

AADC deficiency causes severe motor and intellectual disability as a result of reduced catecholamine levels. Kojima et al. report beneficial effects of gene therapy in six patients with heterogeneous genetic backgrounds. Gene delivery into putamen improved motor function in all patients, plus verbal and cognitive skills in one moderate-phenotype patient.
Keywords: aromatic l-amino acid decarboxylase (AADC) deficiency, adeno-associated virus (AAV) vector, dopamine, putamen, dystonia
Abstract
In patients with aromatic l-amino acid decarboxylase (AADC) deficiency, a decrease in catecholamines and serotonin levels in the brain leads to developmental delay and movement disorders. The beneficial effects of gene therapy in patients from 1 to 8 years of age with homogeneous severity of disease have been reported from Taiwan. We conducted an open-label phase 1/2 study of population including adolescent patients with different degrees of severity. Six patients were enrolled: four males (ages 4, 10, 15 and 19 years) and one female (age 12 years) with a severe phenotype who were not capable of voluntary movement or speech, and one female (age 5 years) with a moderate phenotype who could walk with support. The patients received a total of 2 × 1011 vector genomes of adeno-associated virus vector harbouring DDC via bilateral intraputaminal infusions. At up to 2 years after gene therapy, the motor function was remarkably improved in all patients. Three patients with the severe phenotype were able to stand with support, and one patient could walk with a walker, while the patient with the moderate phenotype could run and ride a bicycle. This moderate-phenotype patient also showed improvement in her mental function, being able to converse fluently and perform simple arithmetic. Dystonia disappeared and oculogyric crisis was markedly decreased in all patients. The patients exhibited transient choreic dyskinesia for a couple of months, but no adverse events caused by vector were observed. PET with 6-[18F]fluoro-l-m-tyrosine, a specific tracer for AADC, showed a persistently increased uptake in the broad areas of the putamen. In our study, older patients (>8 years of age) also showed improvement, although treatment was more effective in younger patients. The genetic background of our patients was heterogeneous, and some patients suspected of having remnant enzyme activity showed better improvement than the Taiwanese patients. In addition to the alleviation of motor symptoms, the cognitive and verbal functions were improved in a patient with the moderate phenotype. The restoration of dopamine synthesis in the putamen via gene transfer provides transformative medical benefit across all patient ages, genotypes, and disease severities included in this study, with the most pronounced improvements noted in moderate patients.
Introduction
Aromatic l-amino acid decarboxylase (AADC) deficiency (OMIM #608643) is an autosomal recessive neurotransmitter disorder caused by defects in the dopa decarboxylase (DDC) gene, which encodes AADC. About 140 patients have been reported worldwide thus far (Hyland and Clayton, 1992; Pons et al., 2004; Ito et al., 2008; Manegold et al., 2009; Brun et al., 2010; Ide et al., 2010; Chen et al., 2014; Atwal et al., 2015; Kojima et al., 2016; Lee et al., 2017; Wassenberg et al., 2017). AADC catalyses the formation of neurotransmitters from l-DOPA and 5-hydroxytryptophan to dopamine and serotonin, respectively. Consequently, the depletion of dopamine and serotonin, as well as norepinephrine and epinephrine, which are synthesized from dopamine, is induced in patients with AADC deficiency. In the CSF and serum of patients with AADC deficiency, the concentrations of these biogenic amines and metabolites are reduced, while those of l-DOPA and 3-O-methyldopa are elevated (Hyland and Clayton, 1992; Wassenberg et al., 2017). The main phenotype of AADC deficiency is movement disorder, including loss of voluntary movement, hypotonia, intermittent oculogyric crisis (OGC) and limb dystonia (Korenke et al., 1997; Brun et al., 2010). OGC presents as a sustained upward or lateral deviation of the eyes in combination with backward and lateral flexion of the neck. Patients also present with autonomic dysfunction characterized by impairment of the sympathetic regulation of heart rate and blood pressure, excessive sweating and hyper-salivation, intellectual disability, hypoglycaemia, epilepsy, emotional instability and sleep disturbance (Hyland and Clayton, 1992; Brun et al., 2010; Arnoux et al., 2013; Helman et al., 2014; Lee et al., 2014; Spitz et al., 2017). The disease onset is in early infancy, and most patients with the severe phenotype are bedridden for life. However, some relatively mild phenotypes have also been reported (Hyland et al., 1992; Swoboda et al., 2003; Tay et al., 2007; Leuzzi et al., 2015). In the guideline for AADC deficiency, cases are classified as severe (80%: no or very limited achievement of developmental milestones, fully dependent), mild (5%: mild developmental delay and intellectual disability, ambulatory without assistance) and moderate (15%: between severe and mild) (Wassenberg et al., 2017). Several drugs have been applied for treatment, including vitamin B6, which is a coenzyme of AADC, l-DOPA, dopamine agonists, and monoamine oxidase B (MAOB) inhibitors. However, only patients with the moderate or mild phenotype respond to drugs—mainly dopamine agonists and MAOB inhibitors (Fiumara et al., 2002; Mastrangelo et al., 2013; Leuzzi et al., 2015)—with drug therapy providing little or no benefit for most severe patients (Hwu et al., 2018).
The successful application of gene therapy in the treatment of four patients with AADC deficiency (age 4–6 years) was reported from Taiwan in 2012 (Hwu et al., 2012), and 10 additional patients (age 1.67–8.42 years) were reported in 2017 (Chien et al., 2017). The prevalence of AADC deficiency in the Taiwanese population is high because of the founder mutation IVS6+4A>T, and most Taiwanese patients present with the severe phenotype (Lee et al., 2009; Hwu et al., 2018). After gene therapy, some previously bedridden patients were able to sit or stand with support. The treatment vector (AAV-hAADC-2) was made by inserting the human AADC gene with cytomegalovirus (CMV) promoter into the type 2 adeno-associated virus (AAV) vector. The vector was then injected into the putamen bilaterally by a stereotactic operation (Hwu et al., 2012). AAV-hAADC-2 was initially developed for clinical studies of gene therapy for Parkinson’s disease (Muramatsu et al., 2010).
