

Abstract
Background:
Traditional drug discovery approaches focus on a limited set of target molecules for treatment against specific indications/diseases. However, drug absorption, dispersion, metabolism, and excretion (ADME) involve interactions with multiple protein systems. Drugs approved for particular indication(s) may be repurposed as novel therapeutics for others. The severely declining rate of discovery and increasing costs of new drugs illustrate the limitations of the traditional reductionist paradigm in drug discovery.
Methods:
We developed the Computational Analysis of Novel Drug Opportunities (CANDO) platform based on a hypothesis that drugs function by interacting with multiple protein targets to create a molecular interaction signature that can be exploited for therapeutic repurposing and discovery. We compiled a library of compounds that are human ingestible with minimal side effects, followed by an ‘all-compounds’ vs ‘all-proteins’ fragment-based multitarget docking with dynamics screen to construct compound-proteome interaction matrices that were then analyzed to determine similarity of drug behavior. The proteomic signature similarity of drugs is then ranked to make putative drug predictions for all indications in a shotgun manner.
Results:
We have previously applied this platform with success in both retrospective benchmarking and prospective validation, and to understand the effect of druggable protein classes on repurposing accuracy. Here we use the CANDO platform to analyze and determine the contribution of multitargeting (polypharmacology) to drug repurposing benchmarking accuracy. Taken together with the previous work, our results indicate that a large number of protein structures with diverse fold space and a specific polypharmacological interactome is necessary for accurate drug predictions using our proteomic and evolutionary drug discovery and repurposing platform.
Conclusion:
These results have implications for future drug development and repurposing in the context of polypharmacology.
Keywords: Shotgun drug discovery, virtual screening, computational proteomics, polypharmacology, multitarget
INTRODUCTION
Traditional approaches to drug discovery focus on a limited set of interactions between individual protein targets and small molecule compounds. The goal generally is to target an essential protein responsible for pathogenesis so as to completely inhibit its function. Almost all current drugs have been developed by this approach. However, the number of novel drugs being discovered every year has been reduced to a handful. Currently less than 30 new drugs approved each year and most of them are analogues to other existing drugs or other patent workarounds (data obtained from <fda.gov>) and the estimated average total costs for developing a novel drug and bringing it to market can be up to $2.6 billion, according to the Tufts Center for the Study of Drug Development <csdd.tufts.edu>. Thus there exists severe limitations for novel drug development as it is typically associated with including long timeframes (10-15 years) and large investment outlays [1-4].
A solution is to repurpose/reposition existing drugs that are relatively benign in terms of side effects for new indications [3, 5-11]. We were one of the first groups to propose shotgun drug repurposing for malaria, using a computational multitarget docking with dynamics approach [9]; we have since validated our predictive models numerous times (see [3, 5-15] for a few examples). Drug repurposing can be made much more efficient and efficacious by considering variations (mutations) in proteins encoded by individual host genomes to identify several candidates for treatment of the same indication/disease paving the path towards exome-specific clinical trial groups, thereby enabling personalized/precision medicine. Systematic exploration of drug repurposing opportunities by sharing preclinical and clinical trials data is hindered by extensive competition within the pharmaceutical industry related to intellectual property issues. We utilize this repurposing paradigm along with a computational platform we have developed that evaluates relationships between compound-proteome interaction signatures to predict genome- and indication-specific drug regimens for particular individuals in a shotgun and holistic manner (i.e., against all indications simultaneously). To verify and improve the accuracy of our platform, we have been working with collaborators worldwide for preclinical and clinical validation. The experimental results obtained are integrated back into the modeling platform to iteratively improve putative drug prediction accuracy. Here, we use this platform to explore drug polypharmacology.
Overview of Drug Discovery
Small molecule drug discovery begins by assaying compounds for in vitro activity against diseases/indications of interest in a high-throughput manner with or without knowledge of molecular targets identified by biochemistry and molecular biology [10, 16, 17]. Compounds observed to be active in a phenotypic (protein or cell) assay (hits) are further tested in vivo, typically in animal models when available, while pharmacokinetic and pharmacodynamic properties are simultaneously measured. Compounds that are successful at this stage are considered leads and go through further biochemical and structural optimization. Once toxicity and efficacy against the disease of interest is established, the candidate molecule is considered for testing in human patients and formally designated as a drug candidate. The candidate drug is then subjected to preclinical and clinical studies [18] and if successful, undergoes commercial production and distribution [19].
Druggable Protein Classes
Many drugs have no known mechanism of action. Even when they are efficacious, the reasons for side effects are not well under-stood and there is no drug without any side effects whatsoever at the maximal clinically approved doses in all patients. Drug discovery focuses on the elucidation of mechanistic interactions between small molecules and particular “druggable” classes of proteins. G-protein-coupled receptors (GPCRs) and protein kinases are among the most popular class of targets for a wide range of indications/diseases [20-25].
GPCRs constitute a large superfamily of transmembrane protein receptors unique to eukaryotes and are the most successful targets in modern medicine. Approximately 36% of marketed pharmaceuticals target GPCRs [20]. However, the endogenous ligands of 100-200 GPCRs remain unidentified. These so-called orphan GPCRs with unidentified natural functions are an important source of drug targets [21].
Protein kinases constitute nearly 2% of all human genes, and play a critical role in cellular signaling pathways [22]. Perturbations to these pathways have been linked to cancer, diabetes, and other inflammatory diseases [23]. These perturbations are often due to mutation, translocation, or upregulation events that cause one or more kinases to become highly active and not respond normally to regulatory signals [24]. As a result, much of the effort in developing treatments for these diseases has focused on shutting down these aberrant kinases with targeted inhibitors [25].
Tyrosine kinase inhibitors (TKIs) in particular have shown promise as powerful therapeutics in the treatment of human cancers. Arguably, the biggest success story has been the discovery of imatinib for the treatment of chronic myeloid leukemia (CML), a white blood cell cancer. CML results from a fusion event that produces an active form of the ABL kinase in 95% of CML patients [26]. Imatinib has been highly effective with minimal toxicity in treating patients with CML. It is thought that these properties of imatinib is due to its high degree of selectivity for ABL; imatinib does not bind to the closely related SRC kinase, even though residues at the ABL-imatinib binding interface are nearly identical [26, 27]. Unfortunately, even when perceived highly selective TKIs like imatinib are available, the emergence of resistance mutations limits the duration of therapeutic benefit.
While the mechanisms of some drugs are somewhat well elucidated as illustrated above, our work indicates that most human ingestible drugs function not only by interacting with multiple proteins but also that these proteins are from different druggable protein classes [15]. Current data suggest that protein functions are distributed unevenly across protein structures [28]. The most broadly populated fold families most often catalyze similar chemical functions, but to different substrates. The underlying structural differences can be subtle as in the case of conserved binding sites that bind ATP in protein kinases. Up to 4% of the proteome of higher organisms such as mammals can be represented by a single fold family such as G-protein like receptors, each of which serves as a receptor to recognize a different distribution of ligands. This pattern of different molecular interactions being carried out by proteins with the same fold makes informatic mapping difficult. Advances in methodological accuracy and efficiency that enable mapping of the structural differences underlying these different functions are now detectable through iterative superposition of the corresponding domain and binding sites [29-31]. Differences between advancements in knowledge-based and ab initio protein structure prediction may be instructive for advancement by bioinformatic mapping and docking [32]. With an explosion of databases of biochemical and structural interactome data, the field appears ripe for system-wide bioinformatic mapping of compound-proteome interactions.
Drug Resistance and Multitargeting
We and others have previously explored the effect of amino acid mutations in protein structures in the context of drug interactions for infectious indications such as HIV and malaria [33-38]. With respect to neoplastic indications, drug resistance is thought to be the reason for treatment failure in over 90% of patients with metastatic cancer [39]. For CML, the observation of a large number of secondary resistance cases (where patients regressed after several months of effective treatment) prioritized the development of a second generation of specific kinase inhibitors [26, 40]. While there can be many factors for this resistance, 90% of CML patients who relapse after initially positive response to imatinib have mutations in the BCR-ABL kinase domain that disrupt imatinib binding [26, 40-42]. Laboratory and clinical studies suggested that these mutations are not random. Particular mutations were observed with high frequency, indicating a simple physical mechanism driving the emergence of resistance [26, 41, 42]. Furthermore, upon administration of second-line BCR-ABL inhibitors, the accumulation of new resistance mutations against these drugs was observed clinically [43]. All these observations suggest a role for underlying multiple pathways acting in a disease specific manner in response to a synthetic agent, like imatinib, to cause specific mutations for drug resistance. This so-called synthetic lethality may be overcome by designing drugs to work in a disease specific polypharmacological manner by taking into account an interactome of multiple disease pathways and drug interactions (Fig. 1).
Fig. (1).
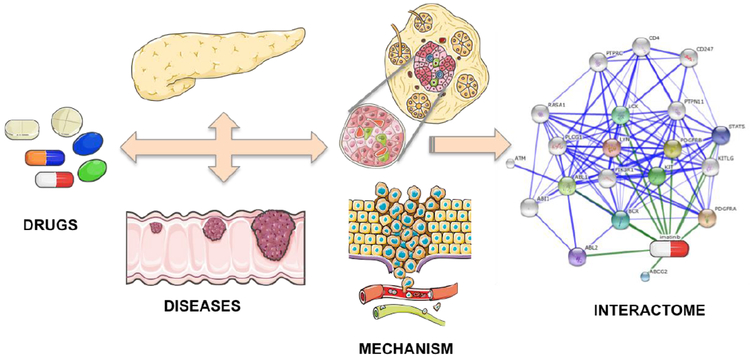
Interactome based therapeutic discovery paradigm used by the CANDO platform. Humans typically ingest a therapeutic drug (a small molecule or a biologic agent) with the goal of targeting particular indications (including infectious, inherited, and neoplastic diseases). The schematics for drug, disease and mechanism were adapted and modified from Servier Medical Art under Creative Commons CC-BY license. The visualization of the network of interaction between imatinib and its interactome was made using the STITCH4 platform [153]. In terms of mechanism, a given drug works by binding to one or more target proteins of interest, but also binds to other proteins causing off-target (neutral) and anti-target (negative) side effects. The CANDO platform makes interaction predictions for "all drugs" (currently a library of 3733 human approved compounds) against "all proteins" (currently a library of 48,278 proteins) to determine its efficacy against whole systems of interest, thereby enabling it to make putative drug predictions for “all” indications (that the library of drugs are currently approved for) simultaneously by performing comparative analyses of drug-proteome interaction signatures. In contrast to traditional drug discovery approaches focused on single targets, our innovative paradigm inverts the traditional one by first compiling a library of compounds/drugs that are safe to ingest with established side effects, and the shotgun virtual screen against whole proteomes/interactomes enables drug predictions for all indications simultaneously.
