

Abstract
Functionalized indoles are recurrent motifs in bioactive natural products and pharmaceuticals. While transition metal-catalyzed carbene transfer has provided an attractive route to afford C3-functionalized indoles, these protocols are viable only in the presence of N-protected indoles, due to competition from the more facile N—H insertion reaction. Herein, a biocatalytic strategy for enabling the direct C—H functionalization of unprotected indoles is reported. Engineered variants of myoglobin provide efficient biocatalysts for this reaction, which has no precedents in the biological world, enabling the transformation of a broad range of indoles in the presence of ethyl α-diazoacetate to give the corresponding C3-functionalized derivatives in high conversion yields and excellent chemoselectivity. This strategy could be exploited to develop a concise chemoenzymatic route to afford the nonsteroidal anti-inflammatory drug indomethacin.
Graphical Abstract
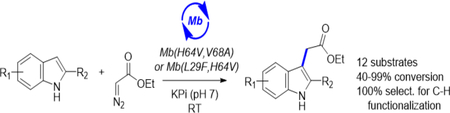
Catalysis without protection: A novel biocatalytic strategy for the synthesis of C3-functionalized indoles via myoglobin-catalyzed carbene transfer is reported. This approach enabled the transformation of a broad range of indole derivatives bearing unprotected N—H groups with high efficiency and excellent chemoselectivity and this transformation could be integrated into a chemoenzymatic scheme for the synthesis of a drug molecule (indomethacin) at the preparative scale.
Functionalized indoles are structural motifs found in many biologically active molecules, including alkaloids, agrochemicals, and drugs.[1] Because of their relevance as ‘privileged scaffolds’ in medicinal chemistry, methodologies for the synthesis of functionalized indoles have received significant attention.[2] Among them, indole functionalization via transition metal-catalyzed carbene transfer provides an attractive route to C3-substituted indoles, which constitute the core structure of various drugs[3] and medicinally important agents.[4] Kerr, Davis, Fox, and Hashimoto reported the successful application of Rh-based catalysts for realizing this transformation with diazo compounds.[5] More recently, other catalytic systems have been introduced to enable these reactions.[6] Invariably, however, the scope of these protocols have been limited to the C—H functionalization of indoles in which the N—H group is masked either through alkylation or via a protecting group,[5–6] due to inherently higher reactivity of this functional group toward carbene insertion. The application of these catalysts to unprotected indoles have indeed resulted in mixtures of N—H, C—H, and double N—H/C—H insertion products.[5a, 6b, 7] These limitations impose the need of additional protection/deprotection steps for the functionalization of indoles using carbene transfer chemistry (Scheme 1).
Scheme 1.

Metal-catalyzed functionalization of N-protected indoles vs. biocatalytic functionalization of unprotected indoles reported here.
In previous work, we established that engineered variants of myoglobin, a small (15 kDa) heme-containing protein, can provide efficient catalysts for carbene transfer reactions involving α-diazoesters, including olefin cyclopropanations[8] and carbene Y—H insertion (Y = N, S)[9] reactions, among others.[10] Biocatalytic carbene transfer reactions using other hemoproteins or metalloprotein scaffolds have also been reported.[11] More recently, we established that myoglobin variants incorporating non-native first-row transition metals (e.g., Co, Mn) are also viable catalysts for promoting carbene C(sp3)—H insertion,[12] a reaction previously achievable only using precious metals (e.g., Ir).[11e] These findings prompted us to investigate the reactivity of myoglobin in the context of the C(sp2)—H functionalization of indoles. Here, we report the successful implementation of a biocatalytic strategy for the chemoselective functionalization of the C3 C—H bond in unprotected indoles (Scheme 1).
