

SUMMARY
The import of superoxide dismutase-2 (SOD2) into mitochondria is vital for the survival of eukaryotic cells. SOD2 is encoded within the nuclear genome and translocated into mitochondria for activation after translation in the cytosol. The molecular chaperone Hsp70 modulates SOD2 activity by promoting import of SOD2 into mitochondria. In turn, the activity of Hsp70 is controlled by co-chaperones, particularly CHIP, which directs Hsp70-bound proteins for degradation in the proteasomes. We investigated the mechanisms controlling the activity of SOD2 to signal activation and maintain mitochondrial redox balance. We demonstrate that Akt1 binds to and phosphorylates the C terminus of Hsp70 on Serine631, which inhibits CHIP-mediated SOD2 degradation thereby stabilizing and promoting SOD2 import. Conversely, increased mitochondrial-H2O2 formation disrupts Akt1-mediated phosphorylation of Hsp70, and non-phosphorylatable Hsp70 mutants decrease SOD2 import, resulting in mitochondrial oxidative stress. Our findings identify Hsp70 phosphorylation as a physiological mechanism essential for regulation of mitochondrial redox balance.
In Brief
SOD2 is synthesized on free cytosolic ribosomes as an inactive precursor and must be imported into mitochondria for activation. Zemanovic et al. report that the phosphorylation-dephosphorylation cycle of Hsp70 is an essential physiological mechanism controlling SOD2 activity.
Graphical Abstract
INTRODUCTION
Superoxide dismutase-2 (SOD2, NP_001019636.1), a member of SOD family of antioxidants, plays an essential role in cells’ protection against mitochondrial oxidative damage (Mukherjee et al., 2011; Smith et al., 2017). SOD2 deficiency can cause severe oxidative stress within the mitochondria and lead to cell death. For example, transgenic mice lacking SOD2 suffer severe cardiopulmonary and neurological damage (Misawa et al., 2006; Li et al., 1995). Conversely, SOD2 overactivity has been linked to increased invasiveness of tumor metastasis (Idelchik et al., 2017; Konzack and Kietzmann, 2014; Hemachandra et al., 2015). Since accumulation of superoxide (O2•−) radical, particularly inside the mitochondria, is often considered as an initial step in the pathogenesis of these diseases, the control of SOD2 activity is therefore critical for maintaining the balance between cell survival and proliferation. However, mechanisms for how the cells upregulate their SOD2 activity in response to stimuli and restrict SOD2 import following its activation to maintain mitochondrial redox balance remain unknown.
Stimulation of cells by a variety of agonists is well known to induce rapid increases in SOD2 activity (Afolayan et al., 2014). Such rapid responses are unlikely to be mediated by only transcriptional events. Since activation of SOD2 occurs exclusively inside the mitochondria (Karnati et al., 2013), mechanisms controlling the mitochondrial import of SOD2 likely account for the resulting increase in SOD2 activity. SOD2 is synthesized on cytosolic ribosomes as a precursor with a mitochondrial targeting sequence (MTS) located at its amino terminus (Pourvali et al., 2016). The mitochondrial targeting sequence contains instructions to import newly made SOD2 across the outer and inner mitochondrial membranes into the mitochondrial matrix, where manganese (Mn2+) is incorporated into SOD2 (Culotta et al., 2006) and the mitochondrial targeting sequence is proteolytically removed by the mitochondrial processing peptidase (MPP), leading to its full activation (Karnati et al., 2013). To deliver SOD2 to mitochondria in the highly crowded cytoplasm, cells rely on inducible Hsp70 (HspA1A, NP_005336), a molecular chaperone that recognizes and binds short hydrophobic sequences on the amino terminus of SOD2. The interaction of Hsp70 prevents aggregation and transports SOD2 to mitochondria in an import-ready form (Afolayan et al., 2017). In addition to the folding events, Hsp70 also directs SOD2 to the ubiquitin-proteasomal system (UPS) for degradation (VanPelt and Page, 2017; Zhan et al., 2017). Given the biological importance of SOD2 to mitochondrial functions, Hsp70 chaperone activity is controlled by a network of co-chaperones. One such co-chaperone is the Carboxyl terminus of Hsp70 interacting protein (CHIP), a U-box containing E3 ligase that targets client proteins for degradation (Shao et al., 2016; Zhan et al., 2017; Paul and Ghosh, 2014; Xiong et al., 2017). The ubiquitin ligase activity of CHIP is mediated by its N-terminal tetratricopeptide (TPR) domain that binds to the extreme EEVD motif in the C-terminal domain of Hsp70 with possible involvements of amino acid residues in the α-helical lid sub-domain and the unstructured tail of the C-terminal domain of Hsp70. Binding of CHIP to Hsp70 promotes ubiquitination and clearance of Hsp70-bound proteins (Connell et al., 2001; Luo et al., 2010). Thus, the ability to shift the refolding activity of Hsp70 to UPS makes CHIP a key determinant of SOD2 activity. Therefore, the activity of SOD2 is critically dependent on the interaction between CHIP and Hsp70. Under steady-state conditions, the balance between refolding activity of Hsp70 and ubiquitin activity of CHIP maintains SOD2 activity. If a higher SOD2 activity is needed, the chaperone activity of Hsp70 must shifts more toward refolding than degradation. However, the cellular mechanism by which CHIP mediates SOD2 activity remains unknown.
Reversible phosphorylation of proteins has been recognized as a key mechanism for regulation of cellular processes. Recently, it was reported that the C terminus of Hsp70 contains phosphorylation sites for kinases such as Casein kinases (Muller et al., 2013). The functional relevance of these phosphorylation sites, however, has only been investigated in cancers. Phosphorylation of the C-terminal residues near the extreme EEVD sequence of Hsp70 was reported to alter CHIP binding affinity. Further, the presence of other phosphorylation sites in Hsp70 has been reported (Melling et al., 2009, Truman et al., 2012); however, whether phosphorylation of Hsp70 influences SOD2 activity in ways that are proportional to activation signals remains unknown.
A central related question to resolve is how cells discern when the O2•−/H2O2 balance has been achieved and to halt the further import of SOD2. Recent reports that the levels of SOD2 are increased in cells treated with H2O2 are inconsistent with redox regulation (Liu et al., 2004; Zou et al., 2017). This inconsistency between SOD2 expression and activity and redox regulation prompted us to examine the mechanisms controlling, not only the induction but also the termination of SOD2 activation. Such knowledge will not only advance our understanding of SOD2 biology, but also identify essential targets for increasing the intrinsic activity of SOD2 in cardiopulmonary diseases and alter diseases progression. Here, we provide evidence that phosphorylation-dephosphorylation cycles of Hsp70 control mitochondrial redox balance by modulating CHIP-mediated degradation of SOD2. We demonstrated that phosphorylation of Hsp70 on Ser631 byAkt1 inhibited CHIP binding affinity and promoted the import of SOD2. With increased mitochondrial H2O2, a product of the SOD2 antioxidant activity, further import of SOD2 is inhibited by inducing the expression of PP2C, a protein phosphatase that deactivates Akt1 kinase and decreased rates of Hsp70 phosphorylation. These findings show that mitochondrial oxidative stress is regulated by the combined actions of Hsp70 phosphorylation and E3 ligase activity of CHIP.
RESULTS
Akt1 Regulates SOD2 Activity
Previously, we reported that Hsp70 transports nascent SOD2 to mitochondria after translation in the cytosol (Afolayan et al., 2014). Therefore, to identify the mechanism regulating the SOD2 import, we investigated potential factors regulating Hsp70 chaperone activity by transfecting HEK293T cells with plasmids expressing human influenza hemagglutinin (HA)-tagged Hsp70 and treated the cells with acetylcholine (ACh), an agonist known to induce mitochondrial ATP synthesis that facilitates SOD2 import. Analysis of the interactome revealed that Hsp70 has a broad substrate spectrum (Figures S1A–S1F; Table S1), and, among the proteins found to associate, Akt1 was particularly intriguing because the kinase is reported to regulate SOD2 activity at the transcriptional level. To test whether Akt1 influences SOD2 activity at posttranslational levels, we performed time-course experiments comparing Akt1-stimulated signaling to mitochondrial SOD2 localization. Healthy bovine pulmonary artery endothelial cells (PAECs) were serum starved for 8 hr and after that treated with platelet-derived growth factor (PDGF), a factor that activates phosphatidylinositol-3 kinase (PI3K) and its downstream kinase, Akt, and assessed the associations between SOD2 and mitochondria using immunofluorescence (IF) methods. Serum-starved PAECs exhibited decreased associations between SOD2 and mitochondria. PDGF treatment increased mitochondrial SOD2 localization within minutes with maximal effect occurring at 20 min after stimulation (Figure 1A). We then tested whether the observed increase in localization resulted in corresponding increases in the import and activation of SOD2 by measuring SOD2-specific activity and rates of H2O2 formation over time. PDGF increased SOD2 activity with maximal activity at 20 min after growth factor stimulation (Figure 1B). Consistent with this finding, we observed correspondingly lower levels of O2•− (Figure 1C) and higher mitochondrial H2O2 formation (Figure 1D). Moreover, increased SOD2 activity coincided with PDGF induction of T308/S473 phosphorylation of Akt1 (Figure S2A). Strikingly, Akt1 and its active products were barely detectable in mitochondria, suggesting that Akt1 influence on SOD2 activity occurs in the cytosol (Figure 1E). To test whether PI3K was involved, we treated PAECs with a PI3K inhibitor, Ly294002, and observed decreased mitochondrial SOD2 protein contents compared to levels in the vehicle-treated controls (Figure 2A). To determine the direct involvement of Akt1, we used a short interfering RNA (siRNA)-targeting native Akt1 proteins. Akt1-siRNA decreased Akt1 proteins (Figure 2B), which led to the accumulation of SOD2 proteins in the cytosol rather than mitochondria compared to scrambled siRNA (Figure 2C). To confirm Akt1’s role, we treated Akt1-siRNA-transfected cells with constructs expressing constitutively active (myristoylated) Akt1 or dominant-negative (DN) Akt1179M. Overexpression of myr-Akt1 increased mitochondrial SOD2 protein contents (Figure 2C) and activity (Figure 2D) compared to DN-Akt. These data, together, indicate that Akt1 regulates SOD2 activity post-translationally.
Figure 1. Akt1 Controls SOD2 Activity.
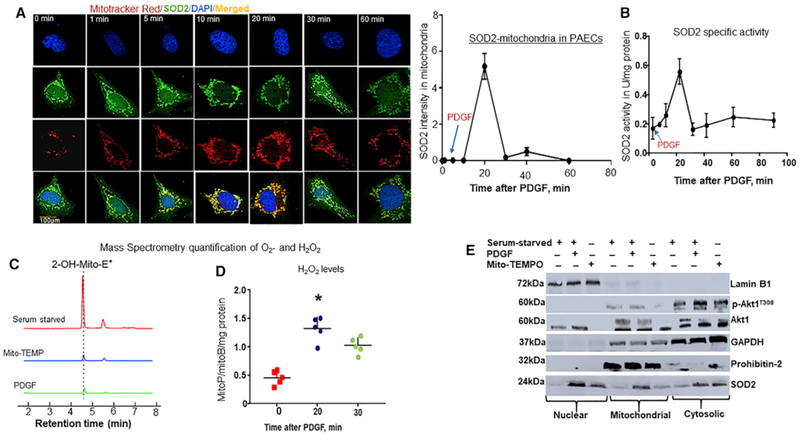
(A) Akt1 influences mitochondrial SOD2 localization. PAECs were serum starved (SS) for 8 hr and afterward treated with PDGF (10 ng/mL) and subjected to immunofluorescence staining at the indicated time points. Photomicrograph shows time-elapsed mitochondrial SOD2 localization (n = 5). Scale bar, 100 μm. Plotted is the ratio of mitochondria to cytosolic SOD2 intensity (n = 6/data point).