In the present study, we performed gene therapy for six patients (4–19 years of age) with AADC deficiency using an AADC-expressing AAV vector similar to that reported in the Taiwanese study (Hwu et al., 2012; Chien et al., 2017). Our patients differed from the Taiwanese patients in that our patients had a variable genetic background, one had the moderate phenotype, and four were older than the Taiwanese patients.
Materials and methods
Study design
This study was planned as an open-label, phase 1/2 trial at Jichi Medical University Hospital (Tochigi, Japan) to investigate the effects of gene therapy in patients with AADC deficiency. The primary objective of this study was to verify the safety of the therapy; the secondary objective was to obtain preliminary data on the clinical response to the gene therapy by assessing the improvement in the motor function, cognitive function, autonomic function and other factors. We recorded adverse events, as well as neurological findings, physical findings, laboratory findings (including the results of CSF catecholamine metabolites analysis) and brain MRI, CT and EEG findings. The study population included patients with AADC deficiency who were unable to stand, who presented motor disturbance and dystonia and in whom the diagnosis of AADC deficiency had been confirmed based on a CSF analysis, AADC enzyme activity or a genetic analysis. We excluded patients with mild-phenotype AADC deficiency who were able to stand and walk independently as well as seriously ill patients. We also excluded patients with normal findings on PET using the non-catecholic tracer 6-[18F] fluoro-l-m-tyrosine (FMT) (FMT-PET), a specific tracer for AADC (Muramatsu et al., 2010), as well as patients in whom brain MRI could not be performed.
Patients
Six patients with characteristic manifestations of AADC deficiency were enrolled from 1 May 2015, to 31 July 2017. The profiles of the patients are listed in Tables 1 and 2. Patients 1, 2 and 4–6, who showed the severe phenotype (Ito et al., 2008; Ide et al., 2010; Kojima et al., 2016), were bedridden without voluntary movement, with OGCs and generalized dystonic attacks. Their mental status was relatively preserved, to the extent that they could visually follow objects, smile and distinguish people and situations. They understood simple words but could not speak. Patient 4 had refractory tonic convulsions. Patient 3 showed the moderate phenotype (Kojima et al., 2016). The patient had been bedridden until 3 years of age before receiving a diagnosis of AADC deficiency. After MAOB inhibitor treatment was started, the patient gained the ability to walk unsteadily with support and to speak several words unclearly (Kojima et al., 2016). However, this improvement stopped (Kojima et al., 2016), so we decided to perform gene therapy in this patient. Blood analyses and biochemical investigations revealed normal findings. The blood AADC enzyme activity was low in all patients. The genetic mutations in each patient are shown in Table 1 and Supplementary Fig. 1. A catecholamine metabolite analysis of the CSF revealed a very high concentration of l-DOPA and very low concentration of homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA) in all patients, findings that were compatible with AADC deficiency (Table 3). In addition, brain structural MRI showed no abnormalities. EEG showed no paroxysmal discharges in the patients, with the exception of Patient 4, who showed focal and diffuse epileptic discharges (Ito et al., 2008; Ide et al., 2010; Kojima et al., 2016).
Table 1.
Patient details
| Patient No. | Gender | Age at GT (year) | Severity | Motor status | Intake | Respiratory support | DDC gene mutations | Follow-up period after GT |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 15 | Severe | Bedridden | GS | Laryngo-tracheal separation | c.329C>A, p.(Ala110Glu) | 2 years 7 months |
| Not detected | ||||||||
| 2 | F | 12 | Severe | Bedridden | GS | – | c.329C>A, p.(Ala110Glu) | 2 years 6 months |
| Not detected | ||||||||
| 3 | F | 5 | Moderate | Walk with support | Oral | – | c.315G>C, p.(Try105Cys) | 2 years 1 months |
| c.385C>T, p.(Pro129Ser) | ||||||||
| 4 | M | 19 | Severe | Bedridden | TF | NIPPV | c.1106A>G, p.(Tyr369Cys) | 2 years |
| IVS6+4A>T | ||||||||
| 5 | M | 10 | Severe | Bedridden | GS | – | IVS6+4A>T | 1 years |
| IVS6+4A>T | ||||||||
| 6 | M | 4 | Severe | Bedridden | Oral | – | c.236A>G, p.(Tyr79Cys) | 6 months |
| c.755A>G, p.(Asp252Gly) |
GT = gene therapy; GS = gastrostomy; TF = nasogastric tube feeding; NIPPV = non-invasive positive pressure ventilation.
Table 2.
The clinical course of gene therapy
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL | 2 y 7 m | BL | 2 y 6 m | BL | 2 y 1 m | BL | 2 y | BL | 1 y | BL | 6 m | |
| Motor | ||||||||||||
| Head control | – | + | – | + | + | + | – | + | – | + | – | + |
| Sit | – | ± | – | ± | + | + | – | ± | – | ± | – | ± |
| Walk | – | ± | – | ± | ± | ++ | – | – | – | ± | – | – |
| Grip | – | + | ± | ++ | + | ++ | – | + | – | + | – | ± |
| Involuntary movement | ||||||||||||
| Dystonia | +++ | – | +++ | – | – | – | + | – | +++ | – | +++ | – |
| OGC | +++ | ± | +++ | ± | ++ | ± | ± | – | +++ | ± | +++ | ± |
| Autonomic function | ||||||||||||
| Sweat | ++ | ± | ++ | – | ± | – | ++ | – | ++ | – | + | ± |
| Saliva | +++ | – | +++ | – | – | – | +++ | – | ++ | + | + | – |
| Diarrhea | + | + | – | – | – | + | – | – | – | – | – | – |
| Sleep disturbance | ++ | – | ++ | ± | – | – | ± | ± | ++ | + | ++ | – |
| Mental status | ||||||||||||
| Words | – | – | – | Bubbling | Few words | Conversation | – | ± | – | – | – | Few words |
| Mood | Unstable | Good | Unstable | Smile | Stable | Very good | Stable | Smile | Unstable | Smile | Unstable | Smile |
| Respiratory dysfunction | ± | – | – | – | – | – | + | – | ± | – | – | – |
| Feeding | GS | Oral and GS | GS | Oral | Oral | Oral | TF | Oraland TF | GS | Oral and GS | Oral | Oral |
BL = baseline; GS = gastrostomy; TF = nasogastric tube feeding. Plus/minus symbol in motor functions indicates that they are able to perform the function with support.
