

Abstract
The leading cause of death in the juvenile population is trauma, and in particular neurotrauma. The juvenile brain response to neurotrauma is not completely understood. Endoplasmic reticulum (ER) stress has been shown to contribute to injury expansion and behavioral deficits in adult rodents and furthermore has been seen in adult postmortem human brains diagnosed with chronic traumatic encephalopathy. Whether endoplasmic reticulum stress is increased in juveniles with traumatic brain injury (TBI) is poorly delineated. We investigated this important topic using a juvenile rat controlled cortical impact (CCI) model. We proposed that ER stress would be significantly increased in juvenile rats following TBI and that this would correlate with behavioral deficits using a juvenile rat model. A juvenile rat (postnatal day 28) CCI model was used. Binding immunoglobulin protein (BiP) and C/EBP homologous protein (CHOP) were measured at 4 h in the ipsilateral pericontusion cortex. Hypoxia-inducible factor (HIF)-1α was measured at 48 h and tau kinase measured at 1 week and 30 days. At 4 h following injury, BiP and CHOP (markers of ER stress) were significantly elevated in rats exposed to TBI. We also found that HIF-1α was significantly upregulated 48 h following TBI showing delayed hypoxia. The early ER stress activation was additionally associated with the activation of a known tau kinase, glycogen synthase kinase-3β (GSK-3β), by 1 week. Tau oligomers measured by R23 were significantly increased by 30 days following TBI. The biochemical changes following TBI were associated with increased impulsive-like or anti-anxiety behavior measured with the elevated plus maze, deficits in short-term memory measured with novel object recognition, and deficits in spatial memory measured with the Morris water maze in juvenile rats exposed to TBI. These results show that ER stress was increased early in juvenile rats exposed to TBI, that these rats developed tau oligomers over the course of 30 days, and that they had significant short-term and spatial memory deficits following injury.
Keywords: Controlled cortical impact, Endoplasmic reticulum stress, Novel object recognition, Elevated plus maze, Morris water maze, Tau oligomers
Introduction
Juvenile traumatic brain injury (jTBI) is the leading cause of death and disability in the juvenile population. Often due to falls, sports, and automobile accidents, jTBI results in behavioral and cognitive dysfunction that can lead to learning difficulty and trouble with day-to-day living as the child reaches adulthood [1, 2]. TBI outcome is dependent upon the age at which the injury occurred. Multiple reports have shown the young, developing brain to respond differently to TBI when compared to the adult brain response [3–11]. What is still unknown is how the acute effects of injury can lead to cognitive dysfunction. For instance, brain injury early in life often leads to issues with increased impulsivity, which can lead to poor decision-making [12–14] and disrupt cognitive flexibility [15, 16].
Endoplasmic reticulum (ER) stress following injury has been shown to contribute to the unfolded protein response [17, 18]. ER stress is disrupted following TBI and reversed by docosahexaenoic acid or salubrinal [19, 20]. The ER stress pathway has also been associated with increased glycogen synthase kinase-3β (GSK-3β) activation [21–23], while it has recently been observed that ER stress occurs following TBI; however, it has not been shown in juvenile models. Therefore, there is a tremendous need to further develop animal models of jTBI. Further, early brain injury could lead to the development of tauopathy, which has been associated with impaired ER stress pathways in adult models of brain injury [24]. This can present problems later in life, as it would lead to an altered trajectory of cognitive development.
One group studying ER stress in juvenile animal models found that neonate mice exposed to hypoxia/ischemia had increased neuronal necrosis due to ER stress activation [25]. This finding was also validated in juvenile rats but furthermore linked to short-term memory deficits as well as poor working memory [26]. The proposed mechanism is that ER stress activates the unfolded protein response and contributes to cell death at an accelerated rate in juveniles because of the rapid growth and axonal track formation that occurs at this time period [27]. By activating the first arm of the ER stress pathway in particular (the protein kinase RNA-like ER kinase pathway), damage to the hippocampus has been seen [28].
We sought to investigate the role of ER stress in juveniles exposed to TBI instead of hypoxia. In the current study animals were injured on postnatal day 28, an age corresponding to 7–10 years of age in humans [29]. Following injury, subjects were tested on a neurocognitive battery of tests aimed to examine impairments in memory, impulsivity/anti-anxiety, and executive functioning. We wanted to see whether deficits would be present similar to those reported in the hypoxia literature. The brains were then processed for known early markers of tauopathy and neurofibrillary tangles [30, 31]. Separate groups of subjects underwent surgery and were euthanized at 4 h, 48 h, and 1 week, and the pericontusion cortex was examined for markers associated with ER stress.
Methods
Subjects
Twenty-eight male Sprague-Dawley rats (postnatal day 28) were used for the duration of this study. All rats were housed in cages of 2 or 3 and had access to food and water ad libitum. The rats remained in a temperature- and light-controlled environment on a 12-h light/12-h dark cycle with lights on at 07:00 h and lights off at 19:00 h. Animals were housed with dams until postnatal day 21 at which point they were weaned and housed with littermates of the same gender. Water and food were provided ad libitum. Injury and treatment groups came from multiple, separate litters, from different parents to avoid litter-specific effects. A timeline for the behavioral and biochemical experimental procedures is outlined in Figure 1.
Fig. 1.
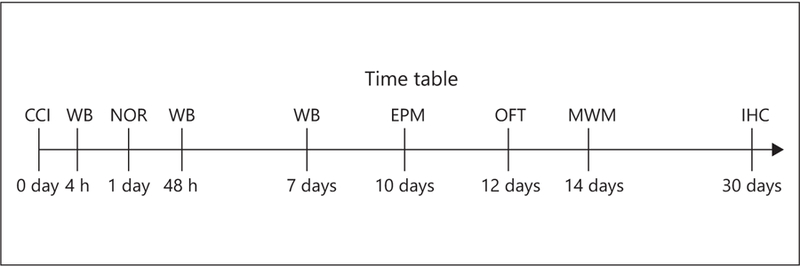
A timeline illustrating time points for the behavioral testing and biochemical assays used in the experiments. Model: CCI, controlled cortical impact; biochemistry: WB, Western blot; IHC, immunohistochemistry; behavior: NOR, novel object recognition; EPM, elevated plus maze; OFT, open field test; MWM, Morris water maze.
Controlled Cortical Impact Surgery
All experimental procedures were approved by the Institutional Animal Care and Use Committee. The controlled cortical impact (CCI) procedure was modified from previous studies [32, 33]. Rats were anesthetized using 5% isofluorane mixed with oxygen. Once anesthetized, the rats received either a CCI or a sham surgery. For the CCI surgery, skin incisions were performed followed by bilateral craniotomy, and an electromagnetically driven CCI device (Leica Impact One) with a 5-mm impactor tip was used to deliver a unilateral injury to the right parietal lobe. The impact was at a velocity of 5.5 m/s with a 200-ms dwell time and a 2-mm deformation of the cortical surface. Each craniotomy was 6 mm in diameter and was centered approximately 2 mm posterior to the bregma and 1 mm from the midline. Bilateral craniotomies were used because they increase contralateral axonal damage and the chances of contralateral hippocampal damage [34–38]. A representative example of the injury is shown in Figure 2a and b. Figure 2c illustrates how the bilateral craniotomy results in mechanical stress upon the contralesional hemisphere. For the sham surgery, rats were anesthetized and had the incision but did not receive craniotomies or an impact. This was done to avoid any potential complications that are associated with the surgical procedure and to control for confounding variables. This is an approach utilized by our laboratory and other previously established research [39, 40]. After either surgery, the rats were placed back in their home cages, and behavioral testing started the following day. For the behavioral testing portion of the study, 12 animals were used for the CCI group and 14 animals were used as shams.