(B) To measure SOD2-specific activity, treated cells were lysed in HEPES buffer, pH 7.4, and lysates were incubated with 1 mM KCN to inhibit Cu/Zn-SOD, and SOD2 activity was measured for 1-hr at different time points. Line graph showing SOD2 activity over time (n = 5/data point). Quantification of mitochondrial reactive oxygen species levels (ROS: O2and H2O2). Treated PAECs were incubated with 5 μM mitoSOX for 30 min or 5 μM MitoB 3-(Dihydroxyl-boronyl-)-benzyl-triphenyl-phosphonium bromide), a mitochondria-targeted ratio metric probe for 3 hr at 37°C.
(C) Liquid chromatography-tandem mass spectrometry (LC-MS/MS) tracing of O2-specific mitoSOX oxidized product, 2-hydroethythidium (2-OH-Mito-E+) retention over time (n = 6).
(D) MitoB and its H2O2 conversion product, MitoP, were extracted after spiking the lysates with deuterated internal standards (ISs) of MitoB and MitoP (100 pmol d15-MitoB and 50 pmol d15-MitoP), and the MitoP-MitoB ratio was quantified by LC-MS/MS relative to deuterated ISs. Scattered plots of MitoP-MitoB ratio over time (n = 6, *p < 0.05 from SS cells).
(E) Subcellular localization of SOD2 in PAECs. PAECs were treated as indicated, and cells were fractionated into nuclear, cytosolic, and mitochondrial fractions, and indicated proteins were measured in cells fractions by WBs using specific loading controls.
Figure 2. Akt1 Regulates SOD2 Activity at Posttranslational Levels.
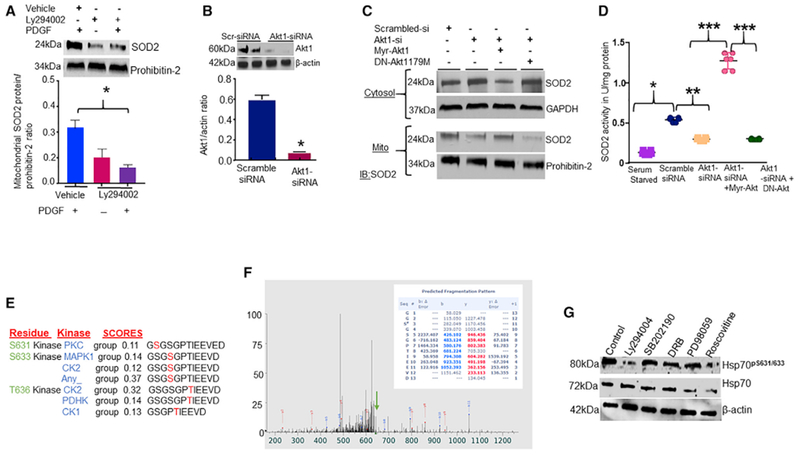
Inhibition of phosphatidylinositol-3 kinase (PI3K)-Akt1 signaling influences SOD2 activity. PAECs were treated with 10 μM PI3K inhibitor Ly294004 for 8 hr and transfected with siRNA targeting native Akt1, and constitutively active (myristoylated)-Akt1 or dominant-negative (DN) Akt1179M mutant was re-expressed in the Akt1-depleted cells. Cells were then fractionated into mitochondrial and cytosolic fractions, and SOD2 protein and activity were measured by WBs and colorimetric assays, respectively.
(A) Bar graph showing the effects of Ly294004 on mitochondrial SOD2 protein levels (n = 4).
(B) Bar graph showing Akt1 protein levels in Akt1-siRNA-transfected cells (n = 5).
(C) Blots showing the effects of Akt1-siRNA, myr-Akt1, or DN Akt1 on levels of SOD2 protein in the cytosolic and mitochondrial fractions (n = 5).
(D) Scattered plots showing SOD2 activity in isolated mitochondria (n = 5, *p < 0.05 from vehicle-treated cells, ** from scramble or SS, ** from Akt1-siRNA, and *** from myr-Akt1/DN-Akt1). Prediction of kinases that could phosphorylate the C terminus of Hsp70 is shown.
(E) The probability of phosphorylation of C-terminal residues of Hsp70 based on software predictions. The scores listed reflect the probability that the kinase phosphorylates the serine or threonine residues in the Hsp70 C terminus.
(F) In vitro phosphorylation of purified Hsp70 proteins. MALDI-mass spectrometry spectra tracing of phosphorylated residues in Hsp70 is shown. Inhibition of CDK minimally influences Ser631 phosphorylation of Hsp70.
(G) Detection of Ser631 phosphorylation in PAECs treated with different protein kinase inhibitors.
Akt1 Phosphorylates Hsp70 at the C-Terminal Ser631
Previously, it was reported that the C terminus of Hsp70 could be phosphorylated in vitro (Muller et al., 2013). Employing sequence analysis, we observed that the serine/threonine residues in the Hsp70 C-terminal domain are not known as potential phosphorylation sites for Akt (Figure 2E). To test whether Akt1 can phosphorylate Hsp70, we performed in vitro kinase reactions using purified Hsp70 and Akt1 proteins. Mass spectrometry analysis of phosphor-Hsp70 revealed that Akt1 is capable of phosphorylating Ser631, Ser633, and T635 residues in Hsp70 (Figures 2F and S2B). Using a probability-based scoring, a validated tool that measures the probability of correct allocation of phospho sites (Beausoleil et al., 2004), we saw that Ser631 is the site most likely to be phosphorylated by Akt1 (Figure S2B; Table 1). Prediction of potential kinases for the C terminus of Hsp70 phospho sites yielded many other kinases including CDK (Figure 2E). To test whether other kinases could phosphorylate Ser631, we screened a series of kinases using specific inhibitors and measured Ser631 phosphorylation by immunoblotting. To detect phospho-Ser631, we generated an antibody that specifically recognized phospho-Ser631/633 of Hsp70 (Figure S2C). Treatment of PAECs with PI3K inhibitor, Wortmann decreased levels of phospho-Hsp70 at Ser631/633. Also, inhibition of CDK reduced phospho-Ser631/633 levels but to a lesser extent (Figure 2G), suggesting that multiple kinases regulate the Hsp70 C terminus phosphorylation.
Table 1.
Akt1 Phosphorylates Hsp70 on Ser631
| ScanF | Reference | Ascore Seq_A | Ascore 1A | Site 1A | Ascore Seq_B | Ascore 1B | Site 1 |
|---|---|---|---|---|---|---|---|
| 8997 | HSPA1A | GGS#GSGPTIEEVD | 13.2 | 631 | GGS#GSGPTIEEVD | 18.9 | 631 |
Table showing a score (Ascores A and B) based allocation of phosphorylation for Ser631 residue in the C-terminal domain of Hsp70 using the Ascore algorithm.
Akt1 Interacts with Hsp70
Since Akt phosphorylates client substrates via direct interactions, we tested whether Akt1 physically interacts with Hsp70 and map the Akt1 binding domain in Hsp70 by overexpressing plasmid constructs expressing GST-tagged truncated Hsp70 that corresponded to published domains in Hsp70 and full-length HA-tagged Akt1 vector in HEK-293T cells. Pull-down studies demonstrated that Akt1 binds to Hsp70 between amino acid 537–638 residues (Figure 3A), corresponding to the substrate-binding domain-α (SBD-α), near the conserved EEVD sequence of Hsp70. To identify where Hsp70 binds in Akt1, we co-expressed a GST-tagged 530Hsp70641 mutant with a series of HA-tagged Akt1 mutants into HEK-293T cells. Pull-down studies showed that Hsp70 bound between amino acids 150–408 in Akt1 (Figure 3B). Collectively, these data show that Akt1 physically interacts with Hsp70 at the SBD-α domain.
Figure 3. Ser631 Phosphorylation of Hsp70 Is a Pre-requisite for Induction of SOD2 Activity in Response to Stimuli.
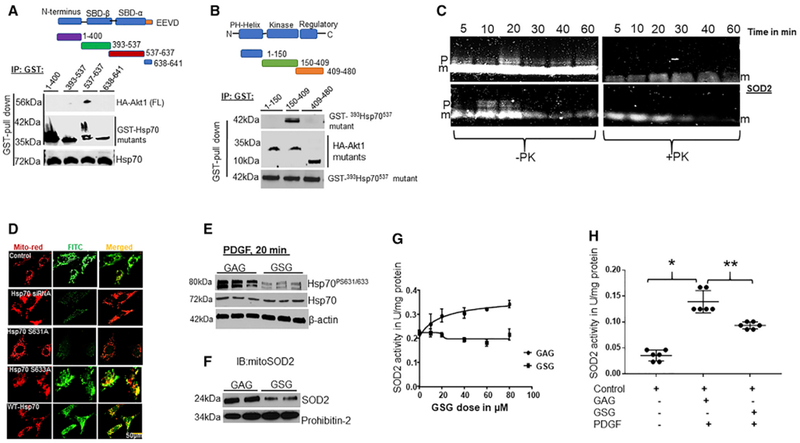
Akt1 Binds to and Phosphorylates Hsp70.
(A) Schematic diagram of Hsp70 fragments used to map Akt1 binding sites in Hsp70 (top). Human GST-Hsp70 mutants (1–400, 393–537, 537–641)and HA-Akt1 (full-length) were co-transfected into HEK293T cells. Lysates were subjected to GST pull-down assays and blotted for Hsp70 fragments and HA epitope (bottom). The expression levels of transfected HA-Akt and GST-Hsp70 fragments were confirmed by WBs.
(B) Schematic diagram of Akt1 fragments used to determine the Hsp70 binding domain in Akt1 (top) (n = 6). HA-Akt1 fragments (1–150,150–408, 408–480) and a GST-393Hsp70537 were co-expressed in HEK-293T and the elute was blotted for HA and Akt1 fragments (bottom) (n = 6). In vitro phosphorylation of Hsp70 influences SOD2 import.
(C) 20 μL in vitro synthesized and FluoroTect GreenLys System BODIPY-FL-labeled-tRNA SOD2 was incubated with isolated bovine heart mitochondria (30 μg) in the presence of (10 μM) purified Hsp70 ± Akt1 (500 U) at 30°C for the indicated time. Detection of the fluorescently labeled SOD2 signal by an infrared scanner (LI-COR, Bioscience, n = 8) (p = precursor SOD2 and m = mature or intermediate SOD2).
(D) Disruption of Ser631 phosphorylation of Hsp70 influences SOD2 activity. PAECs were transfected with Hsp70-siRNA, and constructs expressing non-phosphorylable Hsp70 mutants (Hsp70ΔS631A and Hsp70ΔS633A) were re-expressed in Hsp70-depleted cells, and cells were subjected to IF staining after acetylcholine stimulation. Photomicrograph showing mitochondrial SOD2 localization in PAECs transfected with Hsp70-siRNA or Hsp70-siRNA and cells overexpressing Hsp70 WT or Hsp70 mutants (n = 5). Scale bar, 100 μm. The effect of disruption of Akt1 phosphorylation of Hsp70 by decoy peptide sequence GSG on SOD2 import.