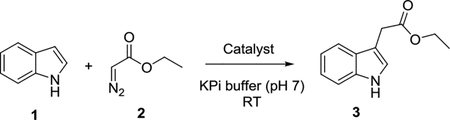
We began these studies by testing the activity of wild-type sperm whale myoglobin (Mb) and its distal histidine variant Mb(H64V)[8a, 9a] toward the conversion of indole (1) to the C3-functionalized product (3) in the presence of ethyl α-diazoacetate (2) as the carbene donor (Table 1). However, neither reactions led to appreciable formation of the desired product 3, a result comparable to that obtained using other hemoproteins or simple hemin as the catalyst (0–1% conversion; Table S1). Since mutation of the amino acid residues within the distal pocket of Mb was previously found to greatly affect its carbene transfer reactivity,[8a, 9] we then extended these studies to a panel of Mb variants, in which each of the active site residues Leu29, Phe43, Val68, and Ile107 is substituted by an amino acid bearing a side chain group of significantly different size (e.g., Leu29 → Ala or Phe; Phe43 → Ile or Ser). While the majority of these Mb variants showed no or only modest improvement in product conversion (0–5%; Table S1), Mb(H64V,V64A) was found to constitute a significantly more efficient catalyst for this reaction, producing 3 in 50% yield (Table 1). Importantly, the Mb(H64V,V68A)-catalyzed reaction proceeds with excellent chemoselectivity, yielding 3 as the only observable product. This result is in stark contrast with that obtained from reactions with Rh2(OAc)4 as the catalyst, from which nearly equal amounts of 3 (45%) and N—H insertion by-product (42%) were formed, along with some double insertion product (13%; Table 1 and Figure S1). Encouraged by these results, the Mb(H64V,V68A)-catalyzed transformation was targeted for optimization. The efficiency of this reaction was found to have a slight dependence on pH (Table S2), with a pH of 9 being optimal (Table 1). However, a more substantial improvement in product yield (54%→85%) was achieved by increasing the indole : EDA ratio from 1:1 to 1:2 (Table 1). Despite the excess diazo reagent and the single-addition protocol, no carbene dimerization (to give diethyl maleate/fumarate) or reaction with water (to give ethyl glycolate) was observed, highlighting the high chemoselectivity of the biocatalyst also with respect to these potential side reactions.
Table 1.
Activity and selectivity of myoglobin variants and other catalysts for the C—H functionalization of indole with ethyl α-diazoacetate (EDA).[a] See also SI Tables S1 and S2.
| Catalyst | EDA equiv | Conv. [b] |
TON | % C-H funct. | |
|---|---|---|---|---|---|
| Hemin | - | 1 | <1% | 1 | n.a. |
| Rh2(OAc)4[c] | - | 1 | 5% | 5 | 45[c] |
| Mb | Protein | 1 | <1% | 1 | n.a. |
| Mb(H64V) | Protein | 1 | <1% | <1 | n.a. |
| Mb(L29A,H64V) | Protein | 1 | 5% | 6 | 100 |
| Mb(H64V,V68A) | Protein | 1 | 50% | 62 | 100 |
| Mb(H64V,V68A)[d] | Protein | 1 | 54% | 68 | 100 |
| Mb(H64V,V68A)[d] | Protein | 2 | 85% | 106 | 100 |
| Mb(H64V,V68A)[e] | Protein | 2 | 34% | 168 | 100 |
| Mb(H64V,V68A) | Cells[f] | 2 | 70% | 18[h] | 100 |
| Mb(H64V,V68A) | Cells[g] | 2 | >99% | 82[h] | 100 |
Reactions conditions: 20 μM Mb variant or hemin (0.8 mol%), 2.5 mM indole, 2.5 or 5 mM EDA, 10 mM dithionite, 16 h. Reported values are mean values from n ≥ 2 experiments (SE <15%).
GC yield using calibration curve with authentic 3.
Using 0.8 mol% catalyst in CH2Cl2; other products: N—H insertion (42%) and double insertion (13%).
pH 9.0.
Using 5 μM protein (0.2 mol%) and pH 9.0.
OD600 = 40.
OD600 = 20. n.a. = not available.
As determined based on the protein concentration in cell lysates using ε410 = 156 mM−1 cm−1.
Under catalyst-limited conditions (0.2 mol%), Mb(H64V,V68A) catalyzes about 168 turnovers (TON). These TON values are lower than those supported by this variant in other carbene transfer reactions with EDA (1,000–10,000),[8a, 9–10] but they compare favourably (3- to 6-fold higher) with those reported using Pd- or Cu-based catalysts for the C3 functionalization of N-protected indoles.[6c, 6d] From time course experiments, the Mb(H64V,V68A)-catalyzed conversion of indole to 3 was determined to proceed with an initial rate of about 10 turnovers/min and to reach completion in less than 15 minutes (Figure S2). Control reactions confirmed that the ferrous form of Mb(H64V,V68A) is responsible for catalysis.