Fig. 2.
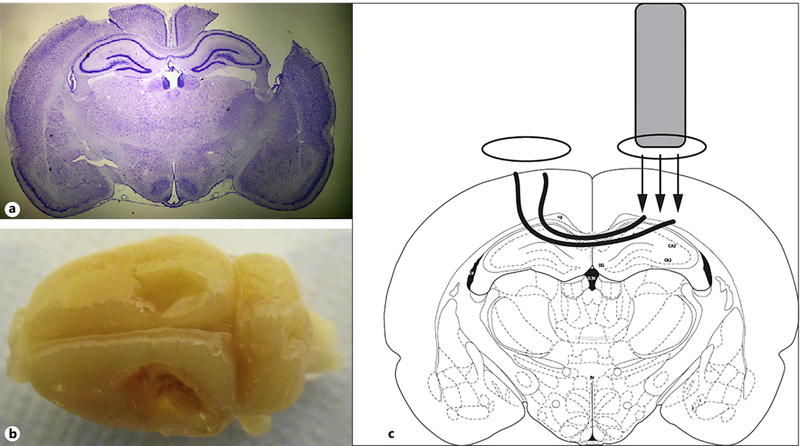
a A representative coronal cresyl violet section illustrating the nature of the injury. b A dorsal image of a CCI injury. c An illustration showing the proposed mechanism of how a bilateral craniotomy results in contralateral damage.
Behavioral Assessments
Novel Object Recognition
The protocol used for the novel object recognition task has been previously established [41]. Prior research has revealed that deficits are present in TBI models during the acute period following injury [42, 43]. The task took place in a clear, Plexiglas box (57.5 cm in length, 36 cm in width, and 38 cm in height) in a room with minimal lighting. Testing began 1 day following surgery. The procedure consisted of 3 phases: habituation, training, and object recognition testing. Habituation took place on day 1, training and testing took place on day 2. In the habituation phase, each rat was placed in an empty box for 20 min to acclimate to the task environment. For the training phase, each rat was placed in the box for 10 min. This time, the box contained 2 identical objects for the rat to explore. After training, the rat was taken out of the box and placed back into the home cage for 1 h, and then testing occurred. For testing, 1 of the 2 objects from training was replaced with a novel object and the rat could explore the box for 5 min. All training and testing were recorded. AnyMaze software was used to analyze the videos and assessed how long the rat attended to each object, as well as how many times the rat interacted with each object. To determine an interaction with an object, a circle (4 cm diameter centered on the object) was drawn around each object, and an interaction was scored each time the animal’s head entered the circle.
Plus Maze
The procedure was modified from a procedure that assessed anxiety-like behaviors in mice [44] and has been used to measure impulsive-like behavior or anti-anxiety in the rodent as indicated by increased time spent in the open arms. Each rat was assessed once on the plus maze task 10 days after the surgical procedure. The plus maze was placed in the center of an open room with minimal light. The apparatus consisted of 4 arms (2 of which have walls and 2 that do not) and was elevated 75.5 cm from the ground. The arms were 51 cm in length and 11.5 cm wide, and the walls were 40.5 cm in height. Each rat could explore the plus maze for 5 min. During those 5 min, the rat’s behavior was recorded. AnyMaze software was used for analysis of the task. The rats were assessed on 2 measures: the number of entries into an open versus a closed arm and the amount of time spent in an open versus a closed arm to quantify impulsive behaviors. The maze was cleaned with 70% ethanol between each rat to eliminate olfactory cues from previous rats.
Open Field
This protocol was modified from an open field test using mice [45]. Each rat was assessed only once on the open field task. Similarly, to the plus maze, this task assessed exploratory and anxiety-like behaviors after injury. Testing for the open field task occurred 12 days after the surgical procedure. Each rat was placed in a clear, circular Plexiglas behavioral apparatus (44.5 cm in diameter; 26.5 cm in height) and allowed to explore the field for 10 min. During the task, behaviors were recorded. After testing, the videos were analyzed using AnyMaze software. Analysis consisted of quantifying the number of times the rat entered the center of the field versus the perimeter, as well as the amount of time the rat spent in each of these 2 areas. Between each test, the open field was cleaned with 70% ethanol to eliminate attenuation to olfactory cues from previous rats.
Morris Water Maze
The Morris water maze test was conducted using a protocol used in prior studies [46]. A polyethylene pool measuring 200 cm × 74 cm was divided into 4 quadrants: northeast, northwest, southeast, and southwest. Inside the pool is a submerged platform measuring 72 cm in height and 13 cm in diameter. Between the pool and the experimenter is a shower curtain colored dark blue to obscure movements of the experimenter. Additionally, overhead light sources were turned off and instead the room was illuminated by two 60-W light bulb lamps obscured by a translucent curtain. The external cues to the pool consisted of 4 items: a painted guitar on the wall, 3 vertical stripes painted on the wall, a pyramid attached to the wall, and the dark blue opaque curtain.
The Morris water maze test was performed 14 days after injury in order to measure spatial memory. Animals were run in groups of 4 with each animal undergoing 4 trials per day for 5 days of training. Animals were placed in the pool by hand facing the wall. Starting locations were randomized, selected from 8 potential starting points, for each trial. Rats were given a time limit of 60 s to successfully find the submerged platform. If the rat was unsuccessful in finding the platform within the time limit, it was guided to the platform by the experimenter. Once on the platform, the animal was given 30 s before being removed from the pool to familiarize themselves with the external cues. Once removed from the pool, animals were kept in single-house cages on top of heating pads between trials. Intertrial intervals were 5 min.
On the following day, 24 h after the final day of training, a probe trial was administered by removing the submerged platform. Each animal could swim for 60 s in search of the platform. One day following the probe trial animals were trained on reversal training. For this the platform location was moved into the quadrant opposite to the one used during training. This was done to test the animal’s ability to demonstrate cognitive flexibility. Subjects were given 4 trials during reversal training Swimming paths was recorded via a camera fixed directly above the pool and were collected using AnyMaze software (San Diego Instruments). Latency to the platform was recorded.
Western Blot
For Western blot, a 1 mm × 1 mm sample (taken anterior to the impact site) of the ipsilateral pericontusion cortex was collected from CCI rats at various time points after injury, and the same approximate region was collected for control rats at various time points following injury (n = 3 per group). The pericontusion cortex is defined as penumbral region surrounding the contusion that is sensitive to further damage [47–49]. 1% SDS was used to prepare the protein samples. The protein assay and Western blot were performed as previously described [50]. Primary antibodies were rabbit anti-binding immunoglobulin protein (BiP) monoclonal antibody (mAB; 1:1,000, No. 3177) and rabbit anti-C/EBP homologous protein (CHOP) mAB (1:1,000, No. 5554) (Cell Signaling); rabbit uncleaved caspase-12 mAB (1:200, No. 21747), mouse cleaved caspase-12 mAB (1:200, No. 515103), rabbit anti-MOX-1 mAB (1:500, No. 398845), and rabbit anti-hypoxia-inducible factor-1α (HIF-1α) mAB (1:500, No. 13515) (Santa Cruz); rabbit p-GSK-3β mAB (1:1,000, No. 5H1L11) (Thermo). A mouse anti-β-actin mAB (1:10,000, No. 3700) (Cell Signaling) was used as an endogenous control to normalize protein loading. Secondary antibodies were IRDye® 800CW, and IRDye® 680RD (LI-COR Bio-sciences). Images were collected and analyzed with an Odyssey fluorescent scanner. Images were converted to gray scale, the values calculated after background subtraction, and then normalized to β-actin to measure relative intensity.