(E) PAECs were incubated with 40 μM GSG or GAG overnight, and phospho-Ser631/633 proteins were measured following PDGF treatment using specific antiserum against phospho-Ser631/633.
(F) Detection of SOD2 proteins in isolated mitochondria after GSG or GAG treatment.
(G) Line graph showing the dose effects of GSG on SOD2 activity in PAECs. The effect of GSG peptide on PDGF induced SOD2 activity.
(H) PAECs were incubated with HBSS, GAG, or GSG and SOD2-specific activity was measured 20 min after PDGF treatment (n = 5; *, **p < 0.05 from HBSS and GSG treatments).
Phosphorylation of the C Terminus of Hsp70 Promotes SOD2 Import
SOD2 is synthesized on free cytosolic ribosomes and post-translationally translocated into mitochondria. To test the effect of Hsp70 phosphorylation on SOD2 import in a homologous system, we synthesized and fluorescently labeled the precursor bovine SOD2 in reticulocyte lysate (lacking mitochondria). We first incubated the fluorescently labeled SOD2 with isolated bovine heart mitochondria in the presence of purified Hsp70 with or without Akt1. With Akt1, we observed an increase in SOD2 signals that peaked 20 min after incubation and thereafter faded away. In the absence of Akt1, we saw a stepwise increase in SOD2 signals (Figure 3C), suggesting that Akt1 promotes a rapid influx of SOD2 into mitochondria. To verify this finding in intact cells, we genetically deleted Ser631 in Hsp70 to generate a non-phosphorylable mutant, Hsp70ΔS631A and examined its effect on SOD2 import. To delineate the role of Ser633 from Ser631, we also generated Hsp70ΔS633A mutant. We knocked down native Hsp70 proteins using siRNA and re-expressed constructs expressing wild-type (WT) or ΔHsp70 mutants and examined for changes in the mitochondrial SOD2 localization. As observed with Hsp70WT, re-expression of Hsp70ΔS633A mutant in Hsp70-depleted cells resulted in increased SOD2 associations with mitochondria. However, re-expression of Hsp70ΔS631A mutant failed to increase levels of mitochondrial SOD2 proteins (Figure 3D). To verify this result using a different approach, we generated a cell-permeable decoy peptide sequence, GSG, encoding Ser631 residue in Hsp70 to block Ser631 phosphorylation. We incubated GSG peptides with PAECs and observed decreases in Ser631 and Ser633 phosphorylation compared to GAG, a scrambled peptide that lacks Ser631 (Figure 3E). These effects are expected given the similarity between amino acids that bracket Ser631 and Ser633 (630GSGSG634). Moreover, GSG treatment significantly decreased the mitochondrial SOD2 protein contents (Figure 3F) and had a dose-dependent effect on SOD2 activity (IC50 = 20 μM, Figure 3G). Although GSG blocked both Ser631 and Ser633 phosphorylation, however, Hsp70ΔS633A mutant had minimal effects on mitochondrial SOD2 localization (Figure 3E). We, therefore, conclude that the decreased mitochondrial SOD2 protein levels observed in GSG-treated cells are primarily due to disruption of Ser631 phosphorylation. To test whether Ser631 phosphorylation mediates the surge in SOD2 activity following PDGF stimulation, we incubated PAECs with GSG before PDGF treatment and measured SOD2 activity 20 min after stimulation. We chose 20 min because the maximal effect of Akt1 occurred at 20 min after PDGF stimulation (Figures 1A and 1B). While PDGF increased phosphorylation of Ser631/633 (Figure S2D), preincubation with GSG abolished the PDGF-mediated increase in SOD2 activity (Figures 3E and 3H). These findings suggest that Ser631 phosphorylation is a pre-requisite for signal activation of SOD2.
Phosphorylation of Hsp70 Increases SOD2 Import by Preventing CHIP-Dependent SOD2 Degradation
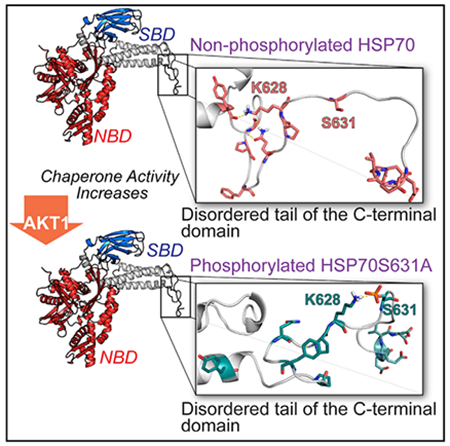
To address the question of how phosphorylation of Hsp70 modulates SOD2 import, we focused our attention on CHIP. Our rationale is based on the knowledge that, when CHIP binds to Hsp70, it stalls refolding of client proteins and facilitates their degradation. To delineate the relationship between phospho-Hsp70 and CHIP, we quantified the amount of CHIP protein bound to phospho- and non-phospho-Hsp70 using a modified ELISA. Purified Hsp70 protein was phosphorylated by Akt1 in in vitro kinase reactions, then phospho- and non-phospho-Hsp70 proteins were adsorbed to GST-coated plates, and recombinant CHIP protein was added. Analysis showed that non-phosphorylated Hsp70 bound higher amounts of CHIP than phospho-Hsp70 (Figure 4A). To determine the impact of phosphorylation on structure, we performed molecular dynamics simulations of Hsp70 and Hsp70ΔS631A mutant and observed that the accessible conformations of the disordered tail of the Hsp70 C terminus were altered when Ser631 was phosphorylated. Importantly, the flexibility of the positively charged K628 and polar Q625 in the parent Hsp70 allows the formation of H-bonds with the amide oxygen of Y611 and amide nitrogen of G613, respectively (Figure 4B). Upon phosphorylation, K628 is tightly bound to the negatively charged phospho-Ser631, promoting the formation of a hydrophobic cluster containing P627, F622, and P619 (Figure 4C). The effect of phosphorylation on the conformation of the disordered tail was confirmed by the different distributions of the intramolecular distances between the residues involved in the above-mentioned intramolecular interaction (Figure 4D). As the functionally significant interaction between the CHIP-TPR domain and Hsp70 C terminus also involves residues from the α-helical lid subdomain and disordered tail (Zhang et al., 2015), such alteration in the conformational space of the tail may likely disrupt the affinity of the Hsp70: CHIP complex. Thus, the data obtained by in silico molecular modeling are consistent with data from the ELISA assays and together demonstrate that CHIP binding to Hsp70 is regulated by phosphorylation.
Figure 4. Ser631 Phosphorylation of Hsp70 Stabilizes SOD2 Proteins.
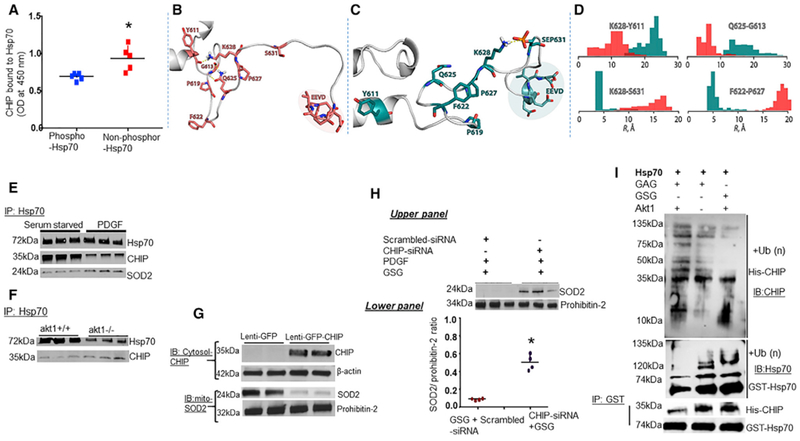
Hsp70 phosphorylation influences CHIP binding affinity.
(A) ELISA analysis of complex formation between Hsp70 and CHIP proteins. GST-coated plates were incubated with Akt1 phosphorylated GST-tagged phosphorylated and non-phosphorylated Hsp70. Purified CHIP protein was tested for interactions with Hsp70. Scattered plot shows the influence of Hsp70 phosphorylation on CHIP association with Hsp70.
Cartoon diagrams of the disordered tail of C-terminal domain of (B) Hsp70 and (C) Hsp70ΔS631Amutant structures obtained from molecular dynamics simulations. Residues involved in the intra-molecular interactions and residues ofthe conserved EEVD domain are shown in stick representation. Coral color corresponds to Hsp70, and teal color corresponds to HspS631.
(D) Effect of the Ser631 phosphorylation on the distribution (shown as histograms) of the inter-atomic distances between residues involved in the intra-molecular interactions shown in panels.
Phosphorylation of Hsp70 prevents CHIP-mediated ubiquitylation of SOD2. Phosphorylation influences ubiquitylation of Hsp70 proteins.
(E) Serum-starved PAECs were treated with PDGF and examined for CHIP association with Hsp70 by IP.
(F) Detection of association between CHIP and Hsp70 in the lungs of akt1-null mice.
(G and H) PAECs were pre-treated with CHIP-siRNA prior to inhibition of Ser631 phosphorylation by GSG and measured SOD2 protein (G) and SOD2 activity (H) in isolated mitochondria.
(I) The effect of phosphorylation on Hsp70 ubiquitylation. GST-tagged puriffed Hsp70 proteins were incubated with GSG or GAG, in the presence or absence of Akt1 and blotted for ubiquitinated Hsp70 and CHIP association with Hsp70.
To investigate whether CHIP is functionally relevant to SOD2 import in vivo, PAECs were serum starved or treated with PDGF and observed that minimal amounts of CHIP eluted with Hsp70 (Figure 4E). Employing akt1-null mice, we saw an increase in the association between Hsp70 and CHIP in lungs of akt1-null mice compared to WT mice (Figure 4F). Further, CHIP overexpression using lentivirus carrying human CHIP gene increased the levels of ubiquitinated-SOD2 proteins compared to controls (Figure S3A) and led to a decrease in mitochondrial SOD2 protein contents (Figure 4G). For confirmation, PAECs were transfected with CHIP-siRNA prior to inhibition of Ser631 phosphorylation with GSG under PDGF stimulation. CHIP-siRNA increased the mitochondrial SOD2 protein and the activity comparable to GFP controls (Figure 4H). Since CHIP association with Hsp70 results in ubiquitination of not only the client proteins but also Hsp70 itself, we tested whether phosphorylation of Ser631 would affect Hsp70 degradation by measuring the levels of ubiquitination of GST-tagged Hsp70 proteins by CHIP and the E2 enzyme UbcH5b in vitro. Incubation of purified Hsp70 proteins with GSG increased the amount of CHIP eluted with Hsp70 and ubiquitinated Hsp70 compared to controls. In contrast, incubation of Akt1 with Hsp70 decreased CHIP binding and ubiquitinated Hsp70 without decreasing CHIP activity (Figure 4I).
