girard J, Guillou H, and Postic C. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest 122: 2176–2194, 2012. 10.1172/JCI41636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berk PD, Wada H, Horio Y, Potter BJ, Sorrentino D, Zhou SL, Isola LM, Stump D, Kiang CL, and Thung S. Plasma membrane fatty acid-binding protein and mitochondrial glutamic-oxaloacetic transaminase of rat liver are related. Proc Natl Acad Sci U S A 87: 3484–3488, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhuiyan J, Pritchard PH, Pande SV, and Seccombe DW. Effects of high-fat diet and fasting on levels of acyl-CoenzymeA binding-protein in liver, kidney, and heart of rat. Metabolism-Clinical and Experimental 44: 1185–1189, 1995. [DOI] [PubMed] [Google Scholar]
- 22.Birkenfeld AL, and Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 59: 713–723, 2014. 10.1002/hep.26672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blaner WS, O’Byrne SM, Wongsiriroj N, Kluwe J, D’Ambrosio DM, Jiang H, Schwabe RF, Hillman EM, Piantedosi R, and Libien J. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta 1791: 467–473, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borgese N, Francolini M, and Snapp E. Endoplasmic reticulum architecture: structures in flux. Curr Opin Cell Biol 18: 358–364, 2006. 10.1016/j.ceb.2006.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, and Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 47: 2726–2737, 2006. 10.1194/jlr.M600299-JLR200 [DOI] [PubMed] [Google Scholar]
- 26.Bovolin P, Schlichting J, Miyata M, Ferrarese C, Guidotti A, and Alho H. Distribution and characterization of diazepam binding inhibitor (DBI) in peripheral tissues of rat. Regul Pept 29: 267–281, 1990. [DOI] [PubMed] [Google Scholar]
- 27.Bradbury MW. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: possible role in steatosis. American journal of physiology Gastrointestinal and liver physiology 290: G194–G198, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Bradbury MW, Stump D, Guarnieri F, and Berk PD. Molecular modeling and functional confirmation of a predicted fatty acid binding site of mitochondrial aspartate aminotransferase. J Mol Biol 412: 412–422, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brady LJ, Brady PS, Romsos DR, and Hoppel CL. Elevated hepatic mitochondrial and peroxisomal oxidative capacities in fed and starved adult obese (ob/ob) mice. Biochem J 231: 439–444, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brasaemle DL, Dolios G, Shapiro L, and Wang R. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem 279: 46835–46842, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Brindley DN. Intracellular translocation of phosphatidate phosphohydrolase and its possible role in the control of glycerolipid synthesis. Prog Lipid Res 23: 115–133, 1984. [DOI] [PubMed] [Google Scholar]
- 32.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, and Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 40: 1387–1395, 2004. 10.1002/hep.20466 [DOI] [PubMed] [Google Scholar]
- 33.Bu SY, and Mashek DG. Hepatic long-chain acyl-CoA synthetase 5 mediates fatty acid channeling between anabolic and catabolic pathways. J Lipid Res 51: 3270–3280, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bu SY, Mashek MT, and Mashek DG. Suppression of long chain acyl-CoA synthetase 3 decreases hepatic de novo fatty acid synthesis through decreased transcriptional activity. J Biol Chem 284: 30474–30483, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buqué X, Martínez MJ, Cano A, Miquilena-Colina ME, García-Monzón C, Aspichueta P, and Ochoa B. A subset of dysregulated metabolic and survival genes is associated with severity of hepatic steatosis in obese Zucker rats. J Lipid Res 51: 500–513, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burrier RE, Manson CR, and Brecher P. Binding of acyl-CoA to liver fatty acid binding protein: effect on acyl-CoA synthesis. Biochim Biophys Acta 919: 221–230, 1987. [DOI] [PubMed] [Google Scholar]
- 37.Cao G, Konrad RJ, Li SD, and Hammond C. Glycerolipid acyltransferases in triglyceride metabolism and energy homeostasis-potential as drug targets. Endocrine, metabolic & immune disorders drug targets 12: 197–206, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Cao JS, Li JL, Li DM, Tobin JF, and Gimeno RE. Molecular identification of microsomal acyl-CoA : glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc Natl Acad Sci U S A 103: 19695–19700, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carr RM, Patel RT, Rao V, Dhir R, Graham MJ, Crooke RM, and Ahima RS. Reduction of TIP47 improves hepatic steatosis and glucose homeostasis in mice. American Journal of Physiology Regulatory, integrative and comparative physiology 302: R996–R1003, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carr RM, and Ahima RS. Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res 340: 187–192, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, Erickson SK, and Farese RV. Identification of a gene encoding an acyl CoA : diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci U S A 95: 13018–13023, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, Voelker T, and Farese RV. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem 276: 38870–38876, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Caviglia JM, Gayet C, Ota T, Hernandez-Ono A, Conlon DM, Jiang H, Fisher EA, and Ginsberg HN. Different fatty acids inhibit apoB100 secretion by different pathways: unique roles for ER stress, ceramide, and autophagy. J Lipid Res 52: 1636–1651, 2011. 10.1194/jlr.M016931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, and Semenkovich CF. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 138: 476–488, 2009. 10.1016/j.cell.2009.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, and Semenkovich CF. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metabolism 1: 309–322, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, and Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol 26: 1063–1076, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen HC, Smith SJ, Ladha Z, Jensen DR, Ferreira LD, Pulawa LK, McGuire JG, Pitas RE, Eckel RH, and Farese RV. Increased insulin and leptin sensitivity in mice lacking acyl CoA : diacylglycerol acyltransferase 1. J Clin Investig 109: 1049–1055, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen MT, Kaufman LN, Spennetta T, and Shrago E. Effects of high fat-feeding to rats on the interrelationship of body weight, plasma insulin, and fatty acyl-coenzyme A esters in liver and skeletal muscle. Metabolism-Clinical and Experimental 41: 564–569, 1992. [DOI] [PubMed] [Google Scholar]
- 49.Chen YQ, Kuo MS, Li SY, Bui HH, Peake DA, Sanders PE, Thibodeaux SJ, Chu SY, Qian YW, Zhao Y, Bredt DS, Moller DE, Konrad RJ, Beigneux AP, Young SG, and Cao GQ. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase. J Biol Chem 283: 10048–10057, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen ZJ, Gropler MC, Norris J, Lawrence JC, Harris TE, and Finck BN. Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arterioscler Thromb Vasc Biol 28: 1738–1744, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chirala SS, Chang H, Matzuk M, Abu-Elheiga L, Mao J, Mahon K, Finegold M, and Wakil SJ. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc Natl Acad Sci U S A 100: 6358–6363, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coburn CT, Knapp FF, Febbraio M, Beets AL, Silverstein RL, and Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem 275: 32523–32529, 2000. [DOI] [PubMed] [Google Scholar]