Immunohistochemistry
Rats were anesthetized with 4% isoflurane, and cardiac perfused with ice-cold 0.9% saline. Brains were rapidly removed and placed into an ice-cold protease/phosphatase inhibitor cocktail mix (HaltTM; Thermo Scientific, Pittsburgh, PA, USA). Tissues were cryoprotected with 30% sucrose, were subsequently flash frozen in liquid nitrogen, and stored at –80 °C. Brain tissue for the CCI and control rats were prepared for cryostat sectioning as previously described [50]. Sections from the ipsilateral pericontusion cortex were stereotaxically selected based on the following dimensions: anterior/posterior 1.94 mm, medial/ lateral 0.5 mm, and dorsal/ventral 3.75 mm. This region was chosen for sections and analysis as it corresponds to the pericontused cortex, an area that is associated with brain plasticity and functional recovery following TBI [47, 49, 51]. Brain slabs were sectioned (20 µm), mounted onto slides, and prepared for staining. Three immediate adjacent sections of 20 μm were cut per each slide. Briefly, brain slices were circumscribed and incubated overnight with primary antibodies: anti-paired helical filament (PHF) mAB (1:500), anti-R23 mAB (1:500), and anti-CP-13 mAB (1:500) kindly gifted from the Peter Davies Laboratory. The next day, an Alexa Fluor® secondary antibody (Invitrogen) was applied to slides for 3 h, and coverslip mounted with Vectashield® 4’,6-diamidino-2-phenylindole nuclear counterstain (Vector). All images were acquired from the ipsilateral pericontusion cortex (10 slides per animal [n = 4 per group]). Antibody-stained fluorescence images were acquired using a confocal microscope (Z1 Axio Observer; Zeiss, Oberkochen, Germany). The ipsilateral pericontusion cortex was analyzed. For antibody-stained fluorescence quantification, 10 distinct cells with clear morphology were randomly selected per slide, outlined, and measured with ImageJ software (NIH) by an observer blinded to the experimental group. Density was adjusted per mean area to give total cell fluorescence normalized to background.
Statistical Analysis
An independent t test was used to analyze data from the novel object recognition task, the plus maze, and the open field task. For the novel object recognition task, the t test assessed the difference between the 2 groups on total number of interactions with each object and the total amount of time of interactions with each object. For the plus maze, the t test assessed the difference between the 2 groups on the total number of times the animal entered an open versus a closed arm, as well as the total amount of time spent in open versus closed arms. For the open field task, the t test assessed the difference between the 2 groups on the total number of entries into the center of the field versus the perimeter, as well as the total amount of time spent in the center versus the perimeter of the field. Differences between the 2 groups on the Morris water maze were analyzed by repeated measures one-way ANOVA for the time needed to locate the hidden platform over the course of 5 days of testing. An independent t test was used to analyze the data from the probe day, whereas a repeated measures one-way ANOVA was used to analyze the trial data for the reversal of the Morris water maze. For the probe day, the t test assessed the difference between the 2 groups on the number of target crossings and the time of the first target crossing. For the reversal, the repeated measures one-way ANOVA assessed the difference between the 2 groups on the amount of time taken to locate the hidden platform. The experimenter conducting the behavioral tests was blinded to the injury group, and the data were analyzed by a separate individual.
For the biochemical studies, one-way ANOVA with Holm-Sidak post hoc analysis was used to compare groups and determine statistically significant differences between groups. The experimenter performing the assays was blinded to the treatment group, and the results were analyzed by an independent evaluator. Relative intensity or corrected total cell fluorescence were used as the variables for analysis. All tests were considered significant if p < 0.05. Data were tested for normality using Shapiro-Wilk tests, and Brown-Forsythe tests were used to test homogeneity of variance. All data are represented as means ± SEM. Data were analyzed using SigmaStat software.
Results
jTBI Leads to Increased Hypoxia without Impacting Endothelial Cell Function
HIF-1α is a transcription factor that responds to decreased levels of cellular oxygen. Following injury there is an acute increase in HIF-1α 48 h after injury that normalizes to sham levels by 1 week after injury (F2, 4 = 18.451, p = 0.01, Holm-Sidak post hoc test p < 0.05; Fig. 3a). MOX-1 is a marker of brain endothelial cell function especially for vessels that course near the skull [52]. Significant activation indicates vascular proliferation in response to injury. Increases are likely due to vascular disruption and ongoing vascular cell proliferation; however, following jTBI there were no significant differences at any time points for the level of MOX-1 (F2, 4 = 3.453, p = 0.135; Fig. 3b).
Fig. 3.
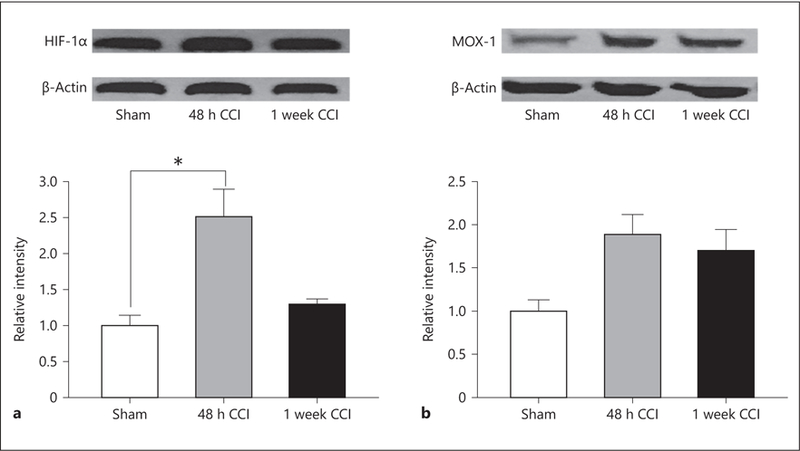
a Hypoxia-inducible factor-1α (HIF-1α) was significantly increased 48 h following CCI (p < 0.05). HIF-1α is activated by ER stress and contributes to tau oligomer formation. b MOX-1 was not significantly elevated following CCI. MOX-1 is a marker of endothelial cell function and responds to endothelial HIF-1α. This confirms that HIF-1α is activated in neurons but not endothelial cells. * p < 0.05.
jTBI Results in Alterations in ER Stress Proteins
BiP and CHOP are proteins associated with ER stress and neurocognitive dysfunction [24]. Survival factors, such as B-cell lymphoma 2, are downregulated by the proapoptotic transcription factor. Increased CHOP leads to cellular death through the promotion of proapoptotic protein transcription [53]. BiP was elevated after 4 h and remained elevated 48 h after injury (F2, 4 = 27.728, p = 0.005, Holm-Sidak post hoc test p < 0.05; Fig. 4a). CHOP was elevated 4 h after injury but was not significantly different from sham animals at 48 h (F2, 4 = 8.319, p = 0.039, Holm-Sidakposthoctest p < 0.05 forshamvs. 4 h; Fig. 4b), though levels were not at sham levels at this time point.
