

Abstract
We employ a single catalyst/oxidant system to enable the asymmetric syntheses of indolines, benzodihydrothiophenes, and indanes by C–H insertion of donor/donor carbenes. This methodology enables the rapid construction of densely substituted five-membered rings that form the core of many drug targets and natural products. Furthermore, oxidation of hydrazones to the corresponding diazo compounds proceeds in situ, enabling a relatively facile one- or two-pot protocol in which isolation of potentially explosive diazo alkanes is avoided. Regioselectivity studies were performed to determine the impact of sterics and electronics in donor/donor metal carbene C–H insertions to form indolines. This methodology was applied to a variety of substrates in high yield, diastereomeric, and enantiomeric ratios and to the synthesis of a patented indane estrogen receptor agonist with anti-cancer activity.
Keywords: asymmetric synthesis, C–H insertion, diastereoselectivity, enantioselectivity, rhodium carbenes
The insertion of metal carbenes into otherwise unreactive C–H bonds is a powerful method for the synthesis of complex molecules. Chiral metal complexes, specifically rhodium tetracarboxylates, can induce high levels of enantioselectivity for both intra- and intermolecular reactions.[1] The asymmetric C–H insertion reactions of metal carbenes flanked by electron withdrawing groups have been well-documented since Teyssie’s seminal publication in 1982.[2] Recently, our group has demonstrated the utility of metal carbenes with two pendant electron donating groups, or “donor/donor” metal carbenes, in the asymmetric synthesis of benzodihydrofurans (Figure 1).[3]
Figure 1.

Additional cores accessible by C–H insertion reactions of donor/donor metal carbenes.
Moreover, the requisite diazo precursor for the donor/donor carbene can be produced in situ from the parent hydrazone. Herein, we apply our methodology to the synthesis of three new classes of products, i.e. indolines, benzodihydrothiophenes and indanes.
Indolines represent a common core structure among natural products and drug discovery candidates.[4] Although these heterocycles can be prepared by a variety of strategies, formation of the C2–C3 bond by C–H insertion offers the opportunity to create two stereogenic centers.[5] Intramolecular C–H insertion has been used previously for the synthesis of indolines, however few examples of substrates containing basic nitrogen have been shown to proceed with high levels of enantioselectivity.[6] We documented the first enantioselective synthesis of an indoline from a donor/donor carbene, albeit with modest enantioselectivity on a substrate lacking a basic nitrogen atom.[3a] Recently, Zhu et. al. reported the enantioselective synthesis of indolines from donor/donor carbenes via a non-diazo approach.[5b]
A variety of indolines were synthesized with high levels of diastereo- and enantioselectivity (Figure 2). Fused indolines derived from cyclic anilines (2a-d) were formed in good to excellent er, dr, and yield. C–H Insertion into primary centers is known to be electronically unfavorable.[2, 7] However, indoline 2e was produced in high yield and modest er. Changing from a methyl to an ethyl substituent has a drastic effect on the enantioselectivity of the reaction, as indoline 2f was produced as primarily one enantiomer. This observation suggests that increased steric bulk at the insertion center plays a role in the high degree of stereoselectivity observed. An indoline derived from an N-tosyl protected amine was also tolerated (2b) allowing for further modification after deprotection. Additionally, indoline 2g derived from a free amine was also synthesized in excellent er, dr and yield. X-ray crystallography of 2c indicated that the relative and absolute configurations of the product were consistent with the sense of induction observed for the formation of benzodihydrofurans.[3a, 8]
Figure 2.

Indolines synthesized by the C–H insertion reaction of donor/donor metal carbenes.
Over the course of our synthesis of indolines, it became apparent that unsymmetrical aniline substrates presented the opportunity to insert into two chemically different C–H bonds. X-ray crystal structures by Fürstner show that the π system of the pendant phenyl group is in conjugation with the carbene center.[9] Additionally, computational experiments in our previous work with benzodihydrofurans have demonstrated that the electron donation afforded by the phenyl ring reduces the electrophilicity of the resultant metal carbene and stabilizes its formation.[3b] These factors motivate a stepwise reaction mechanism unique to our donor/donor system. In contrast, the mechanism of donor/acceptor substituted carbenes is known to be concerted asynchronous.[10] With this mechanistic framework in mind we set out to test the effect of sterics and electronics on the regiochemistry of C–H insertion reactions to form indolines. The insertion begins with a hydride transfer followed by rapid collapse of the resultant zwitterionic intermediate II.[3b] As such, insertion is favored by substituents that stabilize cations at the insertion carbon (Scheme 1). However, steric demand near the insertion carbon can be unfavorable, especially with crowded chiral catalysts.