- 53.Cohen DE. New players on the metabolic stage: How do you like Them Acots? Adipocyte 2: 3–6, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen DE, and Fisher EA. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin Liver Dis 33: 380–388, 2013. 10.1055/s-0033-1358519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coleman RA, and Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res 43: 134–176, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Coleman RA, Lewin TM, and Muoio DM. Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Annu Rev Nutr 20: 77–103, 2000. [DOI] [PubMed] [Google Scholar]
- 57.Cooper DE, Young PA, Klett EL, and Coleman RA. Physiological consequences of compartmentalized acyl-CoA metabolism. J Biol Chem 290: 20023–20031, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cuchel M, and Rader DJ. Microsomal transfer protein inhibition in humans. Curr Opin Lipidol 24: 246–250, 2013. 10.1097/MOL.0b013e32836139df [DOI] [PubMed] [Google Scholar]
- 59.Czaja MJ, Ding WX, Donohue TM Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, and Yin XM. Functions of autophagy in normal and diseased liver. Autophagy 9: 1131–1158, 2013. 10.4161/auto.25063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dahlman I, Kaaman M, Jiao H, Kere J, Laakso M, and Arner P. The CIDEA gene V115F polymorphism is associated with obesity in Swedish subjects. Diabetes 54: 3032–3034, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Day CP, and James OF. Steatohepatitis: a tale of two ““hits””? Gastroenterology 114: 842–845, 1998. [DOI] [PubMed] [Google Scholar]
- 62.Declercq PE, Haagsman HP, Van Veldhoven P, Debeer LJ, Van Golde LM, and Mannaerts GP. Rat liver dihydroxyacetone-phosphate acyltransferases and their contribution to glycerolipid synthesis. J Biol Chem 259: 9064–9075, 1984. [PubMed] [Google Scholar]
- 63.den Boer M, Voshol PJ, Kuipers F, Havekes LM, and Romijn JA. Hepatic steatosis: a mediator of the metabolic syndrome. Lessons from animal models. Arterioscler Thromb Vasc Biol 24: 644–649, 2004. [DOI] [PubMed] [Google Scholar]
- 64.Denechaud PD, Dentin R, Girard J, and Postic C. Role of ChREBP in hepatic steatosis and insulin resistance. FEBS Journal 582: 68–73, 2008. [DOI] [PubMed] [Google Scholar]
- 65.Dentin R, Girard J, and Postic C. Carbohydrate responsive element binding protein (ChREBP) and sterol regulatory element binding protein-1c (SREBP-1c): two key regulators of glucose metabolism and lipid synthesis in liver. Biochimie 87: 81–86, 2005. [DOI] [PubMed] [Google Scholar]
- 66.Ding Y, Wu Y, Zeng R, and Liao K. Proteomic profiling of lipid droplet-associated proteins in primary adipocytes of normal and obese mouse. Acta Biochimica et Biophysica Sinica 44: 394–406, 2012. [DOI] [PubMed] [Google Scholar]
- 67.Diraison F, Dusserre E, Vidal H, Sothier M, and Beylot M. Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. American journal of physiology Endocrinology and metabolism 282: E46–E51, 2002. [DOI] [PubMed] [Google Scholar]
- 68.Diraison F, Moulin P, and Beylot M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non-esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab 29: 478–485, 2003. [DOI] [PubMed] [Google Scholar]
- 69.DiRusso CC, Li H, Darwis D, Watkins PA, Berger J, and Black PN. Comparative biochemical studies of the murine fatty acid transport proteins (FATP) expressed in yeast. J Biol Chem 280: 16829–16837, 2005. [DOI] [PubMed] [Google Scholar]
- 70.Doege H, Baillie RA, Ortegon AM, Tsang B, Wu QW, Punreddy S, Hirsch D, Watson N, Gimeno RE, and Stahl A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid Homeostasis. Gastroenterology 130: 1245–1258, 2006. [DOI] [PubMed] [Google Scholar]
- 71.Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, and Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem 282: 3450–3457, 2007. [DOI] [PubMed] [Google Scholar]
- 72.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Investig 115: 1343–1351, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dowhan W Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem 66: 199–232, 1997. [DOI] [PubMed] [Google Scholar]
- 74.Eberhardt C, Gray PW, and Tjoelker LW. cDNA cloning, expression and chromosomal localization of two human lysophosphatidic acid acyltransferases In: Eicosanoids and Other Bioactive Lipids in Cancer, Inflammation, and Radiation Injury, 4 New York, NY: Springer US, 1999, p. 351–356. [DOI] [PubMed] [Google Scholar]
- 75.Eberhardt C, Gray PW, and Tjoelker LW. Human lysophosphatidic acid acyltransferase - cDNA cloning, expression, and localization to chromosome 9q34.3. J Biol Chem 272: 20299–20305, 1997. [DOI] [PubMed] [Google Scholar]
- 76.Eberlé D, Hegarty B, Bossard P, Ferré P, and Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86: 839–848, 2004. [DOI] [PubMed] [Google Scholar]
- 77.Ehehalt R, Fullekrug J, Pohl J, Ring A, Herrmann T, and Stremmel W. Translocation of long chain fatty acids across the plasma membrane - lipid rafts and fatty acid transport proteins. Mol Cell Biochem 284: 135–140, 2006. [DOI] [PubMed] [Google Scholar]
- 78.Eisinger K, Liebisch G, Schmitz G, Aslanidis C, Krautbauer S, and Buechler C. Lipidomic analysis of serum from high fat diet induced obese mice. Int J Mol Sci 15: 2991–3002, 2014. 10.3390/ijms15022991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ellis JM, Bowman CE, and Wolfgang MJ. Metabolic and tissue-specific regulation of acyl-CoA metabolism. PLos One 10: e0116587, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ersoy BA, Tarun A, D’Aquino K, Hancer NJ, Ukomadu C, White MF, Michel T, Manning BD, and Cohen DE. Phosphatidylcholine transfer protein interacts with thioesterase superfamily member 2 to attenuate insulin signaling. Science Signaling 6: ra64, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, and Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 134: 424–431, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fabbrini E, Sullivan S, and Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 51: 679–689, 2010. 10.1002/hep.23280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Faergeman NJ, and Knudsen J. Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem J 323: 1–12, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Faergeman NJ, Sigurskjold BW, Kragelund BB, Andersen KV, and Knudsen J. Thermodynamics of ligand binding to acyl-coenzyme a binding protein studied by titration calorimetry. Biochemistry 35: 14118–14126, 1996. [DOI] [PubMed] [Google Scholar]
- 85.Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, and Stahl A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. American Journal of Physiology Endocrinology and Metabolism 299: E384–E393, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fan CY, Pan J, Usuda N, Yeldandi AV, Rao MS, and Reddy JK. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem 273: 15639–15645, 1998. [DOI] [PubMed] [Google Scholar]
- 87.Fernández MA, Albor C, Ingelmo-Torres M, Nixon SJ, Ferguson C, Kurzchalia T, Tebar F, Enrich C, Parton RG, and Pol A. Caveolin-1 is essential for liver regeneration. Science 313: 1628–1132, 2006. [DOI] [PubMed] [Google Scholar]
- 88.Ferré P, and Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obesity & Metabolism 12: 83–92, 2010. [DOI] [PubMed] [Google Scholar]
- 89.Filhoulaud G, Guilmeau S, Dentin R, Girard J, and Postic C. Novel insights into ChREBP regulation and function. Trends Endocrinol Metab 24: 257–268, 2013. 10.1016/j.tem.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 90.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JCJ, and Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metabolism 4: 199–210, 2006. [DOI] [PubMed] [Google Scholar]