Fig. 4.
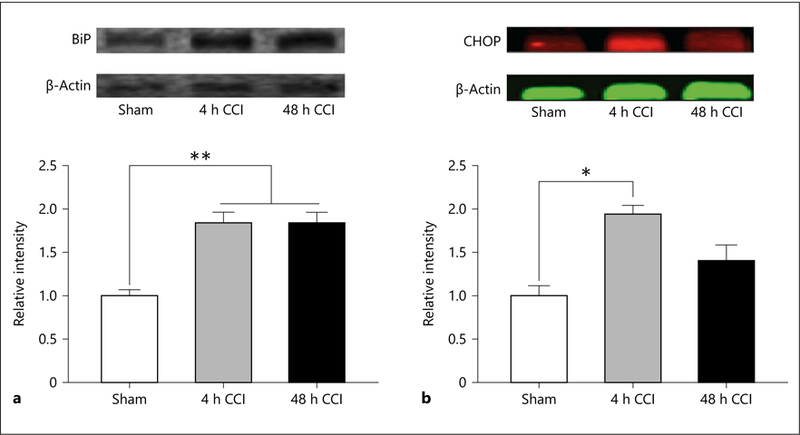
a BiP is a regulator of the 3 arms of the ER stress cascade and was significantly elevated 4 and 48 h following CCI. ** p < 0.01. b CHOP is a marker of the first arm of the ER stress pathway and contributes to activation of phosphorylated GSK-3β and cleavage and activation of caspase-12. CHOP was significantly increased 4 h following CCI. * p < 0.05.
Early Brain Injury Results in Increased Phosphorylated GSK-3β but Not Caspase-12
Increases in caspase-12 have been found to be associated with increased markers of ER stress, such as CHOP [54]. Following early injury there is a nonsignificant trend toward an increase in cleaved caspase-12 that is elevated 1 week after injury (F2, 4 = 5.531, p = 0.071; Fig. 5a).
Fig. 5.
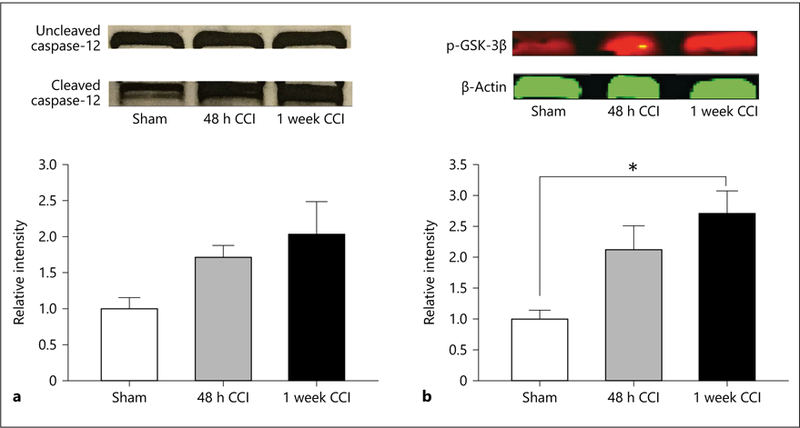
a Cleaved caspase-12 is a key regulator of the apoptotic cascade. A trend towards significance was seen 7 days following CCI (p = 0.071). b Phosphorylated GSK-3β (p-GSK-3β) is the primary contributor towards tau hyperphosphorylation. p-GSK-3β was significantly increased 7 days after CCI. * p < 0.05.
GSK-3β has been found to be associated with increased tauopathy and behavioral dysfunction following brain injury [55]. As shown in Figure 5b, injured animals show an increase in the levels of phosphorylated GSK-3β (p-GSK-3β) that begins to increase 48 h after injury and reaches a significant level 1 week after injury (F2, 4 = 9.855, p = 0.028).
Tauopathy Markers Are Elevated 1 Month after jTBI
In the pericontused cortex at the coordinates outlined above, tissue was examined for early markers of tauopathy 30 days after injury. Immunohistochemistry was used to quantify tau markers. PHF, R23, and CP-13 are all markers of tau conformation change. They are progressive in severity with PHF occurring prior to R23 and R23 prior to CP-13. CP-13 is a precursor for neurofibrillary tangles [56]. Injured subjects demonstrated an increase in PHF when compared to sham subjects (t18 = –5.517, p < 0.001; Fig. 6a). Injured subjects also showed an increase in R23 protein (t18 = –5.435, p < 0.001; Fig. 6b) and an increase in CP-13 (t18 = –3.00, p < 0.01; Fig. 6c).
Fig. 6.
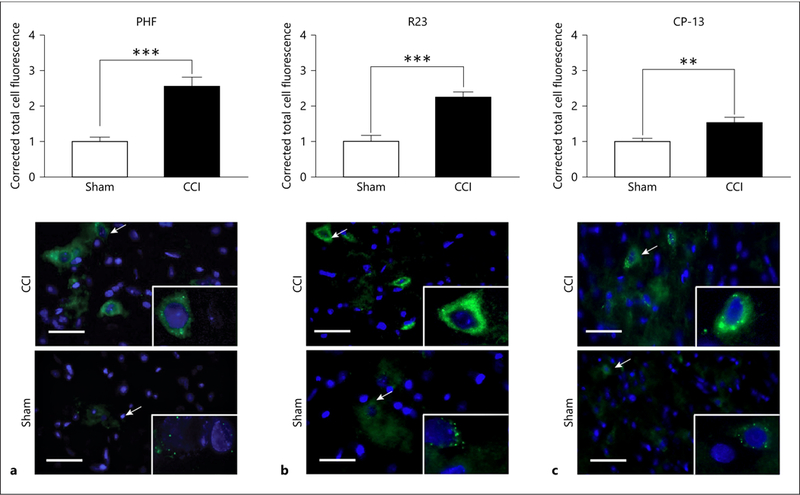
Tau pathology progresses from paired helical filaments to oligomers to neurofibrillary tangle precursors and finally neurofibrillary tangles. We found rapidly progressive tauopathy following CCI in juveniles. a Paired helical filament (PHF) was significantly increased 1 month following CCI. *** p < 0.001. b R23, a marker of tau oligomers, was significantly increased 1 month following CCI. *** p < 0.001. c CP-13, a precursor of tau neurofibrillary tangles, was seen 1 month following CCI. ** p < 0.01. The scale bar is 50 μm, and arrows show the cell(s) that is (are) focused upon at high power. The low-power image is ×20, and the high-power image is ×63.
Object Recognition Memory Deficits Emerge in the Acute Period following Injury
Novel object exploration has been used as a measurement of object recognition functioning. During habituation training to the testing apparatus, CCI subjects explored the apparatus to the same extent as uninjured sham animals, which indicates that the anesthesia did not have an impact upon the subsequent short-term memory testing (p > 0.05). An independent samples 2-tailed t test found that uninjured animals spent a significantly higher percentage of time exploring the novel object compared to CCI animals (t23 = –2.321, p = 0.0295; Fig. 7a). Furthermore, CCI animals spent significantly less time (t26 = –4.526, p = 0.0001; Fig. 7b) and made fewer interactions (t26 = 3.583, p = 0.0014; Fig. 7c) with the novel object. These findings indicate that injured animals experience object recognition memory deficits resulting in them spending less time with novel objects.