Scheme 1.

Mechanism of benzodihydrofuran insertion.
To study the effects of sterics and electronics on the regioselectivity of this chemistry, we synthesized two unsymmetrical hydrazones (5a, b) with both sterically and electronically favorable insertion sites (Table 1). In the presence of Rh2(OAc)4, insertion at the benzylic carbon is favorable, to form 7a (entry 1). Treatment with Rh2(Mes)4 and Rh2(R-PTAD)4, shows a decrease in selectivity for the electronically favorable center as the bulk of the catalyst increases (entries 2, 3). When methyl is contrasted with isopropyl (5b), selectivity for the less substituted carbon is modest (entry 4) and use of the Rh2(Mes)4 catalyst heavily favors methyl insertion (entry 5). Finally, the reaction of 5b with Rh2(R-PTAD)4 also favors methyl insertion to a lesser extent (entry 6). The results indicate that both steric and electronic factors compete for the most favorable insertion pathway.
Table 1.
Regioselectivity of indolines.
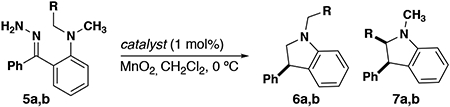
| Entry | R | Catalyst | 6:7 |
|---|---|---|---|
| 1 | Ph | Rh2(OAc)4 | 7:93 |
| 2 | Ph | Rh2(Mes)4[a] | 25:75 |
| 3 | Ph | Rh2(R-PTAD)4 | 51:49 |
| 4 | i-Pr | Rh2(OAc)4 | 61:39 |
| 5 | i-Pr | Rh2(Mes)4[a] | 95:5 |
| 6 | i-Pr | Rh2(R-PTAD)4 | 82:18 |
Rh2(Mes)4 = Rh2(2, 4, 6-trimethylbenzoate)4
Benzodihydrothiophenes form an interesting class of heterocycles for which there are currently few stereoselective synthetic methods.[11] Growing interest in sulfur-containing heterocycles as anti-tumor agents emphasizes the need for new method development in this area.[12] To date, there is only a single report of a benzodihydrothiophene synthesis employing rhodium carbenes with acceptor-substituted diazo compounds.[13] The authors of this study hypothesize that the lack of development of this methodology is due to a propensity of the highly nucleophilic sulfur atom to attack the carbene directly to form an ylide.[14] Due to the reduced electrophilicity of our donor/donor carbenes we are able to synthesize a variety of benzodihydrothiophenes with excellent stereocontrol and high yields.
Similarly to benzodihydrofurans,[3b] 4a, b were synthesized with excellent stereoselectivity and yield (Figure 3). Hydrazone 3c was selectively oxidized to the diazo compound with MnO2 in the presence of a benzylic alcohol. Furthermore, the corresponding diazo-alcohol was tolerated well under our reaction conditions, creating a useful chemical handle for further modification without the use of a protecting group (4c). Sterically crowded insertion centers were also tolerated allowing access to novel alkyl and spirocyclic compounds 4d-f. In contrast, poor selectivity was observed with 4g. This result supports our hypothesis that the degree of stereoselectivity observed corresponds to the presence of steric bulk near the insertion center as shown with indolines. Interestingly, with sufficient activation from the neighboring acetal, six-membered ring formation (4h) was achieved in high yield and er.
Figure 3.

Insertion reactions for the synthesis of benzodihydrothiophenes.