- 91.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, and Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473: 528–531, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fuchs M, Lammert F, Wang DQ, Paigen B, Carey MC, and Cohen DE. Sterol carrier protein 2 participates in hypersecretion of biliary cholesterol during gallstone formation in genetically gallstone-susceptible mice. Biochem J 336 ( Pt 1): 33–37, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fujii H, Ikura Y, Arimoto J, Sugioka K, Iezzoni JC, Park SH, Naruko T, Itabe H, Kawada N, Caldwell SH, and Ueda M. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. Journal of Atherosclerosis and Thrombosis 16: 893–901, 2009. [DOI] [PubMed] [Google Scholar]
- 94.Fujimoto Y, Itabe H, Sakai J, Makita M, Noda J, Mori M, Higashi Y, Kojima S, and Takano T. Identification of major proteins in the lipid droplet-enriched fraction isolated from the human hepatocyte cell line HuH7. Biochimica Et Biophysica Acta-Molecular Cell Research 1644: 47–59, 2004. [DOI] [PubMed] [Google Scholar]
- 95.Fukuda N, and Ontko JA. Interactions between fatty acid synthesis, oxidation, and esterification in the production of triglyceride-rich lipoproteins by the liver. J Lipid Res 25: 831–842, 1984. [PubMed] [Google Scholar]
- 96.Fukushima M, Enjoji M, Kohjima M, Sugimoto R, Ohta S, Kotoh K, Kuniyoshi M, Kobayashi K, Imamura M, Inoguchi T, Nakamuta M, and Nawata H. Adipose differentiation related protein induces lipid accumulation and lipid droplet formation in hepatic stellate cells. In vitro cellular and developmental biology Animal 41: 321–324, 2005. [DOI] [PubMed] [Google Scholar]
- 97.Gale SE, Frolov A, Han X, Bickel PE, Cao L, Bowcock A, Schaffer JE, and Ory DS. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J Biol Chem 281: 11082–11089, 2006. [DOI] [PubMed] [Google Scholar]
- 98.Ganji SH, Tavintharan S, Zhu DM, Xing YD, Kamanna VS, and Kashyap ML. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J Lipid Res 45: 1835–1845, 2004. [DOI] [PubMed] [Google Scholar]
- 99.Goh VJ, and Silver DL. The lipid droplet as a potential therapeutic target in NAFLD. Semin Liver Dis 33: 312–320, 2013. [DOI] [PubMed] [Google Scholar]
- 100.Gong J, Sun Z, and Li P. CIDE proteins and metabolic disorders. Curr Opin Lipidol 20: 121–126, 2009. [DOI] [PubMed] [Google Scholar]
- 101.Gordon DA, Wetterau JR, and Gregg RE. Microsomal triglyceride transfer protein: a protein complex required for the assembly of lipoprotein particles. Trends Cell Biol 5: 317–321, 1995. [DOI] [PubMed] [Google Scholar]
- 102.Gordon GB. Saturated free fatty acid toxicity. II. Lipid accumulation, ultrastructural alterations, and toxicity in mammalian cells in culture. . Exp Mol Pathol 27: 1977. [DOI] [PubMed] [Google Scholar]
- 103.Gossett RE, Frolov AA, Roths JB, Behnke WD, Kier AB, and Schroeder F. Acyl-CoA binding proteins: Multiplicity and function. Lipids 31: 895–918, 1996. [DOI] [PubMed] [Google Scholar]
- 104.Greco D, Kotronen A, Westerbacka J, Puig O, Arkkila P, Kiviluoto T, Laitinen S, Kolak M, Fisher RM, Hamsten A, Auvinen P, and Yki-Järvinen H. Gene expression in human NAFLD. American journal of physiology Gastrointestinal and liver physiology 294: G1281–G1287, 2008. [DOI] [PubMed] [Google Scholar]
- 105.Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, and Mashek DG. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Investig 121: 2102–2110, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grevengoed TJ, Klett EL, and Coleman RA. Acyl-CoA metabolism and partitioning. Annu Rev Nutr 34: 1–30, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gropler MC, Harris TE, Hall AM, Wolins NE, Gross RW, Han XL, Chen ZJ, and Finck BN. Lipin 2 Is a liver-enriched phosphatidate phosphohydrolase enzyme that is dynamically regulated by fasting and obesity in Mice. J Biol Chem 284: 6763–6772, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gross DA, Zhan C, and Silver DL. Direct binding of triglyceride to fat storage-inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. Proc Natl Acad Sci U S A 108: 19581–19586, 2011. 10.1073/pnas.1110817108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo F, Ma Y, Kadegowda AK, Betters JL, Xie P, Liu G, Liu X, Miao H, Ou J, Su X, Zheng Z, Xue B, Shi H, and Yu L. Deficiency of liver comparative gene identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res 54: 2109–2120, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hammond LE, Neschen S, Romanelli AJ, Cline GW, Ilkayeva OR, Shulman GI, Muoio DM, and Coleman RA. Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential in liver for the metabolism of excess acyl-CoAs. J Biol Chem 280: 25629–25636, 2005. 10.1074/jbc.M503181200 [DOI] [PubMed] [Google Scholar]
- 111.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, and Rao MS. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem 275: 28918–28928, 2000. [DOI] [PubMed] [Google Scholar]
- 112.Havel RJ. Postprandial hyperlipidemia and remnant lipoproteins. Curr Opin Lipidol 5: 102–109, 1994. [DOI] [PubMed] [Google Scholar]
- 113.Heacock AM, and Agranoff BW. CDP-diacylglycerol synthase from mammalian tissues. Biochim Biophys Acta 1348: 166–172, 1997. [DOI] [PubMed] [Google Scholar]
- 114.Hellerstein MK, Schwarz JM, and Neese RA. Regulation of hepatic de novo lipogenesis in humans. Annu Rev Nutr 16: 523–557, 1996. [DOI] [PubMed] [Google Scholar]
- 115.Higuchi N, Kato M, Shundo Y, Tajiri H, Tanaka M, Yamashita N, Kohjima M, Kotoh K, Nakamuta M, Takayanagi R, and Enjoji M. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol Res 38: 1122–1129, 2008. 10.1111/j.1872-034X.2008.00382.x [DOI] [PubMed] [Google Scholar]
- 116.Horton JD, Goldstein JL, and Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Investig 109: 1125–1131, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hostetler HA, McIntosh AL, Atshaves BP, Storey SM, Payne HR, Kier AB, and Schroeder F. L-FABP directly interacts with PPAR alpha in cultured primary hepatocytes. J Lipid Res 50: 1663–1675, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang H, Atshaves BP, Frolov A, Kier AB, and Schroeder F. Acyl-coenzyme A binding protein expression alters liver fatty acyl-coenzyme A metabolism. Biochemistry 44: 10282–10297, 2005. [DOI] [PubMed] [Google Scholar]
- 119.Huang H, Starodub O, McIntosh A, Kier AB, Schroeder F, and 9;277(32):29139–51. JBCA. Liver fatty acid-binding protein targets fatty acids to the nucleus. Real time confocal and multiphoton fluorescence imaging in living cells. J Biol Chem 277: 29139–29151, 2002. [DOI] [PubMed] [Google Scholar]
- 120.Hudgins LC, Hellerstein M, Seidman C, Neese R, Diakun J, and Hirsch J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J Clin Investig 97: 2081–2091, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hyslop PA, York DA, and Corina DL. Changes in the composition and fluidity of membranes in obese (ob/ob) mice: a study of hepatic microsomal NADPH-cytochrome P450 oxidoreductase activity. Int J Obes 6: 279–289, 1982. [PubMed] [Google Scholar]
- 122.Iqbal J, and Hussain MM. Intestinal lipid absorption. American Journal of Physiology Endocrinology and Metabolism 296: E1183–1194, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.James O, and Day C. Non-alcoholic steatohepatitis: another disease of affluence. Lancet 353: 1634–1636, 1999. [DOI] [PubMed] [Google Scholar]
- 124.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, and Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem 279: 48968–48975, 2004. [DOI] [PubMed] [Google Scholar]