Increased Mitochondrial H2O2 Limits Further Import of SOD2 by Inducing PP2C
As shown in Figure 1B, induction of SOD2 activity by Akt1 was transient as minimal SOD2 activity was detected in the mitochondria after 20 min of PDGF stimulation. Possible mechanisms for the decreased SOD2 activity include a rapid turnover of activated Akt or increased mitochondrial H2O2 levels. To test the second possibility, we investigated the effect of mitochondrial SOD2 protein levels on Akt1 activation by incubating GSG peptides with PAECs and measured phosphorylation of Akt1 at T308/S473 (Akt1 activity) by western blots (WBs). GSG treatment increased Akt1 activity compared to GAG-treated controls (Figure 5A). Because disruption of Hsp70 phosphorylation by GSG could cause a feedback increase in Akt1 activity, we tested the direct effect of SOD2 by manipulating mitochondrial SOD2 protein levels. We knocked down and overexpressed SOD2 using siRNA and adenovirus carrying human SOD2 gene, respectively. As shown in Figure 5B, SOD2-siRNA decreased mitochondrial H2O2 levels. Further, SOD2-siRNA activated Akt1, and conversely SOD2 overexpression inactivated Akt1 (Figure 5C). This finding suggests that H2O2, the catalytic product of SOD2, plays a critical role in the feedback inhibition of SOD2 activity. We then tested the direct involvement of H2O2 by treating PAECs with polyethylene glycol (PEG)-catalase, a mitochondria-targeted catalase prior to SOD2 overexpression and observed that pre-incubation with PEG-catalase increased Akt1 activity (Figure 5B). We conclude that H2O2 acts as the second messenger in the regulation of Akt1 activity. To identify the signaling molecule downstream of H2O2, we investigated the effects of H2O2 on serine/threonine phosphatases known to dephosphorylate and inactivate the kinases PP2A and PP2C. We transfected PAECs with adenoviral SOD2 and observed that SOD2 has a dose-dependent effect on PP2C expression (Figure 5D). To test the direct effect of H2O2 on PP2C, PAECs were treated with micromolar doses of exogenous H2O2 and observed increased expression levels of PP2C in a dose-dependent manner (Figure 5E). However, we found no changes in the level of PP2A protein either in the SOD2 transfected or H2O2-treated cells, suggesting a specific role for PP2C in SOD2 signaling. To test the selective role of PP2C, we used inhibitors of phosphatases, Calyculin A (inhibits PP1/PP2A), and Sanguinarine chloride (selective for PP2C). Inhibition of PP2C (Figure 5F), in contrast to PP1 and PP2A (Figure 5G), increased Ser631 phosphorylation. These findings demonstrate that increased PP2C expression limits SOD2 import.
Figure 5. Mitochondrial Redox Signaling Involves PP2C.
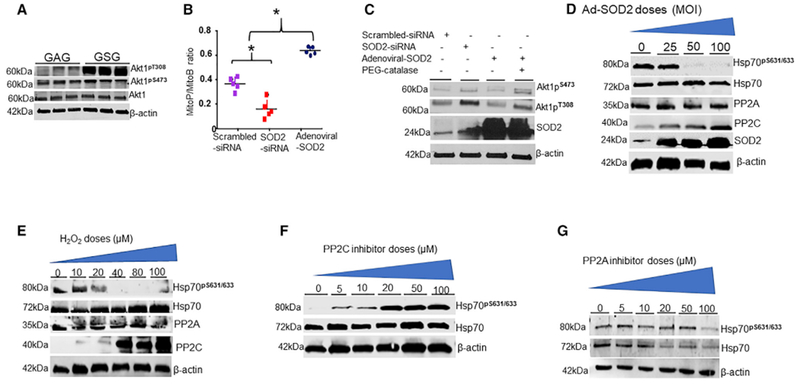
The effects of mitochondrial ROS on Akt1 activation.
(A) PAECs were treated with 40 μM GSG for 2 hr and measured phosphorylated Akt1 on T308/S473 protein levels by IB (n = 4). H2O2 acts as a second messenger in SOD2 signaling. PAECs were transfected with SOD2-siRNA or adenoviral SOD2 (75 multiplicity of infection, MOI).
(B) Detection of mitochondrial H2O2 levels in transfected cells by LC-MS/MS quantification of MitoP-MitoB ratios relative to deuterated internal standards (n = 4) Scattered plot of MitoP-MitoB ratio (n = 5, *p < 0.05 from scramble).
(C) Detection of phosphorylated Akt1 in PAECs transfected with SOD2-siRNA and adenoviral SOD2 (75 MOI) in the presence or absence of 1,000 U polyethylene glycol (PEG)-catalase by WBs. H2O2 influences PP2C expression.
(D) PAECs transfected with varying doses of adenoviral SOD2 were blotted for PP2A and PP2C using indicated antibodies (n = 3). The effect of H2O2 on PP2C expression is shown.
(E) PAECs were treated with exogenous H2O2 for 1 hr, and the levels of PP2A and PP2C proteins were measured by IB (n = 3). Inhibition of PP2C influences Hsp70 phosphorylation.
(F and G) Detection of Ser631 phosphorylation of Hsp70 in PAECs treated with phosphatase inhibitors, Sanguinarine chloride (F) and (G) Calyculin A for 1 hr.
Inactivation of Akt1 Leads to Defects in SOD2 Import In Vivo
To investigate the physiological relevance of Hsp70 phosphorylation on SOD2 activity in vivo, we used two complementary approaches. We treated healthy Sprague-Dawley rats (SDRs) with decoy peptides, GSG or GAG, in the presence or absence of (2-[2, 2, 6,6-tetramethylpiperidin-1-oxyl-4-acylamino]-2-oxoethyl) triphenyl-phosphonium chloride (mito-TEMPO), a specific scavenger of mitochondrial O2•−, by implanting Alzet pumps that would deliver GSG at 2 mg/kg/day for 10 days into SDRs and nursed them in room air. The lungs of GSG and mito-TEMPO-treated SDRs had reduced levels of SOD2 proteins (Figure 6A) and activity (Figure 6B) in their mitochondria compared to levels in the GAG-treated SDRs. Such decreases correlated with higher O2•− (Figure 6C), and lower mitochondrial H2O2 levels (Figure S3B). If Akt1 were the primary kinase that phosphorylates Ser631, then a significant fraction of Hsp70 would be expected to be dephosphorylated in the lungs of akt1-null mice. We generated global akt1-null mice, and, expectedly, the Akt1 protein was absent with slight but significant increases in Akt2 and Akt3 expression in akt1-null mice (Figure S3C). Interestingly, phosphorylated p38MAPK and ERK1/2 levels were markedly increased (Figure 6D). Despite these increases, we observed decreased phosphorylation of Hsp70 on Ser631/633 (Figure 6E) and SOD2 activity (Figure 6F) and increased mitochondrial O2•− levels (Figure S3D). These findings support the idea that Akt1 is responsible for phosphorylation of Hsp70 at Ser631.
Figure 6. The Biologic Relevance of Ser631 Phosphorylation on SOD2 Import In Vivo.
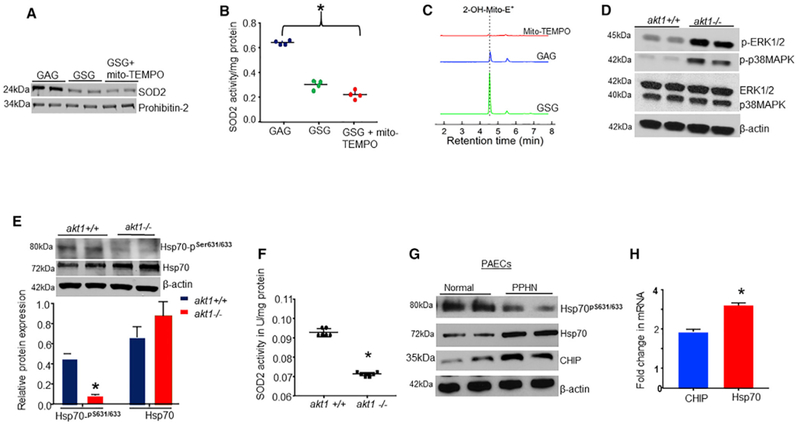
(A–C) Sprague-Dawley rats (SDRs) were treated with GSG or GAG with or without mito-TEMPO, a mitochondrial-targeted specific O2 scavenger (10 μM/day) for 10 days, and the lungs were examined for (A) SOD2 protein levels in lungs mitochondria, (B) SOD2 activity (n = 6, *p < 0.05), and (C) lungs mitochondrial O2•− levels. LC-MS/MS tracing of 2-OH-Mito+E+ overtime in the lungs from treated SDRs. Loss of Akt1 activity is associated with decreased Hsp70 phosphorylation.
(D) Detection of other kinases in the lungs of null and WT akt1 mice by WBs.
(E) Detection of Ser631/633 phosphorylation of Hsp70 in the lungs of akt1-null and WT mice.
(F) Scattered plot showing SOD2 activity in the lungs of akt1-null and WT mice (n = 6, *p < 0.05). The chaperone status of Hsp70 in PAECs from fetal lambs with persistent pulmonary hypertension (PPHN) versus healthy cells.
(G) The levels of Hsp70 proteins, Ser631 phosphorylation, and CHIP expression in PAECs from PPHN and healthy lambs.
(H) The relative expression of Hsp70 and CHIP mRNAs in PAECs from PPHN and healthy lambs quantified by real-time PCR (n = 4, *p < 0.05 from controls).
Ser631 Phosphorylation of Hsp70 Is Decreased in Hypertensive Endothelial Cells
Reduced Akt1 activity has been directly linked to endothelial cell (EC) dysfunction (Iaccarino et al., 2004). Therefore, to assess the physiological relevance of our observations, we examined differences in phosphorylated Hsp70 in PAECs from normal and fetal lambs with pulmonary hypertension (PPHN), a disorder characterized by EC dysfunction. WB analyses revealed that PPHN-PAECs express high Hsp70 protein levels compared to healthy cells (Figure 6G). However, Ser631 phosphorylation of Hsp70 was significantly reduced in PPHN-PAECs (Figure 6G). Previous studies reported that CHIP expression is decreased in conditions with acute oxidative stress (Stankowski et al., 2011). These reports prompted us to investigate whether CHIP expression is different between normal and PPHN-PAECs. Our data showed higher levels of CHIP proteins in PPHN-PAECs compared to healthy controls (Figure 6G). We also interrogated PPHN-PAECs for altered expression of Hsp70 and CHIP mRNAs. These data confirmed that Hsp70 mRNA is overexpressed, with a slight increase in CHIP mRNA expression (Figure 6H). These data indicate that decreased SOD2 activity is a result of the combined actions of increased E3 ligase activity of CHIP and posttranslational modifications of Hsp70.
DISCUSSION
Our findings demonstrate that Hsp70 phosphorylation is an essential post-translational mechanism for regulating mitochondrial SOD2 activity. In addition, we have shown that Hsp70 phosphorylation is itself controlled by PI3K-Akt signaling, which is crucial because this pathway receives input from the cell membrane as well as mitochondria. Our studies show that Ser631 phosphorylation of Hsp70 is essential for increasing mitochondrial delivery and activation of SOD2 in response to physiologic stimuli. Further, upon import and subsequent activation, SOD2 begins to immediately establish a negative feedback system where the resulting decrease in oxidative stress permits Hsp70 dephosphorylation that, in turn, promotes CHIP-dependent ubiquitination and directed degradation of both Hsp70 and SOD2, prompting a two-pronged halt to SOD2 import. The complexity by which these pathways mediate mitochondrial SOD2 activity provides insights into the mechanisms by which the cell maintains such tight controls over redox homeostasis.