Indanes are rigid carbacycles that feature prominently in both natural products and in drug discovery. Unlike their heterocyclic counterparts, the electron-neutral benzene and benzylic carbon are less easily oxidized by enzymes responsible for drug clearance.[15] Enantioselective synthesis of indanes has been achieved primarily by asymmetric reduction of indenes.[16] Hashimoto and Lei have reported the enantioselective synthesis of indanes by C–H insertion of donor/acceptor carbenes.[17] In our seminal publication, we demonstrated that the formation of indanes by C–H insertion of donor/donor metal carbenes was possible in good yield and with a modest degree of stereoselectivity.[3a] In this work, we expand the scope of this chemistry to include a variety of indanes in excellent yield and selectivity.
Aliphatic spirocyclic five- and six-membered rings were particularly effective (Figure 4A, 9a, c). Heteroatoms were also tolerated to generate the corresponding spirocycles 9d, e including a Boc-protected amine as a chemical handle.[8] Disubstitution at the inserting carbon creates a high level of steric crowding, yet stabilizes carbocation formation at the insertion center more readily. As such, these substrates react to form quaternary carbons in high yield and with high levels of stereoselectivity (9b). The absolute configuration of the new stereogenic center was shown to be S upon treatment with Rh2(R-PTAD)4, in agreement with the sense of induction for both benzodihydrofurans and indolines.
Figure 4.

A) Insertion reactions for the synthesis of indanes. B) Synthesis of indane drug 11a.
Substitution of the carbene pendant groups with halogens was also tolerated with excellent selectivity (9f, g). Furthermore, J-values of the relevant protons of these products were consistent with the cis diastereomer observed both in this and in our previous work.[3a] We envisioned 9f as the direct synthetic precursor to a class of indanes patented as an estrogen receptor agonist for the treatment of breast, prostate and ovarian cancers.[18] Treatment of substrate 9f under modified Ullman coupling conditions allowed us to access the first enantioselective synthesis of drug lead 11a[19] in 34% overall yield (Figure 4B).
In conclusion, we have expanded the C–H insertion methodology of donor/donor metal carbenes to the enantioselective synthesis of a variety of indolines, benzodihydrothiophenes, and indanes. Preliminary results suggest the regiochemistry of C–H insertions to form indolines is influenced by competition between electronics that favor carbocation stability and the steric demands of catalyst and substrate. Our methodology allows for the facile synthesis of complex and difficult-to-access benzodihydrothiophenes. Furthermore, insertion into deactivated C–H bonds was accomplished, yielding a variety of indanes. Finally, we demonstrated the utility of this chemistry by carrying out the first highly enantioselective synthesis of patented drug lead 11a in good overall yield.[19]
Experimental Section
Experimental procedures and compound characterization can be found in the Supporting Information (PDF). 1H and 13C NMR spectra for all new compounds and HPLC traces for all enantiomerically enriched insertion products (PDF). X-ray data for compound 2c (CIF) and 9d (CIF).
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Institute Of General Medical Sciences of the National Institutes of Health under Award Number R01GM124234. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Austin Kelly (Franz group, UC Davis) for providing assistance with HPLC traces and Dr. William Long (Agilent Technologies) for obtaining a chiral HPLC trace for compound 4h. We also thank the Kurth group (UC Davis) for use of their IR with help from Zefan Hurley and Lee Dunlap (Olson group, UC Davis). C.N.P. thanks UC Davis for providing a Provost’s Undergraduate Fellowship (PUF). We thank the National Science Foundation (Grant CHE-1531193) for the Dual source X-ray diffractometer.
