

Abstract
The aldehyde dehydrogenase (ALDH) superfamily is a vast group of enzymes that catalyze the NAD+-dependent oxidation of aldehydes to carboxylic acids. ALDH16 is perhaps the most enigmatic member of the superfamily, owing to its extra C-terminal domain of unknown function and the absence of the essential catalytic cysteine residue in certain non-bacterial ALDH16 sequences. Herein we report the first production of recombinant ALDH16, the first biochemical characterization of ALDH16, and the first crystal structure of ALDH16. Recombinant expression systems were generated for the bacterial ALDH16 from Loktanella sp. and human ALDH16A1. Four high resolution crystal structures of Loktanella ALDH16 were determined. Loktanella ALDH16 is found to be a bona fide enzyme, exhibiting NAD+-binding, ALDH activity, and esterase activity. In contrast, human ALDH16A1 apparently lacks measurable aldehyde oxidation activity, suggesting it is a pseudoenzyme, consistent with the absence of the catalytic Cys in its sequence. The fold of ALDH16 comprises three domains: NAD+-binding, catalytic, and C-terminal. The latter is unique to ALDH16 and features a Rossmann fold connected to a protruding β-flap. The tertiary structural interactions of the C-terminal domain mimic the quaternary structural interactions of the classic ALDH superfamily dimer, a phenomenon we call “trans-hierarchical structural similarity”. ALDH16 forms a unique dimer in solution, which mimics the classic ALDH superfamily dimer-of-dimers tetramer. Small-angle X-ray scattering shows that human ALDH16A1 has the same dimeric structure and fold as Loktanella ALDH16. We suggest the Loktanella ALDH16 structure may be considered to be the archetype of the ALDH16 family.
Keywords: X-ray crystallography, small-angle X-ray scattering, Rossmann fold, protein oligomerization, pseudoenzymes
Graphical abstract
Introduction
Aldehyde dehydrogenases (ALDHs) catalyze the NAD(P)+-dependent oxidization of aldehydes to carboxylic acids (Fig. 1A). They are widespread, existing in all organisms. A recent census of the ALDH superfamily uncovered 555 distinct genes, including 32 in archaea, 351 in eubacteria, and 172 in eukarya [1]. The human ALDH superfamily alone comprises 19 enzymes, which function in a multitude of pathways [2, 3], including retinoic acid signaling (ALDH1), detoxification of alcohol (ALDH2), maintaining cellular homeostasis in the eye (ALDH1A1, ALDH3A1), amino acid metabolism (ALDH4A1, ALDH7A1, ALDH18), neuronal function (ALDH2, ALDH5A1, ALDH9A1), and folate metabolism (ALDH1L). Mutations in ALDH genes cause several inherited metabolic disorders, including type II hyperprolinemia, Sjogren-Larsson syndrome, pyridoxine-dependent epilepsy, γ-hydroxybutyric aciduria, and hyperammonemia [2]. Also, some ALDH polymorphisms are associated with an increased risk of alcoholic liver disease [4]. Finally, ALDHs are markers of cancer stem cells and potentially therapeutic targets for cancer treatment [5].
Fig. 1.
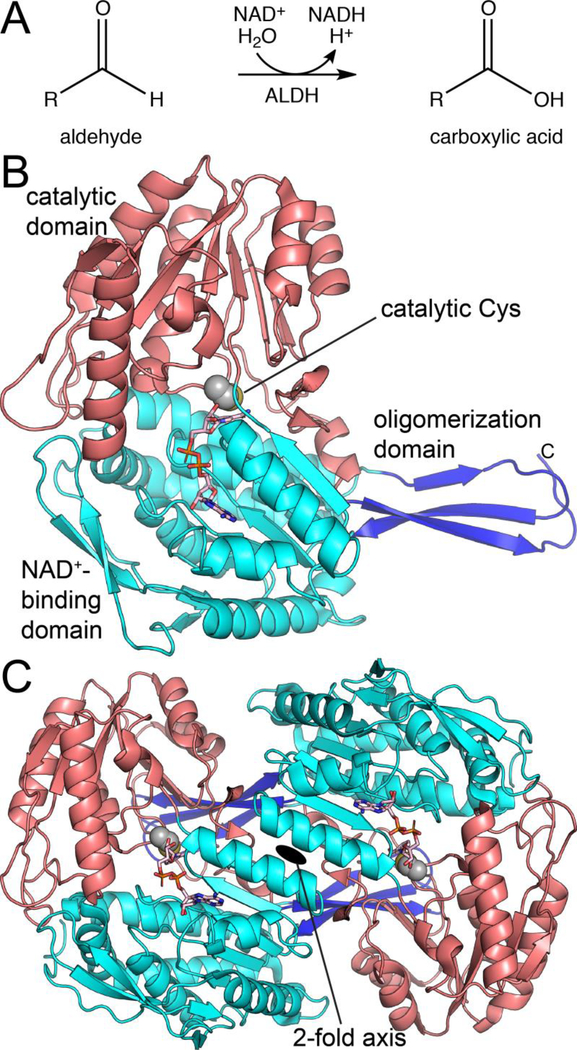
The classical view of the ALDH superfamily. (A) The general reaction catalyzed by ALDHs. (B) Structure of a canonical ALDH, ALDH2 (PDB ID 1O00). NAD+ is shown in pink sticks. (C) Structure of the canonical ALDH dimer, as exemplified by ALDH2 (PDB ID 1O00).
ALDHs share a common protein fold, quaternary structure, and catalytic mechanism. The classical ALDH fold exhibits a three-domain architecture consisting of a Rossmann dinucleotide binding domain, an α/β-catalytic domain, and an oligomerization domain (Fig. 1B). The latter domain mediates a conserved mode of domain-swapped dimerization (Fig. 1C). In many ALDHs, the dimers assemble into higher order oligomers, including tetramers (common) or hexamers (rare). The conserved ALDH catalytic mechanism requires an essential nucleophilic Cys residue, which attacks the carbonyl of the aldehyde substrate, forming hemithioacetal and acyl-enzyme covalent intermediates during the reaction cycle [2]. Numerous crystal structures and biochemical studies have established the three-domain fold, domain-swapped dimer, and conserved catalytic mechanism as hallmarks of the ALDH superfamily.
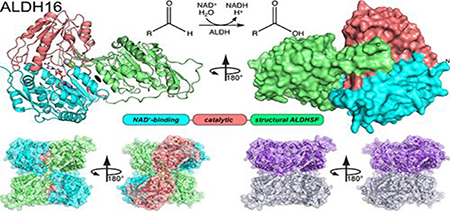
The dogma of a united superfamily was challenged a few years ago when it was noticed that ALDH4 (a.k.a. L-glutamate-γ-semialdehyde dehydrogenase) [6] and ALDH16 [7] deviate from the classical paradigm by exhibiting an atypical domain architecture. Some ALDH4 enzymes and all ALDH16 enzymes have a C-terminal ~250-residue domain not found in other ALDHs (Fig. 2A). Another remarkable feature is that certain ALDH16 members, including human ALDH16A1 (HsALDH16A1), lack the essential catalytic Cys residue, implying they do not possess catalytic activity (Fig. 2B) [7]. These unique features have motivated our structural studies of ALDH4 and ALDH16.
Fig. 2.

The atypical features of the ALDH16 family. (A) Domain diagram of ALDH16. ALDH16 is unique in the ALDH superfamily in having an extra domain near the C-terminus. Abbreviation: ALDHSF, aldehyde dehydrogenase superfamily. (B) ALDH16 phylogenetic tree and an alignment of the catalytic loops of several ALDH16s. Note the absence of the predicted catalytic Cys in some ALDH16 proteins.
We recently described the fold and functions of the C-terminal domain of ALDH4 [8–10]. Those studies utilized the proline catabolic bifunctional enzyme proline utilization A (PutA), which exhibits both proline dehydrogenase and ALDH4 activities. The crystal structures of PutA revealed that the C-terminal domain draws from the repertoire of the ALDH superfamily [8, 9]. Its structure consists of a Rossmann fold that resembles the NAD+-binding domain of classical ALDHs, and a β-flap that resembles the oligomerization domain of typical ALDHs. Because of these structural relationships, we refer to the C-terminal domain of PutA as having an ALDH superfamily fold. Paradoxically, the Rossmann fold of the C-terminal domain does not bind NAD+, nor does the β-flap of the C-terminal domain mediate oligomerization. Instead, the C-terminal domain in PutA functions as an adaptor module whose tertiary structural interactions contribute to ALDH catalytic activity by stabilizing both the NAD+-binding domain and the aldehyde substrate-binding loop [9, 10]. Additionally, the β-flap of the C-terminal domain contributes to substrate channeling by forming part of the tunnel that connects the proline dehydrogenase and ALDH4 active sites.