- 125.Jensen-Urstad AP, and Semenkovich CF. Fatty acid synthase and liver triglyceride metabolism: housekeeper or messenger? Biochim Biophys Acta 1821: 747–753, 2012. 10.1016/j.bbalip.2011.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jo H, Choe SS, Shin KC, Jang H, Lee JH, Seong JK, Back SH, and Kim JB. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 57: 1366–1377, 2013. 10.1002/hep.26126 [DOI] [PubMed] [Google Scholar]
- 127.Jornayvaz FR, and Shulman GI. Diacylglycerol activation of protein kinase Cepsilon and hepatic insulin resistance. Cell Metab 15: 574–584, 2012. 10.1016/j.cmet.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kadereit B, Kumar P, Wang WJ, Miranda D, Snapp EL, Severina N, Torregroza I, Evans T, and Silver DL. Evolutionarily conserved gene family important for fat storage. Proc Natl Acad Sci U S A 105: 94–99, 2008. 10.1073/pnas.0708579105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, and Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Investig 119: 1201–1215, 2009. 10.1172/JCI37007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kang HW, Niepel MW, Han S, Kawano Y, and Cohen DE. Thioesterase superfamily member 2/acyl-CoA thioesterase 13 (Them2/Acot13) regulates hepatic lipid and glucose metabolism. FASEB J 26: 2209–2221, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kantartzis K, Machicao F, Machann J, Schick F, Fritsche A, Haring HU, and Stefan N. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clinical Science 116: 531–537, 2009. [DOI] [PubMed] [Google Scholar]
- 132.Kato M, Higuchi N, and Enjoji M. Reduced hepatic expression of adipose tissue triglyceride lipase and CGI-58 may contribute to the development of non-alcoholic fatty liver disease in patients with insulin resistance. Scand J Gastroenterol 43: 1018–1019, 2008. [DOI] [PubMed] [Google Scholar]
- 133.Kaushik S, and Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22: 407–417, 2012. 10.1016/j.tcb.2012.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaushik S, and Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17: 759–770, 2015. 10.1038/ncb3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kawano Y, and Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol 48: 434–441, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kawano Y, Nishiumi S, Saito M, Yano Y, Azuma T, and Yoshida M. Identification of lipid species linked to the progression of non-alcoholic fatty liver disease. Curr Drug Targets 16: 1293–1300, 2015. [DOI] [PubMed] [Google Scholar]
- 137.Kelder B, Boyce K, Kriete A, Clark R, Berryman DE, Nagatomi S, List EO, Braughler M, and Kopchick JJ. CIDE-A is expressed in liver of old mice and in type 2 diabetic mouse liver exhibiting steatosis. Comparative Hepatology 6: 4, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Khan SA, Wollaston-Hayden EE, Markowski TW, Higgins L, and Mashek DG. Quantitative analysis of the murine lipid droplet-associated proteome during diet-induced hepatic steatosis. J Lipid Res 56: 2260–2272, 2015. 10.1194/jlr.M056812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim KH. Regulation of mammalian acetyl-coenzyme A carboxylase. Annu Rev Nutr 17: 77–99, 1997. [DOI] [PubMed] [Google Scholar]
- 140.Kimmel AR, and Sztalryd C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr Opin Lipidol 25: 110–117, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kimmel AR, and Sztalryd C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu Rev Nutr 36: 471–509, 2016. [DOI] [PubMed] [Google Scholar]
- 142.Kirkby B, Roman N, Kobe B, Kellie S, and Forwood JK. Functional and structural properties of mammalian acyl-coenzyme A thioesterases. Prog Lipid Res 49: 366–377, 2010. [DOI] [PubMed] [Google Scholar]
- 143.Knight BL, Hebbachi A, Hauton D, Brown AM, Wiggins D, Patel DD, and Gibbons GF. A role for PPARalpha in the control of SREBP activity and lipid synthesis in the liver. Biochem J 389: 413–421, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Knudsen J Acyl-Coa-Binding Protein (ACBP) and Its Relation to Fatty Acid-Binding Protein (FABP) - an Overview. Mol Cell Biochem 98: 217–223, 1990. [DOI] [PubMed] [Google Scholar]
- 145.Koga H, Kaushik S, and Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J 24: 3052–3065, 2010. 10.1096/fj.09-144519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kohjima M, Enjoji M, Higuchi N, Kato M, Kotoh K, Yoshimo T, Fujino T, Yada M, Yada R, Harada N, Takayanagi R, and Nakamuta M. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 20: 351–358, 2007. [PubMed] [Google Scholar]
- 147.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, Vogt TF, Hobbs HH, and Cohen JC. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 46: 352–356, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Krammer J, Digel M, Ehehalt F, Stremmel W, Füllekrug J, and Ehehalt R. Overexpression of CD36 and acyl-CoA synthetases FATP2, FATP4 and ACSL1 increases fatty acid uptake in human hepatoma cells. International Journal of Medical Sciences 8: 599–614, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kreuz S, Schoelch C, Thomas L, Rist W, Rippmann JF, and Neubauer H. Acetyl-CoA carboxylases 1 and 2 show distinct expression patterns in rats and humans and alterations in obesity and diabetes. Diabetes Metab Res Rev 25: 577–586, 2009. 10.1002/dmrr.997 [DOI] [PubMed] [Google Scholar]
- 150.Kume K, and Shimizu T. cDNA cloning and expression of murine 1-acyl-sn-glycerol-3-phosphate acyltransferase. Biochem Biophys Res Commun 237: 663–666, 1997. [DOI] [PubMed] [Google Scholar]
- 151.Lafontan M, and Langin D. Lipolysis and lipid mobilization in human adipose tissue. Prog Lipid Res 48: 275–297, 2009. [DOI] [PubMed] [Google Scholar]
- 152.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, and Zechner R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metabolism 3: 309–319, 2006. [DOI] [PubMed] [Google Scholar]
- 153.Lavoie JM, and Gauthier MS. Regulation of fat metabolism in the liver: link to non-alcoholic hepatic steatosis and impact of physical exercise. Cellular and Molecular Life Sciences 63: 1393–1409, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee JH, Wada T, Febbraio M, He J, Matsubara T, Lee MJ, Gonzalez FJ, and Xie W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139: 653–663, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee K, Kerner J, and Hoppel CL. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J Biol Chem 286: 25655–25662, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, and Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15: 3012–3022, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lewin TM, Granger DA, Kim JH, and Coleman RA. Regulation of mitochondrial sn-glycerol-3-phosphate acyltransferase activity: Response to feeding status is unique in various rat tissues and is discordant with protein expression. Arch Biochem Biophys 396: 119–127, 2001. [DOI] [PubMed] [Google Scholar]
- 158.Lewin TM, Wang SL, Nagle CA, Van Horn CG, and Coleman RA. Mitochondrial glycerol-3-phosphate acyltransferase-1 directs the metabolic fate of exogenous fatty acids in hepatocytes. Am J Physiol Endocrinol Metab 288: E835–E844, 2005. [DOI] [PubMed] [Google Scholar]
- 159.Lewis GF, Carpentier A, Adeli K, and Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev 23: 201–229, 2002. [DOI] [PubMed] [Google Scholar]
- 160.Li JZ, Ye J, Xue B, Qi J, Zhang J, Zhou Z, Li Q, Wen Z, and Li P. Cideb regulates diet-induced obesity, liver steatosis, and insulin sensitivity by controlling lipogenesis and fatty acid oxidation. Diabetes 56: 2523–2532, 2007. [DOI] [PubMed] [Google Scholar]