Table 3.
Catecholamine metabolites in CSF
| Patient No. | HVA, ng/ml | 5-HIAA, ng/ml | MHPG, ng/ml | l-DOPA, ng/ml | ||||
|---|---|---|---|---|---|---|---|---|
| BL | 1 m | BL | 1 m | BL | 1 m | BL | 1 m | |
| 1 | 1.1 | 1.2 | <1.0 | <1.0 | <1.0 | <1.0 | 8.2 | 9.8 |
| 2 | 2.1 | 3.9 | <1.0 | <1.0 | <1.0 | <1.0 | 14.5 | 13.2 |
| 3 | 10.7 | 15 | 1.5 | 1.4 | 1.6 | 1.3 | 34.7 | 31.8 |
| 4 | 3.5 | 4.1 | <1.0 | <1.0 | <1.0 | <1.0 | 12.1 | 9.6 |
| 5 | 0.1 | 0.1 | 0.1 | 0.1 | <1.0 | <1.0 | 11.2 | 5 |
| 6 | 2.1 | 6.8 | 1 | 1 | <1.0 | ND | 371 | 173.4 |
| Normal | 4.4–15.1 | 1.8–6.1 | 6.5–51 | 1.2–2.2 | ||||
5-HIAA = 5-hydroxyindoleacetic acid; BL = baseline; l-DOPA, L-3,4-dihydroxyphenylalanine; MHPG = 3-methoxy-4-hydroxyphenylmethyleneglycol; ND = no data.
Before the gene therapy, the main drugs prescribed for patients were as follows: vitamin B6 for Patients 1–5; l-DOPA for Patients 3 and 6; dopamine agonist for Patients 2, 4 and 5; MAOB inhibitor for Patients 3 and 6; and selective serotonin reuptake inhibitor for Patients 1 and 2. In addition to these, carnitine, folic acid, melatonin agonist and gastrointestinal drugs were prescribed for one to three patients, each.
Ethics
This study was approved by the Ethics Committee for Gene Therapy at Jichi Medical University Hospital and the Science Council, Ministry of Health, Labor and Welfare. Conformance with the inclusion criteria was evaluated before each treatment, and the status of the treated patients was reported to the evaluation committee of the Ethics Committee for Gene Therapy at Jichi Medical University Hospital. Written informed consent was obtained from the parents of each patient. The study was conducted in compliance with the Japanese Guidelines on Gene Therapy and is registered with the UMIN Clinical Trials Registry (No. UMIN000017802).
Vector
A recombinant AAV type 2 vector, AAV-hAADC-2, was prepared as described previously (Muramatsu et al., 2010). The expression cassette consisted of a CMV promoter followed by human β-globin intron, human AADC complementary DNA and polyadenylation signal from human growth hormone. Clinical-grade AAV-hAADC-2 was manufactured, and quality control was tested in compliance with the current Good Manufacturing Practices at Takara Bio Inc.
Stereotactic neurosurgery
Patients received infusions of the vector into two target points (one each into the bilateral putamen) by stereotactic surgery that was planned with the dedicated software program included with the StealthStation Treon Navigation System (FrameLink, Medtronic, Ireland) (Muramatsu et al., 2010). Two target points that were sufficiently distant from each other in the dorsolateral direction, as confirmed by CT and MRI, were determined for each putamen. One burr hole was trepanned in each side of the cranial bone, and the vector was injected via a two-track insertion route. At each target point, 50 μl of the vector-containing solution was injected at a rate of 3 μl/min (a total of 200 μl containing 2 × 1011 vector genomes). For each injection, the needle was inserted into the deepest point of each target and then withdrawn 1 mm after each 10 μl injection. Thus, the vector was injected over a length of 5 mm at each point. All procedures were safely performed under general anaesthesia.
Clinical evaluation of the gene therapy
The physical and neurological functions of the patients were evaluated. All patients were videotaped to track their clinical course. The Alberta Infant Motor Scale (AIMS) administered by paediatric neurologists was used to evaluate motor development (Piper et al., 1992). The Kyoto Scale of Psychological Development 2001 was used to evaluate the mental status of the patients (Koyama et al., 2009; Aoki et al., 2016). We used the data for the Cognition-Adaptation and Language-Sociality score; the data from the Motor-Posture score were not used. The scores were rated by specially trained psychotherapists.
PET
To detect the expression of AADC in the brain, FMT-PET was performed according to methods described previously (Muramatsu et al., 2010). Prior to the emission scan, a 10 min transmission scan was obtained for attenuation correction. Subsequently, 0.12 mCi/kg of FMT in saline was infused into an antecubital vein and a 30–90 min static 3D acquisition was started simultaneously using a PET-CT (GEMINI GXL, Philips). Each subject also underwent 3.0-T MRI (Achieva 3.0 T, Philips) using an inversion recovery (IR) proton density (PD)-weighted pulse sequence to enhance the contrast of anatomical structures. The PET and MRI data were co-registered with a fusion processing program (Syntegra, Philips) to produce fusion images. This program provided manual and point-based image registration as well as automated methods of grey value-based image registration, including a mutual information algorithm (Maes et al., 1999). In addition, an adaptive level set of segmentation was used for co-registration of CT and MRI data (Wells et al., 1996; Muramatsu et al., 2010).
Analyses of CSF neurotransmitter metabolites
The levels of HVA, 5-HIAA, dopamine, 3-methoxy-4-hydroxyphenylglycol (MHPG) and l-DOPA in the CSF were measured by high-performance liquid chromatography (HPLC), followed by electrochemical detection of monoamine neurotransmitter metabolites at SRL, Inc.