Herein we turn our attention to the other atypical ALDH superfamily member, ALDH16. Because of its noncanonical features and the lack of either crystal structures or any biochemical characterization, ALDH16 is the least understood member of the ALDH superfamily. ALDH16 is found in animals and some bacteria, but is absent in archaea, fungi and plants [7]. A missense variant in the ALDH16A1 gene is associated with elevated serum uric acid levels and gout [11]. ALDH16A1 may play a role in the autosomal-recessive Mast syndrome (SPG21; MIM 248900) based on immunoprecipitation data implying a protein-protein association with maspardin [12]. Also, recent studies of an ALDH16A1 gene knock-out mouse suggest a role for ALDH16A1 in lipid metabolism and renal function [13]. Although these studies are suggestive of possible roles for ALDH16A1 in human health and disease, more research is clearly needed to understand the functions of ALDH16.
To better understand the molecular function of enigmatic ALDH16 and to provide a foundation for exploring its biological functions, we have determined the first crystal structure of an ALDH16 family member. Recombinant expression systems for both bacterial ALDH16 and HsALDH16A1 were generated. Four crystal structures of a bacterial ALDH16 from Loktanella sp. (LsALDH16) were determined at high resolution limits ranging from 1.5 Å to 2.3 Å. The structures reveal the fold and function of the C-terminal domain, and uncover a fascinating example of trans-hierarchy structural similarity, in which the tertiary structure of ALDH16 mimics the quaternary structure of the classic ALDH dimer. ALDH16 forms a novel dimer, which resembles the classic ALDH tetramer. LsALDH16 is shown to be a genuine ALDH enzyme, whereas HsALDH16A1 appears to be a pseudoenzyme. Small-angle X-ray scattering (SAXS) suggests LsALDH16 and HsALDH16A1 are similar in fold and quaternary structure.
Results and Discussion
The fold of ALDH16
Homolog screening was used to identify an ALDH16 family member that was amenable to high resolution X-ray crystallography. The ALDH16 proteins from human, rat, mouse, bovine, and a few bacteria were produced and screened for crystallization. Diffracting crystals were obtained with the bacterial enzymes from Litoreibacter arenae (LaALDH16) and Loktanella sp. (LsALDH16). Moderate resolution crystals of LaALDH16 were used for molecular replacement phasing and to build an initial model at 3 Å resolution (Table S1 of Supporting Information). The structure of LaALDH16 was then used in molecular replacement to solve high resolution structures of LsALDH16 (Table 1).
Table 1.
Data Collection Statistics for LsALDH16
| apo | NAD+ | NADH | C295A-pNPA | |
|---|---|---|---|---|
| Beamline | ALS 4.2.2 | APS 24-ID-C | ALS 4.2.2 | APS 24-ID-C |
| Space group | P21212 | P21212 | P21212 | P212121 |
| Unit cell parameters (Å, °) | a = 79.6, | a = 79.0, | a = 79.7, | a = 79.4, |
| b = 159.4, | b = 159.0, | b = 159.1, | b = 119.6, | |
| c = 62.7 | c = 62.44 | c = 62.7 | c = 158.7 | |
| Wavelength (Å) | 1.000 | 0.979 | 1.000 | 0.979 |
| Resolution (Å)a | 62.7 – 1.95 (2.00 – 1.95) | 79.5– 1.65 (1.68 – 1.65) | 62.7 – 2.30 (2.38 – 2.30) | 120 – 1.49 (1.51 – 1.49) |
| Observationsa | 374502 (15267) | 550661 | 151496 (14785) | 1628079 (71434) |
| Unique reflectionsa | 57273 (3249) | 92182 | 36080 (3487) | 243674 (11001) |
| Rmerge(I)a | 0.061 (0.255) | 0.101 (0.597) | 0.109 (0.406) | 0.120 (1.610) |
| Rmeas(I)a | 0.073 (0.318) | 0.121 (0.842) | 0.142 (0.528) | 0.142 (1.898) |
| Rpim(I)a | 0.038 (0.185) | 0.065 (0.593) | 0.090 (0.335) | 0.075 (0.994) |
| Mean I/σa | 21.1 (4.7) | 10.0 (1.2) | 9.2 (3.0) | 8.8 (0.9) |
| Mean CC1/2a | 0.998 (0.941) | 0.993 (0.471) | 0.992 (0.848) | 0.998 (0.440) |
| Completeness (%)a | 97.1 (80.0) | 96.8 (73.7) | 99.4 (99.9) | 99.8 (90.8) |
| Multiplicitya | 6.5 (4.7) | 6.0 (2.3) | 4.2 (4.2) | 6.7 (6.5) |
| Wilson B-factor (Å2) | 15.9 | 19.7 | 21.6 | 17.4 |
| No. protein residues | 750 | 751 | 751 | 1504 |
| No. of atoms | ||||
| Protein | 5567 | 5622 | 5555 | 11165 |
| NAD(H) | N/A | 27 | 44 | N/A |
| pNPA | N/A | N/A | N/A | 26 |
| Water | 821 | 748 | 423 | 1224 |
| Rworka | 0.1538 (0.2008) | 0.1686 (0.2767) | 0.1716 (0.2294) | 0.1916 (0.3414) |
| Rfreea,b | 0.1938 (0.2274) | 0.2000 (0.3246) | 0.2235 (0.3117) | 0.2158 (0.3536) |
| RMSD bonds (Å) | 0.007 | 0.006 | 0.007 | 0.005 |
| RMSD angles (°) | 0.805 | 0.810 | 0.827 | 0.807 |
| Ramachandran plotc | ||||
| Favored (%) | 97.46 | 98.13 | 97.33 | 97.73 |
| Outliers (%) | 0.13 | 0.13 | 0.13 | 0.13 |
| Clashscore (PR)c | 1.44 (100) | 1.68 (99) | 1.79 (100) | 1.61 (99) |
| MolProbity score (PR)c | 0.98 (100) | 0.99 (100) | 1.35 (100) | 0.97 (100) |
| Average B-factor (Å2) | ||||
| Protein | 17.5 | 22.4 | 23.7 | 20.4 |
| NAD(H) | N/A | 21.7 | 39.3 | N/A |
| pNPA | N/A | N/A | N/A | 42.8 |
| Water | 29.1 | 34.5 | 27.3 | 29.3 |
| Coordinate error (Å)d | 0.16 | 0.17 | 0.24 | 0.21 |
| PDB ID | 6MVR | 6MVS | 6MVT | 6MVU |
Values for the outer resolution shell of data are given in parentheses.
5 % test set.
From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parentheses.
Maximum likelihood-based coordinate error estimate reported by phenix.refine.
The domain architecture of LsALDH16 consists of a canonical ALDH superfamily catalytic module followed by the additional C-terminal domain (Fig. 3A). The catalytic module consists of a Rossmann fold NAD+-binding domain (residues 1 – 263) and an α/β-catalytic domain (residues 264 – 476), both of which are typical of the ALDH superfamily. Electron density clearly showed the predicted nucleophile Cys295 in the expected active site location, implying that LsALDH16 is a bona fide ALDH enzyme (Fig. 3A).
Fig. 3.
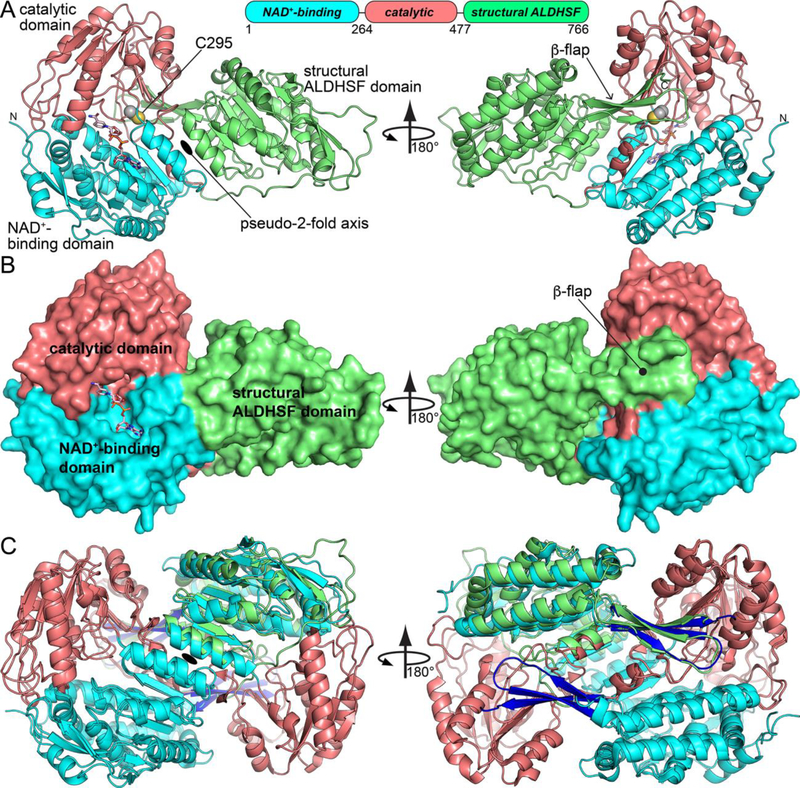
Crystal structure of LsALDH16. (A) Structure of the protomer, highlighting secondary structure and domain architecture. NADH is shown in pink sticks. (B) Surface representation of the LsALDH16 protomer. (C) Superposition of the LsALDH16 protomer with a dimer of ALDH2 showing trans-hierarchical structural symmetry. The coloring of the LsALDH16 domains is the same as in panel A. The color of the ALDH2 domains is the same as in Fig. 1. Note that LsALDH16 is structurally equivalent to three-fourths of a classical ALDH dimer.