- 161.Li LO, Ellis JM, Paich HA, Wang SL, Gong N, Altshuller G, Thresher RJ, Koves TR, Watkins SM, Muoio DM, Cline GW, Shulman GI, and Coleman RA. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J Biol Chem 284: 27816–27826, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, and Lin JD. Genome-wide coactivation analysis of PGC-1α identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metabolism 8: 105–117, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li X, Grundy SM, and Patel SB. Obesity in db and ob animals leads to impaired hepatic very low density lipoprotein secretion and differential secretion of apolipoprotein B-48 and B-100. J Lipid Res 38: 1277–1288, 1997. [PubMed] [Google Scholar]
- 164.Liang JJ, Oelkers P, Guo CY, Chu PC, Dixon JL, Ginsberg HN, and Sturley SL. Overexpression of human diacylglycerol acyltransferase 1, Acyl-CoA : cholesterol acyltransferase 1, or Acyl-CoA : cholesterol acyltransferase 2 stimulates secretion of apolipoprotein B-containing lipoproteins in McA-RH7777 cells. J Biol Chem 279: 44938–44944, 2004. [DOI] [PubMed] [Google Scholar]
- 165.Linden D, William-Olsson L, Rhedin M, Asztely AK, Clapham JC, and Schreyer S. Overexpression of mitochondrial GPAT in rat hepatocytes leads to decreased fatty acid oxidation and increased glycerolipid biosynthesis. J Lipid Res 45: 1279–1288, 2004. [DOI] [PubMed] [Google Scholar]
- 166.Lowe ME. The triglyceride lipases of the pancreas. J Lipid Res 43: 2007–2016, 2002. [DOI] [PubMed] [Google Scholar]
- 167.Madrigal-Matute J, and Cuervo AM. Regulation of Liver Metabolism by Autophagy. Gastroenterology 150: 328–339, 2016. 10.1053/j.gastro.2015.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Magkos F, Su X, Bradley D, Fabbrini E, Conte C, Eagon JC, Varela JE, Brunt EM, Patterson BW, and Klein S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142: 1444–1446 e1442, 2012. 10.1053/j.gastro.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Manmontri B, Sariahmetoglu M, Donkor J, Khalil MB, Sundaram M, Yao Z, Reue K, Lehner R, and Brindley DN. Glucocorticoids and cyclic AMP selectively increase hepatic lipin-1 expression, and insulin acts antagonistically. J Lipid Res 49: 1056–1067, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, Kordari P, Chirala SS, Heird WC, and Wakil SJ. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci U S A 103: 8552–8557, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, and Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 150: 2001. [DOI] [PubMed] [Google Scholar]
- 172.Martin GG, Huang H, Atshaves BP, Binas B, and Schroeder F. Ablation of the liver fatty acid binding protein gene decreases fatty acyl CoA binding capacity and alters fatty acyl CoA pool distribution in mouse liver. Biochemistry 42: 11520–11532, 2003. [DOI] [PubMed] [Google Scholar]
- 173.Martin-Sanz P, Hopewell R, and Brindley DN. Long-chain fatty acids and their acyl-CoA esters cause the translocation of phosphatidate phosphohydrolase from the cytosolic to the microsomal fraction of rat liver. FEBS Lett 175: 284–288, 1984. [DOI] [PubMed] [Google Scholar]
- 174.Mashek DG, McKenzie MA, Van Horn CG, and Coleman RA. Rat long chain acyl-CoA synthetase 5 increases fatty acid uptake and partitioning to cellular triacylglycerol in McArdle-RH7777 cells. J Biol Chem 281: 945–950, 2006. [DOI] [PubMed] [Google Scholar]
- 175.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, Brewer BJ, Reitman ML, and Gonzalez FJ. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Investig 111: 737–747, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, and Gonzalez FJ. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metabolism 7: 302–311, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McArthur MJ, Atshaves BP, Frolov A, Foxworth WD, Kier AB, and Schroeder F. Cellular uptake and intracellular trafficking of long chain fatty acids. J Lipid Res 40: 1371–1383, 1999. [PubMed] [Google Scholar]
- 178.McFie PJ, Banman SL, Kary S, and Stone SJ. Murine diacylglycerol acyltransferase-2 (DGAT2) can catalyse triacylglycerol synthesis and promote lipid droplet formation independent of its localization to the endoplasmic reticulum. J Biol Chem 286: 28235–28246, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Meegalla RL, Billheimer JT, and Cheng D. Concerted elevation of acyl-coenzyme A : diacylglycerol acyltransferase (DGAT) activity through independent stimulation of mRNA expression of DGAT1 and DGAT2 by carbohydrate and insulin. Biochem Biophys Res Commun 298: 317–323, 2002. [DOI] [PubMed] [Google Scholar]
- 180.Memon RA, Fuller J, Moser AH, Smith PJ, Grunfeld C, and Feingold KR. Regulation of putative fatty acid transporters and acyl-CoA synthetase in liver and adipose tissue in ob/ob mice. Diabetes 48: 121–127, 1999. [DOI] [PubMed] [Google Scholar]
- 181.Miquilena-Colina ME, Lima-Cabello E, Sánchez-Campos S, García-Mediavilla MV, Fernández-Bermejo M, Lozano-Rodríguez T, Vargas-Castrillón J, Buqué X, Ochoa B, Aspichueta P, González-Gallego J, and García-Monzón C. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut 60: 1394–1402, 2011. [DOI] [PubMed] [Google Scholar]
- 182.Moffat C, Bhatia L, Nguyen T, Lynch P, Wang M, Wang D, Ilkayeva OR, Han X, Hirschey MD, Claypool SM, and Seifert EL. Acyl-CoA thioesterase-2 facilitates mitochondrial fatty acid oxidation in the liver. J Lipid Res 55: 2458–2470, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, Stevens RD, Bain JR, Newgard CB, Farese RV, Hevener AL, and Farese RV. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metabolism 6: 69–78, 2007. [DOI] [PubMed] [Google Scholar]
- 184.Montagner A, Polizzi A, Fouche E, Ducheix S, Lippi Y, Lasserre F, Barquissau V, Regnier M, Lukowicz C, Benhamed F, Iroz A, Bertrand-Michel J, Al Saati T, Cano P, Mselli-Lakhal L, Mithieux G, Rajas F, Lagarrigue S, Pineau T, Loiseau N, Postic C, Langin D, Wahli W, and Guillou H. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65: 1202–1214, 2016. 10.1136/gutjnl-2015-310798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Motomura W, Inoue M, Ohtake T, Takahashi N, Nagamine M, Tanno S, Kohgo Y, and T. O. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun 340: 1111–1118, 2006. [DOI] [PubMed] [Google Scholar]
- 186.Muoio DM, Seefeld K, Witters LA, and Coleman RA. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem J 338: 783–791, 1999. [PMC free article] [PubMed] [Google Scholar]
- 187.Musso G, Gambino R, and Cassader M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Prog Lipid Res 48: 1–26, 2009 [DOI] [PubMed] [Google Scholar]
- 188.Nagle CA, An J, Shiota M, Torres TP, Cline GW, Liu ZX, Wang S, Catlin RL, Shulman GI, Newgard CB, and Coleman RA. Hepatic overexpression of glycerol-sn-3-phosphate acyltransferase-1 causes insulin resistance. FASEB J 21: A699–A699, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nagle CA, Vergnes L, Dejong H, Wang SL, Lewin TM, Reue K, and Coleman RA. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6(−/−) mice. J Lipid Res 49: 823–831, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Najt CP, Senthivinayagam S, Aljazi MB, Fader KA, Olenic SD, Brock JR, Lydic TA, Jones AD, and Atshaves BP. Liver-specific loss of Perilipin 2 alleviates diet-induced hepatic steatosis, inflammation, and fibrosis. Am J Physiol Gastrointest Liver Physiol 310: G726–738, 2016. 10.1152/ajpgi.00436.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Neschen S, Morino K, Hammond LE, Zhang DY, Liu ZX, Romanelli AJ, Cline GW, Pongratz RL, Zhang XM, Choi CS, Coleman RA, and Shulman GI. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA : glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metabolism 2: 55–65, 2005. [DOI] [PubMed] [Google Scholar]
- 192.Newberry EP, Xie Y, Kennedy S, Han X, Buhman KK, Luo J, Gross RW, and Davidson NO. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J Biol Chem 278: 51664–51672, 2003. [DOI] [PubMed] [Google Scholar]