The quantification of vector-derived genome copies in the blood and urine
Blood and urine samples were obtained before and for 3 days subsequent to surgery, and the vector-derived genome was titrated. DNA was extracted from blood samples using a QIAamp® DNA Blood Midi kit, and 100 ng of DNA was used as a template. DNA was extracted from urine samples using a QIAamp® circulating nucleic acid kit, and 1 μl of sample was used as a template. The primer sequences for AADC were 5′-GGCAACGTGCTGGTCTGTGT-3′ (forward) and 5′-CGTCCCTCAATGCCTTCCATGT-3′ (reverse). Quantitative PCR was carried out as described previously using a Thermal Cycler Dice Real-Time System (TAKARA BIO Inc.).
Titration of neutralizing antibodies against AAV2 capsid in serum
The sera from patients before and 6 months after the operation were measured to quantify the presence of neutralizing antibodies against AAV2 capsid. The procedure for measuring the neutralizing antibodies was performed as described previously (Mimuro et al., 2013, 2014). The titres are shown as the actual dilution factor.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.
Results
Gene therapy
Gene therapy was performed from 29 June 2015 to 14 July 2017. At the time of surgery, the patients were between 4 and 19 years of age. The observation period ranged from 6 months to 2 years 7 months.
Adverse events
All surgeries were completed safely (Table 4). One patient (Patient 6) showed subdural haemorrhaging on brain CT at 3 days after surgery. However, no associated clinical symptoms were observed. No cases of intracerebral haemorrhaging or brain oedema were observed during the study period, and there were no vector-related adverse events. In all patients, transient choreic movements in the extremities and mouth started at around 2 weeks after therapy, increased until 2 months after therapy, and then gradually diminished from 3 to 6 months after therapy. Risperidone was used for two patients (Patients 4 and 5); however, its effectiveness was limited. One patient (Patient 3) showed diarrhoea at 1 year and 3 months after treatment, just after influenza virus infection. Apnoea was not noted in any of the patients.
Table 4.
Adverse events
| Adverse event | Number (%) |
|---|---|
| Transient choreic movements (limb dystonia) | 6 (100) |
| Transient orofacial dyskinesia | 6 (100) |
| Diarrhoea | 1 (16) |
| Subdural haemorrhage with no clinical symptom | 1 (16) |
| Serious adverse events | 0 (0) |
Summary of the clinical effects
The motor functions and evaluation using AIMS
All patients showed improved motor functions (Fig. 1, Table 2 and Supplementary Videos 1–3). The severe patients began to move voluntarily at 1 to 2 months after treatment. All severe patients were able to control their heads from 2 to 8 months and could sit with support from 4 to 8 months. Patients 1, 2 and 5 were able to stand with support from 1.5 years, 4 months and 7 months, respectively. Patient 2 started to walk with a walker from 4 months and gradually became able to walk long distances. Patients 1, 4, 5 and 6 started to move their arms from 2 to 3 months after the treatment. They were able to reach out and grasp objects, although they had difficulty controlling their hand movement well. Patient 2 was able to grasp objects from 4 months and hold food and bring it to her mouth after 1.5 years. Respiration and swallowing were also improved in all patients. Patient 4 was weaned from non-invasive positive pressure ventilation. Four patients had required tube feeding, but all started to eat orally after therapy. Patient 2 was able to eat all foods orally and was weaned from tube feeding. All patients were still hypotonic. The muscle atrophy in the severe patients showed mild improvement but was not resolved.
Figure 1.
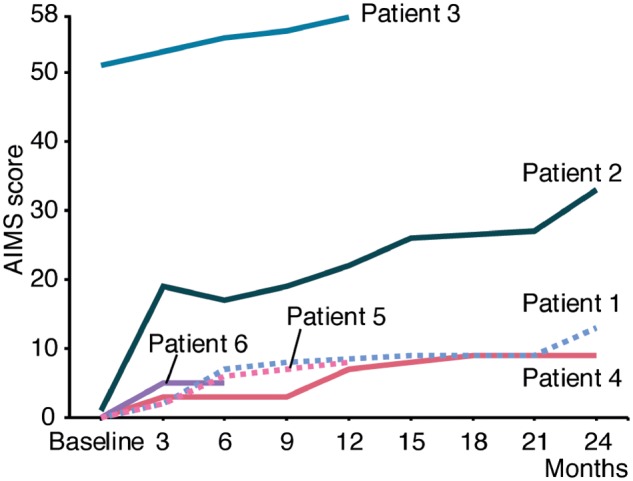
AIMS. The AIMS score of each patient was plotted. Before gene therapy, Patients 1, 4, 5 and 6 had a score of 0, Patient 2 had a score of 1, and Patient 3 had a score of 52. The scores of all patients improved. At 12 months, the score of Patient 3 reached 58, which represents the maximum score on this scale.
Regarding the AIMS scores of the patients (Fig. 1), the scores of Patients 1, 4, 5 and 6 gradually improved from almost 0 to 13 (2 years), 9 (2 years), 8 (1 year) and 5 (6 months), respectively, which was similar to the reported course of the Taiwanese patients (Hwu et al., 2012). Patient 2 showed better improvement than the other patients. Her score increased to 20 after 3 months and 33 at 2 years. On comparing their scores at 6 months, younger patients tended to show superior improvement to older patients with the same or a similar mutation. Patient 2 (age 12 years) improved faster than Patient 1 (age 15 years). Patient 5 (age 10 years) showed better improvement than Patient 4 (age 19 years), despite both having IVS6+4A>T. However, the patients with a missense mutation (Patients 1, 2 and 6) all showed earlier improvement than those with the frameshift mutation IVS6+4A>T (Patients 4 and 5). Patient 3, who had a moderate phenotype, showed marked improvement, being able to walk independently at 6 months, ride a bicycle at 10 months, and play on a swing at 1 year and 6 months after treatment. She was hypotonic but otherwise looked like a healthy young girl.
Dystonia attacks and oculogyric crisis
Dystonia attacks disappeared by 2 months post-treatment in all patients. OGC was decreased markedly after 1 month, but a mild degree of the condition remained. Patients 1, 2, 5 and 6 showed OGC several times a day lasting for several hours before the treatment. After treatment, they suffered from OGC several times a week lasting for several minutes to a few hours. The OGC in Patients 3 and 4 that occurred several times a week lasting for several dozen minutes before the treatment almost completely disappeared and was only observed when the patients were tired, lasting for several minutes.