The cytoplasm is a crowded environment in cells where numerous proteins are in contact with nascent SOD2, potentially predisposing it to aggregation. Though the mitochondrial targeting sequence contains information critical for SOD2 to navigate the cytoplasm and for its translocation into mitochondria (Liu et al., 2010), evidence suggests that SOD2 requires assistance from Hsp70 to prevent its aggregation and to commit the enzyme to mitochondrial pathways (Afolayan et al., 2014). However, mechanisms controlling the delivery of newly made SOD2 to mitochondria in response to stimuli in a timely and proportional manner are unknown. Our data show that phosphorylation induces a structural change in the Hsp70 conformation that potentiates its ability to refold and transport SOD2 to the mitochondria. These observations have led to a proposed model of SOD2 signaling whereby, following cells stimulation, Hsp70 becomes phosphorylated and increases SOD2 import. The transient nature of this response is achieved by rapid dephosphorylation of Hsp70 leading to the eventual termination of SOD2 import and activity (Figure S3E).
Phosphorylation is the most common posttranslational modifications known to regulate cellular functions (Iaccarino et al., 2004). It was recently reported that phosphorylation of the C terminus of Hsp70 on T635 by kinases is associated with high proliferation rates in cancer cells (Muller et al., 2013). Here, we demonstrated that phosphorylation of Hsp70 on Ser631 by Akt1 regulates mitochondrial oxidative stress. Together, these findings, not only show that multiple kinases could phosphorylate Hsp70, but that specific residue in the C terminus of Hsp70 is phosphorylated for selective signaling purposes. Therefore, our findings and that of others indicate a complex regulation of Hsp70 chaperone activity, which is typical for major chaperones such as Hsp70 that is involved in the regulation of diverse signaling pathways. In the present study, our data show that (1) inhibition of Akt1 kinase decreased the rates of Hsp70 phosphorylation, which in turn decreases the ability of Hsp70 to import SOD2; and (2) augmenting Akt1 kinase increased SOD2 import, which is blocked by a Ser631-inhibiting peptide, validating the critical role of Ser631 phosphorylation of Hsp70 in mitochondrial redox signaling. Importantly, the presence of Ser631 only in inducible Hsp70 (Figure S3F) and the site-specific effect of Akt1 have important biological implications in SOD2 signaling: (1) relaying both activation and inhibitory signals via Akt1 to Ser631 in Hsp70 ensures a coordinated regulation of SOD2 activity, and (2) the relative specificity of Ser631 phosphorylation by Akt1 prevents indiscriminate activation of SOD2.
Hydrogen peroxide constitutes a primary signaling molecule in mitochondrial redox sensing (Zou et al., 2017). Previous studies have examined the role of H2O2 in SOD2 signaling. These investigations have yielded mixed results. Studies conducted in rat uterus have reported positive effects of H2O2 on SOD2 activity (Appiah et al., 2009), while others have reported an adverse effect (Kim et al., 2011). The disagreements in these studies may reflect the dose- and time-dependent effects of H2O2 on Akt activation. Indeed, recent studies had demonstrated a transient increase in Akt activity when cells were treated with micromolar doses of H2O2 (Thomas et al., 2007; Lam et al., 2017). In line with this finding, our data show that beyond certain levels, further increases in mitochondrial H2O2 formation decreased SOD2 activity. This effect appears to be mediated by PP2C that de-phosphorylates Hsp70. Upon dephosphorylation, Hsp70 is converted back to its inactive form, which strongly attracts CHIP, and promoted degradation of Hsp70-SOD2 complexes. An increase in SOD2 activity by merely knocking down CHIP proteins prior to disruption of Ser631 phosphorylation demonstrated the effectiveness of this mechanism. Thus, phosphorylation controls the decision to import or target SOD2 for degradation. In vivo, Hsp70 phosphorylation directly impacts SOD2 activity, as ECs stimulated by PDGF displayed increased Hsp70 phosphorylation with minimal amounts of CHIP in complex with Hsp70, indicating that stimulated ECs exhibit a higher Hsp70 refolding activity, in keeping with higher oxidative phosphorylation and the requirement for increased SOD2 activity to remove excess O2•− byproduct. Therefore, phosphorylation-dephosphorylation cycles of Hsp70 serve as an efficient switch that controls the activity of SOD2 appropriate to metabolic and redox states.
Our results identify Akt1 as a central regulator of mitochondrial oxidative stress and vascular function. However, several other mechanisms mediated by Akt1 could explain its mitochondrial protective roles. For example, Akt1 modulates functions of several proteins including endothelial nitric oxide synthase to carry out its cells survival and vascular functions (Lachmandas et al., 2016). Given that Akt1 deficiency induces mitochondrial oxidative stress, which contributes to endothelial dysfunction, induction of Akt1 activity may also contribute to improving vascular function by phosphorylating Hsp70, leading to reduced mitochondrial oxidative stress and improved nitric oxide bioavailability. Based on these findings, it appears that Akt1 deficiency leads to Hsp70 de-phosphorylation, which in turn increases mitochondrial oxidative stress in vascular cells.
In conclusion, our results show that Akt1 integrates activation and inhibition redox signals and couples them to Hsp70 to control SOD2 activity. The sensitivity of Ser631 in Hsp70 to Akt1 and the presence of this residue only in inducible Hsp70 ensure that SOD2 activity remains proportional to activation signals and safeguards the cells from O2−/H2O2 imbalance and its pathological consequences.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Adeleye J. Afolayan, MD (aafolaya@mcw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The Medical College of Wisconsin Animal Care and Use Committee approved all protocols for Animal use. For the generation of PPHN fetal lambs, the pregnant ewe was purchased from May Enterprises Philadelphia and at 124 d gestation (term gestation = 145 d), PPHN was induced by ductal constriction. 8 d later, lambs were delivered by cesarean section. Age-matched sham ligated lambs were used as controls Sprague Dawley rats (SDR) were purchased from Envigo (Madison, WI), while akt1 null mice were purchased from The Jackson Laboratory (strain129P2/Akt1tm1Mbb/J). SDR was treated at 6-week of age. Age-matched SDR served as controls. The number of animals required for this study was based on preliminary observations. All rats were randomly assigned to receive either GSG or GAG peptides. Animals in the GSG-peptide group were further randomly assigned to receive 10mM/day mito-TEMPO or saline concurrently. 2mg/kg/d GSG peptide dose was selected based on preliminary observations. GSG or GAG ± mito-TEMPO was administered continuously using Alzet osmotic pump (size-1003D Cupertino, CA) for 10-d. Animals were nursed in room air for the treatment duration.
METHOD DETAILS
Cell isolation, culture, and treatments
Controls and PPHN-PAECs were isolated from near-term lambs of either gender as before(Konduri et al., 2003). CD31 staining confirmed the identities of the EC phenotype. Passage 4 or less were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) containing 10% (v/v) fetal calf serum and penicillin 100 U/ml, Gentamicin and 1% antifungal in a humidified incubator at 37°C in 21%O2 with 5%CO2. HEK-293T cells were purchased from Cell Application. Cells were grown in high-glucose DMEM supplemented with 10% Fetal Bovine Serum and 1% antibiotic/anti-mycotic (ThermoFisher Scientific). Decoy peptides were dissolved first in dimethyl sulfoxide (DMSO, Sigma-Aldrich) and then added to the culture medium (20–40 μM) immediately before use. 20-100nM siRNAs, the 50MOI adenovirus containing SOD2 or 2μg lentivirus containing CHIP were transfected with FuGENE®6 transfection reagent (Promega).
Domain mappings of Akt1 and Hsp70
2-3 μg HA-tagged Akt1 mutants and GST-tagged-Hsp70 mutants were co-transfected into HEK293T cells. The GST epitope was immuno-precipitated using 1μg/100μg monoclonal antibodies (GeneTex, Irvine CA) and the elute was blotted for HA or GST using specific antibody.
In vitro phosphorylation
1 μg of purified GST-tagged Hsp70, 500 U of Akt1 and 300 μM ATP were incubated at 30°C in a reaction buffer with total volume of 50 μl. The reaction was stopped after 30 minutes by the addition of the Nu-PAGE LDS Sample Buffer (Bio-Rad).
Development of phosphor-specific Hsp70 Ser631/633 antibody
The C-terminal phospho-peptides of Hsp70 CKPEG2AQGPKGG[pS]G[pS]GP-amide with 80–90% purity (21st Century Biochemicals, MA, USA). Immunogen peptides conjugated to carrier protein mix. All peptides were quantified by ESI-MS, HPLC analysis and tandem MS (CID MS/MS) to confirm each peptide sequence. Animals were injected with 5 immunizations and pre-immune serum from each rabbit serum samples were obtained for studies. To verify the specificity of the antibody, we phosphorylated purified Hsp70 proteins in an in vitro kinase reaction and incubated the phosphorylated Hsp70 with an anti-phospho-Ser631/633 antibody with or without its own targeting peptide. WB analyses revealed that the anti-phospho-Ser631 targeting peptide abolished the interaction between recombinant Hsp70 proteins and the anti-phospho-Hsp70-Ser631 antibody.
Kinase and phosphatase inhibitor screen
The kinase screening was performed with phosphatidylinositol 3-kinase inhibitor, Wortmannin (10 mM), mitogen-activated protein kinase)/extracellular signal-regulated kinase inhibitors, PD98059 (100 mM) and CDK inhibitor, Roscovitine (50 mM), CDK9 and CK2 inhibitor, DRB (50 mM) and p38MAPK inhibitor, SB202190 (50 mM). For protein phosphatase screening, varying doses of Calyculin A (inhibits PP1 and PP2A) and Sanguinarine chloride (selective for PP2C) were used.
SOD2 Activity Assay
Specific activity was measured using a colorimetric assay kit (706002, Cayman Chemical, Ann Arbor, MI) as described previously (Afolayan et al., 2014). Briefly, PAECs grown in 60-mm dishes to 80% confluence were pelleted, and the pellets were homogenized in cold 20 mm HEPES buffer (1 mm EGTA, 210 mm mannitol, and 70 mm sucrose). Cell debris was removed by centrifugation at 13,000 rpm at 4°C for 10 min. 1 mM potassium cyanide (151-50-8, Sigma-Aldrich) was added to the lysates during the assay process to inhibit both the cytosolic Cu/Zn-SOD and extracellular SOD (in tissue lysates only). Absorbance was read at 440–460 nm using a microplate reader (SpectraMax®i3X, Molecular Devices). The protein concentration in each well was estimated by Bicinchoninic Acid (BCA) method to calculate SOD2 activity per milligram of protein.
Immunofluorescence (IF) studies
PAECs serum-starved in 0.1% FBS (medium 200, Life Technologies) and exposed or not to 10ng/ml PDGF or acetylcholine for the specified duration, were stained with the cell-permeable mitochondrion-selective dye Mito-Tracker Red (500nM, Life Technologies), fixed, permeabilized, and stained with antibody specific for SOD2 (Enzo-Life, 1:150). Images were acquired using a Carl Zeiss Micro Imaging Microscope. Colocalization between SOD2 and Mito-Tracker was estimated by the software using an algorithm that calculates the Pearson’s correlation coefficient.