Imatinib was discovered by high-throughput screening followed by lead optimization [26], a process that is time-consuming, expensive, and largly ineffective due to high failure rates [44]. Imatinib is also non-specific with imperfect selectivity, in that it also inhibits a number of other kinases [45, 46], and is found to be active against constitutively activated tyrosine kinases such as platelet-derived growth factor receptor (PDGF-R), c-Kit, and macrophage colony stimulating factor receptor (c-fms) [47-50]. The mechanism of action of imatinib appears to be multi-faceted and likely involves the inhibition of multiple kinase targets that regulate various pathogenic functions in distinct tissue and cell compartments (Fig. 1). Imatinib treatment can also impair T-cell function in CML patients [51-53]. Rapid design of inhibitors with desired selectivity profiles, potentially targeting multiple kinases simultaneously to combat resistance [54] will require computational approaches that consider the affinity to the entire kinome simultaneously [55, 56].
The multitarget approach is a necessary one because every drug has to be effective at its site of action and also has to be readily metabolized by the body (for instance, by the cytochrome P450 (CYP450) enzymes, which are responsible for metabolizing the majority of drugs). A majority of small molecule drugs are derived from plant sources [18, 57]. Since these molecules are a result of a dynamic interplay of evolution between plants and other organisms sharing their environment, we hypothesize that interesting or functional small molecules that become drugs have multiple modes of action. Computational screening for multitarget binding and inhibition is effective because it exploits the evolutionary fact that protein structure is more conserved than sequence and function, and provides logical evidence that one compound can be an excellent initial candidate for many different protein targets. We have thus developed a platform that is agnostic to how individual compound-protein interactions are determined (whether predicted or observed) but rather relies on an interaction signature which is either a binary or real value set of numbers that indicates how well a compound interacts with a library of protein structures considered representative of the (current) structural universe.
Enabling Multiple Drug Combinations
In many cases, no single drug is sufficiently effective in the therapeutic range to cure a disease, or even to reduce symptoms or recurrence effectively. Thus multiple drugs can be combined to strengthen the effect. Independent effects due to interaction with multiple targets can decrease therapeutic doses, so that less efficacious and slightly more toxic compounds can be used safely and synergistically to achieve the desired efficacy profile. Similar to the concept of synthetic lethality in cancer, pathogens often develop resistance to single drug therapy, but simultaneous occurrence of multiple mutations that are resistance to a drug cocktail are exponentially less prevalent [58, 59]. We use the concept of multitargeting, or polypharmacology, where a single dose interacts with more than one target and acts synergistically in a disease specific manner, to address these issues.
Perhaps the most successful application of intentional multitarget drug administration is the use of inhibitors against HIV reverse transcriptase, protease, and integrase in the fight against HIV/AIDS [60]. Multidosing is used in a trial-and-error manner where errors usually result in patient suffering and mortality. In order to avoid such situations during human clinical testing or clinical practice, novel approaches have emerged to model synergistic effects of polypharmacology. For example, combinatorial effects have been tested in vitro using an automated robotics and informatics pipeline. There is an effort to identify combinations of compounds that display synergistic effects, such as inhibiting cytokine storm, as well as tumor inhibition, along with low toxicity and higher efficacy. Such complexity is possible due to either a single protein being targeted by multiple inhibitors, or, more likely, inhibition of multiple proteins involved in the same physiologic process [61]. Such interactions can be modeled and predicted by computational approaches. To this end, docking protocols and methods to predict structure activity relationships (SAR) in the context of multitargeting have been integrated into the drug development process, leading to efficiencies in cost and labor [62].
Overview of Docking
Docking refers to physical three dimensional (3D) structural interactions between a receptor (typically, proteins, DNA, RNA, etc.) and a ligand (small molecules, proteins, peptides, etc.) [3, 9, 10, 14, 63-75]. Docking methods are evaluated by measuring the correct pose/binding mode (using RMSD or TMScore of the coordinates of the atoms) or by measuring predicted affinities [10, 64, 65, 70, 71, 76]. More than 20 molecular docking software tools are currently in use for pharmaceutical research. Autodock Vina [77], Gold [78], and Glide [79] are a few examples of commonly used docking software. Each is based on a variety of cheminformatic, forcefield, and atomic bond flexibility algorithms in predicting poses, and meaningful direct comparison of efficiencies and accuracies is difficult. All have been used successfully in virtual high throughput screening to predict hits and leads ([3, 9, 10], are instances of protocols developed by us). However, the use of computational methods for drug discovery is still in its infancy [75, 80]. We have learnt from our previous experiences [81] and developed a hierarchical fragment-based docking with dynamics algorithm [82, 83] using a generalized all atom potential [74] that exploits evolutionary and structural information to predict the >180 million compound-protein interactions analyzed in this study.
Leveraging Docking for Drug Discovery: The CANDO Platform
Docking methods are primarily designed to predict conformational accuracy. In the context of drug discovery (and in our personal experience at translational research), predicting the conformation accurately is not as important as predicting the binding affinity or, more importantly, functional inhibition. These factors are generally not well considered by traditional docking methods [62]. Further, evaluating global effects of a single compound (or a cocktail) on the proteome is necessary to translate the utility of docking methods to medicine.
The Computational Analysis of Novel Drug Opportunities (CANDO) platform (Fig. 2) predicts the rough poses of compound-proteome interactions bioinformatically and hierarchically refines them using fragment-based docking with dynamics simulations of all the atoms in the system, which we have demonstrated is necessary for accurate calculation of binding energies [81]. Also, we have demonstrated that all-atom knowledge-based force fields are much more accurate and consistent than using classical models of physics based force fields for both protein structure prediction and docking [84-90]. We have shown that using docking with dynamics in a multitargeting manner leads to improved hit rates for finding inhibitors of pathogens compared to conventional approaches [9, 10].
Fig. (2).
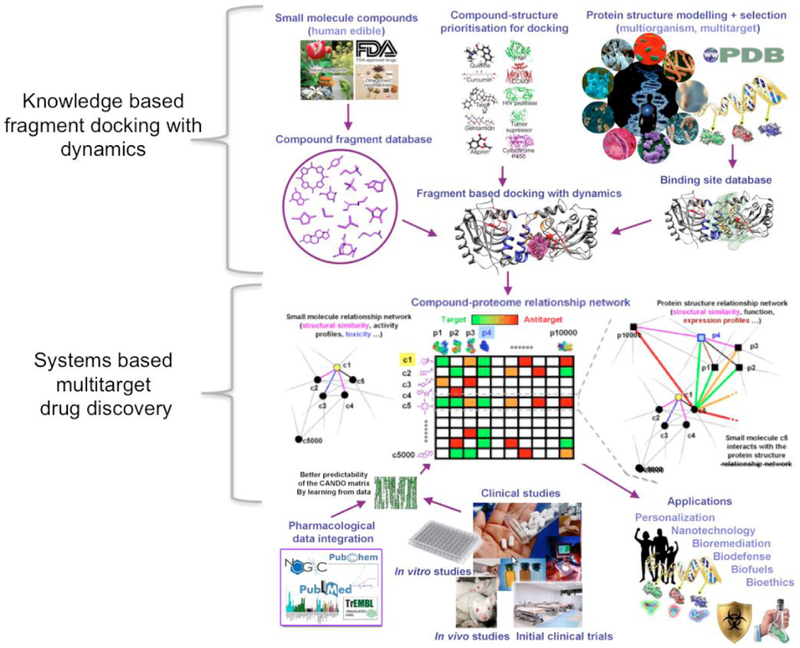
The Computational Analysis of Novel Drug Opportunities (CANDO) platform (http://protinfo.org/cando). The platform consists of a pipeline that generates the compound-proteome interaction matrix and indication-specific protocols to rank compounds that may be repurposed for particular indications/diseases given particular proteomes. The first version (v1) of the platform evaluates interactions between 3,733 human ingestible compounds that are associated with 2030 indications and 48,278 protein structures (46,784 of which are used in this study). Via a process of “contextualization”, where the predicted interactions are evaluated in the context of biomolecular data from a wide variety of sources including molecular docking simulation tools (including our knowledge-based fragment docking with dynamics algorithm), candidate interactions are ranked according to the degree of interaction and similarity for all indications. Predictions made using our systems-based multitarget drug discovery and repurposing platform are subjected to validation via in vitro binding, functional, and cellular assays, in vivo studies (if possible), and off-label use clinical studies. The resulting information is made available to a wide variety of users and applications via the web. The signature comparison and ranking approach used by the CANDO platform yielded benchmarking accuracies of 12-25% for 1439 indications with at least two approved compounds. 58/163 (35%) top ranking predictions had comparable or better activity relative to existing drugs in twelve prospective in vitro studies across ten indications, and represent novel repurposed therapies for indications such as dengue, dental caries, diabetes, herpes, lupus, malaria, and tuberculosis. Our approach can be tailored to specific meta genomes by modeling all the corresponding variant (mutant) protein structures and making predictions of personalized/precision drug regimens for particular individuals. Our blinded shotgun multitarget approach to drug discovery significantly enhances its efficiency, reduces drug development costs, and also provides a holistic framework for multiscale modeling of complex biological systems with broader applications in medicine and engineering.
Our CANDO platform evaluates how all (currently 3,733, as of 20 March 2014) FDA approved and other human ingestible drugs (such as dietary supplements) interact with all (currently 48,278, as of 20 March 2014) protein structures from multiple species as a representative of the protein universe (46,784 of which were used in this study). These compound-proteome interaction signatures were generated using a fragment-based docking with dynamics protocol combined with bio- and chem-informatics approaches. The platform then uses similarity of compound-proteome interaction signatures as indicative of similar functional behavior; nonsimilar signatures (or regions of signatures) are indicative of off- and anti-target [91] interactions, in effect inferring homology of compound/drug behavior at a proteomic level. Similarity can be determined using several metrics, from straightforward summations of identical yes/no interaction predictions and (root mean squared) deviations of interactions scores to sophisticated graph theoretic comparisons that consider the similarity of the underlying (actual or predicted) protein-protein interactions compiled from different publicly available methods and databases [92-95] as well as using methods previously developed by us [96-98]. In an evolutionary context, detecting similarity of interaction signatures enables inferring homology of drug behavior, and our proposed metrics analyze compound targeting behavior in the context of biologically relevant pathways [92, 93, 99]. The signatures are used to rank compounds for all indications. The platform provides an optimized and enriched set of predicted compound-protein interactions, a comprehensive list of indications and compounds that may be readily repurposed, as well as mechanistic understanding of drug behavior at an atomic level. We have successfully used in this approach to validate potential clinical leads for ten indications over twelve studies, including infectious, autoimmune, oncological, neurological and genetic diseases [11, 100-102] and to understand the contribution of druggable protein classes to benchmarking accuracy [15]. The goal of this study is to determine the contribution of multitargeting/polypharmacology to the accuracy of the CANDO platform.