Evaluation of cognitive, social and verbal development
Patient 3 showed a remarkable improvement in her Developmental Quotient score (Fig. 2). Before treatment, her Cognition-Adaptation score was 50, and her Language-Sociality score was 40; after 1 year, they improved to 66 and 84, respectively. She was able to speak words after 3 months, started to make conversation after 6 months, and was able to speak fluently after 1 year. She was able to perform simple arithmetic after 2 years. Patient 6 started to speak several words after 6 months. However, other severe patients spoke no words at all. The Cognition-Adaptation and Language-Sociality scores of Patients 1, 2 and 4 also showed mild improvement. All patients became able to respond, smile, or utter sounds and showed improvement regarding their ability to understand language, particularly Patient 2, who was able to quickly respond to spoken orders.
Figure 2.

The evaluation of the mental development using Kyoto Scale of Psychological Development. The Cognition-Adaptation (C-A) and Language-Sociality (L-S) scores for the Developmental Quotient before and after gene therapy in Patient 3: moderate phenotype (A and B), and Patients 1, 2 and 4: severe phenotype (C and D). (A) Before treatment, the Language-Sociality score of Patient 3 was 42; this improved to 84. A Developmental Quotient score over 70 is in the normal range. (B) The Cognition-Adaptation score of Patient 3 improved from 50 to 66. (C and D) The pretreatment scores of the severe patients were almost 0 but showed mild improvement.
Other symptoms
Autonomic dysfunction, such as hyperhidrosis, improved after 3 to 6 months. Hypersalivation appeared to be improved but was observed under anaesthesia. Thus, the improvement in hypersalivation seems to result from improved swallowing.
All patients had been in an unstable emotional state and cried frequently before the treatment. After treatment, they were always smiling and became emotionally stable. Because we did not imagine this emotional change previously, we were able to not evaluate it systematically. The patients suffered from sleep disturbance, difficulty falling asleep, and difficulty remaining asleep; these factors showed mild improvement after treatment.
Three patients (Patients 1–3) who were followed for over 2 years gained 3.1 kg (2.2–4.4 kg) in body weight (data not shown). Regarding the drugs after the treatment, Patient 2 became drug-free, and the other patients were taking small doses of MAOB inhibitor.
FMT-PET findings
Before treatment, patients showed no signal uptake in the putamen. Patients 1–4 completed FMT-PET at 6 months and 2 years, Patient 5 completed it at 3 months, and Patient 6 completed it at 6 months. All examinations showed high signal intensity in the bilateral putamen, reflecting the expression of the AADC enzyme in the putamen (Fig. 3).
Figure 3.

FMT-PET findings. The expression of AADC was detected by PET after the injection of FMT, the specific tracer for AADC. (A) The co-registered PET and MRI data before treatment. No signal was detected in their brains. (B) At 6 months after treatment (at 3 months after in Patient 5 alone), a high-intensity signal was detected in the bilateral putamen in all patients. (C) The signal in the putamen was maintained after 2 years in all patients.
Analysis of catecholamine metabolites in CSF
Before treatment, the levels of dopamine and the catecholamine metabolites HVA and MHPG as well as the serotonin metabolite 5-HIAA were low, and the l-DOPA level was elevated (Table 3). The level of HVA at 1 month after gene therapy was mildly elevated in Patients 2, 4 and 6 but not in the other patients. Almost normal levels of dopamine and serotonin metabolites were detected in Patient 3 at baseline, which might indicate remnant enzyme activity.
The detection of vector-derived genomes in serum and urine after treatment
No vector sequence was detected in the serum or urine by PCR in any of the patient samples after vector injection.
Neutralizing antibodies against AAV2 capsid
None of the subjects exhibited neutralizing antibodies against AAV2 capsid before operation. All patients had elevated titres (up to 1:56–1:28 000) at 6 months after vector injection.
Discussion
All six of the patients in our study showed marked improvement in their motor function after gene transfer. Although five patients with the severe phenotype had been bedridden before treatment, one patient (Patient 2) became able to walk using a walker, and two patients (Patients 1 and 5) were able to stand with support. The pattern of improvement after gene therapy for elder patients differed from that of normal infantile development. Therefore, the AIMS score and actual motor function of the patients were not correlated in some aspects. However, AIMS scores gradually increased in all patients, suggesting that the AIMS score has some utility for evaluating the motor improvement. The disappearance of dystonia, stabilized respiration and decreased salivation improved the patients’ quality of life.
Differences in the results of our study and the Taiwanese study were (i) that older patients (>8 years of age) also showed improvement, although the treatment was more effective in younger patients than in older ones; (ii) that the genetic background of our patients was heterogeneous and some patients showed better improvement than the Taiwanese patients; and (iii) that the cognitive and verbal functions were markedly improved in one patient with the moderate phenotype (Patient 3). The patients who underwent gene therapy in Taiwan ranged in age from 1 to 8 years (Hwu et al., 2012, 2018). In our study, Patients 1, 2, 4 and 5 were older than the Taiwanese patients, but Patients 1, 4 and 5 showed similar degrees of motor improvement to the Taiwanese patients (Hwu et al., 2012). This indicates that neuronal cells and the dopaminergic tracts can still be activated in older patients, up to at least 19 years of age. However, older patients were slower to recover than younger patients.
Another point to note is the effect of the genetic background. Our patients were genetically heterogeneous (Table 1), and only Patient 5 had IVS6+4A>T homoplasmy like the Taiwanese patients, although one allele of Patient 4 was IVS6+4A>T. The mutation IVS6+4A>T is a splice site mutation resulting in the formation of a truncated DDC protein (Lee et al., 2009). The other mutations were base substitutions inducing amino acid changes; thus, some enzyme activity would be expected, and the position of the mutation might influence the severity of the disease (Supplementary Fig. 1). A relationship between the position of the mutation and the enzyme catalytic activity in AADC has been reported previously (Montioli et al., 2014). Mutations Ala110Glu in Patients 1 and 2, Trp105Cys in Patient 3, Pro129Ser in Patient 3, Tyr369Cys in Patient 4 and Asp252Gly in Patient 6 localized outside of the catalytic domain (Supplementary Fig. 1) were thought to have a milder effect than other mutations (Kojima et al., 2016). The moderate phenotype of Patient 3 may be attributed to the localization of both mutations outside of the catalytic domain (Kojima et al., 2016).