The C-terminal domain (residues 477 – 766) distinguishes ALDH16 from canonical ALDHs. It features a 5-stranded Rossmann fold subdomain (residues 633 – 737) fused to a 3-stranded β-flap substructure (residues 612 – 632, 755 – 766) (Fig. 3A). Thus, the C-terminal domain of LsALDH16 has the ALDH superfamily fold, similar to ALDH4 in PutA.
The Rossmann fold and β-flap of the C-terminal domain resemble the NAD+-binding and oligomerization domains, respectively, of classical ALDHs. For example, the rmsd between the C-terminal domain and ALDH2 (PDB ID 1O00 [14]) is only 1.5 Å over 250 residues, despite only 27% sequence identity. Notably, 95% of the secondary structural elements of the ALDH16 C-terminal domain are also found in the combined NAD+-binding and oligomerization domains of ALDH2, indicating strong structural similarity.
Trans-hierarchical structural similarity
The ALDH16 structure reveals a fascinating example of structural similarity in which the tertiary structure of one protein mimics the quaternary structure of another, a phenomenon that we call “trans-hierarchical structural similarity”. In the present case, the tertiary structural interactions of the C-terminal domain of ALDH16 mimic the quaternary structural interactions in classical ALDH dimers.
To appreciate the trans-hierarchical structural similarity, consider the tertiary structural interactions of the C-terminal domain in LsALDH16. The C-terminal domain makes two sets of tertiary structural interactions with the catalytic module. First, the two Rossmann domains pack against each other and are related by a pseudo 2-fold axis that runs between the αD helices of each Rossmann fold (Fig. 3A, left). For reference, helix αD is the final helix of Rossmann fold. Despite the low sequence identity (30%), the two Rossmann folds of LsALDH16 are structurally similar and superimpose with an rmsd of 1.5 Å over 204 residues. Second, the β-flap of the C-terminal domain interacts with the β-sheet of the catalytic domain via main-chain hydrogen bonds, effectively extending the β-sheet by three strands (Fig. 3A, right). In this location, the β-flap of the C-terminal domain interacts with an active site loop that is known to be involved in substrate binding in ALDHs [15–18]. The large extent of these two sets of tertiary structural interactions is evident in the surface representation of the structure (Fig. 3B).
The tertiary structural contacts of the ALDH16 C-terminal domain are strikingly similar to the quaternary structural interactions in the classical ALDH dimer; consequently, ALDH16 resembles three-fourths of an ALDH dimer (Fig. 3C). The typical ALDH dimer (Fig. 1C) is a domain-swapped assembly featuring a two-point quaternary contact surface that is analogous to the tertiary structural interactions observed in LsALDH16. In particular, the NAD+-binding domains from different protomers in the classic dimer interact across a 2-fold axis near the αD helices, similar to what is seen in LsALDH16 monomer (compare Figs. 1C and 3A). Further, the oligomerization flap in the classical dimer extends the β-sheet of the catalytic domain of the opposite protomer, analogous to the tertiary structural interaction of the β-flap of ALDH16 with the catalytic domain (compare Figs. 1C and 3A). The high degree of trans-hierarchical structural similarity is characterized by an rmsd of only 1.4 Å between the ALDH16 monomer and the 650 equivalent residues in the ALDH2 dimer (Fig. 3C).
The ALDH16 dimer is unique, yet surprisingly familiar
The quaternary structure of ALDH16 in solution was studied using small-angle X-ray scattering (SAXS). SAXS curves from LsALDH16 at different protein concentrations were collected (Fig. 4A, Table 2). The qualitative shape of the scattering curve is constant with increasing concentration, consistently displaying a small bump near q = 0.10 – 0.15 Å−1. The Guinier plots yield a radius of gyration (Rg) in the range of 35 – 36 Å. Calculations of the distance distribution function suggest a maximum particle dimension (Dmax) of 106 – 112 Å and Rg of 35 – 36 Å (Fig. 4B). Because the Rg of a LsALDH16 monomer calculated from the crystal structure is only 28 Å, the data suggest that LsALDH16 forms an oligomer in solution.
Fig. 4.

SAXS analysis of ALDH16. (A) SAXS curves for LsALDH16 collected at three protein concentrations. The dots are experimental data colored as follows: black, 2.0 mg/ml; blue, 4.0 mg/ml; green, 6.0 mg/ml. The SAXS data have been arbitrarily scaled for ease of presentation. The red curves are theoretical SAXS profiles calculated from the crystallographic homodimer with FoXS [59]. The lower inset shows error-weighted residual plots for the FoXS fits. The upper inset shows Guinier plots. (B) SAXS experimental distance distribution functions for LsALDH16. The inset shows the normalized P(r) plots. (C) SAXS curves for HsALDH16A1 collected at three protein concentrations. The dots are experimental data colored as follows: black, 1.0 mg/ml; blue, 1.6 mg/ml; green, 3.2 mg/ml. The SAXS data have been arbitrarily scaled for ease of presentation. The red curves are theoretical SAXS profiles calculated by FoXS from a dimer homology model of HsALDH16A1 based on the LsALDH16 crystallographic dimer. The lower inset shows error-weighted residual plots for the FoXS fits. The upper inset shows Guinier plots. (D) SAXS experimental distance distribution functions for HsALDH16A1. The inset shows the normalized P(r) plots.
Table 2.
SAXS analysis of LsALDH16 and HsALDH16A1
| LsALDH16 | HsALDH16A1 | |||||
|---|---|---|---|---|---|---|
| 2.0 mg/ml | 4.0 mg/ml | 6.0 mg/ml | 1.0 mg/ml | 1.6 mg/ml | 3.2 mg/ml | |
| Guinier Analysis | ||||||
| Rg (Å) | 35.6 ± 1.0 | 36.1 ± 0.4 | 35.0 ± 0.6 | 36.3 ± 1.0 | 37.5 ± 0.9 | 37.6 ± 0.5 |
| qmin(Å−1) | 0.015887 | 0.015330 | 0.018675 | 0.015887 | 0.011985 | 0.014215 |
| qRg range | 0.57 – 1.28 | 0.55 – 1.30 | 0.65 – 1.22 | 0.58 – 1.29 | 0.45 – 1.29 | 0.53 – 1.29 |
| R-square | 0.9972 | 0.9991 | 0.9984 | 0.9899 | 0.9930 | 0.9980 |
| P(r) analysisa | ||||||
| Rg (Å) | 35.5 ± 0.2 | 35.68 ± 0.06 | 35.22 ± 0.04 | 36.65 ± 0.08 | 37.50 ± 0.06 | 37.48 ± 0.06 |
| Dmax (Å) | 109 | 112 | 106 | 109 | 112 | 115 |
| q-range (Å−1) | 0.0159 – 0.3219 | 0.0153 – 0.3208 | 0.0187 – 0.3208 | 0.0159 – 0.3219 | 0.0131 – 0.3219 | 0.0148 – 0.3213 |
| Total estimate | 0.91 | 0.90 | 0.94 | 0.92 | 0.93 | 0.88 |
| Porod Volume (Å3) | 202000 | 204000 | 207000 | 230000 | 236000 | 237000 |
| SAXS mass (kDa)b | 141.5 (0.93) | 151.3 (1.00) | 149.2 (0.98) | 147.1 (0.86) | 156.0 (0.92) | 170.1 (1.00) |
| χ2 from FoXSc | 0.16 | 0.68 | 0.95 | 1.12 | 1.45 | 2.38 |
| SASBDB code | SASDE47 | SASDE57 | SASDE67 | SASDE87 | SASDE97 | SASDEA7 |
From calculations of the distance distribution function using GNOM via PRIMUS.
Molecular mass estimated from the SAXS MoW2 server [58]. The ratio of the experimental molecular mass to the expected value is given in in parentheses. For reference, the dimer masses of LsALDH16 and HsALDH16A1 are 151.6 kDa and 170.3 kDa, respectively.
Goodness-of-fit parameter from FoXS using a dimer model (Fig. 4).
The LsALDH16 oligomer was characterized by comparing the experimental SAXS curve with theoretical ones calculated from oligomer models using FoXS [19]. Analysis of the crystal lattice with PDBePISA [20] suggests a stable dimer with Rg of 34 Å (The P21212) crystallographic asymmetric unit contains one protomer of this dimer). The SAXS curve calculated from the dimer has excellent agreement with the experimental profiles (χ2 = 0.16 – 0.95) (Fig. 4A). The use of dimer-monomer ensembles did not improve the fits. These results suggest that LsALDH16 is predominantly dimeric in solution, and the dimer in solution is the one identified from the crystal lattice by PDBePISA analysis.