Fig. 7.
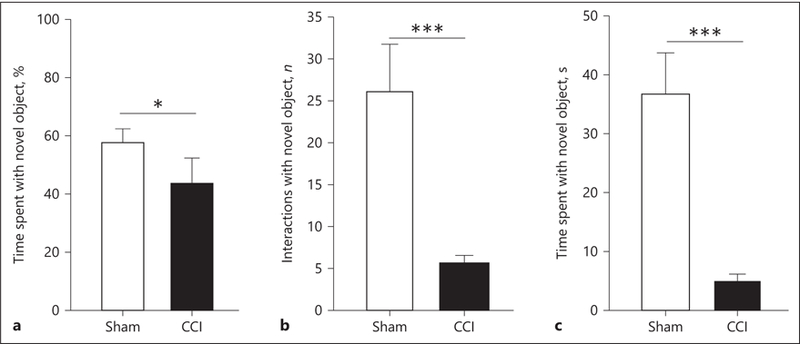
Early CCI injury results in impaired object recognition in the novel object recognition task. a Injured subjects spend significantly less percentages of time with the novel object when compared to control subjects. * p < 0.05. This is reflected by a significantly lowered number of interactions (b) and a diminished amount of time spent with the novel object (c) relative to sham subjects. *** p < 0.001.
jTBI Increases Impulsive-Like Behavior without Increasing Anxiety
Independent samples t tests were run to determine differences between number of entries into open and closed arms in CCI animals and uninjured animals. Analysis found that CCI animals entered open arms a significantly higher amount of times than uninjured animals (t10 = 2.287, p = 0.0452; Fig. 8a). Additionally, another independent samples t test found no significant differences between the number of closed arm entries for CCI animals and uninjured animals (t10 = 1.774, p = 1.06; Fig. 8a).
Fig. 8.
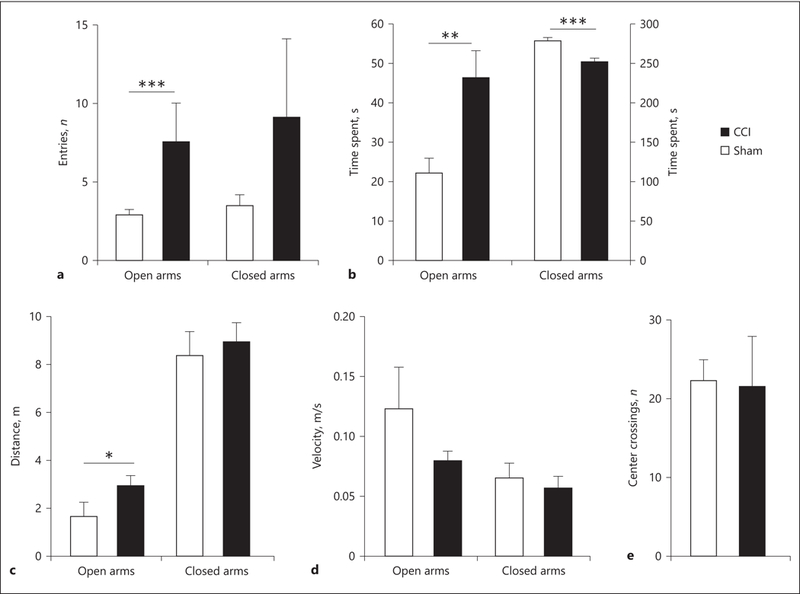
a CCI animals entered open arms significantly more often than sham animals (*** p = 0.001) with no significant differences for entries into closed arms between groups. b CCI animals were shown to spend significantly more time in open arms (** p < 0.01) and significantly less time in the closed arms (*** p < 0.001) than the uninjured animals. c CCI animals also traveled a significantly larger amount of distance in the open arms (* p < 0.05). d There were no differences in the speeds of the 2 groups while they were traveling in the open or closed arms of the maze. Results are indicative that injured animals may be more impulsive spending more time in open arms. e No significant differences were found for number of center crossings between injured and uninjured animals in open field testing.
A separate set of independent samples t tests was run to determine differences in time spent in open and closed arms. t tests found that CCI animals spent a significantly higher amount of time in open arms than uninjured animals (t10 = 2.265, p = 0.0470; Fig. 8b). Analysis also found that CCI animals spent a significantly lower amount of time in closed arms than uninjured animals (t10 = –3.159, p = 0.0102; Fig. 8b). Additional analysis revealed that CCI animals also traveled further distances while in the open arms (p = 0.015) but not in the closed arms (p = 0.668; Fig. 8c). There were no differences between the 2 groups in the speed of the animals while they were exploring the open or closed arms of the maze (p > 0.05; Fig. 8d).
Two-tailed independent samples t tests were run to determine differences between the number of center crossings in an open field in CCI and uninjured animals. Analysis found no significant differences in center crossing numbers between CCI animals and uninjured animals (t13 = –1.888, p = 0.0815; Fig. 8e).
Injury Results in Spatial Memory Deficits 2 Weeks after Injury
A repeated measures ANOVA was used to test for differences between CCI animals and uninjured animals on latency data in the Morris water maze. Significant differences in latency were found between CCI animals and sham animals (F1, 24 = 19.696, p < 0.01). Additionally, analysis found a significant effect due to day on latency. As days passed, the latency to platform decreased (F4, 24 = 32.461, p < 0.01). A significant interaction was found between injury and day on latency (F4, 24 = 2.898, p = 0.026; Fig. 9a). These findings suggest that animals who received an injury did significantly worse on each day of the trial than sham animals. Both groups made significant improvement from day 1 to day 5, which indicates that learning did occur.
Fig. 9.
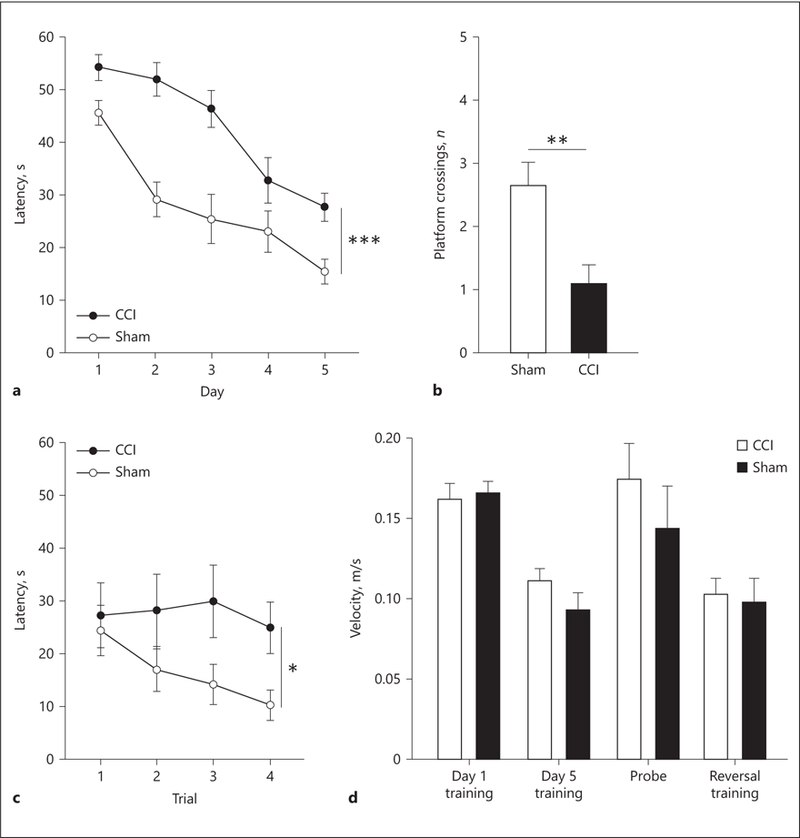
Early injury results in impaired spatial memory and cognitive flexibility. a Significant differences were found in latency to the platform between sham and injured animals. *** p < 0.001. b During the probe trial, 24 h following the last day of training, uninjured animals crossed the platform location significantly more times than injured animals. ** p < 0.01. c 24 h following the probe trial, reversal trials were done. Significant differences were found in latency to the platform between sham and injured animals. * p < 0.05. d These differences were not due to differences in the subjects’ motor ability as no differences were observed for the swim speeds throughout all phases of testing.