APPROACH
We first describe how compound-proteome interaction signatures are computed and used to rank compounds in an indication-specific manner. We then describe how we retrospectively benchmark drug discovery (as opposed to docking) and repurposing platforms, which is necessary for the machine learning approach to optimize parameters and to make predictions of putative drugs. We finally end with a description of how the CANDO platform was used to analyze and assess the extent to which multitargeting or polypharmacology plays a role in benchmarking (repurposing) accuracy. Our goal is not only to provide a description of the CANDO platform and its accuracy but also to determine contributions to drug behavior in the context of polypharmacology and how that information can be leveraged to develop better drug discovery and repurposing platforms and eventually lead to making more accurate predictions that can be translated to the clinic.
CANDO Platform and Pipeline
The CANDO platform is comprised of a unique computational multitarget fragment-based docking with dynamics protocol to implement a comprehensive and efficient drug discovery pipeline with higher efficiency, lowered cost, and increased success rates, compared to current approaches (Fig. 2). The project is funded by 2010 NIH Director’s Pioneer Award and builds upon on our extensive work on drug discovery [3, 9-11, 13, 14, 33-36, 74, 81, 103-105], but now applied at the level of the whole proteome/interactome. We completed the first version (v1) of the CANDO platform on 20 March 2014 that resulted in compound-protein interaction matrices representing all compounds interacting with all multiorganism protein structures representative of the protein universe. The compound library used for screening consists of 3,733 human ingestible compounds including FDA approved drugs and supplements. The initial 39,553 protein structure library (total 48,278 structures) comprises of 14,595 human proteins (8,841 of these are high-confidence models constructed using a combination of one or more protein structure prediction methods including I-TASSER [106, 107], HHBLITS [108], MODELLER [109], KobaMIN [84, 86] and Protinfo CM [110] and 24,958 nonredundant protein structures from all organisms (other than the pathogens explicitly considered) obtained from the protein data bank (PDB).
Based on in-house benchmarking, we have set up a structural modeling pipeline using HHBLITS, I-TASSER and KobaMIN for protein modeling and use COFACTOR [29] and our in-house docking program for initial modeling of small molecule protein interactions. HHBLITS/ITASSER are used to select templates for modeling. I-TASSER iteratively refines the models and templates and our program KobaMIN refines the final models. Protein structure prediction methods are assessed in a blind fashion every two years at the Critical Assessment of Structure Prediction (CASP) experiments. The methods in our modeling pipeline have been among the top performing methods at past CASP experiments. This pipeline has been applied to all human proteins that can be modeled with high accuracy (~60% of the proteome). The benchmarking of individual components (structure prediction, binding site prediction and analysis, docking, potential functions) used in the CANDO platform has been done extensively and published by us and by others [74, 84-90, 106-132].
The pipeline initially uses a bioinformatic docking approach to predict interactions between all the protein structures and all the small molecule compounds. Additional proteins and compounds are added as needed and the CANDO v1 matrix currently consists of an additional 8,745 structures from 17 pathogens (≈ 20% are high confidence models) resulting in final dimensions of 3,733 compounds × 48,278 protein structures or a total of 180,221,774 predicted interactions between proteins and small molecules (the actual number of equivalent docking calculations is greater by almost a factor of 3, representing the average the number of domains and binding sites per protein).
Indication-Specific Interaction Signature Analysis and Comparison Approach to Ranking Compounds
The CANDO pipeline uses similarity of interaction signatures across all proteins as indicative of similar functional behavior and nonsimilar signatures (or regions of signatures) as indicative of off-and anti-target [91] interactions, in effect inferring homology of compound/drug behavior at a proteomic level. The main metric to determine similarity is the root mean square deviation (RMSD) of the pair of real value vectors that comprise an interaction signature. Compound-proteome interaction signatures are compared to each other and ranked using protocols that are disease/indication-specific, i.e., compounds approved for particular indications (if available) are used to generate rankings of other compounds that may have similar interaction signatures. Similar compound-proteome interaction signatures are further clustered and protein (target, anti-target, and off-target) specific weighting is introduced wherever information about involvement of particular proteins whose modulation (typically inhibition) is implicated in pathogenesis.
A consensus of the top-ranked indication-specific compound-proteome interaction signatures are evaluated in the context of additional biological information that includes all-atom fragment-based docking with dynamics simulations using knowledge-based potentials developed by us [74, 84, 85, 87] as well as by others [75], the scientific literature, pathways, gene structure and expression data and other information. The top compounds produced by these integrated rankings are considered to be putative repurposed drugs for a particular disease/indication. The platform thus provides an optimized and enriched set of compound-protein interactions, a complete and comprehensive list of indications and compounds that may be readily repurposeable, as well as a mechanistic understanding of drug behavior at three dimensional (3D) atomic and molecular scales.
The top ranking predictions are validated at the bench using a barrage of approaches. Based on our extensive experience in predicting drug hits and leads, some of which have been patented and are being licensed commercially <www.faqs.org/patents/app/20110112031>, obtaining atomic level mechanistic understanding is limited in use and approaches that predict functional inhibition are typically most useful in the context of drug discovery.
Machine Learning Approaches to Iteratively Refine Predictions
Our approach is to validate the predictions made and obtain bench results that will then iteratively feedback into our pipeline to improve successive predictions. The goal is to compare the results from bench validation for a given set of proteins to the predictions and re-parameterize our platform using machine learning. This enables us to determine which parameter choices have the greatest individual accuracy and also the choices that will lead to compounds becoming viable drugs. Machine learning approaches such as neural networks and support vector machines (SVMs) are capable of weighting the contribution of individual proteins and compounds and particular regions of the interaction signatures as needed to optimize predictive value. In past work, we have used both neural network and SVM packages written by others to develop methods to predict protein function ([121, 133, 134], for instance).
The benchmarking is coupled with machine learning to determine the various parameter weightings. Our current accuracies using the “All” protein set (48,278 structures in CANDO v1) range from ~12% to up to ~50% depending on the cutoff (top10 to top100) or the number of indications considered (i.e, all 1439 indications with two or more approved compounds or only those ~700 reporting a nonzero result) using only the compound-centric approach to prediction (i.e., all proteins weighted equally). This suggests there is significant room for improvement particularly in the number of indications applicable to the CANDO platform (i.e., the coverage). The greater the coverage, the higher the number of accurate predictions that can be made for all the 2030 indications associated with set of the 3,733 compounds based on mappings obtained from the Comprehensive Toxicology Database <ctdbase.org>.
Benchmarking Accuracy of Drug Discovery Platforms
Benchmarking computational platforms for drug discovery is different from benchmarking the ability of software to correctly dock or bind small molecules to proteins. For the latter, the overall accuracy is based on comparisons of predictions to different types of bench validation studies. For the former, however, the goal is to determine the ability of a platform to recognize the likelihood that any given prediction of a small molecule will be effective as a drug for a particular indication.
Initially, a leave-one-out benchmark experiment is performed to calibrate the machine learning approach for all indications with more than one approved compound (1439 such indications for the CANDO v1 library). The indication assignment for one of the compounds is “left out” and the interaction signature similarity comparison approach is used to determine the rank of another compound with the same indication and checking whether the compound without the assigned indication is among the top ranked. Each of the compounds is thus compared and ranked by similarity to all the other compounds in our library. The rank(s) of the compound(s) associated with the same indication is used to determine the performance of our approach. The lower the (average) rank, the better the performance. Similarity is determined using several metrics, such as straightforward summations of identical yes/no interaction predictions and (root mean squared) deviations of interactions scores.
The sensitivity and specificity of this approach is then evaluated using receiver operator characteristic [135] curves. As a computational control, fully randomized CANDO matrices (where all the rows and columns, representing compounds and proteins respectively, are moved to randomly selected locations) are used to perform the same benchmarking analysis to determine the type of results that could be obtained by chance. This allows us to assign a probability value that assesses the likelihood of obtaining any particular prediction relative to a random control.
This benchmark test is made more rigorous by using more sophisticated jack-knifing to leave out related compounds, and different variations of these bootstrapping methods are used to minimize overtraining and other biases inherent in the data sources or methods. In the end, a machine learning model based on neural networks and/or SVMs is obtained so that given a CANDO matrix and a compound-indication mapping table as input, the assignments of compounds to indications by interaction signature comparison is optimized. The initial best machine learning models are further iteratively optimized as prospective predictions are validated and the corresponding bench results are fed back into the machine learning process
The benchmarking described above allows us to (i) better weight the parameters used for various aspects of the structure modeling pipeline; (ii) determine which docking methods, interaction types, and indications are likely to be most accurate and enable assignment of confidence values to each of our predictions (as well as an overall p value); and (iii) optimize accuracy of the entire platform for ranking putative therapeutics. The judicious application of this approach while rigorously minimizing overtraining enables us to bootstrap a platform initially based on docking to one targeted towards drug discovery. The different validations for particular compound-protein interactions are coordinated to maximize the amount of information available for particular high-confidence subsets.
Contribution of Multitargeting/Polypharmacology to Benchmarking Accuracy
To assess the contribution of multitargeting/polypharmacology on CANDO benchmarking accuracy, we compared the distributions and trends of compound-protein interactions for all the 3,733 human ingestible compounds in our library for different interaction score cutoffs and using different proteome sets (Fig. 3). Polypharmacology or multitargeting can be defined by multiple interactions of drugs with all known targets for an indication/disease and/or the interactions with multiple targets of different indications/diseases. Our CANDO methodology was developed to identify similarity of compound behavior using multiorganism compound-proteome interaction signatures as a surrogate for compound perturbation of biological function. Our hypothesis is that the biological functions of compounds/drugs are related to the identification of multitargeting profiles that includes target, off-target and anti-target interactions, and that this information needs to be properly utilized for efficient drug discovery and repurposing. The multiorganism proteome data sets considered are labeled ALL for all 46,784 protein structures used in this study, HUMAN for the 14,595 proteins obtained from a reference Homo sapiens proteome that includes 5754 solved structures from the PDB and 8839 computationally modeled structures (described previously), and NRPDB for a set of 24,958 non-redundant solved PDB structures obtained from eukaryotic, prokaryotic, archaea and viral organismal proteomes. We then assess the relationship between the degree of predicted multitargeting/polypharmacology to the benchmarking accuracies obtained for the indications associated with the human use drugs (Fig. 4). Finally, we provide lists of representative indications and respective benchmarking accuracies corresponding to five compounds with the highest degree of predicted multitargeting and five with the lowest (Fig. 5).
Fig. (3).