We also performed SAXS on HsALDH16A1, which has 37% sequence identity to LsALDH16. The SAXS curve and distance distribution of HsALDH16A1 are strikingly similar to those of LsALDH16 (Figs. 4C and 4D). The Rg of 36 – 38 Å is very close to that of LsALDH16 (35 – 36 Å). The slightly higher Rg may reflect the longer polypeptide chain length of HsALDH16A1 of 802 residues compared to 766 for LsALDH16. These results suggest HsALDH16A1 may have the same oligomeric structure as LsALDH16. We tested this idea by calculating theoretical scattering curves from a model of an HsALDH16A1 dimer built using SWISS-MODEL with the LsALDH16 crystallographic dimer serving as the template for homology modeling. The SAXS curve calculated from the HsALDH16A1 dimer model exhibits very good agreement with the experimental data (χ2 = 1.1 – 2.4) (Fig. 4C, Table 2). These results suggest that human and bacterial ALDH16 share a common oligomeric structure and likely have the same protein fold.
The ALDH16 dimer is unique in the ALDH superfamily. The dimer is shaped like a cube, with the two protomers occupying the upper and lower halves of the cube (Fig. 5A, top). When viewed down the 2-fold axis from one direction, the two catalytic domains are arranged diagonally on the face of the cube (Fig. 5A, bottom left). From the opposite direction, the NAD+-binding and C-terminal domains are arranged on the diagonals, forming an “X” (Fig. A, bottom right).
Fig. 5.
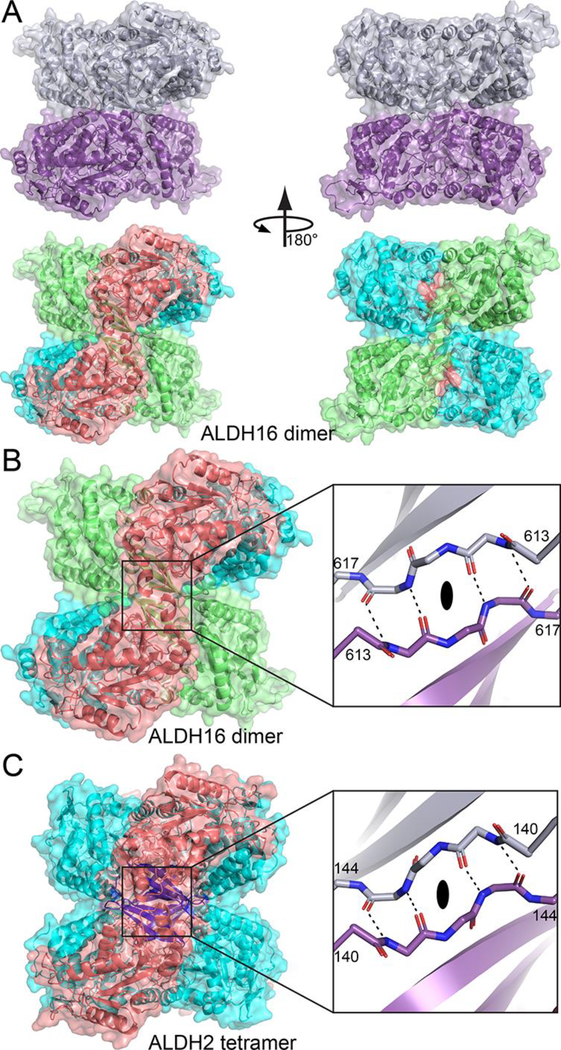
A novel ALDH dimer, which mimics the classic ALDH tetramer. (A) The LsALDH16 dimer viewed down the 2-fold axis. In the top part, the two chains have different colors. In the bottom part, the protein is color-coded by domains as in Fig. 3A: NAD+-binding, cyan; catalytic, deep salmon; and C-terminal domain, lime. (B) Close-up view of the intermolecular anti-parallel β-sheet formed by the β-flaps of the C-terminal domains. The two chains have different colors in the inset. (C) The classic ALDH tetramer, as exemplified by ALDH2, viewed down a 2-fold axis (PDB ID 1O00). On the left, the protein is colored by domains as in Fig. 1: NAD+-binding, cyan; catalytic, deep salmon; and oligomerization, blue. The inset shows a close-up view of the intermolecular anti-parallel β-sheet formed by the oligomerization domains of chains B and C. An identical interaction is formed by chains A and D.
The C-terminal domain is prominent in the dimer interface. The β-flaps of C-terminal domains from the two protomers meet at the center of the dimer to form an intermolecular β-sheet (Fig. 5B). Another large part of the dimer interface involves the α-helices of the C-terminal domain packing against α-helices of the NAD+-binding domain of the opposite protomer (Fig. 5B). In all, the dimer interface buries 2500 Å2 and includes 65 residues, 34 hydrogen bonds and 16 ion pairs.
Although unique, the ALDH16 dimer is surprisingly familiar. The ALDH16 dimer is reminisent of the tetramer formed by most other ALDHs, including ALDH1 [21–24], ALDH2 [25, 26], ALDH5A1 [27], ALDH7A1 [17, 28], and many others [29–37]. The classical tetramer is a dimer-of-dimers assembly in which the oligomerization flaps of different dimers meet in the center of the oligomer to form intermolecular β-sheet interactions (Fig. 5C). This interaction is strikingly similar to the intermolecular β-sheet of the ALDH16 dimer (Fig. 5B). A consequence of these structural similarities is that the ALDH16 dimer and classic ALDH tetramer are similar in both size and shape (Figs. 5B and 5C).
LsALDH16 is a bona fide ALDH enzyme, whereas human ALDH16A1 appears to be a pseudoenzyme
The absence of the predicted catalytic Cys residue in some ALDH16 sequences raised the question of whether ALDH16 is an enzyme [7]. We therefore measured the cofactor binding properties and catalytic activities of an ALDH16 that has the predicted catalytic Cys in its sequence (LsALDH16) and one that lacks the predicted catalytic Cys (HsALDH16A1).
Cofactor binding was measured using fluorescence thermal shift assays [38]. LsALDH16 exhibits a melting temperature (Tm) of 59 °C (Fig. 6A). The Tm was shifted by +2 °C when NAD+ was added, which suggests NAD+ binds to folded LsALDH16. The shift was +1°C in the presence of NADH, also indicating binding. In contrast, neither NAD+ nor NADH induced a positive shift in Tm for HsALDH16A1 (Fig. 6B). In fact, negative shifts were observed with HsALDH16A1, which may indicate that NAD(H) binds more tightly to the denatured protein than the folded protein [39]. These results are consistent with weak or no binding of the cofactor to HsALDH16A1.
Fig. 6.

Cofactor binding to ALDH16. (A) Fluorescence thermal shift data for LsALDH16 in the absence of cofactor (black), the presence of 5 mM NAD+ (red), or the presence of 5 mM NADH (green). (B) Fluorescence thermal shift data for HsALDH16A1 in the absence of cofactor (black), the presence of 5 mM NAD+ (red), or the presence of 5 mM NADH (green). The data in panels A and B represent the average from three experiments and are plotted with negative of the first derivatives of relative fluorescence units as a function of temperature. (C) Electron density evidence for NAD+ bound to LsALDH16. (D) Electron density evidence for NADH bound to LsALDH16. The cages in panels C and D represent polder omit maps (3σ) [60].
The structures of LsALDH16 complexed with NAD+ and NADH were determined to identify which of the two Rossmann folds binds the cofactor. Strong electron density for NAD(H) appeared only in the Rossmann fold of the catalytic module, suggesting the Rossmann fold of the C-terminal domain plays a purely structural role (Figs. 6C and 6D). Density for the ADP fragment of NAD+ is strong, while that of the nicotinamide ribose is rather weak, indicating conformational flexibility (Fig. 6C). Lack of density for the nicotinamide ribose has been observed in other ALDH structures, including ALDH2 [14], ALDH4A1 [15], and ALDH7A1 [17]. In contrast, electron density was strong for the entire NADH molecule (Fig. 6D). The ADP moiety of NAD(H) binds in the canonical site of the Rossmann NAD+-binding domain. The adenine ring is wedged between α-helices C and D of the Rossmann fold, and the cofactor forms hydrogen bonds with Lys188, Glu191, Trp164 and Ser240 (Figs. 6C and 6D). All four hydrogen-bonding residues are highly conserved in bacterial ALDH16 sequences; however, they are replaced by nonpolar residues incapable of hydrogen bonding in HsALDH16A1 (Fig. S1), consistent with the lack of cofactor binding observed with HsALDH16A1.
While the ADP fragment of NADH displays the canonical pose, the nicotinamide riboside is flipped out of the active site onto the surface of the protein, 22 Å away from catalytic Cys295 (Fig. 3A, left). The nicotinamide riboside hydrogen bonds with a neighboring molecule in the crystal lattice, suggesting the flipped-out conformation may be an artifact of crystal packing. An alternative interpretation is that the unusual conformation of the catalytic loop (described in the next section) prevents the nicotinamide ribose from binding in the active site.