Given the feasibility of performing Mb-catalyzed carbene transfer reactions directly in whole cells,[8b, 13] we next investigated the viability of this approach for the biocatalytic C—H functionalization of indole. Accordingly, whole-cell biotransformations were carried out using Mb(H64V,V68A)-expressing E. coli cells (C41(DE3))[14] in the presence of the optimized indole:EDA ratio of 1:2. Interestingly, a progressive improvement in product yields was observed upon decreasing the cell density in these reactions (Table S2), a phenomenon we tentatively attributed to the sequestration or utilization of the indole substrate by the cells. Importantly, under optimal conditions (OD600 = 20), quantitative conversion of the indole substrate to the desired C—H functionalization product 3 was achieved (Table 1). The TON values measured in this reaction was comparable to that obtained with purified protein (82 vs. 106), indicating a nearly optimal utilization of the intracellularly expressed biocatalyst.
Using the whole cell protocol, the scope of the reaction was then probed using a panel of indole derivatives. As summarized in Scheme 2, a variety of 5- (4a-7a), 6- (8a), and 7-substituted indoles (9a) could be efficiently processed by the Mb variant to give the corresponding C3-functionalized products 4b-9b in quantitative conversions (>99%). 9b was isolated in 61% yield. As an exception, 5-methoxy-indole (7a) was converted to 7b with only moderate efficiency using the standard protocol (28%). Significantly improved conversion of this substrate was obtained however using purified Mb(H64V,V68A) and sequential catalyst additions (77%). For each of the indole substrates, the desired carbene C—H insertion product was the only detectable product, indicating that the metalloprotein catalyst maintains excellent chemoselectivity toward C—H functionalization over the more facile carbene N—H insertion reaction regardless of the position and electronic effect of the substituent on the indole ring.
Scheme 2.

Substrate scope for Mb(H64V,V68A)- and Mb(L29F,H64V)-catalyzed C—H functionalization of indoles. Reaction conditions: 2.5 mM indole derivative, 5 mM EDA, whole cells at OD600 >= 20 in 50 mM phosphate buffer (pH 7), RT. [a] Using 75 μM Mb(H64V,V68A)in 50 mM phosphate buffer (pH 9) containing 10 mM Na2S2O4 (sequential catalyst addition protocol). [b] As in [a] but using 85 μM Mb(L29F,H64V). Reported conversions are mean values from n ≥ 2 experiments (SE <10%)
Next, we examined the ability of Mb(H64V,V68A) to accept C2-substitued (10a), N-methylated (11a), and doubly substituted indole derivatives such as 12a, 13a, and 14a (Scheme 2). Although the corresponding C—H functionalization product (10b-14b) was obtained in each case, the yields were generally modest (5–26%), denoting a scope limitation of this Mb variant across this subset of indole derivatives. To identify a better catalyst for these substrates, the original panel of Mb variants was screened against 14a, which bears substitutions at both the C2 and C5 positions and was recognized as a potential precursor for a drug molecule (vide infra). Interestingly, many of these Mb variants displayed significantly higher activity (2- to 5-fold) on this substrate compared to Mb(H64V,V68A) (Table S3) in spite of their inferior performance in the context of indole (Table S1), thus denoting a clear preference of these biocatalysts for bulkier indole substrates. Among them, Mb(F43S,H64V) and Mb(L29F,H64V) emerged as the most promising catalysts for this reaction, furnishing 14b in 31% and 41% yield, respectively (Table S3). The latter reaction could be further optimized to give 14b in 80% (Scheme 2). Gratifyingly, in addition to 14a, Mb(L29F,H64V) was found to exhibit improved activity also toward 2-methyl-, 5-methyl-N-methyl-, and 5-chloro-N-methyl-indole, yielding the corresponding products 10b, 12b, and 13b, respectively, in 50–85% conversion (Scheme 2). For 12b, this corresponds to a nearly 8-fold higher catalytic efficiency compared to Mb(H64V,V68A) (85% vs. 10%). While being complementary in terms of substrate scope, Mb(L29F,H64V) shares with Mb(H64V,V68A) excellent chemoselectivity for the C—H functionalization reaction (no detectable N—H insertion product) and comparable kinetics (initial rate: 7–10 TON/min−1).