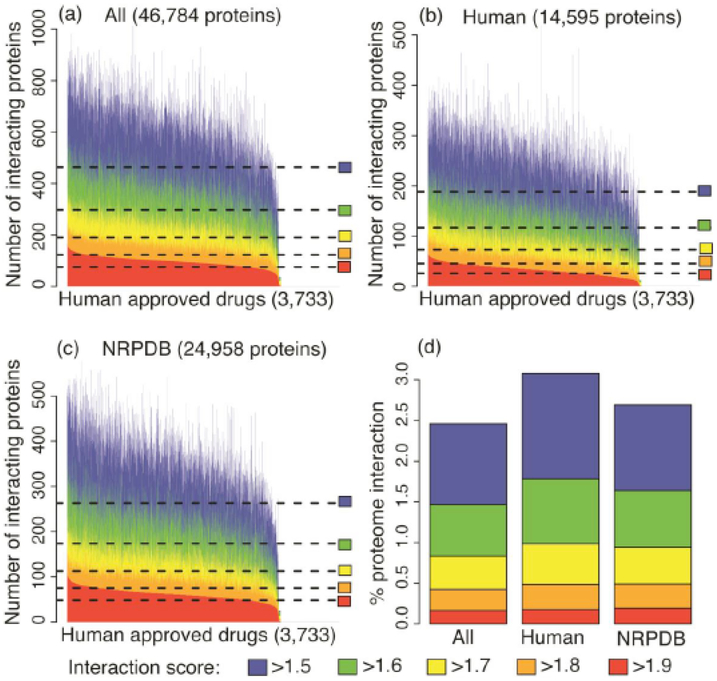
Extent of polypharmacology predicted by the CANDO platform. Panels (a)-(c) indicate the number of proteins predicted to interact with each of the 3733 human use drugs in the CANDO library for different universal proteome sets considered (All: 46,784 proteins; Human: 14,595 proteins; and NRPDB: 24,958 proteins) at different interaction score cutoffs (ranging from >1.5 to to >1.9; with scores ranging from 0-2.0). The data at the most stringest cutoffs (signifying the highest likelihood of an accurate prediction) indicate that the fractions of the proteome involved in strong interactions with our drug library is similar across all the sets considered. A two-sample Kolmogorov-Smirnov test, which compares two distributions with the null-hypothesis that they are not similar, was performed (the lower the p-value, the greater the likelihood of the null hypothesis) indicating that these distributions are very different at each score cutoff (All to Human p-value 2.2e-16 for all cutoffs and All to NRPDB p-value 2.2e-16 for cutoffs >1.9 through >1.6, and 11e-15 for the >1.5 cutoff). While a number of predicted interactions may be incorrect or may not be functionally important, the current data is what is used to obtain the benchmarking and prospective validation results obtained in [11, 15]. Taken together with those results, the above figures illustrate that a large number of protein structures with a broad distribution of the fold space with significant and specific predicted polypharmacology is necessary for making accurate predictions using our proteomic and evolutionary drug discovery and repurposing platform.
Fig. (4).
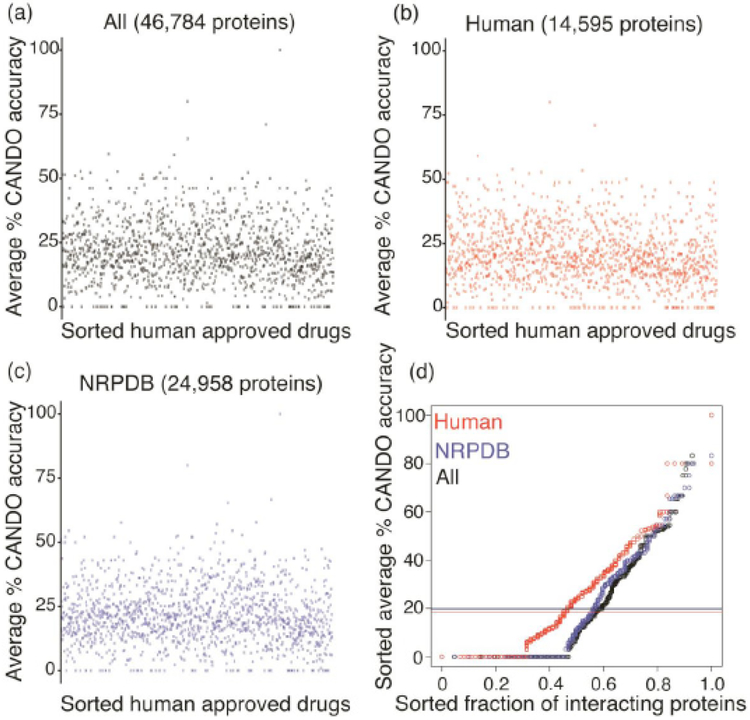
Average benchmarking accuracies for all indications associated with each drug using the CANDO platform, sorted by predicted drug multitargeting/polypharmacy. Panels (a)-(c) show the average accuracies for all the indications associated with each of the 3,733 drugs in the CANDO library shown in the same order as in Figure 3, i.e., sorted by the number of proteins they are predicted to strongly interact with, for each of the three proteomes evaluated. Panel (d) shows the average of the average accuracies for the sorted distribution of average top10 CANDO percent accuracy to the sorted distribution of normalized fraction of interacting proteins for the three proteomes. Visually and otherwise, the benchmarking (repurposing) accuracies obtained for each indication based on similarities of drug-proteome interaction signatures does not appear to correlate with the number of proteins predicted to strongly interact with each of the drugs. This indicates that the benchmarking accuracies obtained using the CANDO platform, as well as any results from prospective validation, are not a simple function of the degree of predicted interactions but rather rely on the specific score distributions within the drug-proteome interaction signatures. The above panels provide evidence for eliminating a source of potential bias for the CANDO benchmarking accuracies and for prospective validations. Further analysis is needed to tease out in detail why current human use drugs behave the way they do. Our initial analysis of polypharmacology provides a starting point to identify the most promiscuous and the most specific drugs for all the indications that they are currently approved for (and any potential future ones), and how that plays a role in their efficacy.
Fig. (5).
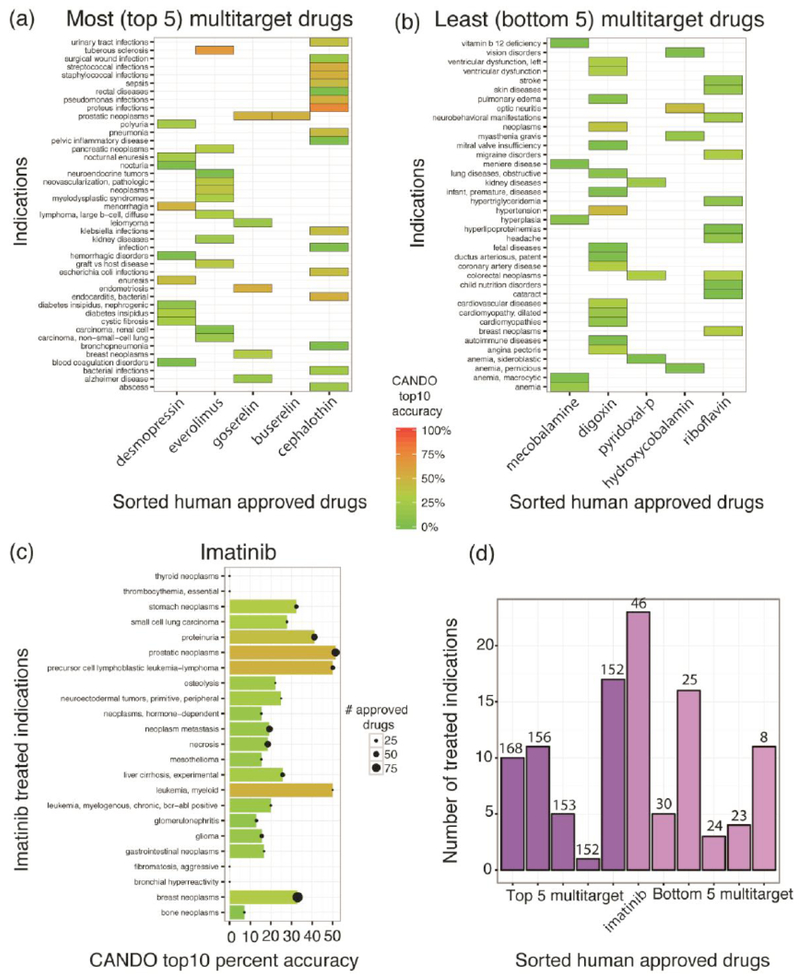
Relationship between indication type and polypharmacology. Panels (a, b) show the top five most and bottom 5 least multitargeting/polypharmacological drugs predicted by the CANDO platform sorted by decreasing order of predicted number of protein interactions. The indications treated by these compounds are listed and the CANDO top10 percent accuracy (retrieval accuracy of the CANDO platform of known drugs for a particular indication in the top 10 ranked list of predictions obtained using compound-proteome signature similarity) is highlighted using colored tiles. There is no simple correlation between multitargeting/polypharmacology and CANDO accuracy for all 3,733 compounds (Fig. 4) but the most multitargeting drugs treat diseases such as B-cell lymphoma, graft vs host disease, lung, kidney, breast, and prostate cancers along with several infectious indications. In contrast, the least multitargeting/polypharmacological drugs are either vitamin supplements or active forms of Vitamin B group, and treat hypertension, vitamin deficiency, nutrition and anemic disorders, and visually contribute lower to drug retrieval accuracy than most multitarget drugs. Panel (c) shows the indications treated by imatinib, one of the few well-known polypharmacological drugs. Imatinib treats neoplastic diseases, most of which show higher CANDO drug retrieval accuracy than the least polypharmacological drugs, shown in panels (b, c). The black filled circles on each bar represent the number of drugs known to treat the indications also treated by imatinib. Panel (d) shows the relationship between the number of treated indications by most and least multitargeting drugs from panels (a) and (b) respectively, in comparison to imatinib. The bars are colored by the number of predicted interacting proteins that is also indicated on the top of each bar of the sorted list of multitargeting drugs. There seems to be no relation between the number of indications treated by multitargeting drugs and the extent of multitargeting, defined as the number of predicted protein interactions. Altogether, these results indicate the complex nature of polypharmacology and its contribution to drug repurposing, which extends beyond interaction with multiple proteins: imatinib with much lower predicted number of protein interactions treats neoplastic indications similar to the most multitargeting compounds and exhibits a greater repurposing potential based on the number of indications it is approved for treatment.
RESULTS AND DISCUSSION
Benchmarking the CANDO Platform
The individual modeling components (structure prediction, binding site prediction and analysis, docking, potential functions) used in the CANDO platform have been benchmarked extensively by us and by others (see methods). The platform itself has been benchmarked using a compound-centric leave-one-out procedure for all indications with at least one approved compound [11, 15]. Briefly summarizing: There are 749/1439 indications where the predictions identify a related compound with the same indication in the top 1% of the ranks (on average). Consistent predictions are produced regardless of the comparison method, metric, or matrix used. In contrast, when randomized compound-proteome matrices are constructed (by randomly swapping the rows and columns representing the compound-proteome interaction signatures) and used for making predictions, only 10-20 out of 1439 indications “work” (using the same criteria as before), with largely inconsistent sets of predictions.