Immunoprecipitation and Immunoblotting
For western blotting, protein lysates were prepared from cells, lungs in MOPs protein extraction buffer containing Protease Inhibitor (Roche) and phosphatase Inhibitor Cocktails (Sigma). The protein concentration of the extracts was determined using Bradford reagent (Bio-Rad). Equal amounts of protein were separated by SDS gel electrophoresis, transferred to nitrocellulose membranes and incubated with either 5% non-fat dry milk or 10% bovine serum in TBS-T buffer, then membranes were incubated overnight at 4°C with primary antibodies: SOD2 (Enzo life, 1:1000), for phospho-Ser631/633 (Ecelone,1:1000), Hsp70 (Enzo-life, 1:1000), p38MAPK, ERK1/2, and Akt (Santa Cruz, 1:1000), in 3% BSA. Next, membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Sigma 1:10000) for 1hr at 25°C in 5% non-fat milk or 3% BSA. Antibodies were revealed using ECL reagents (Perkin-Elmer) and using the imaging films (Bio-Rad) and iBrightFL1000 (Invitrogen by Thermo-Fisher). Protein expression was quantified using the ImageJ software.
MALDI-TOF-mass spectrometry
2-3μg HA-tagged Hsp70 expression plasmid was transfected into HEK293T cells. After 24-hr, cells were treated with ACh and HA epitope was immunoprecipitated using a mouse monoclonal antibody. Following SDS-PAGE analyses, gels were stained with Coomassie blue stain, the Hsp70 differential bands were excised and cut into 1 mm3 piece. Gel pieces were first treated with 45 mM dithiothreitol (DTT) for 20 min, followed by treatment with 100 mM iodoacetamide for 20 min. After destaining with 50% acetonitrile in 50 mM ammonium bicarbonate, the gel bands were digested with sequencing-grade trypsin in 25 mM ammonium bicarbonate overnight at 37°C. Peptides were extracted by gel dehydration (60% aceto-nitrite, 0.1% TFA), the extracts were dried by vacuum centrifugation, and peptides were reconstituted in 0.1% formic acid. Extracted peptides were separated by reverse phase liquid chromatography and analyzed by tandem mass spectrometry (LC-MS/MS). For LC-MS/MS analysis, the peptide extracts were first loaded into a capillary reverse phase analytical column (100 μm inner diameter × 30cm length) using a Famos autosampler (LC Packings San Francisco, CA). The analytical column was packed with 20cm of C18 reverse phase material (Jupiter, 3 μm beads, 300 A, Phenomenex), directly into a laser-pulled emitter tip. Peptides were gradient-eluted at a flow rate of 500 nL/min, and the mobile phase solvents consisted of 0.1% formic acid, 99. 9% water (solvent A) and 0.1% formic acid, 99.9% acetonitrile (solvent B). A 90-min gradient was performed, consisting of the following: 0–10 min, 2% B; 10–50 min, 2%−40% B; 50–60 min, 40%−95% B; 60–65 min, 95% B; 65–70 min 95%−2% B; 70–90 min, 2% B. Eluting peptides were mass analyzed on an LTQ Orbitrap Velos Pro ion mass spectrometer (Thermo-Scientific), each equipped with a Nanoelectrospray ionization source. For the majority of LC-MS/MS analyses, the instruments were operated using a data-dependent method with dynamic exclusion enabled. Full scan (m/z 300-2000) spectra were acquired with the Orbitrap (resolution 60,000). The top 5 most abundant ions in each MS scan were selected for fragmentation via collision-induced dissociation in the LTQ ion trap. An isolation width of 2 m/z activation time of 10 or 30 ms, and 35% normalized collision energy were used to generate MS2 spectra. Dynamic exclusion settings allowed for a repeat count of 2 within a duration of 10 s, and the exclusion duration time was set to 15 s. For selected LC-MS/MS analyses, the LTQ Orbitrap Velos was operated using a method consisting of data-dependent and targeted scan events, for which specific m/z values corresponding to Hsp70 peptides of interest were provided in the data acquisition method to facilitate targeted MS spectra despite the intensity of peptide precursors. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, SEQUEST (Thermo-Scientific). All databases include a reversed version of all the sequence, and the data was filtered to between a 1 and 2% peptide false discovery rate.
Phospho-proteomics analysis by MS/MS
Purified GST-Hsp70 proteins (Sigma) was incubated with 500U Akt1 (Sigma) in in-vitro kinase reactions. The reaction was separated by 4%−12% SDS-PAG, and the band containing the phosphorylated Hsp70 was digested, washed and dehydrated, and IMAC enrichment followed by phosphor-peptide desalting was performed. Briefly, Peptides were dissolved in 120 μL of IMAC-binding buffer, transferred to the IMAC beads (100ul of IAC beads in 1 mL of IMAC binding and spinning to remove the liquid) and incubated for 60 min, with vigorous shaking, at 25°C. During this time, Stage Tips were prepared by packing two disks of Empore 3M C18 material onto 250 μL pipette tips. After the 60-min incubation IMAC beads were transferred to the top of the Stage Tips and spin down. The beads were retained on the Stage Tip, and the solution passed through. As the buffer contains 40%acetonitrilee (ACN), non-phosphorylated peptides, which are not retained in the IMAC resin, were retained by the C18 material. 50 μL of IMAC binding buffer were washed with 40 μL of 1% FA, followed by another wash with 70 μL of 500 mM K2HPO4, pH 7.2. This step was repeated twice. At this point phospho-peptides were eluted from IMAC resin and retained on the C18, which are then washed with 40 mL of 1% FA to remove phosphate salts. Phospho-peptides were then eluted from stage Tips into vials for MS analyses with 40 μL of 50%ACN 0.5% HAcO, and the samples were dried by vacuum centrifugation. Samples were then resuspended in 10 μL of 5% ACN, 4% FA and 1 μL of each sample was injected onto the LC-MS/MS system on an LTQ Orbitrap. All MS/MS spectra collected were searched with the SEQUEST algorithm against a composite human database with forward and reversed sequences.
For database searches and data analyses, MS/MS spectra were searched against a target-decoy human database using the SEQUEST algorithm (version 27), with 50 ppm precursor mass tolerance, tryptic enzyme specificity with two missed cleavages. Dynamic modifications were 79.96633 Da on Serine, Threonine and Tyrosine phosphorylation and a maximum of four modifications of any one type and five total modifications were allowed per peptide. SEQUEST XCorr and delta n’ score cutoffs were empirically determined for the entire dataset, and mass deviation (in p.p.m.) and peptide solution charge filters were determined for each sample using decoy matches as a guide and aiming to maximize the number of peptide spectra matches, although maintaining an estimated FDR of ≤ 1%. Identified phosphor-peptides passing our filtering criteria were submitted to the Ascore algorithm for precise site localization.
Mitochondrial ROS quantification
Mitochondrial O2and H2O2 were measured using liquid chromatography-mass spectrometry (LC-MS/MS, Shimadzu Nexera UHPLC system). For mitochondrial O2 measurement, samples from lungs, distal PAs and cells were incubated with mitoSOX (5 μM in fresh culture medium at 37°C for 30 min). Changes in O2 levels were assessed by measuring O2 specific mitoSOX oxidation product-2dihydroethidiumm (2-OH-Mito-E+) by LC-MS/MS and adjusted for protein conc. For H2O2 measurement, 3-borono phenylmethyl-triphenyl-phosphonium, mono-bromide (MitoB), a ratio-metric mass spectrometric probe was used. PAs and cells were incubated with MitoB (5μM in saline at 37°C for 3-6-hr). Changes in mitochondrial H2O2 levels were assessed by LC-MS/MS after spiking the homogenate with ISs (100pmol d15-MitoB and 50 pmol d15-MitoP in 10 μL ethanol).
Nuclear and cytoplasmic extractions
Harvested cells were washed with PBS. Ice-cold cytoplasmic extraction reagent (CERI, ThermoFisher Scientific) was added to the cell pellets, vortexed vigorously for 15 s to entirely suspend the cell pellet and incubated on ice for 10 min. Ice-cold CER II, ThermoFisher) reagent was added to the cell lysates, vortexed and incubated on ice for 5 min. The cell lysates were centrifuged for 5 min at maximum speed (~16,000×g, and the supernatant (cytoplasmic extract) was transferred to a pre-chilled tube. The insoluble pellets were re-suspended in ice-cold nuclear extraction reagent (NER, ThermoFisher), incubated on ice and vortexed for 15 s every 10 min, for a total of 40 min. Reactions were separated at maximum speed (~16,000×g) for 10 min, and the supernatant (nuclear extract) was stored at −80° C until use.
Mitochondrial isolations
Cell fractionation was performed as previously described. PAECs, grown in 100-mm dishes, were pelleted, and the pellets were washed in 10 mL of cold 1 × PBS, resuspended in mitochondrial isolation buffer (20 mM HEPES buffer (pH 7.2) containing 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose with EDTA), and homogenized on ice by passage in a 1 mL syringe with a 27-gauge needle 10 times. Next, lysates were treated as indicated and separated into cytosolic and mitochondrial fractions by differential centrifugations. The cytosolic and mitochondrial lysates were used for further studies. For isolation of bovine heart mitochondria, harvested heart from normal fetal lambs was cooled and washed in ice-cold isolation buffer (10 mM HEPES, 200 mM mannitol, 50mM sucrose, 1 mM EGTA, pH 7.2), ventricular tissue was excised, minced and homogenized using tissue homogenizer at low speed. The suspension was centrifuged at low speed, 8000×g for 10 min at 4°C. The pellets were re-suspended in isolation buffer and centrifuged at 850×g for 10 min, and the obtained supernatant was centrifuged again at a higher speed to collect the mitochondria. The mitochondria were washed in SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPs-KOH, pH 7.2) and kept on ice for import assays.
In vitro mitochondrial import assays
The mitochondrial import assay was performed as described elsewhere with minor modifications(Mokranjac, 2016). Briefly, cell-free translation of bovine recombinant SOD2 proteins was performed using the TNT-coupled transcription /translation rabbit reticulocyte lysate system (Promega, Madison WI) according to the manufacturer’s directions. Rapidly thawed TnT® Quick Master Mix was in a 1.5 micro-centrifuge tube was placed on ice. 2 μg of SOD2 plasmid (TOPGene, Canada) containing the T7 promoter were mixed with 1 μL Methionine, and 2-3 μL FluoroTectGreen Lys-tRNA. The total reaction volume was adjusted to 50 μL with nuclease-free water. Reactions were incubated for 90 min at 30°C. The result of translation was analyzed by SDS-PAGE analysis of translation products using Odyssey infrared scanner (LI-COR Biosciences, Nebraska). Afterward, 40 μL of the reaction was incubated for 60 min at 30°C with 20 μM purified Hsp70. Later, the control- and Hsp70-treated-FluoroTect-labeled recombinant SOD2 were diluted in import buffer (10 mM MOPs–KOH pH 7.2, 5 mM MgCl2, 80 mM KCl, 2 mM KHPO4 and 250 mM Sucrose) containing 2 mM ATP, 0.4 mM ADP and 1 mM DTT in the presence or absence of 500U Akt1. Bovine heart mitochondria (30 μg) were added to the translated proteins such that the volume of mitochondria made up 40% of the reaction volume and the exogenous protein constituted 35% of the total reaction volume. The translocation reaction was allowed to incubate for the indicated time at 30°C with intermittent gentle mixing. Mitochondria were re-isolated by centrifugation, suspended in the import buffer in the presence of 50 μg/ml proteinase K for 10 min on ice and mixed with SDS to terminate proteolytic reactions. Mitochondria were collected by centrifugation and imported proteins were separated by gel electrophoresis followed by infrared scanning using Odyssey LI-COR infrared scanner. In some experiments, the SOD2 recombinant protein was not added to the import reaction before analysis of protein import into mitochondria.