References
- [1].a) Davies HML, Beckwith REJ, Chem. Rev 2003, 103, 2861–2904; [DOI] [PubMed] [Google Scholar]; b) Davies HML, Hansen T, J. Am. Chem. Soc 1997, 119, 9075–9076; [Google Scholar]; c) Davies HML, Morton D, Chem. Soc. Rev 2011, 40, 1857–1869; [DOI] [PubMed] [Google Scholar]; d) Doyle MP, Duffy R, Ratnikov M, Zhou L, Chem. Rev 2010, 110, 704–724; [DOI] [PubMed] [Google Scholar]; e) Doyle MP, Ratnikov M, Liu Y, Org. Biomol. Chem 2011, 9, 4007–4016; [DOI] [PubMed] [Google Scholar]; f) Egger J, Carreira EM, Nat. Prod. Rep 2014, 31, 449–455; [DOI] [PubMed] [Google Scholar]; g) Zhu S, Zhu D, Chen L, Zhang H, Ma Z, Jiang H, Angew. Chem. Int. Ed 2018, 10.1002/anie.201805676; Angew. Chem. 2018. [DOI] [Google Scholar]
- [2].a) Demonceau A, Noels AF, Hubert AJ, Teyssié P, Chem. Commun 1981, 0, 688–689; [Google Scholar]; b) Demonceau AN, Hubert AF, Teyssie, P. AJ, Bull. Soc. Chim. Belg 1984, 93, 945–948. [Google Scholar]
- [3].a) Soldi C, Lamb KN, Squitieri RA, González-López M, Di Maso MJ, Shaw JT, J. Am. Chem. Soc 2014, 136, 15142–15145; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lamb KN, Squitieri RA, Chintala SR, Kwong AJ, Balmond EI, Soldi C, Dmitrenko O, Castiñeira Reis M, Chung R, Addison JB, Fettinger JC, Hein JE, Tantillo DJ, Fox JM, Shaw JT, Chem. Eur. J 2017, 23, 11843–11855. [DOI] [PubMed] [Google Scholar]
- [4].a) Zhang Y-B, Zhang X-L, Chen N-H, Wu Z-N, Ye W-C, Li Y-L, Wang G-C, Org. Lett 2017, 19, 424–427; [DOI] [PubMed] [Google Scholar]; b) Ngantchou I, Nyasse B, Denier C, Blonski C, Hannaert V, Schneider B, Bioorg. Med. Chem. Lett 2010, 20, 3495–3498; [DOI] [PubMed] [Google Scholar]; c) Dejon L, Mohammed H, Du P, Jacob C, Speicher A, MedChemComm 2013, 4, 1580–1583. [Google Scholar]
- [5].a) Solé D, Mariani F, Bennasar ML, Fernández I, Angew. Chem. Int. Ed 2016, 55, 6467–6470; Angew. Chem. 2016, 128, 6577–6580; [DOI] [PubMed] [Google Scholar]; b) Zhu D, Ma J, Luo K, Fu H, Zhang L, Zhu S, Angew. Chem. Int. Ed 2016, 55, 8452–8456; Angew. Chem. 2016, 128, 8592–8596. [DOI] [PubMed] [Google Scholar]
- [6].a) Garner R, Tetrahedron Lett. 1968, 9, 221–224; [Google Scholar]; b) Krogsgaard-Larsen N, Begtrup M, Herth MM, Kehler J, Synthesis 2010, 2010, 4287–4299; [Google Scholar]; c) Lee S, Lim H-J, Cha KL, Sulikowski GA, Tetrahedron 1997, 53, 16521–16532; [Google Scholar]; d) Lim H-J, Sulikowski GA, J. Org. Chem 1995, 60, 2326–2327; [Google Scholar]; e) Mahoney SJ, Fillion E, Chem. Eur. J 2012, 18, 68–71; [DOI] [PubMed] [Google Scholar]; f) Reddy Annapureddy R, Zhou CY, Guo Z, Wei J, Che CM, Angew. Chem. Int. Ed 2014, 53, 14175–14180; Angew. Chem. 2014, 126, 14399–14404; [DOI] [PubMed] [Google Scholar]; g) Santi M, Mueller STR, Folgueiras-Amador AA, Uttry A, Hellier P, Wirth T, Eur. J. Org. Chem 2017, 2017, 1889–1893; [Google Scholar]; h) Sulikowski GA, Lee S, Tetrahedron Lett. 1999, 40, 8035–8038. [Google Scholar]
- [7].Davies HML, Hansen T, Churchill MR, J. Am. Chem. Soc 2000, 122, 3063–3070. [Google Scholar]
- [8].CCDC 1566698 (2c) and CCDC 1565066 (9d) contain the supplementary crystallographic data for this paper. These data are provided free of charge by The Cambridge Crystallographic Data Centre.