- 193.Newberry EP, Xie Y, Kennedy SM, Luo J, and Davidson NO. Protection against Western diet-induced obesity and hepatic steatosis in liver fatty acid-binding protein knockout mice. Hepatology 44: 1191–1205, 2006. [DOI] [PubMed] [Google Scholar]
- 194.Ohsaki Y, Cheng J, Suzuki M, Fujita A, and Fujimoto T. Lipid droplets are arrested in the ER membrane by tight binding of lipidated apolipoprotein B-100. J Cell Sci 121: 2415–2422, 2008. [DOI] [PubMed] [Google Scholar]
- 195.Oishi K, Amagai N, Shirai H, Kadota K, Ohkura N, and Ishida N. Genome-wide expression analysis reveals 100 adrenal gland-dependent circadian genes in the mouse liver. DNA Research 12: 191–202, 2005. [DOI] [PubMed] [Google Scholar]
- 196.Okada K, LeClair KB, Zhang Y, Li Y, Ozdemir C, Krisko TI, Hagen SJ, Betensky RA, Banks AS, and Cohen DE. Thioesterase superfamily member 1 suppresses cold thermogenesis by limiting the oxidation of lipid droplet-derived fatty acids in brown adipose tissue. Mol Metab 5: 340–351, 2016. 10.1016/j.molmet.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Okumura T Role of lipid droplet proteins in liver steatosis. J Physiol Biochem 67: 629–636, 2011. [DOI] [PubMed] [Google Scholar]
- 198.Ong KT, Mashek MT, Bu SY, Greenberg AS, and Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 53: 116–126, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ota T, Gayet C, and Ginsberg HN. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J Clin Investig 118: 316–332, 2008. 10.1172/JCI32752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Owen MR, Corstorphine CC, and Zammit VA. Overt and latent activities of diacylglycerol acyltransferase in rat liver microsomes: Possible roles in very-low-density lipoprotein triacylglycerol secretion. Biochem J 323: 17–21, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Park C, and Cuervo AM. Selective autophagy: talking with the UPS. Cell Biochem Biophys 67: 3–13, 2013. 10.1007/s12013-013-9623-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pessayre B, and Fromenty B. NASH: a mitochondrial disease. J Hepatol 42: 928–940, 2005. [DOI] [PubMed] [Google Scholar]
- 203.Pohl J, Ring A, and Stremmel W. Uptake of long-chain fatty acids in HepG2 cells involves caveolae: analysis of a novel pathway. J Lipid Res 43: 1390–1399, 2002. [DOI] [PubMed] [Google Scholar]
- 204.Pol A, Luetterforst R, Lindsay M, Heino S, Ikonen E, and Parton RG. A caveolin dominant negative mutant associates with lipid bodies and induces intracellular cholesterol imbalance. J Cell Biol 152: 1057–1070, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Postic C, Dentin R, Denechaud PD, and Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr 27: 179–192, 2007. 10.1146/annurev.nutr.27.061406.093618 [DOI] [PubMed] [Google Scholar]
- 206.Rao MS, and Reddy JK. Peroxisomal β-oxidation and steatohepatitis. Semin Liver Dis 21: 43–55, 2001. [DOI] [PubMed] [Google Scholar]
- 207.Rasmussen JT, Faergeman NJ, Kristiansen K, and Knudsen J. Acyl-CoA-binding protein (ACBP) can mediate intermembrane acyl-CoA transport and donate acyl-CoA for b-oxidation and glycerolipid synthesis. Biochem J 299: 165–170, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, Blaner WS, Goldberg IJ, Schwabe RF, Chua SC Jr., and Huang LS. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem 283: 13087–13099, 2008. 10.1074/jbc.M800533200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, and Cuervo AM. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A 109: E705–714, 2012. 10.1073/pnas.1113036109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, and Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40: 1461–1465, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rosendal J, Ertbjerg P, and Knudsen J. Characterization of ligand binding to acyl-CoA-binding protein. Biochem J 290: 321–326, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ruhanen H, Perttilä J, Hölttä-Vuori M, Zhou Y, Yki-Järvinen H, Ikonen E, Käkelä R, and Olkkonen VM. PNPLA3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res 55: 739–746, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Saltiel AR, and Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799–806, 2001. [DOI] [PubMed] [Google Scholar]
- 214.Samuel VT, and Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126: 12–22, 2016. 10.1172/JCI77812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, and Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120: 1183–1192, 2001. [DOI] [PubMed] [Google Scholar]
- 216.Sanyal AJ, and Pacana T. A Lipidomic Readout of Disease Progression in A Diet-Induced Mouse Model of Nonalcoholic Fatty Liver Disease. Trans Am Clin Climatol Assoc 126: 271–288, 2015. [PMC free article] [PubMed] [Google Scholar]
- 217.Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, Zhang XM, Cline GW, Yu XX, Geisler JG, Bhanot S, Monia BP, and Shulman GI. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Investig 116: 817–824, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Schadinger SE, Bucher NL, Schreiber BM, and Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. American journal of physiology Endocrinology and metabolism 288: E1195–E1205, 2005. [DOI] [PubMed] [Google Scholar]
- 219.Schneider JL, Suh Y, and Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20: 417–432, 2014. 10.1016/j.cmet.2014.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Schoonjans K, Watanabe M, Suzuki H, Mahfoudi A, Krey G, Wahli W, Grimaldi P, Staels B, Yamamoto T, and Auwerx J. Induction of the acyl-Coenzyme-A synthetase gene by fibrates and fatty-acids is mediated by a peroxisome proliferator response element in the C-promoter. J Biol Chem 270: 19269–19276, 1995. [DOI] [PubMed] [Google Scholar]
- 221.Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, and McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61: 1896–1907, 2015. 10.1002/hep.27667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Schroeder F, Petrescu AD, Huang H, Atshaves BP, McIntosh AL, Martin GG, Hostetler HA, Vespa A, Landrock D, Landrock KK, Payne HR, and Kier AB. Role of fatty acid binding proteins and long chain fatty acids in modulating nuclear receptors and gene transcription. Lipids 43: 1–17, 2008. [DOI] [PubMed] [Google Scholar]
- 223.Schwarz JM, Linfoot P, Dare D, and Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr 77: 43–50, 2003. [DOI] [PubMed] [Google Scholar]
- 224.Schwarz JM, Neese RA, Turner S, Dare D, and Hellerstein MK. Short-term alterations in carbohydrate energy intake in humans. Striking effects on hepatic glucose production, de novo lipogenesis, lipolysis, and whole-body fuel selection. J Clin Investig 96: 2735–2743, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Seedorf U, Raabe M, Ellinghaus P, Kannenberg F, Fobker M, Engel T, Denis S, Wouters F, Wirtz KWA, Wanders RJ, Maeda N, and Assmann G. Defective peroxisomal catabolism of branched fatty acyl coenzyme A in mice lacking the sterol carrier protein-2/sterol carrier protein-x gene function. Genes Dev 12: 1189–1201, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Seessle J, Liebisch G, Schmitz G, Stremmel W, and Chamulitrat W. Palmitate activation by fatty acid transport protein 4 as a model system for hepatocellular apoptosis and steatosis. Biochim Biophys Acta 1851: 549–565, 2015. 10.1016/j.bbalip.2015.01.004 [DOI] [PubMed] [Google Scholar]
- 227.Semenkovich CF. Regulation of fatty acid synthase (FAS). Prog Lipid Res 36: 43–53, 1997. [DOI] [PubMed] [Google Scholar]
- 228.Shen LL, Liu H, Peng J, Gan L, Lu L, Zhang Q, Li L, He F, and Jiang Y. Effects of farnesoid X receptor on the expression of the fatty acid synthetase and hepatic lipase. Mol Biol Rep 38: 553–559, 2011. [DOI] [PubMed] [Google Scholar]