Details concerning the mechanisms underlying the effects of gene transfer, including what circuits of dopamine systems are activated and how the dopamine that is produced works, remain to be elucidated. The most prevalent feature of AADC deficiency is the loss of voluntary movement. Dopamine plays an important role in the proper regulation of voluntary movement. The main pathway for dopaminergic control of the motor function system is from the substantia nigra to the striatum (Bolam et al., 2000; Gerfen et al., 2011). Others are from the ventral tegmentum to the frontal area and nucleus accumbens, which are associated with higher brain functions and satisfaction, respectively. Cortico-basal ganglia loops, such as motor, prefrontal, limbic and oculomotor loops, have been proposed as functions of the basal ganglia (Alexander et al., 1990; Hikosaka et al., 2000; Middleton and Strick, 2000). In the motor loop, the motor area in the cerebral cortex projects into the putamen (Gerfen et al., 2011). In patients with AADC deficiency, dopamine depletion in the striatum leads to the failure of the motor loop function, resulting in the loss of voluntary movement (Gerfen et al., 2011). For the vector injection site, we targeted the putamen, which is the site that receives dopaminergic projections from the substantia nigra, in order to ensure improvement in the motor function. In preclinical studies in a non-human primate model of Parkinson disease, more than 85–90% of transduced cells were medium spiny neurons (Muramatsu et al., 2002; Daadi et al., 2006). We suspect that dopamine was synthesized and secreted from these postsynaptic neurons in the putamen and then functioned as a neurotransmitter to stimulate postsynaptic dopaminergic receptor directly or that it was secreted from the presynaptic terminal after the uptake by dopamine transporters. The mechanism underlying dystonia in patients with AADC deficiency is not well understood. Patients with several diseases associated with low levels of dopamine, such as Segawa disease, show dystonia (Segawa et al., 1976). An imbalance between the direct and indirect pathways caused by dopamine depletion has been proposed to be associated with the course of dystonia (Wichmann et al., 1998; Vitek et al., 1999; Sanger, 2003). Providing a sufficient amount of dopamine to the striatum would help control dystonia. However, the most prominent adverse event observed after the gene therapy was transient dyskinesia: choreic movements that were observed in all patients after their dystonia improved. In contrast to dystonia, excessive dopamine is considered to be involved in the course of dyskinesia (Wichmann et al., 1998; Sanger, 2003). Because the l-DOPA level was excessive and the dopamine receptors were assumed to be hypersensitive based on the low level of dopamine before gene transfer, a transient hyper-dopamine state was induced. Hypersensitivity of DRD2 might strongly but transiently activate the pathways. Furthermore, as dopamine receptors are reported to exist all over the dendrites and not in the postsynaptic membrane alone, these receptors might be stimulated, thus allowing a sufficient dopamine effect to be obtained (Yung et al., 1995; Uchigashima et al., 2016). Limbic and oculomotor loops were also considered to be activated since patients showed improvement in their emotion as well as saccadic eye movements and OGC. Dopamine is reported to play a role in synaptic plasticity, modulating glutamatergic neurons in order to enhance the synaptic connection to create a neuronal network (Calabresi et al., 2007; Shen et al., 2008). The loss of dopamine resulted in poor synaptic and neuronal network formation. Patients with some remnant enzyme activity may—to some extent—have a synaptic and neuronal network that shows a stronger and faster response to AADC induction.
Another promising result of our study was the high degree of improvement observed in the cognitive and verbal function of Patient 3. Our target of gene therapy was to improve the motor function of patients, and improvement in cognitive and verbal function were not expected. After gene therapy, the patient’s Language-Sociality score improved to the normal range, and she became able to converse normally. The contribution of dopamine to the higher brain function is mainly achieved by projection from the ventral tegmentum to the frontal area, which is associated with the executive function. This projection was not expected to be activated. The prefrontal cortex and striatum (mainly caudate and partially rostral part of putamen) contribute to cognition, memory and other functions via their connection as the prefrontal loop (Middleton and Strick, 2000). Furthermore, the frontal area of the putamen is rich in neural connectivity between many areas of the cortex. The dopamine produced following gene therapy likely activated these networks.
The AADC enzyme expression after gene transfer was clearly visualized by an FMT-PET analysis, and the enzyme was still expressed at almost the same intensity at 2 years after gene transfer treatment. This confirmed that the transduced DDC gene and production of AADC enzyme were maintained for long periods in neuronal cells with no cell division (Bankiewicz et al., 2006). The long-term enzyme expression was also observed in the post-mortem analysis of a monkey; AAV2-AADC that had been injected into the putamen was still detectable after 15 years (Sehara et al., 2017). The level of catecholamine and serotonin metabolites in the CSF did not change markedly after the gene transfer therapy, except for a mild elevation of HVA in Patients 2–4 and 6. However, this HVA elevation was mild and not confirmed to be related to the increase in dopamine synthesis. The slightness of this change may have been because of the small number of gene copies injected into a restricted area of the brain or because the analysis was performed too soon (1 month after injection) to reflect the gene transfer.
Although the present patients were older than the previously studied Taiwanese patients, they were treated with the same dose of vector and showed similar improvements in their motor performance and putaminal tracer uptake on PET. These findings provide independent confirmation of the safety, tolerability and potential efficacy of AADC gene therapy. Future studies focusing on the optimal vector dose and defining the relationship between the vector dose and clinical effects are necessary. In conclusion, these data indicate that the AAV vector-mediated gene transfer of AADC is safe and that it may benefit patients with AADC deficiency.
Supplementary Material
Acknowledgements
We thank the patients and their families as well as all of the staff working in Jichi Children Medical Center Tochigi and Jichi Medical University Hospital. We also thank Jun-ichi Saito and Genta Akutsu (Utsunomiya Central Clinic) for their expert technical support with the imaging sessions and Dr. Chizuru Seiwa for helping with the clinical assessment of Patients 1 and 2. We thank Yasushi Saga and Ryota Watano for their peri-operative support in accordance with the Cartagena Act. We thank Naomi Takino and Mika Ito for their technical help on vector preparation.