Computational details
A hybrid homology model of full-length human Hsp70 (i.e., residues 1-641) has been constructed in YASARA software from three template-based models (PDB codes: 4FL9, 4PO2 and 5BN8, overall Z-score = −0.028). Molecular dynamics simulations have been performed in Amber package (Salomon-Ferrer et al., 2012) using ff14SB force field (Maier et al., 2015; Krieger et al., 2009) with 8 Å cutoff and particle mesh Ewald algorithm with periodic boundary conditions. The simulation box was set at 10 Å from the protein along each axis and filled with TIP3P water models and Na+ to neutralize the cell. The resulting Hsp70 structure was gradually heated using the modified Berendsen thermostat from 0 to 298 K with constant volume followed by the equilibration with constant pressure (isotropic position scaling) until the target solvent density was reached (Maier et al., 201). The sample structure from this simulation was used as the starting geometry for the production 50 ns simulations. An analogous procedure was used for the simulation of the phosphorylated Hsp70S631. The data from these trajectories were used to construct histograms of the distance distributions in Figure 6C.
Enzyme-linked immunosorbent assays
ELISA was used to measure complex formation between purified CHIP proteins and phosphorylated and non-phosphorylated Hsp70 proteins. Purified GST-tagged Hsp70 proteins were phosphorylated by adding 300 μM ATP with 500U Akt1 proteins with 2 μM Hsp70 in vitro kinase reactions for 30-min at 25°C. Hsp70 phosphorylation was verified by WBs and once verified, phosphorylate, and non-phosphorylated -Hsp70 were diluted into PBS. Then, 100 μL aliquots of 50nM phospho-Hsp70 or non-phospho-Hsp70 solutions were added into the GST-coated wells of 96 microtiter plates. Hsp70 was allowed to adhere to the walls of wells during a 1-hr incubation at 25°C. After 1-hr, wells were gently washed three times to remove unbound Hsp70 with 50 mM phosphate, pH 7.4, 150 mM NaCl, 0.02% Triton X-100 (PBST). Then, the wells were blocked via 1-hr incubation with 200 μL of 1% bovine serum albumin (BSA) in PBS. CHIP (0.4 μg) was diluted into reactions in PBS with 0.02% Triton X-100, 0.2% BSA, and 1 mM ATP and then added to each well. After a 1-hr incubation at 25°C, the wells were washed three times with PBST. CHIP proteins retained in the wells were detected with a 1:5000 dilution of rabbit CHIP in PBST. CHIP antibody was incubated for 1-hr at 25°C. Unbound antibody was removed with three washes with PBST. Horseradish peroxidase-coupled secondary goat anti-rabbit serum was then added to detect the CHIP antibody retained in the wells. Three more washes with PBST were performed to wash away any unbound secondary antibody. H202 (0.05% final concentration) to a solution containing 0.22mg/ml 2, 2′-azinobis-(3-ethyl-benzothiazoline-6-sulfonate) in 50 mM sodium citrate, pH 4.0. This 2, 2′-azinobis-(3-ethyl-benzothiazoline-6-sulfonate solution was added to the wells of the microtiter plate, and color formation was measured at 450 nm with a microplate reader (Bio-Rad).
In vitro-Ubiquitination assays
E1/E2 Ubiquitin reaction mixtures were pre-charged by incubating 40 mM UbcH5b, 100 mM Ubiquitin, and 0.5 mM E1 for 30 min at 37°C in 50 mM HEPES (pH 7.0), 50 mM NaCl, 20 mM ATP, 40 mM MgCl2, and 0.5 mM dithiothreitol (DTT). Ubiquitination of Hsp70 constructs used pre-charged E1/E2. Ubiquitin reaction mixture was added to a solution of 4 mM GST-wild-type (WT)-Hsp70 or Hsp70 ± Akt1 and decoy peptides, GSG or GAG and 4 mM CHIP in 50 mM NaCl, 50 mM HEPES (pH 7.0). CHIP auto-ubiquitination utilized pre-charged E1/E2 Ubiquitin reaction mixture added to 4 mM CHIP in 50 mM NaCl, 50 mM HEPES (pH 7.0), 0.5 mM DTT. Ubiquitination reactions were incubated at 37°C and stopped at 1-hr by adding 2× SDS sample buffer containing 20 mM DTT and 50 mM EDTA. Quenched reactions were subjected to SDS-PAGE and immunoblotting using anti-CHIP or anti-GST primary antibodies.
Detection of Ubiquitinated-SOD2 in vivo
Polyubiquitin-modified SOD2 was isolated from cell lysates using the Ubiquitin Enrichment method (ThermoFisher, Scientific). ~0.15mg of protein from cells treated with 20 μM GSG or 2 μg Lentivirus CHIP was incubated with the anti-SOD2 polyclonal antibody (Enzo-Life: 1μg/100μg protein) Poly-ubiquitin affinity resin was added to the spin columns containing SOD2 complexes and incubated at 4°C for 2 hr. The spin column was centrifuged, washed with wash buffer and SDS-PAGE sample-loading buffer was added and heated at 95°C for 10 min. Proteins were separated by western blotting.
Quantification of Hsp70 and CHIP mRNAs
RNA was extracted via the TRIzol (Sigma) method from lung tissue, and the contaminated DNA was removed by the TURBO DNA-free Kit (Ambion) in a 37°C water bath for 60 min. cDNA was synthesized from the extracted RNA using the iScript cDNA synthesis kit (Bio-Rad). The PCR primers were designed using Primer3 as previously described and sequences are as follows: inducible Hsp70 (forward 5′-TTCATCAAG CCCCGCC TTT-3′ and reverse 5′ACTCCAGTTTCCTCCAGCGCGA-3′), and CHIP (stub1) (forward 5′-GGAACCGTAGGT GTTCGAGG-3′ and reverse 5′-GTTCTGAGGGA GGGTGAAGC-3′). Real-time RT-PCR was performed using the ViiA7 real-time PCR detection system (Applied Biosystems, Life Technologies).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests and graphs were done with GraphPad Prism 7.0. Values are expressed as fold change or mean ± standard error of the mean. Unpaired Student t tests were used for comparison between two groups, and one-way analysis of variance followed by a Tukey-Kramer post-test was used for > 2 groups. A significance level inferior to 5% (p < 0.05) was considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
The accession number for the mass spectrometry analysis of the Hsp70 interactome as well as phosphor-proteomic analysis of the C-terminal domain of Hsp70 by Akt1 reported in this paper is PRIDE: PXD011409, PXD011422.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SOD2 | Enzo Life Sciences | Cat#ADI-SOD-110-F;RRID:AB_2750987 |
| Rabbit polyclonal anti-CHIP (Stub1) | GeneTex | Cat#GTX55810;RRID:AB_2750988 |
| Rabbit polyclonal anti-PP2A-Cα/β | Santa Cruz | Cat# sc-14020;RRID:AB_2169625 |
| Rabbit polyclonal anti-PP2Cα/β | Santa Cruz | Cat#sc-48829;RRID:AB_2169776 |
| Mouse monoclonal anti-Akt1 | GeneTex | Cat#GTX49160;RRID:AB_2750991 |
| Rabbit polyclonal anti-Akt2 | GeneTex | Cat#GTX128457;RRID:AB_2750992 |
| Goat polyclonal anti-Akt3 | Santa Cruz | Cat#11520;RRID:AB_2750993 |
| Rabbit monoclonal anti-phosp-Akt1T308 | GeneTex | Cat#GTX79150;RRID:AB_11163439 |
| Rabbit polyclonal anti-phospho-Akt1S473 | Cell signaling | Cat#4060P;RRID:AB_2750995 |
| Mouse monoclonal anti-HspA1A | GeneTex | Cat#GTX110079;RRID:AB_11171898 |
| Rabbit polyclonal anti-phospho- and total ERK1/2 | Cell Signaling | Cat#4696;RRID:AB_390780 |
| Rabbit monoclonal anti-phospho-and p38MAPK | Cell signaling | Cat#9790;RRID:AB_10831196 |
| Mouse monoclonal anti-phospho-Hsp70 Serine 631/633 | 21st Century, Boston | Custom:RRID:AB_2750999 |
| Bacterial and Virus Strains | ||
| Adenovirus-Human SOD2 | Blood Research Institute (BRI), WI | NA |
| Lentivirus-Human CHIP | BRI | NA |
| Lentivirus-Human Akt1 | BRI | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human recombinant SOD2 | Abcam | Cat#Ab84639 |
| Human recombinant Akt1 | My Bio Source | Cat#MBS203190 |
| Human recombinant (His-tagged) HspA1A | Sigma-Aldrich | Cat#SRP5190 |
| PDGF-BB | Sigma-Aldrich | Cat#P8147-1VL |
| DEPMPO-biotin | Cayman | Cat#0414736-11 |
| Mito-TEMPO | Cayman | Cat#0486522-4 |
| MitoB | Cayman | Cat#04865934-11 |
| MitoB-d15 | Cayman | Cat#0491693-3 |
| MitoP-d15 | Cayman | Cat#0486828-1 |
| MitoP | Cayman | Cat#0486829-1 |
| Sanguinarine chloride (PP2C inhibitor) | R and D systems | Cat#2302 |
| BMS 265246 | R and D systems | Cat#5654 |
| Roscovitine (Seliciclib, CYC202, CDK inhibitor) | Selleck hem | Cat#S1153 |
| Calyculin A (PP1 and PP2a inhibitor) | Sigma-Aldrich | Cat#C5552 |
| SB 202190 (p38MAPKinase inhibitor) | Sigma-Aldrich | Cat#S7067 |
| PD-098,059 (MAPK/ERK inhibitor) | Sigma-Aldrich | Cat#P215 |
| Wortmannin (PI3K inhibitor) | Cell Signaling | Cat#9901 |
| Acetylcholine | Sigma | Cat#A6625-25G |
| Hydrogen peroxide | EMD | Cat#HX0635-1 |
| MITOSOX RED | Life Technologies | Cat#M36008 |
| Potassium cyanide | Sigma | Cat#60178-100G |
| YGRKKRRQRRR-GSG (custom) | Biomatik | #SP0005-95-01199 |
| YGRKKRRQRRR-GAG (custom) | Biomatik | #SP0005-95-01199 |
| Critical Commercial Assays | ||
| Ubiquitin Enrichment Kit | ThermoFisher | Cat#89899 |
| SOD2 dismutase kit | Cayman | Cat#706002 |
| Mitochondrial isolation kit | Life Technologies | Cat#89874 |
| Subcellular protein fractionation kit | ThermoFisher | Cat#78840 |
| TNT® T7Quick Coupled T/T System | Promega | Cat#L1171 |
| FluoroTecGreenLys in vitro Translation labeling system | Promega | Cat#L5001 |
| Pierce anti-GST coated clear strip plates | ThermoFisher | Cat#151144 |
| Deposited Data | ||
| Mass spectrometry raw data | This paper | PRIDE: PXD011409 |
| Mass spectrometry phosphorylation data | This paper | PRIDE: PXD011422 |
| Experimental Models: Cell Lines | ||
| HEK293T cells | Konduri Lab (Gift) | NA |
| Pulmonary artery endothelial cells | Konduri Lab | NA |
| Experimental Models: Organisms/Strains | ||
| Akt1 Homozygous floxed mice (Female) | The Jackson Lab | Cat#JAX026474 |
| Akt1 Homozygous floxed mice (Male) | The Jackson Lab | Cat#JAX026474 |
| Sprague Dawley rat female | Envigo | 2830671 |
| Ductal ligation model of PPHN | Konduri Lab | NA |
| Oligonucleotides | ||
| forward 5-GGAACCGTAGGT GTTCGAGG-3′(CHIP) | QIAGEN | Cat#GS207 |
| Reverse 5-GTTCTGAGGGA GGGTGAAGC-3′(CHIP) | QIAGEN | Cat#SI04364136 |
| forward 5′-TTCATCAAGCCCCGCC TTT-3′(Hsp70) | Sigma | #3021492665-30/1 |
| Reverse 5′-ACTCCAGTTTCCTCCAGCGCGA-3′ (Hsp70 | Sigma | 3021492665-40/0 |
| Recombinant DNA | ||
| pBabe puroL myr Akt K179M T308A S473A | Addgene | Cat#1072 |
| pBabe puroL MYR Akt T308A, S473A | Addgene | Cat#9014 |
| pET24a Akt1 | TOPGene Tech. Inc. | NA |
| pET15b-SOD2 for in vitro transcription and translation | TOPgene Tech. Inc. | NA |
| pcDNA3.1-GST-Hsp70 | TOPgene Tech. Inc. | NA |
| pcDNA3.1-GST-Hsp70 mutants (1-400, 396-537 & 537-641) | TOPgene Tech. Inc. | NA |
| pcDNA3.1-HA-Akt1 mutants (1-150, 150-409, & 409- | TOPgene Tech Inc. | NA |
| pET21-Hsp70S631A, pET21-Hsp70S633A | TOPgene Tech Inc. | NA |
| Software and Algorithms | ||
| Ascore software | Beausoleil et al., 2006 | NA |
| YASARA software | Salomon-Ferrer et al., 2012 | NA |
| ImageJ | https://imagej.nih.gov/ij | NA |
| GraphPad Prism 7 | https://www.graphpad.com/ | NA |
| Other | ||
| Osmotic Pump | Durect Corporation | Cat#0004317 |
Highlights.