To investigate whether LsALDH16 uses NAD+ in catalysis, we measured ALDH activity using hexanal as the aldehyde substrate and NAD+ as the hydride acceptor. ALDH activity was evident with LsALDH16 (Fig. S2A of Supporting Information), and fitting of initial rate data to the Michaelis-Menten model yielded kinetic parameters of kcat of 3.3 s−1, Km of 21.3 μM and kcat/Km of 15.5 × 104 (M−1s−1) (Fig. 7A, Table 3). These values are comparable to those of the plant ALDH2, RF2C (39% identical to LsALDH16, [26]). Mutation of the presumed catalytic Cys295 to Ala (C295A) abolished ALDH activity (Fig. 7A). These results show that LsALDH16 is a bona fide ALDH enzyme.
Fig. 7.

Catalytic activity data showing that LsALDH16 is a bona fide enzyme. (A) ALDH activity of LsALDH16 using hexanal as the substrate in the presence of 2.5 mM NAD+ (black). Data for the C295A variant of LsALDH16 are shown in red. (B) Esterase activity of LsALDH16 using pNPA as the substrate in the absence of NAD+ (black). Data for the C295A variant of LsALDH16 are shown in red. Inset 1 shows polder omit electron density (3σ) for pNPA bound to C295A. Inset 2 shows that pNPA occupies the NAD+ adenine site (pNPA in pink, NAD+ in gray). In the activity plots in panels A and B, the data are shown as the mean ± standard error from triplicate measurements. The kinetic parameters are listed in Table 3.
Table 3.
Kinetic parameters of LsALDH16
| Km (μM) | V max (μmol·mg −1 s−1) | k cat (s −1) | k cat /Km (M −1 s −1) | Ki(μM) | |
|---|---|---|---|---|---|
| Hexanal | 21.3 ± 6.2 | 41.0 ± 2.9 × 10 −3 | 3.3 ± 0.2 | 15.5 ± 4.6 ×10 4 | N/A |
| pNPA | 1740 ± 340 | 131.6 ± 20.4 × 10 −3 | 10.6 ± 1.6 | 0.6 ± 0.2 × 10 4 | 1290 ± 270 |
In contrast, ALDH activity was not observed with HsALDH16A1 (Fig. S2B). We also performed assays of HsALDH16A1 using other aldehyde substrates, including propanal, nonanal, acetaldehyde and aminoadipate semialdehyde, but activity was not observed. These results suggest that HsALDH16A1 is a pseudoenzyme [40], i.e., a protein that is structurally homologous to active enzymes but lacks catalytic activity.
Some ALDHs also exhibit esterase activity, which requires the catalytic Cys but does not require a hydride acceptor [2]. We tested the esterase activity of LsALDH16 and HsALDH16A1 using p-nitrophenylacetate (pNPA) as the substrate in the absence of NAD+. LsALDH16 exhibits esterase activity, and fitting of the initial rate data to a substrate inhibition model yielded the kinetic parameters kcat of 10.6 s−1, Km of 1.7 mM, and Ki of 1.2 mM (Fig. 7B, Table 3). The 1.5 Å resolution structure of LsALDH16 C295A complexed with pNPA shows electron density for pNPA in NAD+ adenine site (Fig. 7B, inset). In contrast, esterase activity assays with HsALDH16A1 were negative. These results further support the idea that LsALDH16 is a genuine ALDH enzyme, whereas HsALDH16A1 appears to be a pseudoenzyme.
Inactivation of LsALDH16 by 4-diethylaminobenzaldehyde (DEAB, Fig. 8A) was also studied. DEAB is a covalent inactivator of some ALDHs [41, 42]. The mechanism of inactivation begins with the attack of the catalytic Cys on the aldehyde of DEAB to produce a hemithioacetal intermediate, followed by hydride transfer to NAD+ resulting in the acyl-enzyme intermediate. In the normal catalytic cycle, the acyl-enzyme is hydrolyzed to produce the carboxylic acid product, but for some ALDHs with DEAB, the reaction stalls at the acyl-enzyme intermediate, leaving the catalytic Cys covalently modified. To test for inactivation, LsALDH16 was incubated with DEAB and NAD+ for various times, and then the ALDH activity in the presence of hexanal and NAD+ was measured. Enzyme activity decreased with increasing incubation time, a hallmark of covalent inactivation (Fig. 8A). Further, mass spectral analysis of LsALDH16 that had been incubated with DEAB and NAD+ yielded a shift of 175 Da, consistent with the known mechanism of inactivation (Fig. 8B). These results further support the idea that bacterial ALDH16 is an ALDH enzyme.
Fig. 8.

Inactivation of LsALDH16 by DEAB. (A) Time-dependence of the loss of catalytic activity in the presence of DEAB. LsALDH16 was incubated with DEAB and NAD+, and then aliquots were taken at various time points and assayed for activity using hexanal as the substrate and NAD+ as the cofactor. (B) Mass spectrometry evidence for the covalent modification of LsALDH16 by DEAB. The observed mass shift of 175 Da is consistent with previous studies of the inactivation of ALDHs by DEAB [41, 42].
Conformation of the catalytic loop and composition of the aldehyde substrate pocket
The catalytic loop of LsALDH16 adopts a noncanonical conformation. The conformation of the loop is unambiguously defined by the electron density maps and exhibits the same conformation in all four LsALDH16 structures (Fig. 9A). Comparison with ALDH2 shows that catalytic Cys295 is displaced by 4 Å from the canonical position, and the loop of LsALDH16 occupies the space normally reserved for the nicotinamide ribose of NAD+ (Fig. 9B). Thus, it appears that the active site observed in the crystal is incompatible with the active conformation of the cofactor. Because LsALDH16 displays catalytic activity, the catalytic loop presumably isomerizes into the active conformation in solution in the presence of cofactor and substrate.
Fig. 9.

The active site of LsALDH16. (A) 1.65 Å resolution electron density for the catalytic loop of the NAD+ complex (polder omit, 3σ). (B) Comparison of the catalytic loops of LsALDH16 (gray) and ALDH2 (pink). (C) Residues in the predicted aldehyde binding site of LsALDH16. The surface represents the region likely occupied by aldehyde substrates as deduced from structures of ALDHs complexed with products and inhibitors (PDB 6ALJ, 6B5H, 5FHZ, and 4ZUL). Residues in blue line the predicted aldehyde cavity. Note the preponderance of nonpolar residues lining the cavity.
The active site of LsALDH16 was analyzed to gain insight into the composition of the aldehyde binding site. The characterization of the aldehyde pocket was based on structures of ALDHs complexed with inhibitors or products, including ALDH1A2 complexed with inhibitors (PDB 6ALJ and 6B5H, [23]), ALDH1A3 complexed with the product retinoic acid (PDB 5FHZ, [24]), and ALDH7A1 complexed with the product α-aminoadipate [17]. These ligands define a volume of space that corresponds to the aldehyde pocket of ALDHs (Fig. 9C).
The residues of the aldehyde pocket of LsALDH16 are predominantly nonpolar (Fig. 9C). Among them are those of the aromatic box, a conserved feature of substrate recognition by ALDHs [18]. Comparison to other ALDHs identifies the triad of Phe166, Trp173, and Phe453 as the aromatic box of LsALDH16. In addition to the box, eight other nonpolar and aromatic residues line the predicted aldehyde pocket. The aromatic box and several other residues in the aldehyde pocket are highly conserved by bacterial ALDH16s. This analysis suggests that bacterial ALDH16 may exhibit a preference for nonpolar aldehyde substrates.
Concluding remarks
We demonstrated that bacterial ALDH16 is a bona fide ALDH enzyme, whereas HsALDH16A1 likely is a pseudoenzyme. The former result is consistent with the fact that bacterial ALDH16 sequences contain key conserved residues known to function directly in catalysis, including the catalytic Cys, an Asn residue that hydrogen bonds to the oxyanion intermediate, and the Glu residue that activates an active site water molecule for hydrolysis of the of acyl-enzyme intermediate (Cys295, Asn165, and Glu261 in LsALDH16). The lack of measureable aldehyde oxidation activity with HsALDH16A1 is consistent with the fact that all three of the aforementioned critical residues are absent in mammalian (and zebrafish) ALDH16 sequences.
It appears that HsALDH16A1 is a pseudoenzyme. An implication of this finding is that future investigations into the biological roles of HsALDH16A1 might concentrate on the mechanisms typically ascribed to pseudoenzymes [40], such as modulating the activity of other proteins, including genuine ALDH enzymes, via protein-protein interaction. For example, a rare missense mutation in ALDH16A1 (Pro to Arg) is associated with increased serum uric acid levels and gout [11]. It is hypothesized that ALDH16A1 binds to HPRT1, a key enzyme in uric acid metabolism, and the mutation disrupts this protein-protein interaction, diminishing HPRT1 enzymatic activity, leading to hyperuricemia [7]. In a different context, the interaction of ALDH16A1 with maspardin has been proposed to play a role in the pathogenesis of mast syndrome (SPG21), an autosomal-recessive form of hereditary spastic paraplegia characterized by dementia and other brain abnormalities [12].