Using fully randomized matrices as computational controls automatically accounts for normalization issues to account for different number of approved drugs for each indication. Random controls are essential as the leave-one-out benchmarking method is more likely to work by chance on indications with tens of approved compounds compared to those with only a few approved compounds. Our benchmarking results indicate higher average accuracies for indications with larger number of approved drugs. Moreover, compared to random controls, the increase in benchmarking accuracy across all indications is much higher than expected by chance: The average of average accuracies obtained using matrices based on variations of the 48,278 proteins is 15.1% and the maximum average accuracy achieved (by filtering for number of compounds) is 49.8%; in contrast the best performing random matrix (out of a 1000) produced an average accuracy of 0.4% and the maximum accuracy achieved when measured as a function of the number of compounds is 0.9%. Based on the benchmarking, we can determine the indications for which the current version of the CANDO platform is most relevant, which enables us to assign confidence values to each of our predictions.
Multitargeting in Human Approved Drugs Using the CANDO Platform
The benchmarking accuracy is a measure of drug repurposeability. To quantify the level of multitargeting/polyphamacology for each compound, we use multiple cutoff scores (>1.5, >1.6, >1.7, >1.8, >1.9) based on our scoring scheme [15] that correspond to moderate to high compound-protein interaction prediction accuracies. All the 3,733 compounds are sorted by their degree of multitargeting based on the highest cutoff score of >1.9 and plotted against the number of interacting proteins (Fig. 3).
Using the ALL (46,784) proteome set we find that the most predicted multitargeting/polypharmacological drugs interact with 464, 298, 190, 123, and 76 proteins using interaction score cutoffs >1.5, >1.6, >1.7, >1.8, and >1.9 respectively (Fig. 3a). If we consider only human proteins (Fig. 3b), the most predicted multitargeting drugs interact with 189, 117, 73, 45, and 26 proteins with interaction scores >1.5, >1.6, >1.7, >1.8 and >1.9 respectively. Similarly, for NRPDB (Fig. 3c), we get 263, 174, 113, 75, and 48 interacting proteins for the most predicted multitargeting drugs. Our results indicate that even if our interaction prediction method is wrong half the time, there are still a significant number of drugs approved for human use that are highly polypharmacological (Fig. 3). This suggests that this is an essential feature for a small molecule compound becoming a drug as almost all human approved and ingestible compounds exhibit some degree of polypharamacological signal. Some of the most promiscuous drugs using the interaction profile for the ALL proteome set include eptifibatide, a drug used during heart attack or angioplasty to avoid blood clots [136, 137] and belongs to the same class of drugs as aspirin, and the second most promiscuous drug desmopressin, a synthetic replacement for vasopressin, the hormone that is used to treat the symptoms of diabetes insipidus by reducing urine production, and has been suggested as a therapeutic for various other indications (see Fig. 5a) [138, 139].
Next, we assessed the similarity of distributions of multitargeting/polypharmacology for all 3,733 compounds between different proteomes: ALL, HUMAN and NRPDB. Visually the distributions of multitargeting/polypharmacology looks very similar for the different proteomes (Figs. 3a,b,c). Moreover, on average, all drugs interact with a similar (~2.5-3%) fraction of interacting proteins relative to the proteome size (Fig. 3d). However, a two-sample Kolmogorov-Smirnov (K-S) statistical test using a two-sided alternative hypothesis (which is a statistical measure of assessing similarity or difference between two distributions with lower p-value indicating greater difference) shows that ALL is very different from NRPDB as well as from HUMAN, with p-values of 2.2e-16 for all score cutoffs shown in Fig. 3 with an exception of a slightly higher p-value, but still significantly different distribution between ALL and NRPDB for the >1.5 cutoff. Thus, we conclude that almost all human approved drugs are polypharmacological but the particular distributions of interacting proteins are selective and specific. This could be the result of an evolutionary feature that allows for small molecules with a fairly limited set of atoms (tens to hundreds) to confer selectivity and specificity in the context of whole proteomes, and in the context of simultaneously competing with other molecules for binding and performing their biological function.
Relationship Between Multitargeting and Retrospective Drug Discovery Using the CANDO Platform
From the results described in the previous section and Fig. (3), we concluded that most, if not all, human approved drugs are multitargeting, and offered an evolutionary explanation for how small molecule ligands (drugs or otherwise) derive selectivity and specificity in a proteomic context. Here we wanted to assess the degree to which multitargeting/polypharmacology was responsible for CANDO benchmarking accuracy, i.e., whether the drug repurposing signal in CANDO is dominated by the corresponding polypharmacological signal.
Fig. (4) compares the entire distribution of average CANDO benchmarking accuracies for all 1439 indications as a function of polypharmacology, with the order of compounds sorted by the degree of interactions based on the interaction score cutoff of >1.9 (i.e., same order as in Fig. 3). The CANDO accuracy shown is based on the top10 criterion, i.e. the accuracy of retrospectively identifying a known drug-indication association in a ranked list of the top 10 predictions made by the platform. We used the most stringent criterion (top10) to evaluate drug-indication accuracies in a manner that makes them readily verifiable with straightforward bench experiments, as used by us to prospectively verify predictions for 12 different indications.
Fig. (4a-c) shows that multitargeting/polypharmacology does not correlate with CANDO benchmarking accuracy for different proteome sets considered. For the sorted list of compounds, the average CANDO accuracy over all compounds is lower for HUMAN compared to NRPDB and ALL proteins set (Fig. 4d, horizontal lines). The Q-Q plot (Fig. 4d) shows that the distribution of average CANDO benchmarking accuracies for more multitargeting/polypharmacological drugs is not similar between HUMAN-NRPDB and HUMAN-ALL proteome sets. Furthermore, the sorted distribution of the fraction of the maximum number of interacting proteins for each proteome set correlates with average CANDO benchmarking accuracy (Fig. 4d). This suggests that drug discovery is not a simple function of multitargeting/polypharmacology (Fig. 4a-c) but taken together with the results in Fig. 4d and our prior work [11, 15] indicates that the specific distribution of polypharmacology is important for drug repurposing accuracy.
Our results suggest that multitargeting/polypharmacology is a necessary but not sufficient condition for drug discovery. Polypharmacology of approved drugs is likely a real phenomenon, as detailed in the introduction sections and the predictions observed in Fig. (3). That is, most drugs likely work by interacting with a significant number of proteins (tens or hundreds): examples include multi-kinase inhibitor drugs like imatinib, among others. CANDO exploits this feature in the form of interaction signatures for drug discovery and repurposing. Furthmore, polypharmacology refers to positive interactions (binding, inhibition, etc.) to multiple targets, whereas the CANDO platform considers both interacting and non-interacting proteins to define the signature of a compound as a vector of real value scores that quantify strength of interactions. Our findings thus indicate that for drug efficacy, it is not only the significant numbers of protein interactions that are important even in the context of multitargeting/polypharmacology, but also the much larger number of non-interacting proteins and their distribution. Hence, multitargeting/polypharmacology, in its simplest form as shown in Fig. (3) that does not consider targets, off-targets, and anti-targets, is not sufficient for effective drug discovery and repurposing. A complete description of the polypharmacological signal appears to be essential for defining when small molecules become drugs, how they derive specificity against an indication/disease, and how they play a selective and specific role in modulating metabobolic, regulatory, and signaling pathways.
A simple test of using a binary interaction matrix (interaction on or off) results in 50% lower accuracy compared to using the real value interaction scores for benchmarking the CANDO platform (holding all other conditions the same). Furthermore, the real value interaction scores may account for an evolutionary phenomenon where multiple small molecules compete in terms of binding to a single site to influence molecular function, as well as binding to homologous protein molecules across multi-organism proteomes cooperatively to influence biological function. Taken together, this suggests that quantifying the nature of interaction as a competitive/cooperative polypharmacological signal is a better surrogate for polypharmacology compared to using binary yes/no interactions of compounds with proteins.
Nature of Polypharmacological Interactions to Identify Drugs Efficiently Using the CANDO Platform
Small molecule drugs work by considering both positive and negative interactions, specific for the indication of interest; defined as the target, off-target and anti-target interactions in the context of one or more indications (Figs. 1 and 2). Thus a simple model of polypharmacology as multitargeting, as shown in Fig. (3), is a very rough proxy, and it is not sufficient for drug discovery and repurposing, as indicated by the data in Fig. (4). At the simplest level, two drugs binding to the same target do not necessarily work synergistically -- if they bind to the same site (mutually exclusively), they are additive [140] and if they bind to distinct sites independently (i.e., without affecting each other's binding), they should be synergistic. So the effects of two compounds on a given target will depend on where they bind, its binding affinity, and mode of action due to binding (agonist/antagonist). Moreover, if one compound binds to multiple proteins, it depends on where it binds, with what affinity, and what action is performed in the context of the indication/disease. Finally, there are many small molecules in a biological system exerting their influence on both molecular and holistic function simultaneously as well as sequentially. If polypharmacology in its simplest form is considered only to be the strongest positive interactions to a small number of targets, then its applicability for computational drug repurposing is limited. However, a holistic approach that considers positive, negative, and neutral interactions across whole proteomes is likely to be useful, as these interactions are derived from evolutionary methods that use structural homology to derive interaction scores that can likely be used as a surrogate for binding affinity. The benchmarking results obtained using the CANDO platform indicate the use of real value interaction scores over binary on/off interaction scores.
A specific interaction signature for a compound that specifies binding efficacy, nature of interaction, and the secondary effect of neighboring interactions between interacting proteins themselves, defines a continuum of interactions across multiple protein classes. Such an interaction signature is a surrogate for polypharmacology that is useful for drug discovery and repurposing. These conclusions suggest a model for how a small molecule with few atoms derives selectivity and specificity for particular indications, and to modulate functions of biological systems. Our answers are obtained by exploring the evolutionary nature of the biomolecular interactome (interaction between and within small molecules, proteins, DNA, RNA, etc.). The nature of predicted polypharmacology of small molecule interactions with multiple proteins in the context of an indication/disease illustrates the limitations of the traditional model for drug discovery, and indicates that the interactome based paradigm as implemented by CANDO is a useful model for effective drug discovery and repurposing.
Most and Least Polypharmacological Drugs Target Different Indications
The nature of poylpharmacology as it applies to drug discovery and repurposing is complex and not a simple function of multitargeting as explained in the previous sections. In order to understand the effect of multitargeting and polypharmacology on drug repurposing, we examine the indications/diseases that are treated by the most and least multitargeting drugs and draw comparisons with imatinib, one of the few well-known polypharmacological drugs (Fig. 5). The five most multitargeting drugs in our study include, desmopressin, everolimus, goserelin, buserelin, and cephalothin, which are predicted to strongly interact with over 150 proteins. These drugs are known to treat several complex indications/diseases with multiple etiologies (Fig. 5a), including reducing urine production to treat the symptoms of diabetic insipidus or nocturia (desmopressin), different forms of lung, kidney, breast and prostate cancers (everolimus, goserelin, buserelin), and grampositive microorganism infections (cephalothin). As an example, the anti-cancer agent everolimus has been shown to be effective against advanced breast cancer resistant to endocrine therapy [141, 142]; a successful randomized Phase III BOLERO-2 trial of adding everolimus to exemestane showed its effetiveness in the treatment of postmenopausal hormone receptor-positive advanced breast cancer [143-145]. Such examples validate the importance of multitargeting/polypharmacology to combat synthetic lethality in cancer and provide guidance for multiple drug combination therapies.