Animals were tested on a probe day to measure spatial memory and platform acquisition. During the probe test the platform was removed from the pool. The number of platform crossings was analyzed for injured and uninjured animals. An independent samples t test showed a significant difference in platform crossings between CCI animals and uninjured animals. Uninjured animals crossed the platform significantly more than the injured animals (t24 = –3.415, p < 0.001; Fig. 9b). This difference indicates that animals who were not injured had a better spatial memory than injured animals.
Animals were also tested on reversal days where the platform was placed 180º from its normal location. A repeated measures ANOVA showed significant effects of injury on reversal latency (F1, 24 = 5.227, p = 0.031; Fig. 9c). Additional analysis showed no significant effects on reversal latency due to trial number (F3, 24 = 1.245, p = 0.30), or interaction of injury and trial number (F3, 24 = 0.939, p = 0.426). These findings suggest that animals who received an injury had significant deficits in cognitive flexibility in the task. Swim speed was analyzed for all phases of testing and revealed no significant differences in speed throughout testing (p values >0.05; Fig. 9d), which indicates that the differences observed were not due to motor deficits or impairments in the ability to swim or perform the task. Taken together, injured subjects did not remember spatial cues to find the platform, which indicates longterm spatial memory deficits.
Discussion
This is the first study examining how jTBI results in the activation of biochemical mechanisms, such as ER stress. It is possible that these mechanisms tie directly to worsened behavioral outcomes but further studies are warranted to test this hypothesis. We found that ER stress is activated early following CCI in juvenile rats and was associated with subsequent cellular neuroinflammation and an increase in precursor neurofibrillary tangle markers. This biochemical progression was found to correlate with object recognition deficits on novel object recognition, increased impulsivity/anti-anxiety on elevated plus maze, and cognitive deficits on Morris water maze. Future studies are warranted to elucidate the exact mechanisms by which biochemical changes can contribute to behavioral deficits. The benefit of this pilot study is that we mapped a time course of injury expansion and found behavioral deficits that parallel those seen in human jTBI patients [57]. Currently, limited treatment options exist for this select group of patients, and little is known about how TBI in juveniles differs from TBI in adults. This study provides increased understanding about the pathophysiology of TBI in juveniles. When coupled with matching data from human pediatric TBI patients, these findings may serve as an initial framework that could be used to discover novel pharmacological treatment approaches for this population going forward.
CHOP and BiP were used to measure ER stress activation. CHOP is a component of the ER stress cascade while BiP is a regulator of all 3 arms of the pathway [58]. Interestingly, we found that CHOP and BiP were both significantly increased 4 h after injury, but BiP remained elevated at 48 h. This is an important finding because CHOP directly activates p-GSK-3β and the proapoptotic marker caspase-12 [54]. We found a significant increase in p-GSK-3β 1 week following CCI and a trend towards significance for caspase-12. Future studies will increase the number of experimental groups to further tease out this relationship. p-GSK-3β has been shown to be the primary contributor of tau hyperphosphorylation [59], and apoptosis has been postulated as a proposed mechanism for pathological tau to enter the extracellular milieu [60]. Once tau becomes phosphorylated, neuroinflammation must persist for the formation of tau oligomers [19]. The continued activation of BiP allowed for the activation of the second arm of the ER stress pathway. This may be the triggering event for acute cellular hypoxia and neuroinflammation, but further studies are warranted to prove this connection and investigate these pathways [61]. The timing of ER stress activation correlated with the upregulation of HIF-1α at 48 h. HIF-1α is a regulator of multiple intracellular pathways and has recently been shown to contribute to tau oligomer formation [62].
We then measured the progression of tauopathy with a series of molecular markers at 1 month. PHF, R23, and CP-13 mark a progression in tauopathy and are all markers of tau conformation change [56]. PHF is a precursor to tau oligomers, R23 is a marker for tau oligomers, and CP-13 is a precursor for neurofibrillary tangles. These markers are progressive in severity with PHF occurring prior to R23 and R23 prior to CP-13. Taken together our results support similar findings in adult models [24]. However, there are some notable differences in that HIF-1α plays a more notable role in oligomer formation in juveniles and the progression of injury is quicker than that seen in adults. Juveniles exposed to TBI have previously been shown to be at increased risk for engaging in potentially harmful behaviors such as excess alcohol consumption and are at a significantly increased risk for cognitive and behavioral disorders [63, 64].
In this study, we found that behavioral deficits can develop early after injury. The jTBI rats spent a lower percentage of time exploring the novel object showing acute deficits in object recognition. Furthermore, the jTBI rats had more impulsive-like or anti-anxiety behavior in that they spent more time and had more entries into the open arms of the elevated plus maze. This interpretation of less anxiety is based on the finding that rodents with anxiety-like features spend little time in the open arms of the elevated plus maze. Rodents are nocturnal creatures and typically choose to spend time in dark enclosed areas, which are safer than bright open places. This is a significant finding in that jTBI patients have been shown to have increased impulsive behavior in their adolescent and early adult years leading to frequent incarceration [65]. Another significant finding is that CCI caused deficits on latency to platform over the period of training and caused is that CCI caused deficits on latency to platform over the period of training and caused jTBI rats to spend significantly less time exploring where the platform had been on the probe trial. Future studies will directly compare rodent TBI serum samples to human pediatric serum samples and correlate biochemical changes to behavioral deficits in rodents and clinical outcomes in humans.
Conclusion
jTBI remains a significant challenge for treatment and preventing cognitive and behavioral deficits. In this seminal paper, we mapped for the first time the biochemical sequence of events following injury in juvenile rats. Interestingly, ER stress was activated early, HIF-1α activation occurred at 48 h, and early tauopathy was seen at 30 days. These biochemical events occurred on a similar timeline to what was observed with object recognition deficits, impulsivity or anti-anxiety, and cognitive decline measured with behavioral assays. Going forward, this foundation of understanding the injury pathophysiology in juveniles will serve as a framework for discovering innovative treatments for this patient population.