Mb-catalyzed carbene transfer reactions are believed to proceed via the formation of an electrophilic heme-bound carbene intermediate,[8a, 15] which can react with various nucleophiles such as olefins,[8a] amine,[9a] thiols,[9b] or phosphines[10]. Based on the ability of this reactive intermediate to give rise to sulfonium ylides upon reaction with thiol/thioether nucleophiles[9b, 16] and the well established nucleophilicity of indoles at the C3 site,[17] a plausible mechanism for the Mb-catalyzed indole C—H functionalization is proposed in Scheme 3. According to it, reaction of the catalytically active ferrous Mb (I) with the diazo reagent leads to the heme-carbene intermediate II, which undergoes nucleophilic attack by the indole substrate to yield the zwitterionic species III. From this intermediate, the final product can then arise from a [1,2]-proton shift from the C3 atom to the ester α-carbon atom or, alternatively, through dissociation of the zwitterionic enolate followed by protonation by the solvent. To discriminate between these pathways, the enzymatic reaction was performed with 1-methyl-3-deutero-indole (11a-3-d). Insightfully, the product of this reaction showed complete loss of the deuterium label (Figure S4), thus supporting the involvement of solvent-mediated protonation. This result, along with the absence of a kinetic isotope effect (KIE) in reactions with 11a-3-d vs. 11a (Figure S4–S5), also rule out a carbene C—H insertion process, as in the latter case retention of the deuterium label in the product and a positive KIE[18] would be expected. On the other hand, no observable formation of a C2/C3 cyclopropane intermediate disfavours, without necessarily excluding, an alternative scenario involving a cyclopropanation/fragmentation pathway.[6b] Overall, the proposed mechanism shares similarities with those proposed for related reactions promoted by Rh-[5c] and non-heme Fe-based catalysts[6a] but differs from that reported for indole functionalization with diazo reagents with Cu-complexes.[6b]
Scheme 3.

Proposed mechanism for the Mb-catalyzed C—H functionalization of indoles. The relative position of the Phe29 residue in Mb(L29F,H64V) is shown (grey).
By comparison with Mb and Mb(H64V) (Table 1), the enhanced reactivity of Mb(H64V,V68A) toward in this reaction can be largely attributed to the V68A mutation. This amino acid substitution expands the active site in proximity of heme cofactor, which could facilitate attack of the heme-bound carbene by the indole (II→III), as previously observed for other nucleophiles.[9] At the same time, the diminished reactivity of Mb(H64V,V68A) toward C2-substituted indoles (i.e., 10b and 14a; Scheme 2) can be rationalized based on increased steric interactions between the C2 substituent and the porphyrin ring in the hemoprotein (Scheme 3). Interestingly, this substrate scope limitation was effectively overcome using Mb(L29F,H64V). From the structure-activity data gathered for closely related Mb variants (e.g., Mb(H64V) and Mb(L29A,H64V); Table S3), the superior reactivity of Mb(L29F,H64V) toward C2-substituted or other bulky indole substrates appears to depend upon the presence of a Phe residue at position 29. A molecular model of Mb(L29F,H64V) shows that Phe29 projects its side-chain phenyl group above the heme cofactor (Figure S3). This arrangement is well suited to potentially favor the interaction (e.g., via π stacking) and/or affect the orientation of the indole substrate during attack to the heme-carbene intermediate (II→III, Scheme 3), possibly reducing steric clashes between the C2 substituent and heme ring. Similar protein/substrate interactions could also help dictate the high chemoselectivity of the biocatalyst toward C3 functionalization over N—H carbene insertion, although further studies are clearly warranted to elucidate these aspects in more detail.