The least multitargeting drugs in our study include mecobalamine (vitamin supplement), digoxin (treats heart problems/atrial fibrillation), pyridoxal-p (active form of vitamin B6), hydroxy-cobalamin (treats cyanide poisting) and riboflavin (part of vitamin B group), mainly used to treat vitamin and nutritional deficiency, anemia, hypertension, among other indications (Fig. 5b). On average, the bottom 5 (least) multitargeting drugs as predicted by the CANDO platform still interacts with more than 20 putative biological targets. However, the least multitargeting drug, riboflavin, is a vitamin supplement and visually contributes lower to drug retrieval benchmarking accuracy than most multitarget drugs (Fig. 5a, 5b). Interestingly, imatinib, which is one of the few well-known polypharmacological drugs and predicted to interact with only 46 proteins, is mainly used to treat neoplastic diseases (Fig. 5c) as with most multitargeting drugs (over 150 interacting proteins), but cannot combat drug resistance. Moreover, imatinib by itself displays greater drug repurposing potential, as it has been approved to treat over 20 indications/diseases, compared to the most and least multitargeting drugs taken together (Fig. 5d). This may suggest that imatinib, a multiple tyrosine kinase inhibitor, interacts with multiple pathways either directly or indirectly via the kinase-signaling cascade, and develops a continuum of interactions between targets, off-targets and anti-targets, both within and across multiple druggable protein classes to achieve synergistic effects for multiple indications/diseases.
SUMMARY AND CONCLUSION
Structure-based rational drug discovery is typically limited to, and by, screening a compound library against one protein structure associated with a particular disease (target) to identify inhibitor leads that eventually will lead to drugs that treat the indication/disease. The most effective drugs in humans (e.g. Aspirin® or Gleevec®) inevitably interact with and bind to multiple proteins, a feature that traditional models based on single target drugs fail to take into account. Imatinib mesylate, also known as Gleevec®, is a 2-phenylaminopyrimidine derivate designed as a specific inhibitor of the ABL protein tyrosine kinases (v-ABL, BCR-ABL, and c-ABL) [146, 147] but has been one of the widely used polypharmacological drugs. Imatinib’s activity against cells bearing the BCR-ABL translocation has yielded remarkable results in treating CML with minimal side-effects reported after 5 years of continuous administration [26, 148, 149]. In addition, imatinib was found to be active against other constitutively activated tyrosine kinases [47-50]. The inhibitory activity against c-Kit and PDGF-R has enabled the development of effective treatments for gastrointestinal stromal tumors [150], eosinophilic disorders and systemic mast cell disease [151, 152]. We present a paradigm where drug efficacy is viewed as a process, considering interactions with multiple biomolecules as well as the noninteractions. This interactome based drug discovery and repurposing paradigm is implemented by the Computational Analysis of Novel Drug Opportunities (CANDO) platform which not only enables predictions of novel putative therapeutic leads, but also aids in mechanistic analysis and virtual surgery using small molecules to determine the importance of druggable protein classes [15], and the contribution of polypharmacology (this work), to benchmarking accuracy.
The traditional process of drug discovery is focused on individual compound-protein interactions and requires many rounds of screening, modeling, and synthesis in a trial-and-error approach that is costly, time-consuming, and often ineffective and unsafe in humans. This traditional trials approach, used to develop almost all the drugs to date, has yielded a 95% failure rate before or during clinical use. Existing drugs with known safety profiles that interact with therapeutic targets can be repurposed and rapidly deployed for use in mono and multi-drug therapies. We developed the CANDO platform to evaluate how all approved and other human ingestible drugs interact with all structures representative of the protein universe, and use the resulting information to make predictions of drug efficacy against all indications in a shotgun manner. This approach is highly efficient, producing significantly more drug leads per computing cycle than more conventional methodologies by taking advantage of statistical multiplier effects in much the same manner seen in whole genome shotgun sequencing. The CANDO platform uses signatures of interactions across a representative set of protein structures and determines similarity of compound/drug behavior at a proteomic level (a form of homology modeling). The signatures are used to rank compounds for each indication/disease and provide an optimized and enriched set of compound-protein interactions, a complete and comprehensive list of compounds that may be readily repurposed, as well as a mechanistic understanding of drug behavior at a proteomic level.
The CANDO platform outputs a matrix containing confidence scores measuring the strength of interactions between all human use drugs and a library of protein structures we have constructed as a representation of the protein universe. This evolutionary based drug discovery platform can also be used to predict the small molecule/protein interactions that are implicated in specific indications/diseases, as well as specific biological functions. The CANDO platform can also be used to identify sets of compounds that affect different pathways or different components in the same pathway. The interaction network information can also be used to determine whether different components act antagonistically or synergistically to guide experiments and identify multiple mechanisms of the same drug.
These structural and evolutionary models of drug discovery are largely static in nature, and are designed for general purpose applications. We can use network-driven methods that integrate heterogeneous temporal and spatial genomics data to specify predictive signatures that identify new candidate therapeutic targets and drugs in a dynamic fashion. This may be done by formulating the problem as one of statistical model selection in which subsets of variables are chosen to build robust models, and adopt the Bayesian paradigm to coherently and rigorously combine different data sources. This leads to a paradigm of disease driven drug discovery, where compounds are designed by accounting for interactions with targets, anti-targets, and off-targets, and the relevant polypharmacology in the context of particular indications is used to identify a drug.
Our work provides a molecular framework to contextualize drug discovery with a paradigm superior to hit-and-trial methodologies currently used by traditional approaches. Altogether, these efforts by us and others to fully quantify the nature and extent of multitargeting/polypharmacology in the context of specific indications will hopefully be realized in developing new and repurposed therapies to efficiently combat diseases with multiple etiologies (such as cancers, infectious and autoimmune diseases).
ACKNOWLEDGEMENTS
Research reported in this publication was supported by a NIH Director's Pioneer Award (1DP1OD006779) to Ram Samudrala and a Clinical and Translational Sciences Award (UL1TR001412) from the National Center for Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Gaurav Chopra gratefully acknowledges support from a Purdue University startup package and JDRF (3-PDF-2014-205-A-N).
Biography

Gaurav Chopra

Ram Samudrala
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- [1].Bonander N, Bill RM. Relieving the first bottleneck in the drug discovery pipeline: using array technologies to rationalize membrane protein production. Expert Rev Proteomics 2009; 6(5): 501–5. [DOI] [PubMed] [Google Scholar]
- [2].Gillespie SH, Singh K. XDR-TB, what is it; how is it treated; and why is therapeutic failure so high? Recent Pat Antiinfect Drug Discov 2011; 6(2): 77–83. [DOI] [PubMed] [Google Scholar]
- [3].Horst JA. Computational Multitarget Drug Discovery, in Polypharmacology in Drug Discovery. USA: John Wiley & Sons, Inc; 2012; pp. 263–301. [Google Scholar]
- [4].Sacks LV, Behrman RE. Challenges, successes and hopes in the development of novel TB therapeutics. Future Med Chem 2009; 1(4): 749–56. [DOI] [PubMed] [Google Scholar]
- [5].Swamidass SJ. Mining small-molecule screens to repurpose drugs. Brief Bioinform 2011; 12(4): 327–35. [DOI] [PubMed] [Google Scholar]
- [6].Ekins S, Williams AJ, Krasowski MD, Freundlich JS. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov Today 2011; 16(7–8): 298–310. [DOI] [PubMed] [Google Scholar]
- [7].Xu K, Cote TR. Database identifies FDA-approved drugs with potential to be repurposed for treatment of orphan diseases. Brief Bioinform 2011; 12(4): 341–5. [DOI] [PubMed] [Google Scholar]
- [8].Ren J, Xie L, Li WW, Bourne PE. SMAP-WS: a parallel web service for structural proteome-wide ligand-binding site comparison. Nucleic Acids Res 2010; 38(Web Server issue): W441–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jenwitheesuk E, Samudrala R. Identification of potential multitarget antimalarial drugs. JAMA 2005; 294(12): 1490–1. [DOI] [PubMed] [Google Scholar]
- [10].Jenwitheesuk E, Horst JA, Rivas KL, Van Voorhis WC, Samudrala R. Novel paradigms for drug discovery: computational multitarget screening. Trends Pharmacol Sci 2008; 29(2): 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Minie M, Chopra G, Sethi G, et al. CANDO and the infinite drug discovery frontier. Drug Discov Today 2014; 19(9): 1353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Costin JM, Jenwitheesuk E, Lok SM, et al. Structural optimization and de novo design of dengue virus entry inhibitory peptides. PLoS Negl Trop Dis 2010; 4(6): e721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nicholson CO, Costin JM, Rowe DK, et al. Viral entry inhibitors block dengue antibody-dependent enhancement in vitro. Antiviral Res 2011; 89(1): 71–4. [DOI] [PubMed] [Google Scholar]
- [14].Horst JA, Pieper U, Sali A, et al. Strategic protein target analysis for developing drugs to stop dental caries. Adv Dent Res 2012; 24(2): 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sethi G, Chopra G, Samudrala R. Multiscale modelling of relationships between protein classes and drug behavior across all diseases using the CANDO platform. Mini Rev Med Chem 2015; 15(8): 705–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun X, Vilar S, Tatonetti NP. High-throughput methods for combinatorial drug discovery. Sci Transl Med 2013; 5(205): 205rv1. [DOI] [PubMed] [Google Scholar]
- [17].Goldman AD, Baross JA, Samudrala R. The enzymatic and metabolic capabilities of early life. PLoS One 2012; 7(9): e39912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pink R, Hudson A, Mouries MA, Bendig M. Opportunities and challenges in antiparasitic drug discovery. Nat Rev Drug Discov 2005; 4(9): 727–40. [DOI] [PubMed] [Google Scholar]
- [19].Lombardino JG, Lowe JA 3rd, The role of the medicinal chemist in drug discovery--then and now. Nat Rev Drug Discov 2004; 3(10): 853–62. [DOI] [PubMed] [Google Scholar]
- [20].Xiao X, Min JL, Wang P, Chou KC. iGPCR-drug: a web server for predicting interaction between GPCRs and drugs in cellular networking. PLoS One 2013; 8(8): e72234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Oh DY, Kim K, Kwon HB, Seong JY. Cellular and molecular biology of orphan G protein-coupled receptors. Int Rev Cytol 2006; 252: 163–218. [DOI] [PubMed] [Google Scholar]