Glossary
Abbreviations
- AADC
aromatic l-amino acid decarboxylase
- AAV
adeno-associated virus
- AIMS
Alberta Infant Motor Scale
- FMT
6-[18F] fluoro-l-m-tyrosine
- HVA
homovanillic acid
- OGC
oculogyric crisis
Funding
This research was supported by Japan Agency for Medical Research and Development (AMED) under Grant Number JP17ek0109168.
Competing interests
S.M. and T.S. own equity in a gene therapy company (Gene Therapy Research Institution) that commercializes the use of AAV vectors for gene therapy applications. To the extent that the work in this manuscript increases the value of these commercial holdings, they have a conflict of interest. The other authors declare no conflicts of interest in association with the present study.
References
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 1990; 13: 266–71. [DOI] [PubMed] [Google Scholar]
- Aoki S, Hashimoto K, Ikeda N, Takekoh M, Fujiwara T, Morisaki N et al. . Comparison of the Kyoto scale of psychological development 2001 with the parent-rated Kinder Infant Development Scale (KIDS). Brain Dev 2016; 38: 481–90. [DOI] [PubMed] [Google Scholar]
- Arnoux JB, Damaj L, Napuri S, Serre V, Hubert L, Cadoudal M et al. . Aromatic L-amino acid decarboxylase deficiency is a cause of long-fasting hypoglycemia. J Clin Endocrinol Metab 2013; 98: 4279–84. [DOI] [PubMed] [Google Scholar]
- Atwal PS, Donti TR, Cardon AL, Bacino CA, Sun Q, Emrick L et al. . Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol Genet Metab 2015; 115: 91–4. [DOI] [PubMed] [Google Scholar]
- Bankiewicz KS, Forsayeth J, Eberling JL, Sanchez-Pernaute R, Pivirotto P, Bringas J et al. . Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther 2006; 14: 564–70. [DOI] [PubMed] [Google Scholar]
- Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. J Anat 2000; 196 (Pt 4): 527–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun L, Ngu LH, Keng WT, Ch’ng GS, Choy YS, Hwu WL et al. . Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology 2010; 75: 64–71. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci 2007; 30: 211–9. [DOI] [PubMed] [Google Scholar]
- Chen PW, Lee NC, Chien YH, Wu JY, Wang PC, Hwu WL. Diagnosis of aromatic L-amino acid decarboxylase deficiency by measuring 3-O-methyldopa concentrations in dried blood spots. Clin Chim Acta 2014; 431: 19–22. [DOI] [PubMed] [Google Scholar]
- Chien Y-H, Lee N-C, Tseng S-H, Tai C-H, Muramatsu S-I, Byrne BJ et al. . Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: an open-label, phase 1/2 trial. Lancet Child Adolescent Health 2017; 1: 265–73. [DOI] [PubMed] [Google Scholar]
- Daadi MM, Pivirotto P, Bringas J, Cunningham J, Forsayeth J, Eberling J et al. . Distribution of AAV2-hAADC-transduced cells after 3 years in Parkinsonian monkeys. Neuroreport 2006; 17: 201–4. [DOI] [PubMed] [Google Scholar]
- Fiumara A, Brautigam C, Hyland K, Sharma R, Lagae L, Stoltenborg B et al. . Aromatic L-amino acid decarboxylase deficiency with hyperdopaminuria. Clinical and laboratory findings in response to different therapies. Neuropediatrics 2002; 33: 203–8. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 2011; 34: 441–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman G, Pappa MB, Pearl PL. Widening phenotypic spectrum of AADC deficiency, a disorder of dopamine and serotonin synthesis. JIMD Rep 2014; 17: 23–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka O, Takikawa Y, Kawagoe R. Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol Rev 2000; 80: 953–78. [DOI] [PubMed] [Google Scholar]
- Hwu WL, Muramatsu S, Tseng SH, Tzen KY, Lee NC, Chien YH et al. . Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci Transl Med 2012; 4: 134ra61. [DOI] [PubMed] [Google Scholar]
- Hwu WL, Chien YH, Lee NC, Li MH. Natural history of aromatic L-amino acid decarboxylase deficiency in Taiwan. JIMD Rep 2018; 40: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland K, Clayton PT. Aromatic L-amino acid decarboxylase deficiency: diagnostic methodology. Clin Chem 1992; 38: 2405–10. [PubMed] [Google Scholar]
- Hyland K, Surtees RA, Rodeck C, Clayton PT. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology 1992; 42: 1980–8. [DOI] [PubMed] [Google Scholar]
- Ide S, Sasaki M, Kato M, Shiihara T, Kinoshita S, Takahashi JY et al. . Abnormal glucose metabolism in aromatic L-amino acid decarboxylase deficiency. Brain Dev 2010; 32: 506–10. [DOI] [PubMed] [Google Scholar]
- Ito S, Nakayama T, Ide S, Ito Y, Oguni H, Goto Y et al. . Aromatic L-amino acid decarboxylase deficiency associated with epilepsy mimicking non-epileptic involuntary movements. Dev Med Child Neurol 2008; 50: 876–8. [DOI] [PubMed] [Google Scholar]
- Kojima K, Anzai R, Ohba C, Goto T, Miyauchi A, Thony B et al. . A female case of aromatic l-amino acid decarboxylase deficiency responsive to MAO-B inhibition. Brain Dev 2016; 38: 959–63. [DOI] [PubMed] [Google Scholar]
- Korenke GC, Christen HJ, Hyland K, Hunneman DH, Hanefeld F. Aromatic L-amino acid decarboxylase deficiency: an extrapyramidal movement disorder with oculogyric crises. Eur J Paediatr Neurol 1997; 1: 67–71. [PubMed] [Google Scholar]
- Koyama T, Osada H, Tsujii H, Kurita H. Utility of the Kyoto Scale of Psychological Development in cognitive assessment of children with pervasive developmental disorders. Psychiatry Clin Neurosci 2009; 63: 241–3. [DOI] [PubMed] [Google Scholar]
- Lee HF, Tsai CR, Chi CS, Chang TM, Lee HJ. Aromatic L-amino acid decarboxylase deficiency in Taiwan. Eur J Paediatr Neurol 2009; 13: 135–40. [DOI] [PubMed] [Google Scholar]