References
- 1.Pinto PS, Poretti A, Meoded A, Tekes A, Huisman TA: The unique features of traumatic brain injury in children. Review of the characteristics of the pediatric skull and brain, mechanisms of trauma, patterns of injury, complications and their imaging findings. Part 1. J Neuroimag 2012;22:e1–e17. [DOI] [PubMed] [Google Scholar]
- 2.Pinto PS, Meoded A, Poretti A, Tekes A, Huisman TA: The unique features of traumatic brain injury in children. Review of the characteristics of the pediatric skull and brain, mechanisms of trauma, patterns of injury, complications, and their imaging findings. Part 2. J Neuroimag 2012;22:e18–e41. [DOI] [PubMed] [Google Scholar]
- 3.Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Manvelyan H, Whetstone W, Noble-Haeusslein LJ: Age is a determinant of leukocyte infiltration and loss of cortical volume after traumatic brain injury. Dev Neurosci 2010;32:454–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu BR, Liu CL, Ouyang Y, Blomgren K, Siesjo BK: Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab 2000; 20:1294–1300. [DOI] [PubMed] [Google Scholar]
- 5.Semple BD, Noble-Haeusslein LJ, Jun Kwon Y, Sam PN, Gibson AM, Grissom S, Brown S, Adahman Z, Hollingsworth CA, Kwakye A, Gimlin K, Wilde EA, Hanten G, Levin HS, Schenk AK: Sociosexual and communication deficits after traumatic injury to the developing murine brain. PLoS One 2014;9:e103386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jantzie LL, Talos DM, Jackson MC, Park HK, Graham DA, Lechpammer M, Folkerth RD, Volpe JJ, Jensen FE: Developmental expression of N-methyl-D-aspartate (NMDA) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb Cortex 2015;25:482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giza CC, Prins ML, Hovda DA, Herschman HR, Feldman JD: Genes preferentially induced by depolarization after concussive brain injury: effects of age and injury severity. J Neurotrauma 2002;19:387–402. [DOI] [PubMed] [Google Scholar]
- 8.Giza CC, Maria NS, Hovda DA: N-methyl-D-aspartate receptor subunit changes after traumatic injury to the developing brain. J Neurotrauma 2006;23:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osteen CL, Giza CC, Hovda DA: Injury-induced alterations in N-methyl-D-aspartate receptor subunit composition contribute to prolonged 45calcium accumulation following lateral fluid percussion. Neuroscience 2004; 128:305–322. [DOI] [PubMed] [Google Scholar]
- 10.Gaudet CM, Lim YP, Stonestreet BS, Threlkeld SW: Effects of age, experience and inter-alpha inhibitor proteins on working memory and neuronal plasticity after neonatal hypoxia-ischemia. Behav Brain Res 2016; 302:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prins ML, Hovda DA: Traumatic brain injury in the developing rat: effects of maturation on Morris water maze acquisition. J Neurotrauma 1998;15:799–811. [DOI] [PubMed] [Google Scholar]
- 12.Max JE, Schachar RJ, Levin HS, Ewing-Cobbs L, Chapman SB, Dennis M, Saunders A, Landis J: Predictors of secondary attention-deficit/hyperactivity disorder in children and adolescents 6 to 24 months after traumatic brain injury. J Am Acad Child Adolescent Psychiatry 2005;44:1041–1049. [DOI] [PubMed] [Google Scholar]
- 13.Max JE, Lindgren SD, Knutson C, Pearson CS, Ihrig D, Welborn A: Child and adolescent traumatic brain injury: correlates of disruptive behaviour disorders. Brain Injury 1998;12:41–52. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt AT, Hanten GR, Li X, Vasquez AC, Wilde EA, Chapman SB, Levin HS: Decision making after pediatric traumatic brain injury: trajectory of recovery and relationship to age and gender. Int J Dev Neurosci 2012;30:225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks BL, Daya H, Khan S, Carlson HL, Mikrogianakis A, Barlow KM: Cognition in the emergency department as a predictor of recovery after pediatric mild traumatic brain injury. J Int Neuropsychol Soc 2016;22:379–387. [DOI] [PubMed] [Google Scholar]
- 16.Plourde V, Brooks BL: Is computerized cognitive testing useful in children and adolescents with moderate-to-severe traumatic brain injury? J Int Neuropsychol Soc 2017;23:304–313. [DOI] [PubMed] [Google Scholar]
- 17.Larner SF, Hayes RL, Wang KK: Unfolded protein response after neurotrauma. J Neurotrauma 2006;23:807–829. [DOI] [PubMed] [Google Scholar]
- 18.Begum G, Harvey L, Dixon CE, Sun D: ER stress and effects of DHA as an ER stress inhibitor. Transl Stroke Res 2013;4:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Logsdon AF, Lucke-Wold BP, Nguyen L, Matsumoto RR, Turner RC, Rosen CL, Huber JD: Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res 2016;1643:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, Moore AN: Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma 2015;32:1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Golpich M, Amini E, Hemmati F, Ibrahim NM, Rahmani B, Mohamed Z, Raymond AA, Dargahi L, Ghasemi R, Ahmadiani A: Glycogen synthase kinase-3 beta (GSK-3beta) signaling: implications for Parkinson’s disease. Pharmacol Res 2015;97:16–26. [DOI] [PubMed] [Google Scholar]
- 22.Kovacs K, Decatur C, Toro M, Pham DG, Liu H, Jing Y, Murray TG, Lampidis TJ, Merchan JR: 2-Deoxy-glucose downregulates endothelial AKT and ERK via interference with N-linked glycosylation, induction of endoplasmic reticulum stress, and GSK3beta activation. Mol Cancer Ther 2016;15:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meares GP, Mines MA, Beurel E, Eom TY, Song L, Zmijewska AA, Jope RS: Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER) stress-induced CHOP expression in neuronal cells. Exp Cell Res 2011;317: 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM, Robson MJ, Omalu BI, Huber JD, Rosen CL: Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg 2016;124: 687–702. [DOI] [PubMed] [Google Scholar]
- 25.Chavez-Valdez R, Flock DL, Martin LJ, Northington FJ: Endoplasmic reticulum pathology and stress response in neurons precede programmed necrosis after neonatal hypoxia-ischemia. Int J Dev Neurosci 2016;48: 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai XH, Li XC, Jin SW, Liang DS, Wen ZW, Cao HC, Mei HF, Wu Y, Lin ZD, Wang LX: Endoplasmic reticulum stress plays critical role in brain damage after chronic intermittent hypoxia in growing rats. Exp Neurol 2014;257:148–156. [DOI] [PubMed] [Google Scholar]
- 27.Bueter W, Dammann O, Leviton A: Endoplasmic reticulum stress, inflammation, and perinatal brain damage. Pediatr Res 2009;66: 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y, Yi W, Qin J, Zhao Y, Zhang J, Chang X: Carbon monoxide offers neuroprotection from hippocampal cell damage induced by recurrent febrile seizures through the PERK-activated ER stress pathway. Neurosci Lett 2015;585:126–131. [DOI] [PubMed] [Google Scholar]
- 29.Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ: Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 2013; 106–107: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Von Bergen M, Barghorn S, Muller SA, Pickhardt M, Biernat J, Mandelkow EM, Davies P, Aebi U, Mandelkow E: The core of tau-paired helical filaments studied by scanning transmission electron microscopy and limited proteolysis. Biochemistry 2006;45:6446–6457. [DOI] [PubMed] [Google Scholar]
- 31.Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, Bailes JE, Lee JM, Rosen CL, Huber JD: Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl Res 2015;166:509–528. e501. [DOI] [PubMed] [Google Scholar]