To further assess the synthetic value of the present method, the synthesis of the non-steroidal anti-inflammatory drug indomethacin (16) was targeted via a chemoenzymatic route integrating Mb-catalyzed indole C—H functionalization (Scheme 4). Starting from commercially available 2-methyl-5-methoxy-indole (14a), a preparative-scale (1 mmol) reaction with Mb(L29F,H64V)-expressing cells afforded ~0.1 g of the desired C3-functionalized product 14b in 40% isolated yield. N-acylation of this intermediate with p-chlorobenzoyl chloride, followed by hydrolysis afforded the target drug. This chemoenzymatic sequence furnishes a more concise route than that originally reported for the synthesis of this molecule (3 vs. 5 steps)[3b] and a viable alternative to more recent syntheses involving precious metal catalysts.[19] More importantly, these results provide a proof-of-principle demonstration of the scalability of the Mb-catalyzed transformation disclosed here.
Scheme 4.

Chemoenzymatic synthesis of indomethacin.
In summary, we have reported the first example of a biocatalytic strategy for carbene-mediated functionalization of indoles. This approach could be applied to the C3 functionalization of unprotected indoles, a goal previously unattainable using synthetic transition metal catalysts due to competition from the inherently more favourable carbene N—H insertion reaction. Using two engineered Mb variants with complementary substrate profile, a broad range of variously substituted indole derivatives, including C2-substituted and doubly substituted indoles, could be processed with high efficiency and excellent chemoselectivity. This newly developed Mb-catalyzed transformation could be further leveraged to implement a concise chemoenzymatic route for the synthesis of a drug molecule. This study expands the range of abiotic transformations accessible through engineered and artificial metalloenzymes.[20]
Supplementary Material
Acknowledgements
This work was supported by the U.S. National Institute of Health grant GM098628. A.T. acknowledges support from the Ford Foundation Graduate Fellowship Program. LC-MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
References
- [1].a) Sharma V, Kumar P, Pathak D, J. Heterocyclic Chem 2010, 47, 491–502; [Google Scholar]; b) Kochanowska-Karamyan AJ, Hamann MT, Chem. Rev 2010, 110, 4489–4497; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang MZ, Chen Q, Yang GF, Eur. J. Med. Chem 2015, 89, 421–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Chen JB, Jia YX, Org. Biomol. Chem 2017, 15, 3550–3567; [DOI] [PubMed] [Google Scholar]; b) Bandini M, Eichholzer A, Angew. Chem. Int. Ed 2009, 48, 9608–9644. [DOI] [PubMed] [Google Scholar]
- [3].a) Shaw E, J. Am. Chem. Soc 1955, 77, 4319–4323; [Google Scholar]; b) Shen TY, Lucas S, Sarett LH, Rosegray A, Nuss GW, Willett JD, Ellis RL, Holly FW, Matzuk AR, Wilson AN, Winter CA, Windholz TB, et al. , J. Am. Chem. Soc 1963, 85, 488–489. [Google Scholar]
- [4].a) Rawson DJ, Dack KN, Dickinson RP, James K, Bioorg. Med. Chem. Lett 2002, 12, 125–128; [DOI] [PubMed] [Google Scholar]; b) Batt DG, Qiao JX, Modi DP, Houghton GC, Pierson DA, Rossi KA, Luettgen JM, Knabb RM, Jadhav PK, Wexler RR, Bioorg. Med. Chem. Lett 2004, 14, 5269–5273. [DOI] [PubMed] [Google Scholar]
- [5].a) Gibe R, Kerr MA, J. Org. Chem 2002, 67, 6247–6249; [DOI] [PubMed] [Google Scholar]; b) Lian YJ, Davies HML, J. Am. Chem. Soc 2010, 132, 440-+; [DOI] [PubMed] [Google Scholar]; c) DeAngelis A, Shurtleff VW, Dmitrenko O, Fox JM, J. Am. Chem. Soc 2011, 133, 1650–1653; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Goto T, Natori Y, Takeda K, Nambu H, Hashimoto S, Tetrahedr. Asymm 2011, 22, 907–915. [Google Scholar]