SOD2 is synthesized in the cytosol and imported into mitochondria for activation
Inducible Hsp70 transports newly made SOD2 to mitochondria for subsequent import
Phosphorylation of the C terminus of Hsp70 by Akt1 facilitates SOD2 import
Hsp70 dephosphorylation promotes SOD2 degradation to terminate SOD2 import
ACKNOWLEDGMENTS
The authors acknowledge financial support from the NIH (K08-AJA080271), HL057268 (G.G.K.), Muma Endowed Chair in Neonatology (G.G.K.), and Children’s Research Institute (A.J.A. and G.G.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, Pritchard K, and Konduri GG (2014). Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am. J. Physiol. Lung Cell. Mol. Physiol 306, L351–L360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afolayan AJ, Alexander M, Holme RL, Michalkiewicz T, Rana U, Teng RJ, Zemanovic S, Sahoo D, Pritchard KA Jr., and Konduri GG (2017). Domain mapping of heat shock protein 70 reveals that glutamic acid 446 and arginine 447 are critical for regulating superoxide dismutase 2 function. J. Biol. Chem 292, 2369–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appiah I, Milovanovic S, Radojicic R, Nikolic-Kokic A, Orescanin-Dusic Z, Slavic M, Trbojevic S, Skrbic R, Spasic MB, and Blagojevic D (2009). Hydrogen peroxide affects contractile activity and anti-oxidant enzymes in rat uterus. Br. J. Pharmacol 158, 1932–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Ville´ n J, Li J, Cohn MA, Cantley LC, and Gygi SP (2004). Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 101, 12130–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Ville´ n J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 24, 1285. [DOI] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Ho¨ hfeld J, and Patterson C (2001). The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96. [DOI] [PubMed] [Google Scholar]
- Culotta VC, Yang M, and O’Halloran TV (2006). Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 1763, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemachandra LP, Shin DH, Dier U, Iuliano JN, Engelberth SA, Uusitalo LM, Murphy SK, and Hempel N (2015). Mitochondrial superoxide dismutase has a protumorigenic role in ovarian clear cell carcinoma. Cancer Res. 75, 4973–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Ciccarelli M, Sorriento D, Cipolletta E, Cerullo V, Iovino GL, Paudice A, Elia A, Santulli G, Campanile A, et al. (2004). AKT participates in endothelial dysfunction in hypertension. Circulation 109, 2587–2593. [DOI] [PubMed] [Google Scholar]
- Idelchik MDPS, Begley U, Begley TJ, and Melendez JA (2017). Mitochondrial ROS control of cancer. Semin. Cancer Biol. 47, 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnati S, L€uers G, Pfreimer S, and Baumgart-Vogt E (2013). Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem. Cell Biol. 140, 105–117. [DOI] [PubMed] [Google Scholar]
- Kim SY, Bae S, Choi KH, and An S (2011). Hydrogen peroxide controls Akt activity via ubiquitination/degradation pathways. Oncol. Rep 26, 1561–1566. [DOI] [PubMed] [Google Scholar]
- Konduri GG, Ou J, Shi Y, and Pritchard KA Jr. (2003). Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 285, H204–H211. [DOI] [PubMed] [Google Scholar]
- Konzack A, and Kietzmann T (2014). Manganese superoxide dismutase in carcinogenesis: Friend or foe? Biochem. Soc. Trans 42, 1012–1016. [DOI] [PubMed] [Google Scholar]
- Krieger E, Joo K, Lee J, Lee J, Raman S, Thompson J, Tyka M, Baker D, and Karplus K (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 77 (Suppl 9), 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmandas E, Beigier-Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, Ammerdorffer A, Boutens L, de Jong D, Kanneganti TD, et al. (2016). Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur. J. Immunol 46, 2574–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam HC, Baglini CV, Lope AL, Parkhitko AA, Liu HJ, Alesi N, Malinowska IA, Ebrahimi-Fakhari D, Saffari A, Yu JJ, et al. (2017). p62/SQSTM1 cooperates with hyperactive mTORC1 to regulate glutathione production, maintain mitochondrial integrity, and promote tumorigenesis. Cancer Res. 77, 3255–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet 11, 376–381. [DOI] [PubMed] [Google Scholar]
- Liu GH, Xie DH, Zhu GH, Wu WJ, and Ge SL (2004). [Effect of EGCG on H2O2-induced MnSOD gene expression in cultured spiral ganglion cells]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 29, 682–685. [PubMed] [Google Scholar]
- Liu YM, Li XD, Guo X, Liu B, Lin AH, Ding YL, and Rao SQ (2010). SOD2 V16A SNP in the mitochondrial targeting sequence is associated with noise induced hearing loss in Chinese workers. Dis. Markers 28, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Zhong J, Chang R, Hu H, Pandey A, and Semenza GL (2010). Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J. Biol. Chem. 285, 3651–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier JA,Martinez C, Kasavajhala K,Wickstrom L, Hauser KE, and Simmerling C (2015). ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melling CW, Thorp DB, Milne KJ, and Noble EG (2009). Myocardial Hsp70 phosphorylation and PKC-mediated cardioprotection following exercise. Cell Stress Chaperones 14, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa H, Nakata K, Matsuura J, Moriwaki Y, Kawashima K, Shimizu T, Shirasawa T, and Takahashi R (2006). Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiol. Dis 23, 169–177. [DOI] [PubMed] [Google Scholar]
- Mokranjac D (2016). Mitochondrial protein import: An unexpected disulfide bond. J. Cell Biol. 214, 363–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Forde R, Belton A, and Duttaroy A (2011). SOD2, the principal scavenger of mitochondrial superoxide, is dispensable for embryogenesis and imaginal tissue development but essential for adult survival. Fly (Austin) 5, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, and Vojtesek B (2013). C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 32, 3101–3110. [DOI] [PubMed] [Google Scholar]
- Paul I, and Ghosh MK (2014). The E3 ligase CHIP: Insights into its structure and regulation. BioMed Res. Int 2014, 918183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourvali K, Abbasi M, and Mottaghi A (2016). Role of superoxide dismutase 2 gene Ala16Val polymorphism and total antioxidant capacity in diabetes and its complications. Avicenna J. Med. Biotechnol 8, 48–56. [PMC free article] [PubMed] [Google Scholar]
- Shao M, Li L, Song S, Wu W, Peng P, Yang C, Zhang M, Duan F, Jia D, Zhang J, et al. (2016). E3 ubiquitin ligase CHIP interacts with C-type lectinlike receptor CLEC-2 and promotes its ubiquitin-proteasome degradation. Cell. Signal 28, 1530–1536. [DOI] [PubMed] [Google Scholar]
- Smith MR, Fernandes J, Go YM, and Jones DP (2017). Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun 482, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R, Case DA, and Walker RC (2012). An overview of the Amber biomolecular simulation package. https://onlinelibrary.wiley.com/doi/abs/10.1002/wcms.1121. [Google Scholar]
- Stankowski JN, Zeiger SL, Cohen EL, DeFranco DB, Cai J, and McLaughlin B (2011). C-terminus of heat shock cognate 70 interacting protein increases following stroke and impairs survival against acute oxidative stress. Antioxid. Redox Signal. 14, 1787–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Kotamraju S, Zielonka J, Harder DR, and Kalyanaraman B (2007). Hydrogen peroxide induces nitric oxide and proteosome activity in endothelial cells: A bell-shaped signaling response. Free Radic. Biol. Med 42, 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, Perrett S, Prodromou C, Jones GW, and Kron SJ (2012). CDKdependent Hsp70 Phosphorylation controls G1 cyclin abundance and cellcycle progression. Cell 151, 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanPelt J, and Page RC (2017). Unraveling the CHIP: Hsp70 complex as an information processor for protein quality control. Biochim. Biophys. Acta Proteins Proteom 1865, 133–141. [DOI] [PubMed] [Google Scholar]
- Xiong W, Liu S, Cai W, Wen J, Fu Y, Peng J, and Zheng Z (2017). The carboxyl terminus of heat shock protein 70-interacting protein (CHIP) participates in high glucose-induced cardiac injury. Free Radic. Biol. Med. 106, 339–344. [DOI] [PubMed] [Google Scholar]
- Zhan S, Wang T, and Ge W (2017). Multiple functions of the E3 ubiquitin ligase CHIP in immunity. Int. Rev. Immunol. 36, 300–312. [DOI] [PubMed] [Google Scholar]
- Zhang H, Amick J, Chakravarti R, Santarriaga S, Schlanger S, McGlone C, Dare M, Nix JC, Scaglione KM, Stuehr DJ, et al. (2015). A bipartite interaction between Hsp70 and CHIP regulates ubiquitination of chaperoned client proteins. Structure 23, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Ratti BA, O’Brien JG, Lautenschlager SO, Gius DR, Bonini MG, and Zhu Y (2017). Manganese superoxide dismutase (SOD2): Is there a center in the universe of mitochondrial redox signaling? J. Bioenerg. Biomembr 49, 325–333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the mass spectrometry analysis of the Hsp70 interactome as well as phosphor-proteomic analysis of the C-terminal domain of Hsp70 by Akt1 reported in this paper is PRIDE: PXD011409, PXD011422.