- [9].a) Werlé C, Goddard R, Fürstner A, Angew. Chem. Int. Ed 2015, 54, 15452–15456; Angew. Chem. 2015, 127, 15672–15676; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Werlé C, Goddard R, Philipps P, Farès C, Fürstner A, J. Am. Chem. Soc 2016, 138, 3797–3805. [DOI] [PubMed] [Google Scholar]
- [10].Nakamura E, Yoshikai N, Yamanaka M, J. Am. Chem. Soc 2002, 124, 7181–7192. [DOI] [PubMed] [Google Scholar]
- [11].a) He Z, Shrives Harry J, Fernández-Salas José A, Abengózar A, Neufeld J, Yang K, Pulis Alexander P, Procter David J, Angew. Chem. Int. Ed 2018, 57, 5759–5764; Angew. Chem. 2018, 130, 5861–5866; [DOI] [PubMed] [Google Scholar]; b) Li W, Schlepphorst C, Daniliuc C, Glorius F, Angew. Chem. Int. Ed 2016, 55, 3300–3303; Angew. Chem. 2016, 128, 3361–3364; [DOI] [PubMed] [Google Scholar]; c) Tosatti P, Pfaltz A, Angew. Chem. Int. Ed 2017, 56, 4579–4582; Angew. Chem. 2017, 129, 4650–4653; [DOI] [PubMed] [Google Scholar]; d) Urban S, Beiring B, Ortega N, Paul D, Glorius F, J. Am. Chem. Soc 2012, 134, 15241–15244. [DOI] [PubMed] [Google Scholar]
- [12].a) Harayama Y, Yoshida M, Kamimura D, Wada Y, Kita Y, Chem. Eur. J 2006, 12, 4893–4899; [DOI] [PubMed] [Google Scholar]; b) Tohma H, Harayama Y, Hashizume M, Iwata M, Kiyono Y, Egi M, Kita Y, J. Am. Chem. Soc 2003, 125, 11235–11240; [DOI] [PubMed] [Google Scholar]; c) Wada Y, Harayama Y, Kamimura D, Yoshida M, Shibata T, Fujiwara K, Morimoto K, Fujioka H, Kita Y, Org. Biomol. Chem 2011, 9, 4959–4976; [DOI] [PubMed] [Google Scholar]; d) Zou Y, Hamann MT, Org. Lett 2013, 15, 1516–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Skerry PS, Swain NA, Harrowven DC, Smyth D, Bruton G, Brown RCD, Chem. Commun 2004, 0, 1772–1773. [DOI] [PubMed] [Google Scholar]
- [14].Aggarwal V, Richardson J, Sci. Synth 2004, 27, 21–104. [Google Scholar]
- [15].Vilums M, Heuberger J, Heitman Laura H, A. P IJ, Med. Res. Rev 2015, 35, 1097–1126. [DOI] [PubMed] [Google Scholar]
- [16].a) Hao X-D, Chang J, Qin B-Y, Zhong C, Chu Z-B, Huang J, Zhou W-J, Sun X, Eur. J. Med. Chem 2015, 102, 26–38; [DOI] [PubMed] [Google Scholar]; b) Schrems MG, Neumann E, Pfaltz A, Angew. Chem. Int. Ed 2007, 46, 8274–8276; [DOI] [PubMed] [Google Scholar]; c) Zhang Z, Wang J, Li J, Yang F, Liu G, Tang W, He W, Fu J-J, Shen Y-H, Li A, Zhang W-D, J. Am. Chem. Soc 2017, 139, 5558–5567. [DOI] [PubMed] [Google Scholar]
- [17].a) Hong B, Li C, Wang Z, Chen J, Li H, Lei X, J. Am. Chem. Soc 2015, 137, 11946–11949; [DOI] [PubMed] [Google Scholar]; b) Saito H, Oishi H, Kitagaki S, Nakamura S, Anada M, Hashimoto S, Org. Lett 2002, 4, 3887–3890. [DOI] [PubMed] [Google Scholar]
- [18].Govek SP, Smith ND, Seragon Pharmaceuticals, Inc., USA; Aragon Pharmaceuticals, Inc. Indane estrogen receptor modulators and uses thereof, 2014, US8853423B2. [Google Scholar]
- [19].Malik MS, Rastogi SN, Indian J. Chem. Sect. B: Org. Chem. Incl. Med. Chem 1981, 20B, 174–175. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.