- [22].Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002; 298(5600): 1912–34. [DOI] [PubMed] [Google Scholar]
- [23].Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat Rev Genet 2010; 11(1): 60–74. [DOI] [PubMed] [Google Scholar]
- [24].Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer 2004; 4(3): 177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vieth M, Sutherland JJ, Robertson DH, Campbell RM. Kinomics: characterizing the therapeutically validated kinase space. Drug Discov Today 2005; 10(12): 839–46. [DOI] [PubMed] [Google Scholar]
- [26].Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood 2005; 105(7): 2640–53. [DOI] [PubMed] [Google Scholar]
- [27].Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 2007; 15(3): 299–311. [DOI] [PubMed] [Google Scholar]
- [28].Osadchy M, Kolodny R. Maps of protein structure space reveal a fundamental relationship between protein structure and function. Proc Natl Acad Sci USA 2011; 108(30): 12301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Roy A, Zhang Y. Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure 2012; 20(6): 987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang K, Horst JA, Cheng G, Nickle DC, Samudrala R. Protein meta-functional signatures from combining sequence, structure, evolution, and amino acid property information. PLoS Comput Biol 2008; 4(9): e1000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang K, Samudrala R. FSSA: a novel method for identifying functional signatures from structural alignments. Bioinformatics 2005; 21(13): 2969–77. [DOI] [PubMed] [Google Scholar]
- [32].Vyas VK, Ukawala RD, Ghate M, Chintha C. Homology modeling a fast tool for drug discovery: current perspectives. Indian J Pharm Sci 2012; 74(1): 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jenwitheesuk E, Samudrala R. Heptad-repeat-2 mutations enhance the stability of the enfuvirtide-resistant HIV-1 gp41 hairpin structure. Antivir Ther 2005; 10(8): 893–900. [PubMed] [Google Scholar]
- [34].Jenwitheesuk E, Samudrala R. Prediction of HIV-1 protease inhibitor resistance using a protein-inhibitor flexible docking approach. Antivir Ther 2005; 10(1): 157–66. [PubMed] [Google Scholar]
- [35].Wang K, Jenwitheesuk E, Samudrala R, Mittler JE. Simple linear model provides highly accurate genotypic predictions of HIV-1 drug resistance. Antivir Ther 2004; 9(3): 343–52. [PubMed] [Google Scholar]
- [36].Wang K, Samudrala R, Mittler JE. HIV-1 genotypic drug-resistance interpretation algorithms need to include hypersusceptibility-associated mutations. J Infect Dis 2004; 190(11): 2055–6; author reply 2056. [DOI] [PubMed] [Google Scholar]
- [37].Howell DP, Samudrala R, Smith JD. Disguising itself--insights into Plasmodium falciparum binding and immune evasion from the DBL crystal structure. Mol Biochem Parasitol 2006; 148(1): 1–9. [DOI] [PubMed] [Google Scholar]
- [38].Bockhorst J, Lu F, Janes JH, et al. Structural polymorphism and diversifying selection on the pregnancy malaria vaccine candidate VAR2CSA. Mol Biochem Parasitol 2007; 155(2): 103–12. [DOI] [PubMed] [Google Scholar]
- [39].Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol 2005; 205(2): 275–92. [DOI] [PubMed] [Google Scholar]
- [40].Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer 2007; 7(5): 345–56. [DOI] [PubMed] [Google Scholar]
- [41].Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293(5531): 876–80. [DOI] [PubMed] [Google Scholar]
- [42].Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2(2): 117–25. [DOI] [PubMed] [Google Scholar]
- [43].Gruber FX, Ernst T, Porkka K, et al. Dynamics of the emergence of dasatinib and nilotinib resistance in imatinib-resistant CML patients. Leukemia 2012; 26(1): 172–7. [DOI] [PubMed] [Google Scholar]
- [44].Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 2010; 9(3): 203–14. [DOI] [PubMed] [Google Scholar]
- [45].Fabian MA, Biggs WH 3rd, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 2005; 23(3): 329–36. [DOI] [PubMed] [Google Scholar]
- [46].Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008; 26(1): 127–32. [DOI] [PubMed] [Google Scholar]
- [47].Carroll M, Ohno-Jones S, Tamura S, et al. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood 1997; 90(12): 4947–52. [PubMed] [Google Scholar]
- [48].Dewar AL, Cambareri AC, Zannettino AC, et al. Macrophage colony-stimulating factor receptor c-fms is a novel target of imatinib. Blood 2005; 105(8): 3127–32. [DOI] [PubMed] [Google Scholar]
- [49].Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ.. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000; 96(3): 925–32. [PubMed] [Google Scholar]
- [50].Okuda K, Weisberg E, Gilliland DG, Griffin JD.. ARG tyrosine kinase activity is inhibited by STI571. Blood 2001; 97(8): 2440–8. [DOI] [PubMed] [Google Scholar]
- [51].Boissel N, Rousselot P, Raffoux E, et al. Defective blood dendritic cells in chronic myeloid leukemia correlate with high plasmatic VEGF and are not normalized by imatinib mesylate. Leukemia 2004; 18(10): 1656–61. [DOI] [PubMed] [Google Scholar]
- [52].Gao H, Lee BN, Talpaz M, et al. Imatinib mesylate suppresses cytokine synthesis by activated CD4 T cells of patients with chronic myelogenous leukemia. Leukemia 2005; 19(11): 1905–11. [DOI] [PubMed] [Google Scholar]
- [53].Mattiuzzi GN, Cortes JE, Talpaz M, et al. Development of Varicella-Zoster virus infection in patients with chronic myelogenous leukemia treated with imatinib mesylate. Clin Cancer Res 2003; 9(3): 976–80. [PubMed] [Google Scholar]
- [54].Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol 2005; 12(6): 621–37. [DOI] [PubMed] [Google Scholar]
- [55].Sciabola S, Stanton RV, Wittkopp S, et al. Predicting kinase selectivity profiles using Free-Wilson QSAR analysis. J Chem Inf Model 2008; 48(9): 1851–67. [DOI] [PubMed] [Google Scholar]
- [56].Huang D, et al. Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 2010; 26(2): 198–204. [DOI] [PubMed] [Google Scholar]
- [57].Huang D, Zhou T, Lafleur K, Nevado C, Caflisch A.. A global approach to analysis and interpretation of metabolic data for plant natural product discovery. Nat Prod Rep 2013; 30(4): 565–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Goldberg DE, Siliciano RF, Jacobs WR Jr., Outwitting evolution: fighting drug-resistant TB, malaria, and HIV. Cell 2012; 148(6): 1271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 2010; 74(3): 417–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hirschel B, Francioli P. Progress and problems in the fight against AIDS. N Engl J Med 1998; 338(13): 906–8. [DOI] [PubMed] [Google Scholar]
- [61].Borisy AA, Elliott PJ, Hurst NW, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA 2003; 100(13): 7977–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kar S, Roy K. How far can virtual screening take us in drug discovery? Expert Opin Drug Discov 2013; 8(3): 245–61. [DOI] [PubMed] [Google Scholar]
- [63].Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol Pharmacol 2000; 57(2): 213–8. [PubMed] [Google Scholar]
- [64].Cross JB, Thompson DC, Rai BK, et al. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model 2009; 49(6): 1455–74. [DOI] [PubMed] [Google Scholar]
- [65].Biesiada J, Porollo A, Velayutham P, Kouril M, Meller J.. Survey of public domain software for docking simulations and virtual screening. Hum Genom 2011; 5(5): 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Brewerton SC, The use of protein-ligand interaction fingerprints in docking. Curr Opin Drug Discov Dev 2008; 11(3): 356–64. [PubMed] [Google Scholar]
- [67].Huang SY, Zou X. Ensemble docking of multiple protein structures: considering protein structural variations in molecular docking. Proteins 2007; 66(2): 399–421. [DOI] [PubMed] [Google Scholar]
- [68].Huang SY, Zou X. Efficient molecular docking of NMR structures: application to HIV-1 protease. Protein Sci 2007; 16(1): 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004; 47(7): 1739–49. [DOI] [PubMed] [Google Scholar]
- [70].Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004; 56(2): 235–49. [DOI] [PubMed] [Google Scholar]
- [71].Ou-Yang SS. Computational drug discovery. Acta Pharmacol Sin 2012; 33(9): 1131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Deng Z, Chuaqui C, Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem 2004; 47(2): 337–44. [DOI] [PubMed] [Google Scholar]
- [73].Pouliot Y, Chiang AP, Butte AJ. Predicting adverse drug reactions using publicly available PubChem BioAssay data. Clin Pharmacol Ther 2011; 90(1): 90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bernard B, Samudrala R. A generalized knowledge-based discriminatory function for biomolecular interactions. Proteins 2009; 76(1): 115–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Yuriev E, Ramsland PA. Latest developments in molecular docking: 2010–2011 in review. J Mol Recognit 2013; 26(5): 215–39. [DOI] [PubMed] [Google Scholar]
- [76].Grigoryan AV, Wang H, Cardozo TJ. Can the energy gap in the protein-ligand binding energy landscape be used as a descriptor in virtual ligand screening? PLoS One 2012; 7(10): e46532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010; 31(2): 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins 2003; 52(4): 609–23. [DOI] [PubMed] [Google Scholar]
- [79].Tubert-Brohman I, Sherman W, Repasky M, Beuming T. Improved docking of polypeptides with Glide. J Chem Inf Model 2013; 53(7): 1689–99. [DOI] [PubMed] [Google Scholar]
- [80].Damm-Ganamet KL, Smith RD, Dunbar JB Jr, Stuckey JA, Carlson HA. CSAR benchmark exercise 2011–2012: evaluation of results from docking and relative ranking of blinded congeneric series. J Chem Inf Model 2013; 53(8): 1853–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Jenwitheesuk E, Samudrala R. Improved prediction of HIV-1 protease-inhibitor binding energies by molecular dynamics simulations. BMC Struct Biol 2003; 3: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chopra G A knowledge-based fragment docking with dynamics protocol for protein-ligand docking. unpublished. [Google Scholar]
- [83].Konc J, Samudrala R, Chopra G. Candock: Computational analytics on evolutionary interaction network based docking. unpublished. [Google Scholar]
- [84].Chopra G, Kalisman N, Levitt M. Consistent refinement of submitted models at CASP using a knowledge-based potential. Proteins 2010; 78(12): 2668–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Chopra G, Summa CM, Levitt M. Solvent dramatically affects protein structure refinement. Proc Natl Acad Sci USA 2008; 105(51): 20239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rodrigues JP, Levitt M, Chopra G. KoBaMIN: a knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res 2012; 40(Web Server issue): W323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Samudrala R, Moult J. An all-atom distance-dependent conditional probability discriminatory function for protein structure prediction. J Mol Biol 1998; 275(5): 895–916. [DOI] [PubMed] [Google Scholar]
- [88].Samudrala R, Moult J. A graph-theoretic algorithm for comparative modeling of protein structure. J Mol Biol 1998; 279(1): 287–302. [DOI] [PubMed] [Google Scholar]
- [89].Samudrala R, Xia Y, Huang E, Levitt M.. Ab initio protein structure prediction using a combined hierarchical approach. Proteins 1999; Suppl 3: 194–8. [DOI] [PubMed] [Google Scholar]
- [90].Xia Y, Huang ES, Levitt M, Samudrala R. Ab initio construction of protein tertiary structures using a hierarchical approach. J Mol Biol 2000; 300(1): 171–85. [DOI] [PubMed] [Google Scholar]