The extra domain of ALDH16 contains a Rossmann fold subdomain and protruding β-flap, and is thus structurally similar to the NAD+-binding and oligomerization domains of classical ALDHs. The tertiary structure of ALDH16 resembles the quaternary structure of the classic ALDH dimer, a relationship we refer to as trans-hierarchical structural similarity. Essentially, ALDH16 mimics three-fourths of an ALDH dimer. Also, ALDH16 forms a unique dimer not observed previously in the ALDH superfamily. Nevertheless, the ALDH16 dimer mimics the familiar ALDH tetramer, both in terms of the specific intermolecular β-sheet hydrogen bonding pattern at the center of the oligomer and the overall shape of the assembly.
We suggest the structure reported here may be considered to be the archetype of the ALDH16 family. This is based on SAXS analysis suggesting that LsALDH16 and HsALDH16A1 (37% sequence identity) are similar in oligomeric structure, and mostly likely, protein fold. Thus, our high resolution structure of LsALDH16 provides an accurate template for modeling other ALDH16 proteins, which should facilitate future studies of the molecular and biological functions of ALDH16 enzymes and pseudoenzymes.
Materials and methods
Expression clones
A codon-optimized gene encoding ALDH16 from Loktanella sp. 3ANDIMAR09 (LsALDH16, NCBI RefSeq WP_056035394.1, 766 residues) was obtained from GenScript in the pET24b expression vector. The gene was designed so that the expressed protein includes an N-terminal His6-tag and tobacco etch virus protease (TEVP) recognition motif with the sequence MHHHHHHSSGVDLGTENLYFQ/S, where / denotes the cleavage site. A stop codon was inserted after the gene to prevent expression of the C-terminal His tag encoded by pET24b. Treatment with TEVP produces the full-length 766-residue LsALDH16 protein preceded by Ser0. The C295A mutant variant of LsALDH16 was generated using the QuikChange II site-directed mutagenesis kit (Agilent).
An expression plasmid encoding ALDH16 from Litoreibacter arenae (LaALDH16, NCBI RefSeq WP_021099445, 766 residues) was designed similarly. LaALDH16 and LsALDH16 are 73% identical in amino acid sequence.
A plasmid containing the cDNA of human ALDH16A1 (HsALDH16A1, GenBank ID AAH14895.2, 802 residues) was purchased from Thermo-Fisher. The cDNA was subcloned into pET28a between restriction sites NdeI and XhoI. We engineered the pET28a vector to have a TEVP cleavage in place of the thrombin cleavage site. Thus, the expressed protein includes the full-length HsALDH16A1 sequence and an N-terminal His6-tag and TEVP recognition site. Treatment with TEVP results in the HsALDH16A1 polypeptide preceded by Gly-Ser-His. HsALDH16A1 and LsALDH16 are 37% identical in amino acid sequence.
Protein expression and purification
LsALDH16 was expressed in Escherichia coli BL21(DE3) codon plus cells, grown to an OD600 of 0.6 at 37 °C, chilled to 18 °C, and induced overnight with 500 mM isopropyl β-D-thiogalactopyranoside. Cells were harvested via centrifugation at 3,500g, resuspended in binding buffer (50 mM Hepes pH 7.5, 300 mM NaCl, 20 mM imidazole and 5% glycerol (w/v)), and lysed by sonication. Unbroken cells and cell debris were removed via centrifugation at 16,500g. Cleared lysate was loaded onto a column containing 3 mL Ni-NTA (QIAGEN) pre-equilibrated with binding buffer. Following extensive washing of the resin with the binding buffer, the bound protein was eluted using the binding buffer containing 250 mM imidazole. The His6-tag was cleaved by first incubating the protein sample with TEVP for 2 h at 28 °C, and then dialyzing the sample, which still contained the added TEVP, overnight at 4 °C into a buffer containing 50 mM Tris-HCl (pH 8.0), 10 mM NaCl, 2.5% glycerol and 0.5 mM tris(2-carboxyethyl)phosphine. The target protein was separated from the His-tag and uncleaved protein using the Ni-NTA column, with the cleaved LsALDH16 appearing in the flow-through. The flow-through was loaded onto a 1-ml HiTrap Q anion exchange column (GE) using a buffer consisting of 50 mM Tris-HCl (pH 8.0) and 2.5% glycerol, and a linear gradient of NaCl eluted LsALDH16 at ~200 mM NaCl. The protein concentration was estimated using the bicinchoninic acid method (Pierce kit) with bovine serum albumin as the standard. Fractions containing LsALDH16 were identified by SDS–PAGE and further purified via size exclusion chromatography on a FPLC equipped with a HiLoad 16/60 Superdex 200 pg column equilibrated with 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2.5% glycerol and 0.5 mM tris(2-carboxyethyl)phosphine. NAD+ was not added during the purification. LaALDH16 and HsALDH16A1 were expressed and purified as described above for LsALDH16. The molecular masses of HsALDH16A1 and LsALDH16 were 85394.7 and 80792.3 Da by electrospray ionization mass spectroscopy, consistent with the expected sizes for the recombinant proteins of 85395.7 and 80793.2 Da, respectively.
Protein crystallization
Optimized crystals of wild-type LsALDH16 and C295A were obtained by hanging-drop vapor diffusion using the protein concentrated to 6 mg/ml in a storage buffer consisting of 20 mM Tris-HCl at pH 8.0, 100 mM NaCl, 2.5% glycerol and 0.5 mM tris(2-carboxyethyl)phosphine. For crystallization of the cofactor complexes, a stock of NAD+ or NADH was first added to the protein sample at a final concentration of 5 mM, mixed and allowed to equilibrate for 30 minutes on ice. The sample was centrifuged for 5 minutes and the supernatant was used for setting up crystallization experiments. Crystals were grown from drops composed of the protein sample, reservoir, and a microseed solution mixed in a volume ratio of 1:1:0.2, where the reservoir solution comprised 20% (w/v) PEG 3350, 200 mM ammonium sulfate and 100 mM Bis-Tris at pH 5.5, and the microseed solution was made from crushed crystals diluted 1:100 in reservoir. Clear, colorless crystals grew to 200 × 200 × 50 μm3 over a period of a week at room temperature. Crystals of C295A complexed with p-nitrophenylacetate (pNPA) were prepared by soaking the apo crystals with the reservoir solution supplemented with 10 mM pNPA for about 30 minutes. All LsALDH16 crystals were cryo-protected with 15% glycerol and flash-cooled in liquid N2.
Crystals of LaALDH16 were also grown and used to determine the initial phases of the LsALDH16 structures. These crystals were obtained in a condition from a Morpheus screen (Molecular Dimensions) containing 60 mM CaCl2/MgCl2, 100 mM HEPES/MOPS at pH 7.5, 20% PEG 500 MME and 10 % PEG 20,000. LaALDH16 crystals were cryo-protected with 15% glycerol and flash-cooled in liquid N2.
Diffraction data collection and structure determination
Diffraction data were collected in shutterless mode from single crystals at the MBC beamline 4.2.2 at the Advanced Light Source, Berkeley, CA, or at the NE-CAT 24-ID-C beamline at the Advanced Photon Source, Argonne, IL. Each dataset consisted of a wedge of 180° of data collected with a Taurus-1 (4.2.2) or a Pilatus 6MF (24-ID-C) detector over a period of 180 seconds. Images were written to disk every 0.2 seconds, so that the data set consisted of 900 images with an effective oscillation width of 0.2°. The data were indexed, integrated, and scaled with the XDS package [43]. Intensities were merged and converted to amplitudes with AIMLESS [44]. Data processing statistics for LaALDH16 and LsALDH16 are provided in Tables S1 and 1, respectively.
The space group of the wild-type LsALDH16 crystals is P21212 with unit cell parameters a = 80 Å, b = 159 Å, and c = 63 Å, and one protein molecule per asymmetric unit. The C295A-pNPA complex has space group P212121 with unit cell parameters of a = 79 Å, b = 120 Å, and c = 159 Å, and two molecules arranged as a dimer in the asymmetric unit. For LaALDH16, the space group is P2 with unit cell parameters of a = 98 Å, b = 63 Å, and c = 134 Å, β = 109°, and two protein molecules in the asymmetric unit.
Initial phases were determined by molecular replacement with the program PHASER [45]. The initial solution of ALDH16 was first determined from a 2.95 Å resolution dataset of LaALDH16 and subsequently used to solve the apo structure of LsALDH16. Domain search models for the LaALDH16 dataset were generated with SWISS-MODEL [46] using the sequence of LaALDH16 and a structural template from chain A of the bifunctional proline catabolic enzyme proline utilization A from Sinorhizobium meliloti (PDB entry 5kf6 [8]). Sinorhizobium meliloti PutA contains a proline dehydrogenase domain, an L-glutamate-γ-semialdehyde dehydrogenase (ALDH4) domain, and a C-terminal domain that is homologous to the C-terminal domain of ALDH16. The PutA ALDH4 domain is 33% identical to the LaALDH16 ALDH module, while PutA C-terminal domain is 31% identical to the LaALDH16 C-terminal domain. The two models generated by SWISS-MODEL were input to PHASER as separate “ensembles”. PHASER generated a solution in space group P2 consisting of two copies each of the ALDH and C-terminal domains.