- 229.Shen X, Zhang K, and Kaufman RJ. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat 28: 79–92, 2004. 10.1016/j.jchemneu.2004.02.006 [DOI] [PubMed] [Google Scholar]
- 230.Shin DH, Paulauskis JD, Moustaïd N, and Sul HS. Transcriptional regulation of p90 with sequence homology to Escherichia coli glycerol-3-phosphate acyltransferase. J Biol Chem 266: 23834–23839, 1991. [PubMed] [Google Scholar]
- 231.Shindou H, Hishikawa D, Harayama T, Yuki K, and Shimizu T. Recent progress on acyl CoA: lysophospholipid acyltransferase research. J Lipid Res 50: S46–S51, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Simard JR, Kamp F, and Hamilton JA. Measuring the adsorption of Fatty acids to phospholipid vesicles by multiple fluorescence probes. Biophys J 94: 4493–4503, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ. Autophagy regulates lipid metabolism. Nature 458: 1131–1135, 2009. 10.1038/nature07976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Siniossoglou S Phospholipid metabolism and nuclear function: roles of the lipin family of phosphatidic acid phosphatases. Biochim Biophys Acta 1831: 575–581, 2013. [DOI] [PubMed] [Google Scholar]
- 235.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, and Hobbs HH. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 61: 108–118, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Smagris E, Gilyard S, BasuRay S, Cohen JC, and Hobbs HH. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem 291: 10659–10676, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Smathers RL, and Petersen DR. The human fatty acid-binding protein family: evolutionary divergences and functions. Human Genomics 5: 170–191, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, and Farese RV. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat Genet 25: 87–90, 2000. [DOI] [PubMed] [Google Scholar]
- 239.Stahl A, Gimeno RE, Tartaglia LA, and Lodish HF. Fatty acid transport proteins: a current view of a growing family. Trends Endocrinol Metab 12: 266–273, 2001. [DOI] [PubMed] [Google Scholar]
- 240.Stamps AC, Elmore MA, Hill ME, Kelly K, Makda AA, and Finnen MJ. A human cDNA sequence with homology to non-mammalian lysophosphatidic acid acyltransferases. Biochem J 326: 455–461, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Stolz A, Ernst A, and Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501, 2014. 10.1038/ncb2979 [DOI] [PubMed] [Google Scholar]
- 242.Stone SJ, Levin MC, Zhou P, Han J, Walther TC, and Farese RVJ. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J Biol Chem 284: 5352–5361, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Stone SJ, Myers HM, Watkins SM, Brown BE, Feingold KR, Elias PM, and Farese RV. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J Biol Chem 279: 11767–11776, 2004. [DOI] [PubMed] [Google Scholar]
- 244.Strable MS, and Ntambi JM. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol 45: 199–214, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Straub BK, Stoeffel P, Heid H, Zimbelmann R, and Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology 47: 1936–1946, 2008. [DOI] [PubMed] [Google Scholar]
- 246.Stremmel W, Diede HE, Rodilla-Sala E, Vyska K, Schrader M, Fitscher B, and Passarella S. The membrane fatty acid-binding protein is not identical to mitochondrial glutamic oxaloacetic transaminase (mGOT). Mol Cell Biochem 98: 191–199, 1990. [DOI] [PubMed] [Google Scholar]
- 247.Stremmel W, Pohl J, Ring A, and Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids 36: 981–989, 2001. [DOI] [PubMed] [Google Scholar]
- 248.Stremmel W, Staffer S, Wannhoff A, Pathil A, and Chamulitrat W. Plasma membrane phospholipase A2 controls hepatocellular fatty acid uptake and is responsive to pharmacological modulation: implications for nonalcoholic steatohepatitis. FASEB J 28: 159–170, 2014. [DOI] [PubMed] [Google Scholar]
- 249.Stremmel W, Strohmeyer G, Borchard F, Kochwa S, and Berk PD. Isolation and partial characterization of a fatty acid binding protein in rat liver plasma membranes. Proc Natl Acad Sci U S A 82: 4–8, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Su W, Wang Y, Jia X, Wu W, Li L, Tian X, Li S, Wang C, Xu H, Cao J, Han Q, Xu S, Chen Y, Zhong Y, Zhang X, Liu P, Gustafsson JA, and Guan Y. Comparative proteomic study reveals 17beta-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 111: 11437–11442, 2014. 10.1073/pnas.1410741111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Subramanian S, and Chait A. Hypertriglyceridemia secondary to obesity and diabetes. Biochim Biophys Acta 1821: 819–825, 2012. [DOI] [PubMed] [Google Scholar]
- 252.Takeuchi K, and Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab 296: E1195–E1209, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Thompson BR, Lobo S, and Bernlohr DA. Fatty acid flux in adipocytes: The in’s and out’s of fat cell lipid trafficking. Mol Cell Endocrinol 318: 24–33, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Thumser AE, and Wilton DC. The binding of cholesterol and bile salts to recombinant rat liver fatty acid-binding protein. Biochem J 320 ( Pt 3): 729–733, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Thumser AE, and Wilton DC. The binding of natural and fluorescent lysophospholipids to wild-type and mutant rat liver fatty acid-binding protein and albumin. Biochem J 307 ( Pt 1): 305–311, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Tillander VE, Alexson SE, and Cohen DE. Deactivating fatty acids: acyl-CoA thioesterase-mediated control of lipid metabolism. Trends Endocrinol Metab in press: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Tiwari S, Siddiqi S, and Siddiqi SA. CideB protein is required for the biogenesis of very low density lipoprotein (VLDL) transport vesicle. J Biol Chem 288: 5157–5165, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Tong FM, Black PN, Coleman RA, and DiRusso CC. Fatty acid transport by vectorial acylation in mammals: Roles played by different isoforms of rat long-chain acyl-CoA synthetases. Arch Biochem Biophys 447: 46–52, 2006. [DOI] [PubMed] [Google Scholar]
- 259.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, and Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93: 241–252, 1998. [DOI] [PubMed] [Google Scholar]
- 260.Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, Xu E, Hammerschmidt P, Bronneke HS, Trifunovic A, LoSasso G, Wunderlich FT, Kornfeld JW, Bluher M, Kronke M, and Bruning JC. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20: 678–686, 2014. 10.1016/j.cmet.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 261.Vasconcellos R, Alvarenga EC, Parreira RC, Lima SS, and Resende RR. Exploring the cell signalling in hepatocyte differentiation. Cell Signal 28: 1773–1788, 2016. 10.1016/j.cellsig.2016.08.011 [DOI] [PubMed] [Google Scholar]
- 262.Vergnes L, Beigneux AP, Davis R, Watkins SM, Young SG, and Reue K. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J Lipid Res 47: 745–754, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Visser ME, Kastelein JJ, and Stroes ES. Apolipoprotein B synthesis inhibition: results from clinical trials. Curr Opin Lipidol 21: 319–323, 2010. 10.1097/MOL.0b013e32833af4c1 [DOI] [PubMed] [Google Scholar]
- 264.Viswakarma N, Yu S, Naik S, Kashireddy P, Matsumoto K, Sarkar J, Surapureddi S, Jia Y, Rao MS, and Reddy JK. Transcriptional regulation of Cidea, mitochondrial cell death-inducing DNA fragmentation factor alpha-like effector A, in mouse liver by peroxisome proliferator-activated receptor alpha and gamma. J Biol Chem 282: 18613–18624, 2007. [DOI] [PubMed] [Google Scholar]