- Lee LK, Cheung KM, Cheng WW, Ko CH, Lee HH, Ching CK et al. . A rare cause of severe diarrhoea diagnosed by urine metabolic screening: aromatic L-amino acid decarboxylase deficiency. Hong Kong Med J 2014; 20: 161–4. [DOI] [PubMed] [Google Scholar]
- Lee WT, Lin JH, Weng WC, Peng SS. Microstructural changes of brain in patients with aromatic L-amino acid decarboxylase deficiency. Hum Brain Mapp 2017; 38: 1532–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzzi V, Mastrangelo M, Polizzi A, Artiola C, Van Kuilenburg AB, Carducci C et al. . Report of two never treated adult sisters with aromatic L-amino Acid decarboxylase deficiency: a portrait of the natural history of the disease or an expanding phenotype? JIMD Rep 2015; 15: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes F, Vandermeulen D, Suetens P. Comparative evaluation of multiresolution optimization strategies for multimodality image registration by maximization of mutual information. Med Image Anal 1999; 3: 373–86. [DOI] [PubMed] [Google Scholar]
- Manegold C, Hoffmann GF, Degen I, Ikonomidou H, Knust A, Laass MW et al. . Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis 2009; 32: 371–80. [DOI] [PubMed] [Google Scholar]
- Mastrangelo M, Caputi C, Galosi S, Giannini MT, Leuzzi V. Transdermal rotigotine in the treatment of aromatic L-amino acid decarboxylase deficiency. Mov Disord 2013; 28: 556–7. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Strick PL. Basal ganglia and cerebellar loops: motor and cognitive circuits. Brain Res Brain Res Rev 2000; 31: 236–50. [DOI] [PubMed] [Google Scholar]
- Mimuro J, Mizukami H, Hishikawa S, Ikemoto T, Ishiwata A, Sakata A et al. . Minimizing the inhibitory effect of neutralizing antibody for efficient gene expression in the liver with adeno-associated virus 8 vectors. Mol Ther 2013; 21: 318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimuro J, Mizukami H, Shima M, Matsushita T, Taki M, Muto S et al. . The prevalence of neutralizing antibodies against adeno-associated virus capsids is reduced in young Japanese individuals. J Med Virol 2014; 86: 1990–7. [DOI] [PubMed] [Google Scholar]
- Montioli R, Dindo M, Giorgetti A, Piccoli S, Cellini B, Voltattorni CB. A comprehensive picture of the mutations associated with aromatic amino acid decarboxylase deficiency: from molecular mechanisms to therapy implications. Hum Mol Genet 2014; 23: 5429–40. [DOI] [PubMed] [Google Scholar]
- Muramatsu S, Fujimoto K, Ikeguchi K, Shizuma N, Kawasaki K, Ono F et al. . Behavioral recovery in a primate model of Parkinson’s disease by triple transduction of striatal cells with adeno-associated viral vectors expressing dopamine-synthesizing enzymes. Hum Gene Ther 2002; 13: 345–54. [DOI] [PubMed] [Google Scholar]
- Muramatsu S, Fujimoto K, Kato S, Mizukami H, Asari S, Ikeguchi K et al. . A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther 2010; 18: 1731–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper MC, Pinnell LE, Darrah J, Maguire T, Byrne PJ. Construction and validation of the Alberta Infant Motor Scale (AIMS). Can J Public Health 1992; 83 (Suppl 2): S46–50. [PubMed] [Google Scholar]
- Pons R, Ford B, Chiriboga CA, Clayton PT, Hinton V, Hyland K et al. . Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology 2004; 62: 1058–65. [DOI] [PubMed] [Google Scholar]
- Sanger TD. Childhood onset generalised dystonia can be modelled by increased gain in the indirect basal ganglia pathway. J Neurol Neurosurg Psychiatry 2003; 74: 1509–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa M, Hosaka A, Miyagawa F, Nomura Y, Imai H. Hereditary progressive dystonia with marked diurnal fluctuation. Adv Neurol 1976; 14: 215–33. [PubMed] [Google Scholar]
- Sehara Y, Fujimoto KI, Ikeguchi K, Katakai Y, Ono F, Takino N et al. . Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson’s disease. Hum Gene Ther Clin Dev 2017; 28: 74–79. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science 2008; 321: 848–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz MA, Nguyen MA, Roche S, Heron B, Milh M, De Lonlay P et al. . Chronic Diarrhea in L-Amino Acid Decarboxylase (AADC) deficiency: a prominent clinical finding among a series of ten french patients. JIMD Rep 2017; 31: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda KJ, Saul JP, Mckenna CE, Speller NB, Hyland K. Aromatic L-amino acid decarboxylase deficiency: overview of clinical features and outcomes. Ann Neurol 2003; 54 (Suppl 6): S49–55. [DOI] [PubMed] [Google Scholar]
- Tay SK, Poh KS, Hyland K, Pang YW, Ong HT, Low PS et al. . Unusually mild phenotype of AADC deficiency in 2 siblings. Mol Genet Metab 2007; 91: 374–8. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Ohtsuka T, Kobayashi K, Watanabe M. Dopamine synapse is a neuroligin-2-mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc Natl Acad Sci USA 2016; 113: 4206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek JL, Chockkan V, Zhang JY, Kaneoke Y, Evatt M, Delong MR et al. . Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballismus. Ann Neurol 1999; 46: 22–35. [DOI] [PubMed] [Google Scholar]
- Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N et al. . Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis 2017; 12: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells WM, Grimson WL, Kikinis R, Jolesz FA. Adaptive segmentation of MRI data. IEEE Trans Med Imaging 1996; 15: 429–42. [DOI] [PubMed] [Google Scholar]
- Wichmann T, Delong MR. Models of basal ganglia function and pathophysiology of movement disorders. Neurosurg Clin N Am 1998; 9: 223–36. [PubMed] [Google Scholar]
- Yung KK, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience 1995; 65: 709–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.