- 32.Dash PK, Zhao J, Kobori N, Redell JB, Hylin MJ, Hood KN, Moore AN: Activation of alpha 7 cholinergic nicotinic receptors reduce blood-brain barrier permeability following experimental traumatic brain injury. J Neurosci 2016;36:2809–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL: A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 1991;39:253–262. [DOI] [PubMed] [Google Scholar]
- 34.Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, Moore AN: Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma 2015;32:1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mao H, Jin X, Zhang L, Yang KH, Igarashi T, Noble-Haeusslein LJ, King AI: Finite element analysis of controlled cortical impact-induced cell loss. J Neurotrauma 2010;27:877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao H, Yang KH, King AI, Yang K: Computational neurotrauma – design, simulation, and analysis of controlled cortical impact model. Biomech Model Mechanobiol 2010;9: 763–772. [DOI] [PubMed] [Google Scholar]
- 37.Mao H, Zhang L, Yang KH, King AI: Application of a finite element model of the brain to study traumatic brain injury mechanisms in the rat. Stapp Car Crash J 2006;50:583–600. [DOI] [PubMed] [Google Scholar]
- 38.Meaney DF, Ross DT, Winkelstein BA, Brasko J, Goldstein D, Bilston LB, Thibault LE, Gennarelli TA: Modification of the cortical impact model to produce axonal injury in the rat cerebral cortex. J Neurotrauma 1994;11: 599–612. [DOI] [PubMed] [Google Scholar]
- 39.Martens KM, Vonder Haar C, Hutsell BA, Hoane MR: A discrimination task used as a novel method of testing decision-making behavior following traumatic brain injury. J Neurotrauma 2012;29:2505–2512. [DOI] [PubMed] [Google Scholar]
- 40.Cole JT, Yarnell A, Kean WS, Gold E, Lewis B, Ren M, McMullen DC, Jacobowitz DM, Pollard HB, O’Neill JT, Grunberg NE, Dalgard CL, Frank JA, Watson WD: Craniotomy: true sham for traumatic brain injury, or a sham of a sham? J Neurotrauma 2011;28:359– 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bevins RA, Besheer J: Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study “recognition memory.” Nat Protoc 2006;1:1306–1311. [DOI] [PubMed] [Google Scholar]
- 42.Eakin K, Baratz-Goldstein R, Pick CG, Zindel O, Balaban CD, Hoffer ME, Lockwood M, Miller J, Hoffer BJ: Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One 2014; 9:e90617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sta Maria NS, Reger ML, Cai Y, Baquing MAT, Buen F, Ponnaluri A, Hovda DA, Harris NG, Giza CC: D-Cycloserine restores experience-dependent neuroplasticity after traumatic brain injury in the developing rat brain. J Neurotrauma 2017;34:1692–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Komada M, Takao K, Miyakawa T: Elevated plus maze for mice. J Vis Exp 2008;22:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng JS, Craft R, Yu GQ, Ho K, Wang X, Mohan G, Mangnitsky S, Ponnusamy R, Mucke L: Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One 2014;9:e115765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dash PK, Mach SA, Moore AN: The role of extracellular signal-regulated kinase in cognitive and motor deficits following experimental traumatic brain injury. Neuroscience 2002; 114:755–767. [DOI] [PubMed] [Google Scholar]
- 47.Harish G, Mahadevan A, Pruthi N, Sreenivasamurthy SK, Puttamallesh VN, Keshava Prasad TS, Shankar SK, Srinivas Bharath MM: Characterization of traumatic brain injury in human brains reveals distinct cellular and molecular changes in contusion and pericontusion. J Neurochem 2015;134:156–172. [DOI] [PubMed] [Google Scholar]
- 48.Harris NG, Mironova YA, Hovda DA, Sutton RL: Chondroitinase ABC enhances pericontusion axonal sprouting but does not confer robust improvements in behavioral recovery. J Neurotrauma 2010;27:1971–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harris NG, Mironova YA, Hovda DA, Sutton RL: Pericontusion axon sprouting is spatially and temporally consistent with a growth-permissive environment after traumatic brain injury. J Neuropathol Exp Neurol 2010;69:139– 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, Naser ZJ, Knotts CM, Huber JD, Rosen CL: Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol 2015;52:1119–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris NG, Verley DR, Gutman BA, Thompson PM, Yeh HJ, Brown JA: Disconnection and hyper-connectivity underlie reorganization after TBI: a rodent functional connectomic analysis. Exp Neurol 2016;277:124–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, Kaley G: Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res 2002;90:1159–1166. [DOI] [PubMed] [Google Scholar]
- 53.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ: Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001;21:1249– 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL: Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci 2014;8:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I: Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci 2004; 117:1653–1663. [DOI] [PubMed] [Google Scholar]
- 56.D’Abramo C, Acker CM, Jimenez HT, Davies P: Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS One 2013;8:e62402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pikstra AR, Metting Z, Fock JM, van der Naalt J: The juvenile head trauma syndrome – deterioration after mild TBI: diagnosis and clinical presentation at the emergency department. Eur J Paediatr Neurol 2017;21:344–349. [DOI] [PubMed] [Google Scholar]
- 58.Logsdon AF, Lucke-Wold BP, Rosen CL, Huber JD: Disparity among neural injury models and the unfolded protein response. J Neurol Disord Stroke 2014;2:1074. [PMC free article] [PubMed] [Google Scholar]
- 59.Licht-Murava A, Paz R, Vaks L, Avrahami L, Plotkin B, Eisenstein M, Eldar-Finkelman H: A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci Signal 2016; 9:ra110. [DOI] [PubMed] [Google Scholar]
- 60.Kriegel J, Papadopoulos Z, McKee AC: Chronic traumatic encephalopathy: is latency in symptom onset explained by tau propagation? Cold Spring Harb Perspect Med 2018; 8:a024059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rong LL, Huang HC, Yu L, Li JZ: Hypoxia stimulates expression of connective tissue growth factor through p38 signaling pathway in human renal interstitial fibroblasts (in Chinese). Beijing Da Xue Xue Bao 2005;37:378– 381. [PubMed] [Google Scholar]
- 62.Salomon-Zimri S, Glat MJ, Barhum Y, Luz I, Boehm-Cagan A, Liraz O, Ben-Zur T, Offen D, Michaelson DM: Reversal of ApoE4-driven brain pathology by vascular endothelial growth factor treatment. J Alzheimers Dis 2016;53:1443–1458. [DOI] [PubMed] [Google Scholar]
- 63.Weil ZM, Karelina K, Gaier KR, Corrigan TE, Corrigan JD: Juvenile traumatic brain injury increases alcohol consumption and reward in female mice. J Neurotrauma 2016;33:895– 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mychasiuk R, Hehar H, Esser MJ: A mild traumatic brain injury (mTBI) induces secondary attention-deficit hyperactivity disorder-like symptomology in young rats. Behav Brain Res 2015;286:285–292. [DOI] [PubMed] [Google Scholar]
- 65.Gordon WA, Spielman LA, Hahn-Ketter AE, Sy KT: The relationship between traumatic brain injury and criminality in juvenile offenders. J Head Trauma Rehabil 2017;32:393– 403. [DOI] [PubMed] [Google Scholar]