- [6].a) Cai Y, Zhu SF, Wang GP, Zhou QL, Adv. Synth. Cat 2011, 353, 2939–2944; [Google Scholar]; b) Delgado-Rebollo M, Prieto A, Perez PJ, Chemcatchem 2014, 6, 2047–2052; [Google Scholar]; c) Gao X, Wu B, Yan Z, Zhou YG, Org. Biomol. Chem 2016, 14, 8237–8240; [DOI] [PubMed] [Google Scholar]; d) Gao X, Wu B, Huang WX, Chen MW, Zhou YG, Angew. Chem. Int. Ed 2015, 54, 11956–11960. [DOI] [PubMed] [Google Scholar]
- [7].Keller H, Langer E, Lehner H, Monatsh. Chem 1977, 108, 123–131. [Google Scholar]
- [8].a) Bordeaux M, Tyagi V, Fasan R, Angew. Chem. Int. Ed 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bajaj P, Sreenilayam G, Tyagi V, Fasan R, Angew. Chem. Int. Ed 2016, 55, 16110–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Sreenilayam G, Fasan R, Chem. Commun 2015, 51, 1532–1534; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tyagi V, Bonn RB, Fasan R, Chem. Sci 2015, 6, 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tyagi V, Fasan R, Angew. Chem. Int. Ed 2016, 55, 2512–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Coelho PS, Brustad EM, Kannan A, Arnold FH, Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]; b) Reynolds EW, Schwochert TD, McHenry MW, Watters JW, Brustad EM, Chembiochem 2017, 18, 2380–2384; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gober JG, Ghodge SV, Bogart JW, Wever WJ, Watkins RR, Brustad EM, Bowers AA, ACS Chem. Biol 2017, 2, 1726–1731; [DOI] [PubMed] [Google Scholar]; d) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC, Nat. Commun 2015, 6, 7789; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dydio P, Key HM, Nazarenko A, Rha JY, Seyedkazemi V, Clark DS, Hartwig JF, Science 2016, 354, 102–106; [DOI] [PubMed] [Google Scholar]; f) Kan SBJ, Lewis RD, Chen K, Arnold FH, Science 2016, 354, 1048–1051; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Rioz-Martinez A, Oelerich J, Segaud N, Roelfes G, Angew. Chem. Int. Ed 2016, 55, 14136–14140; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Weissenborn MJ, Low SA, Borlinghaus N, Kuhn M, Kummer S, Rami F, Plietker B, Hauer B, Chemcatchem 2016, 8, 1636–1640; [Google Scholar]; i) o MW, Vargas DA, Lehnert N, Inorg. Chem 2017, 56, 5623–5635; [DOI] [PubMed] [Google Scholar]; j) Ohora K, Meichin H, Zhao LM, Wolf MW, Nakayama A, Hasegawa J, Lehnert N, Hayashi T, J. Am. Chem. Soc 2017, 139, 17265–17268; [DOI] [PubMed] [Google Scholar]; k) Chen K, Huang XY, Kan SBJ, Zhang RK, Arnold FH, Science 2018, 360, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sreenilayam G, Moore EJ, Steck V, Fasan R, Adv. Synth. Cat 2017, 359, 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tinoco A, Steck V, Tyagi V, Fasan R, J. Am. Chem. Soc 2017, 139, 5293–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bordeaux M, Singh R, Fasan R, Bioorg. Med. Chem 2014, 22, 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wei Y, Tinoco A, Steck V, Fasan R, Zhang Y, J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, Fasan R, Angew. Chem. Int. Ed 2016, 55, 13562–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sundberg RJ, in Heterocyclic Scaffolds II:. Topics in Heterocyclic Chemistry, Vol. 26 (Ed.: Gribble G), Springer, Berlin, Heidelberg, 2010, pp. 47–115. [Google Scholar]
- [18].Babu SD, Hrytsak MD, Durst T, Can. J. Chem 1989, 67, 1071–1076. [Google Scholar]
- [19].a) Kasaya Y, Hoshi K, Terada Y, Nishida A, Shuto S, Arisawa M, Eur. J. Med. Chem 2009, 4606–4613; [Google Scholar]; b) Mukai C, Takahashi Y, Org. Lett 2005, 7, 5793–5796. [DOI] [PubMed] [Google Scholar]
- [20].a) Brandenberg OF, Fasan R, Arnold FH, Curr. Opin. Biotech 2017, 47, 102–111; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schwizer F, Okamoto Y, Heinisch T, Gu YF, Pellizzoni MM, Lebrun V, Reuter R, Kohler V, Lewis JC, Ward TR, Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.