The initial solution from molecular replacement was modified in COOT [47, 48] by deleting parts of the model that had poor fit to the electron density map. Iterative rounds of model building and restrained refinement were carried out with COOT, PHENIX AutoBuild [49] and phenix.refine [50]. To solve the apo LsALDH16 structure, a search model was generated with CHAINSAW [51] using the sequence of LsALDH16 and a template from the monomer A of the LaALDH16 structure (73% sequence identity). These calculations showed that the space group of apo LsALDH16 is P21212 with one protein molecule in an asymmetric unit. The model was completed after iterative rounds of model building and restrained refinement. Subsequently, the apo LsALDH16 structure was used to solve the other LsALDH16 structures. The restraint files for ligands were generated in PHENIX eLBOW [52]. Refined structures were validated with MolProbity [53] and the PDB validation server. Refinement statistics are provided in Table 1. We note that the C295A-pNPA data set exhibits translational non-crystallographic symmetry as evidenced by an off-origin peak in the native Patterson map with height of 47% relative to the origin peak. Nevertheless, the structure could be solved by molecular replacement and refined to acceptable statistics (Table 1).
Small-angle X-ray scattering (SAXS)
Shutterless SAXS data collection was performed with a Pilatus detector at beamline 12.3.1 of the Advanced Light Source through the SIBYLS Mail-in High Throughput SAXS program [54]. Samples of LsALDH16 and HsALDH16A1 were passed through a 16/60 Superdex 200 prep grade size exclusion chromatography column that had been equilibrated with 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2.5% glycerol, and 0.5 mM Tris(3-hydroxypropyl)phosphine. The proteins were dialyzed overnight against a buffer of 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 2.0% glycerol, and 0.5 mM Tris(3-hydroxypropyl)phosphine and concentrated. Protein concentration was estimated with the bicinchoninic acid assay. For each protein, thirty-two images were collected in 10 sec at three different protein concentrations (Table 2). Images for background subtraction were collected similarly from the dialysate. The SAXS FrameSlice web tool (http://sibyls.als.lbl.gov/ran)was used to analyze the data for radiation damage and to identify the best buffer replicate for subtraction. Data averaging and merging were also carried out with FrameSlice as follows. For each sample, the Guinier region averaged over the first 3 images was merged with the Porod and wide-q region averaged over frames 7 – 12 images. PRIMUS [55] was used to inspect the merged data and to derive the SAXS parameters. The maximum particle dimension was estimated from calculations of the pair distribution function using GNOM [56] via PRIMUS.
Thermal shift assays
Differential scanning fluorimetry was performed using a bench top real-time PCR instrument (QuantStudio™ 3 System). Melting curves were generated by slowly warming ALDH16 (at 0.5 mg/mL) through a thermal cycle (4 to 95 °C) in 0.3°C increments for 60 seconds in the buffer containing 20 mM Tris-HCl, 150 mM NaCl, and SYPRO Orange (5X) dye, while monitoring increased fluorescence of the dye as it binds to denatured protein. For samples containing the cofactor, the protein was pre-incubated with 5 mM cofactor on ice before the measurement.
Enzyme activity assays
ALDH activity was measured by monitoring the production of NADH at 340 nm (molar extinction coefficient of 6220 M−1cm−1) at room temperature using hexanal as the substrate. Assays were performed both in a standard cuvette and a microplate reader. For example, the progress curve shown in Fig. S2A was generated from a 1-mL reaction performed in a cuvette containing 1 μM enzyme, 5 mM NAD+, 1 mM hexanal in 100 mM sodium pyrophosphate at pH 8. For the determination of Michaelis-Menten kinetic parameters, the assays were performed in 96-well plates in a Biotek Epoch 2 UV-Vis spectrophotometer with hexanal as the variable substrate. The reaction mixture (200-μl) contained 15 nM enzyme, 2.5 mM NAD+, and hexanal in 100 mM sodium pyrophosphate at pH 8. The C295A mutant was measured similarly but at much the higher enzyme concentration of 140 nM; activity was not observed with C295A, as expected (Fig. 7A). The initial velocity for each substrate concentration was extracted from the first 60 seconds for each progress curve. The data were fit to a Michaelis-Menten model using Origin (2018) and values are shown as the mean ± standard error from triplicate measurements.
DEAB is a mechanism-based covalent inactivator of some ALDHs [41, 42]. The mechanism-based inactivation of LsALDH16 by DEAB was examined by monitoring the rate of loss of enzyme activity after treatment with DEAB using hexanal as the substrate, as we described previously for ALDH7A1 [41]. The enzyme was first equilibrated with 2.5 mM NAD+ for an hour and then 200 μM DEAB was added to initiate inactivation. Aliquots were removed at 0, 10, 20, and 30 minutes after the start of inactivation, diluted 1:100 into a 1-mL reaction mixture containing 5 mM NAD+ and 1 mM hexanal in 100 mM sodium pyrophosphate at pH 8, and the residual activity was measured. Data were fit to a one phase exponential decay model using Graphpad Prism (v 5.0) and the data points represent the average of three independent experiments (each n = 3).
Esterase activity was measured using p-nitrophenylacetate (pNPA) as the substrate in the absence of NAD+ by monitoring the rate of p-nitrophenolate production spectrophotometrically by absorbance at 405 nm. Michaelis-Menten kinetic parameters were determined similarly as the ALDH activity using a serial dilution of pNPA (eight 2-fold dilutions starting from 5.5 mM) in 200-μl reactions in a 96-well plate. The reaction mixture contained 15 nM enzyme and varying concentrations of pNPA in 100 mM sodium pyrophosphate at pH 8. The C295A mutant was studied similarly using 140 nM enzyme and exhibited no measurable esterase activity, as expected.
Mass Spectrometry
The masses of apo HsALDH16A1, apo LsALDH16 and DEAB-inactivated LsALDH16 were determined using mass spectrometry. Protein samples were vortexed briefly at the lowest speed, and then diluted in 1% formic acid to 1 pmol/μl. A 0.5-uL injection was loaded onto a C8 column and separated with a LC gradient of about 18.5 minutes. The mass was acquired on Agilent 6520 QTOF and the data were summed across the peaks of elution and deconvoluted for intact mass.
Accession numbers
The coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under the following accession numbers: 6MVR (apo), 6MVS (NAD+), 6MVT (NADH), 6MVU (C295A-pNPA). The SAXS curves have been deposed in the Small Angle Scattering Biological Data Bank [57] under the accession numbers listed in Table 2.
Supplementary Material
Highlights.
ALDH16 is enigmatic, differing from other ALDHs in having an extra domain
We report the first structure and biochemical characterization of ALDH16
Bacterial ALDH16 is a genuine enzyme; human ALDH16A1 is a pseudoenzyme
The fold ALDH16 mimics the quaternary structure of the classic ALDH dimer
ALDH16 forms a unique dimer, which surprisingly resembles the classic ALDH tetramer
The structure reported here serves as the archetype of the ALDH16 family
Acknowledgements
Research reported in this publication was supported by the NIGMS of the National Institutes of Health under award number R01GM093123. We thank Dr. Ritcha Mehra-Chaudhary for help with crystallization of LaALDH16, Drs. Jay Nix and Narayanasami Sukumar for help with X-ray diffraction data collection at ALS and APS, respectively, and Katherine Burnett for collecting SAXS data through the SIBYLS mail-in program. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. Additional support for the SIBYLS beamline comes from the National Institutes of Health project ALS-ENABLE (P30 GM124169) and a High-End Instrumentation Grant S10OD018483.