- [91].Bird JJ, Brown DR, Mullen AC, Helper T cell differentiation is controlled by the cell cycle. Immunity 1998; 9(2): 229–237. [DOI] [PubMed] [Google Scholar]
- [92].Cheng F, Zhou Y, Li W, Liu G, Tang Y. Prediction of chemical-protein interactions network with weighted network-based inference method. PLoS One 2012; 7(7): e41064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Cheng F, Liu C, Jiang J, et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput Biol 2012; 8(5): e1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Bader GD, Betel D, Hogue CW. BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res 2003; 31(1): 248–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 1991; 9(1): 56–68. [DOI] [PubMed] [Google Scholar]
- [96].McDermott J, Guerquin M, Frazier Z, Chang AN, Samudrala R. et al. BIOVERSE: enhancements to the framework for structural, functional and contextual modeling of proteins and proteomes. Nucleic Acids Res 2005; 33(Web Server issue): W324–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].McDermott J, Bumgarner R, Samudrala R. Functional annotation from predicted protein interaction networks. Bioinformatics 2005; 21(15): 3217–26. [DOI] [PubMed] [Google Scholar]
- [98].McDermott J, Samudrala R. Enhanced functional information from predicted protein networks. Trends Biotechnol 2004; 22(2): 60–2; discussion 62–3. [DOI] [PubMed] [Google Scholar]
- [99].Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci 2005; 26(4): 178–82. [DOI] [PubMed] [Google Scholar]
- [100].Chopra G, Samudrala R. Multiscale modeling of complex molecular and physiological systems with application to shotgun drug repurposing for myriad diseases. unpublished. [Google Scholar]
- [101].Chopra G Discovery and verification of a novel broad spectrum herpes virus inhibitor. unpublished. [Google Scholar]
- [102].Chopra G Systems biology based approach to repurpose FDA human approved drugs for treatment of autoimmune diabetes in NOD mice. unpublished. [Google Scholar]
- [103].Jenwitheesuk E, Samudrala R. Virtual screening of HIV-1 protease inhibitors against human cytomegalovirus protease using docking and molecular dynamics. AIDS 2005; 19(5): 529–31. [DOI] [PubMed] [Google Scholar]
- [104].Jenwitheesuk E, Samudrala R. Identification of potential HIV-1 targets of minocycline. Bioinformatics 2007; 23(20): 2797–9. [DOI] [PubMed] [Google Scholar]
- [105].Jenwitheesuk E, Wang K, Mittler JE, Samudrala R. Improved accuracy of HIV-1 genotypic susceptibility interpretation using a consensus approach. AIDS 2004; 18(13): 1858–9. [DOI] [PubMed] [Google Scholar]
- [106].Xu D, Zhang J, Roy A, Zhang Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins 2011; 79 (Suppl 10): 147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 2008; 9: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods 2012; 9(2): 173–5. [DOI] [PubMed] [Google Scholar]
- [109].Yang Z, Lasker K, Schneidman-Duhovny D, et al. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol 2012; 179(3): 269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hung LH, Ngan SC, Liu T, Samudrala R. PROTINFO: new algorithms for enhanced protein structure predictions. Nucleic Acids Res 2005; 33(Web Server issue): W77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Khoury GA, Liwo A, Khatib F, et al. WeFold: a coopetition for protein structure prediction. Proteins 2014; 82(9): 1850–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Samudrala R, Pedersen JT, Zhou HB, Luo R, Fidelis K, Moult J. Confronting the problem of interconnected structural changes in the comparative modeling of proteins. Proteins 1995; 23(3): 327–36. [DOI] [PubMed] [Google Scholar]
- [113].Samudrala R, Moult J. Handling context-sensitivity in protein structures using graph theory: bona fide prediction. Proteins 1997; (Suppl 1): 43–9. [DOI] [PubMed] [Google Scholar]
- [114].Samudrala R, Moult J. Determinants of side chain conformational preferences in protein structures. Protein Eng 1998; 11(11): 991–7. [DOI] [PubMed] [Google Scholar]
- [115].Samudrala R, Xia Y, Levitt M, Huang ES. A combined approach for ab initio construction of low resolution protein tertiary structures from sequence. Pac Symp Biocomput 1999: 505–16. [DOI] [PubMed] [Google Scholar]
- [116].Huang ES, Samudrala R, Ponder JW. Ab initio fold prediction of small helical proteins using distance geometry and knowledge-based scoring functions. J Mol Biol 1999; 290(1): 267–81. [DOI] [PubMed] [Google Scholar]
- [117].Samudrala R, Huang ES, Koehl P, Levitt M. Constructing side chains on near-native main chains for ab initio protein structure prediction. Protein Eng 2000; 13(7): 453–7. [DOI] [PubMed] [Google Scholar]
- [118].Huang ES, Samudrala R, Park BH. Scoring functions for ab initio protein structure prediction. Methods Mol Biol 2000; 143: 223–45. [DOI] [PubMed] [Google Scholar]
- [119].Samudrala R, Levitt M. Decoys 'R' Us: a database of incorrect conformations to improve protein structure prediction. Protein Sci 2000; 9(7): 1399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Samudrala R, Levitt M. A comprehensive analysis of 40 blind protein structure predictions. BMC Struct Biol 2002; 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hung LH, Samudrala R. Accurate and automated classification of protein secondary structure with PsiCSI. Protein Sci 2003; 12(2): 288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hung LH, Samudrala R. PROTINFO: Secondary and tertiary protein structure prediction. Nucleic Acids Res 2003; 31(13): 3296–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wang K, Fain B, Levitt M, Samudrala R. Improved protein structure selection using decoy-dependent discriminatory functions. BMC Struct Biol 2004; 4: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ngan SC, Hung LH, Liu T, Samudrala R. Scoring functions for de novo protein structure prediction revisited. Methods Mol Biol 2008; 413: 243–81. [DOI] [PubMed] [Google Scholar]
- [125].Liu T, Guerquin M, Samudrala R. Improving the accuracy of template-based predictions by mixing and matching between initial models. BMC Struct Biol 2008; 8: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kittichotirat W, Guerquin M, Bumgarner RE, Samudrala R. Protinfo PPC: a web server for atomic level prediction of protein complexes. Nucleic Acids Res 2009; 37(Web Server issue): W519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Liu T, Horst JA, Samudrala R. A novel method for predicting and using distance constraints of high accuracy for refining protein structure prediction. Proteins 2009; 77(1): 220–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Horst J, Samudrala R. Diversity of protein structures and difficulties in fold recognition: the curious case of protein G. F1000 Biol Rep 2009; 1: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hung LH, Samudrala R. Accelerated protein structure comparison using TM-score-GPU. Bioinformatics 2012; 28(16): 2191–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Hung LH, Guerquin M, Samudrala R. GPU-Q-J, a fast method for calculating root mean square deviation (RMSD) after optimal superposition. BMC Res Notes 2011; 4: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Moughon SE, Samudrala R. LoCo: a novel main chain scoring function for protein structure prediction based on local coordinates. BMC Bioinformatics 2011; 12: 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Laurenzi A, Hung LH, Samudrala R. Structure prediction of partial-length protein sequences. Int J Mol Sci 2013; 14(7): 14892–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].McDermott JE, Corrigan A, Peterson E, et al. Computational prediction of type III and IV secreted effectors in gram-negative bacteria. Infect Immun 2011; 79(1): 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Samudrala R, Heffron F, McDermott JE. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog 2009; 5(4): e1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Viré E, Brenner C, Deplus R, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006; 439(7078): 871–4. [DOI] [PubMed] [Google Scholar]
- [136].Granada JF, Kleiman NS. Therapeutic use of intravenous eptifibatide in patients undergoing percutaneous coronary intervention: acute coronary syndromes and elective stenting. Am J Cardiovasc Drugs 2004; 4(1): 31–41. [DOI] [PubMed] [Google Scholar]
- [137].O'Shea JC, Tcheng JE. Eptifibatide: a potent inhibitor of the platelet receptor integrin glycoprotein IIb/IIIa. Expert Opin Pharmacother 2002; 3(8): 1199–210. [DOI] [PubMed] [Google Scholar]
- [138].Chang YL, Lin AT, Chen KK. Short-term effects of desmopressin on water and electrolyte excretion in adults with nocturnal polyuria. J Urol 2007; 177(6): 2227–9; discussion 2230. [DOI] [PubMed] [Google Scholar]
- [139].Carraro A, Fano M, Cuttica M, Bernareggi V, Giusti M, Giordano G. Long-term treatment of central diabetes insipidus with oral DDAVP. Minerva Endocrinol 1992; 17(4): 189–93. [PubMed] [Google Scholar]
- [140].Breitinger HG. Drug Synergy – Mechanisms and Methods of Analysis, Toxicity and Drug Testing. In: Acree W, Ed. Toxicity and Drug Testing. INTECH Open Access Publisher; 2012. [Google Scholar]
- [141].Shtivelband MI. Everolimus in hormone receptor-positive advanced breast cancer: targeting receptor-based mechanisms of resistance. Breast 2013; 22(4): 405–10. [DOI] [PubMed] [Google Scholar]
- [142].Milani A, Geuna E1, Mittica G1, Valabrega G1. Overcoming endocrine resistance in metastatic breast cancer: Current evidence and future directions. World J Clin Oncol 2014; 5(5): 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Yardley DA, Noguchi S, Pritchard KI, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther 2013; 30(10): 870–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Hortobagyi GN. Everolimus plus exemestane for the treatment of advanced breast cancer: a review of subanalyses from BOLERO-2. Neoplasia 2015; 17(3): 279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Beaver JA, Park BH. The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol 2012; 8(6): 651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Deininger MW, Goldman JM, Lydon N, Melo JV. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood 1997; 90(9): 3691–8. [PubMed] [Google Scholar]
- [147].Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996; 2(5): 561–6. [DOI] [PubMed] [Google Scholar]
- [148].Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001; 344(14): 1031–7. [DOI] [PubMed] [Google Scholar]
- [149].Kantarjian HM, Cortes JE, O'Brien S, et al. Long-term survival benefit and improved complete cytogenetic and molecular response rates with imatinib mesylate in Philadelphia chromosome-positive chronic-phase chronic myeloid leukemia after failure of interferon-alpha. Blood 2004; 104(7): 1979–88. [DOI] [PubMed] [Google Scholar]
- [150].Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344(14): 1052–6. [DOI] [PubMed] [Google Scholar]
- [151].Pardanani A Imatinib for systemic mast-cell disease. Lancet 2003; 362(9383): 535–6. [DOI] [PubMed] [Google Scholar]
- [152].Pardanani A, Reeder T, Porrata LF, et al. Imatinib therapy for hypereosinophilic syndrome and other eosinophilic disorders. Blood 2003; 101(9): 3391–7. [DOI] [PubMed] [Google Scholar]
- [153].Kuhn M, Szklarczyk D, Pletscher-Frankild S, et al. STITCH 4: integration of protein-chemical interactions with user data. Nucleic Acids Res 2014; 42(Database issue): D401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]