- 265.Volmer R, van der Ploeg K, and Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A 110: 4628–4633, 2013. 10.1073/pnas.1217611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Wakil SJ, and Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res 50: S138–S143, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Walther TC, and Farese RVJ. Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 81: 687–714, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wang C, Zhao Y, Gao X, Li L, Yuan Y, Liu F, Zhang L, Wu J, Hu P, Zhang X, Gu Y, Xu Y, Wang Z, Li Z, Zhang H, and Ye J. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 61: 870–882, 2015. [DOI] [PubMed] [Google Scholar]
- 269.Wang H, Bell M, Sreenivasan U, Hu H, Liu J, Dalen K, Londos C, Yamaguchi T, Rizzo MA, Coleman R, Gong D, Brasaemle D, and Sztalryd C. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem 286: 15707–15715, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Wang J, Bie J, and Ghosh S. Intracellular cholesterol transport proteins enhance hydrolysis of HDL-delivered cholesteryl esters and facilitate preferential elimination of resulting cholesterol into bile. J Lipid Res In press: 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wang S, Lee DP, Gong N, Schwerbrock NMJ, Mashek DG, Gonzalez-Baro MR, Stapleton C, Li LO, Lewin TM, and Coleman RA. Cloning and functional characterization of a novel mitochondrial N-ethylmaleimide-sensitive glycerol-3-phosphate acyltransferase (GPAT2). Arch Biochem Biophys 465: 347–358, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Waterman IJ, and Zammit VA. Activities of overt and latent diacylglycerol acyltransferases (DGATs I and II) in liver microsomes of ob/ob mice. Int J Obes 26: 742–743, 2002. [DOI] [PubMed] [Google Scholar]
- 273.Watkins PA. Very-long-chain acyl-CoA synthetases. J Biol Chem 283: 1773–1777, 2008. 10.1074/jbc.R700037200 [DOI] [PubMed] [Google Scholar]
- 274.Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 25: 161–168, 2014. 10.1097/MOL.0000000000000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wendel AA, Cooper DE, Ilkayeva OR, Muoio DM, and Coleman RA. Glycerol-3-phosphate acyltransferase (GPAT)-1, but not GPAT4, incorporates newly synthesized fatty acids into triacylglycerol and diminishes fatty acid oxidation. J Biol Chem 288: 27299–27306, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wendel AA, Lewin TM, and Coleman RA. Glycerol-3-phosphate acyltransferases: Rate limiting enzymes of triacylglycerol biosynthesis. Biochimica Et Biophysica Acta-Molecular and Cell Biology of Lipids 1791: 501–506, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.West J, Tompkins CK, Balantac N, Nudelman E, Meengs B, White T, Bursten S, Coleman J, Kumar A, Singer JW, and Leung DW. Cloning and expression of two human lysophosphatidic acid acyltransferase cDNAs that enhance cytokine-induced signaling responses in cells. DNA Cell Biol 16: 691–701, 1997. [DOI] [PubMed] [Google Scholar]
- 278.Wilfling F, Haas JT, Walther TC, and Farese RVJ. Lipid droplet biogenesis. Curr Opin Cell Biol 29: 39–45, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, and Weiss EJ. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 157: 570–585, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wu JW, Wang SP, Alvarez F, Casavant S, Gauthier N, Abed L, Soni KG, Yang G, and Mitchell GA. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 54: 122–132, 2011. [DOI] [PubMed] [Google Scholar]
- 281.Wurie HR, Buckett L, and Zammit VA. Diacylglycerol acyltransferase 2 acts upstream of diacylglycerol acyltransferase 1 and utilizes nascent diglycerides and de novo synthesized fatty acids in HepG2 cells. FEBS Journal 279: 3033–3047, 2012. [DOI] [PubMed] [Google Scholar]
- 282.Xu W, Wu L, Yu M, Chen FJ, Arshad M, Xia X, Ren H, Yu J, Xu L, Xu D, Li JZ, Li P, and Zhou L. Differential Roles of Cell Death-inducing DNA Fragmentation Factor-alpha-like Effector (CIDE) Proteins in Promoting Lipid Droplet Fusion and Growth in Subpopulations of Hepatocytes. J Biol Chem 291: 4282–4293, 2016. 10.1074/jbc.M115.701094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Yang YY, Pritchard PH, Bhuiyan J, Seccombe DW, and Moghadasian MH. Overexpression of Acyl-CoA binding protein and its effects on the flux of free fatty acids in McA-RH 7777 cells. Lipids 36: 595–600, 2001. [DOI] [PubMed] [Google Scholar]
- 284.Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, and Li P. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metabolism 9: 177–190, 2009. [DOI] [PubMed] [Google Scholar]
- 285.Yen CLE, Monetti M, Burri BJ, and Farese RV. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. J Lipid Res 46: 1502–1511, 2005. [DOI] [PubMed] [Google Scholar]
- 286.Yen CLE, Stone SJ, Koliwad S, Harris C, and Farese RV. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49: 2283–2301, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Yeon JE, Choi KM, Baik SH, Kim KO, Lim HJ, Park KH, Kim JY, Park JJ, Kim JS, Bak YT, Byun KS, and Lee CH. Reduced expression of peroxisome proliferator-activated receptor-alpha may have an important role in the development of non-alcoholic fatty liver disease. J Gastroenterol Hepatol 19: 799–804, 2004. [DOI] [PubMed] [Google Scholar]
- 288.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, and Reddy JK. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem 278: 498–505, 2003. [DOI] [PubMed] [Google Scholar]
- 289.Zammit VA. Hepatic triacylglycerol synthesis and secretion: DGAT2 as the link between glycaemia and triglyceridaemia. The Biochemical Journal 451: 1–12, 2013. [DOI] [PubMed] [Google Scholar]
- 290.Zhang L, Miyaki K, Nakayama T, and Muramatsu M. Cell death-inducing DNA fragmentation factor alpha-like effector A (CIDEA) gene V115F (G–>T) polymorphism is associated with phenotypes of metabolic syndrome in Japanese men. Metabolism 57: 502–505, 2008. [DOI] [PubMed] [Google Scholar]
- 291.Zhang L, and Wang HH. The essential functions of endoplasmic reticulum chaperones in hepatic lipid metabolism. Dig Liver Dis 48: 709–716, 2016. 10.1016/j.dld.2016.03.016 [DOI] [PubMed] [Google Scholar]
- 292.Zhang Y, Li Y, Niepel MW, Kawano Y, Han S, Liu S, Marsili A, Larsen PR, Lee CH, and Cohen DE. Targeted deletion of thioesterase superfamily member 1 promotes energy expenditure and protects against obesity and insulin resistance. Proc Natl Acad Sci U S A 109: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Zhou H, and Liu R. ER stress and hepatic lipid metabolism. Front Genet 5: 112, 2014. 10.3389/fgene.2014.00112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, Ren S, Evans RM, and Xie W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281: 15013–15020, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Zhou SL, Gordon RE, Bradbury M, Stump D, Kiang CL, and Berk PD. Ethanol up-regulates fatty acid uptake and plasma membrane expression and export of mitochondrial aspartate aminotransferase in HepG2 cells. Hepatology 27: 1064–1074, 1998. [DOI] [PubMed] [Google Scholar]
- 296.Zhuravleva E, Gut H, Hynx D, Marcellin D, Bleck CK, Genoud C, Cron P, Keusch JJ, Dummler B, Esposti MD, and Hemmings BA. Acyl coenzyme A thioesterase Them5/Acot15 is involved in cardiolipin remodeling and fatty liver development. Mol Cell Biol 32: 2685–2697, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Zoltowska M, Ziv E, Delvin E, Lambert M, Seidman E, and Levy E. Both insulin resistance and diabetes in Psammomys obesus upregulate the hepatic machinery involved in intracellular VLDL assembly. Arterioscler Thromb Vasc Biol 24: 118–123, 2004. [DOI] [PubMed] [Google Scholar]