Abbreviations:
- ALDH
aldehyde dehydrogenase
- DEAB
4-diethylaminobenzaldehyde
- HsALDH16A1
human aldehyde dehydrogenase 16A1
- LaALDH16
Litoreibacter arenae aldehyde dehydrogenase 16
- LsALDH16
Loktanella sp. aldehyde dehydrogenase 16
- pNPA
p-nitrophenylacetate; PutA, proline utilization A
- SAXS
small-angle X-ray scattering
- TEVP
tobacco etch virus protease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sophos NA, Vasiliou V. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact. 2003;143-144:5–22. [DOI] [PubMed] [Google Scholar]
- [2].Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, et al. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol Rev. 2012;64:520–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chem Biol Interact. 2013;202:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol. 2008;4:697–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ma I, Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011;7:292–306. [DOI] [PubMed] [Google Scholar]
- [6].Singh RK, Tanner JJ. Unique structural features and sequence motifs of proline utilization A (PutA). Front Biosci (Landmark Ed). 2012;17:556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vasiliou V, Sandoval M, Backos DS, Jackson BC, Chen Y, Reigan P, et al. ALDH16A1 is a novel non-catalytic enzyme that may be involved in the etiology of gout via protein-protein interactions with HPRT1. Chem Biol Interact. 2013;202:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Luo M, Gamage TT, Arentson BW, Schlasner KN, Becker DF, Tanner JJ. Structures of Proline Utilization A (PutA) Reveal the Fold and Functions of the Aldehyde Dehydrogenase Superfamily Domain of Unknown Function. J Biol Chem. 2016;291:24065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Korasick DA, Gamage TT, Christgen S, Stiers KM, Beamer LJ, Henzl MT, et al. Structure and characterization of a class 3B proline utilization A: Ligand-induced dimerization and importance of the C-terminal domain for catalysis. J Biol Chem. 2017;292:9652–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Luo M, Christgen S, Sanyal N, Arentson BW, Becker DF, Tanner JJ. Evidence that the c-terminal domain of a type b puta protein contributes to aldehyde dehydrogenase activity and substrate channeling. Biochemistry. 2014;53:5661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sulem P, Gudbjartsson DF, Walters GB, Helgadottir HT, Helgason A, Gudjonsson SA, et al. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat Genet. 2011;43:1127–30. [DOI] [PubMed] [Google Scholar]
- [12].Hanna MC, Blackstone C. Interaction of the SPG21 protein ACP33/maspardin with the aldehyde dehydrogenase ALDH16A1. Neurogenetics. 2009;10:217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Charkoftaki G, Chen Y, Han M, Sandoval M, Yu X, Zhao H, et al. Transcriptomic analysis and plasma metabolomics in Aldh16a1-null mice reveals a potential role of ALDH16A1 in renal function. Chem Biol Interact. 2017;276:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Perez-Miller SJ, Hurley TD. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry. 2003;42:7100–9. [DOI] [PubMed] [Google Scholar]
- [15].Srivastava D, Singh RK, Moxley MA, Henzl MT, Becker DF, Tanner JJ. The three-dimensional structural basis of type II hyperprolinemia. J Mol Biol. 2012;420:176–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pemberton TA, Tanner JJ. Structural basis of substrate selectivity of Delta(1)-pyrroline-5-carboxylate dehydrogenase (ALDH4A1): Semialdehyde chain length. Arch Biochem Biophys. 2013;538:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Luo M, Tanner JJ. Structural basis of substrate recognition by aldehyde dehydrogenase 7A1. Biochemistry. 2015;54:5513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Riveros-Rosas H, Gonzalez-Segura L, Julian-Sanchez A, Diaz-Sanchez AG, Munoz-Clares RA. Structural determinants of substrate specificity in aldehyde dehydrogenases. Chem Biol Interact. 2013;202:51–61. [DOI] [PubMed] [Google Scholar]
- [19].Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016;44:W424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–68. [DOI] [PubMed] [Google Scholar]
- [21].Morgan CA, Hurley TD. Development of a high-throughput in vitro assay to identify selective inhibitors for human ALDH1A1. Chem Biol Interact. 2015;234:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tsybovsky Y, Krupenko SA. Conserved catalytic residues of the ALDH1L1 aldehyde dehydrogenase domain control binding and discharging of the coenzyme. J Biol Chem. 2011;286:23357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen Y, Zhu JY, Hong KH, Mikles DC, Georg GI, Goldstein AS, et al. Structural Basis of ALDH1A2 Inhibition by Irreversible and Reversible Small Molecule Inhibitors. ACS Chem Biol. 2018;13:582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Moretti A, Li J, Donini S, Sobol RW, Rizzi M, Garavaglia S. Crystal structure of human aldehyde dehydrogenase 1A3 complexed with NAD(+) and retinoic acid. Sci Rep. 2016;6:35710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure of mitochondrial aldehyde dehydrogenase: the genetic component of ethanol aversion. Structure. 1997;5:701–11. [DOI] [PubMed] [Google Scholar]
- [26].Koncitikova R, Vigouroux A, Kopecna M, Andree T, Bartos J, Sebela M, et al. Role and structural characterization of plant aldehyde dehydrogenases from family 2 and family 7. Biochem J. 2015;468:109–23. [DOI] [PubMed] [Google Scholar]
- [27].Kim YG, Lee S, Kwon OS, Park SY, Lee SJ, Park BJ, et al. Redox-switch modulation of human SSADH by dynamic catalytic loop. EMBO J. 2009;28:959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Korasick DA, White TA, Chakravarthy S, Tanner JJ. NAD(+) promotes assembly of the active tetramer of aldehyde dehydrogenase 7A1. FEBS Lett. 2018;592:3229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Do H, Lee CW, Lee SG, Kang H, Park CM, Kim HJ, et al. Crystal structure and modeling of the tetrahedral intermediate state of methylmalonate-semialdehyde dehydrogenase (MMSDH) from Oceanimonas doudoroffii. J Microbiol. 2016;54:114–21. [DOI] [PubMed] [Google Scholar]
- [30].Kopecna M, Vigouroux A, Vilim J, Koncitikova R, Briozzo P, Hajkova E, et al. The ALDH21 gene found in lower plants and some vascular plants codes for a NADP(+) -dependent succinic semialdehyde dehydrogenase. Plant J. 2017;92:229–43. [DOI] [PubMed] [Google Scholar]
- [31].Huo L, Davis I, Liu F, Andi B, Esaki S, Iwaki H, et al. Crystallographic and spectroscopic snapshots reveal a dehydrogenase in action. Nature communications. 2015;6:5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Johansson K, El-Ahmad M, Ramaswamy S, Hjelmqvist L, Jornvall H, Eklund H. Structure of betaine aldehyde dehydrogenase at 2.1 A resolution. Protein Sci. 1998;7:2106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Di Costanzo L, Gomez GA, Christianson DW. Crystal structure of lactaldehyde dehydrogenase from Escherichia coli and inferences regarding substrate and cofactor specificity. J Mol Biol. 2007;366:481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McClerklin SA, Lee SG, Harper CP, Nwumeh R, Jez JM, Kunkel BN. Indole-3-acetaldehyde dehydrogenase-dependent auxin synthesis contributes to virulence of Pseudomonas syringae strain DC3000. PLoS Pathog. 2018;14:e1006811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Son HF, Park S, Yoo TH, Jung GY, Kim KJ. Structural insights into the production of 3-hydroxypropionic acid by aldehyde dehydrogenase from Azospirillum brasilense. Sci Rep. 2017;7:46005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zarzycki J, Sutter M, Cortina NS, Erb TJ, Kerfeld CA. In Vitro Characterization and Concerted Function of Three Core Enzymes of a Glycyl Radical Enzyme - Associated Bacterial Microcompartment. Sci Rep. 2017;7:42757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Coitinho JB, Pereira MS, Costa DM, Guimaraes SL, Araujo SS, Hengge AC, et al. Structural and Kinetic Properties of the Aldehyde Dehydrogenase NahF, a Broad Substrate Specificity Enzyme for Aldehyde Oxidation. Biochemistry. 2016;55:5453–63. [DOI] [PubMed] [Google Scholar]
- [38].Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–21. [DOI] [PubMed] [Google Scholar]
- [39].Cimmperman P, Baranauskiene L, Jachimoviciute S, Jachno J, Torresan J, Michailoviene V, et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys J. 2008;95:3222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Murphy JM, Mace PD, Eyers PA. Live and let die: insights into pseudoenzyme mechanisms from structure. Curr Opin Struct Biol. 2017;47:95–104. [DOI] [PubMed] [Google Scholar]
- [41].Luo M, Gates KS, Henzl MT, Tanner JJ. Diethylaminobenzaldehyde is a covalent, irreversible inactivator of ALDH7A1. ACS Chem Biol. 2015;10:693–7. [DOI] [PubMed] [Google Scholar]
- [42].Morgan CA, Parajuli B, Buchman CD, Dria K, Hurley TD. N,N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes. Chem Biol Interact. 2015;234:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kabsch W XDS. Acta crystallographica Section D, Biological crystallography. 2010;66:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].French S, Wilson K, IUCr. On the treatment of negative intensity observations. Acta Crystallographica Section A: Crystal Physics, Diffraction, Theoretical and General Crystallography. 1978;34:517–25. [Google Scholar]
- [45].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, et al. Phaser crystallographic software. Journal of applied crystallography. 2007;40:658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. [DOI] [PubMed] [Google Scholar]
- [47].Emsley P, Cowtan K, IUCr. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–32. [DOI] [PubMed] [Google Scholar]
- [48].Emsley P, Lohkamp B, Scott WG, Cowtan K, IUCr. Features and development of Coot. Acta crystallographica Section D, Biological crystallography. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta crystallographica Section D, Biological crystallography. 2008;64:61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta crystallographica Section D, Biological crystallography. 2012;68:352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Stein N, IUCr. CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. Journal of applied crystallography. 2008;41:641–3. [Google Scholar]
- [52].Moriarty NW, Grosse-Kunstleve RW, Adams PD, IUCr. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta crystallographica Section D, Biological crystallography. 2009;65:1074–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010;66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, et al. High-Throughput SAXS for the Characterization of Biomolecules in Solution: A Practical Approach. Totowa, NJ: Humana Press, Totowa, NJ; 2014. p. 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI, IUCr. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of applied crystallography. 2003;36:1277–82. [Google Scholar]
- [56].Svergun DI, IUCr. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of applied crystallography. 1992;25:495–503. [Google Scholar]
- [57].Valentini E, Kikhney AG, Previtali G, Jeffries CM, Svergun DI. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015;43:D357–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Crystallogr. 2010;43:101–9. [Google Scholar]
- [59].Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, et al. High-Throughput SAXS for the Characterization of Biomolecules in Solution: A Practical Approach. Methods Mol Biol 2014. p. 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol. 2017;73:148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.