

Abstract
Engineered polymer vesicles, termed as polymersomes, confer a flexibility to control their structure, properties, and functionality. Self-assembly of amphiphilic copolymers leads to vesicles consisting of a hydrophobic bilayer membrane and hydrophilic core, each of which is loaded with a wide array of small and large molecules of interests. As such, polymersomes are increasingly being studied as carriers of imaging probes and therapeutic drugs. Effective delivery of polymersomes necessitates careful design of polymersomes. Therefore, this review article discusses the design strategies of polymersomes developed for enhanced transport and efficacy of imaging probes and therapeutic drugs. In particular, the article focuses on overviewing technologies to regulate the size, structure, shape, surface activity, and stimuli-responsiveness of polymersomes and discussing the extent to which these properties and structure of polymersomes influence the efficacy of cargo molecules. Taken together with future considerations, this article will serve to improve the controllability of polymersome functions and accelerate the use of polymersomes in biomedical applications.
Keywords: Self-assembly, drug delivery, biomedical, vesicles, bilayer membrane
Graphical Abstract
Polymersomes, as engineered polymer vesicles, are well-received as efficient carriers for the delivery of diagnostic and therapeutic molecules. Chemical modifications to self-assembling polymers allow customization of nano-sized carriers for a wide variety of clinical applications. This review highlights the critical design features and discusses the current efforts to engineer polymersomes for the next generation of nanomedicine.

1. Introduction
Since the late nineties, liposomes have been developed to reproduce bi-layered structure of biological cell membrane by using lipids with varied functional and charged groups. This bi-layered structure protects and transports delicate therapeutic proteins and nucleic acids. One commercialized liposomal carrier is Doxil®, which preferentially accumulates in disease sites and minimizes the off-target interactions of highly potent diagnostic probes and drugs.[1,2] Overall, these carriers are valued to increase the efficacy of molecular cargos. However, the interaction of liposome components with the immune system has limited their translational potential. The fast liposomal clearance has been largely attributed to the opsonization of liposomes by serum proteins.[3] To evade the detection of the immune system, liposomes are often modified with poly(ethylene glycol) to sterically inhibit both electrostatic and hydrophobic interactions with plasma proteins.[4,5]
To improve the stability and functionality of synthetic vesicles, amphiphilic polymers have attracted attentions as a customizable alternative building block to lipids. These amphiphilic polymers in the form of diblock, triblock or graft copolymers self-assemble to polymeric vesicles called polymersomes. The resulting polymersomes present hollow spheres that contain aqueous core surrounded by a bilayer membrane. The bilayer membrane is composed of hydrated hydrophilic coronas facing the inner core and outer aqueous medium. Hydrophobic segments of the amphiphilic polymer form the middle part of the membrane and serve as protective shell that separates the core from the outside medium. The size, morphology, rigidity and stimulus-responsiveness of polymersomes can be further tailored by tuning the hydrophilic-to-hydrophobic ratio of copolymers. Polymersomes also present greater structural and colloidal stability than liposomes in aqueous media and tunable mechanical properties.
With the capacity to control structure and properties of the polymersomes, there have been increasing efforts to use the polymersomes as a new generation of nanomedicine for clinical diagnosis and therapies. In particular, compartmentalization of polymersomes made them favorable tools for a series of biomedical applications including diagnosis and controlled drug delivery. The aqueous core encapsulates hydrophilic drugs, enzymes, growth factor, peptides, and nucleotides while the membrane integrates hydrophobic drugs or imaging probes within its hydrophobic bilayer. The external surface of the membrane may be modified to display surface moieties such as oligopeptides and proteins to promote uptake by or confer adhesion to target cells.
We propose that a comprehensive understanding of engineering principles underlying assembly/disassembly, physicochemical interactions, and bio-functionalization of polymersomes would greatly serve to expedite the translation of polymersomes into clinical settings. To this end, this review article provides an overview of the technologies developed (Figure 1) to regulate the size (Section 2), structure (Section 3), shape (Section 4), surface activity of polymersomes (Section 5), and responsiveness to environment (Section 6). In each section, we will discuss the extent that structure and properties of the polymersomes modulate physicochemical interactions and subsequent biological transports and engraftment to target tissues. Also, with polymersomes loaded with bioimaging contrast agents or drug molecules, we will discuss the extent to which structure and properties of polymersomes modulate the contrast-to-noise level of diagnostic images and therapeutic efficacy of drug cargos. Both in vitro and in vivo studies used to evaluate functions of polymersomes will be examined carefully to propose future direction of polymersome design. Overall, this review article serves to expedite the usage of polymersomes in clinical settings.
Figure 1.

Overview of the design criteria of polymersomes for biomedical applications. These include the size, membrane structure, shape, surface activity of polymersomes and their responsiveness towards internal and external stimuli.
2. Controlling polymersome size
The microvascular system of most normal organs is non-sinusoidal that may be either fenestrated or non-fenestrated. Fenestrated blood capillaries are endothelium with pore sizes between 6–12 nm.[6] This excludes particles above 12 nm in diameter. In contrast, blood vessels in the tumor are typically leaky and torturous, where the distance between endothelial cells of the tumor vasculature ranges from 380 nm to 780 nm. Additionally, tumor site is characterized with poor lymphatic drainage. Thus, polymersomes smaller than the gap between endothelial cells of the tumor vasculature are able to extravasate tumor vasculature and accumulate in target tumor, often called the enhanced permeability and retention (EPR) effect.[7,8] However, polymersomes smaller than gap between endothelial cells of fenestrated blood vessels encounter filtration through the kidney and accumulation in the liver.[9,10]
Also, polymersome size plays an important role in the intracellular uptake.[11] Given that many of the drug targets are localized in the subcellular compartments, an efficient translocation of the particles across the cell membrane is essential to attain desired therapeutic outcomes.[12] Endocytosis provides a means of cellular uptake of polymersomes that cannot enter the cells by passive diffusion.[13,14] Polymersomes initiate endocytosis by forming contacts with cell surface receptors. The receptor-ligand binding signals the cellular membrane to wrap around the polymersomes that leads to polymersome internalization. Note that 50-60 nm is the optimal diameter for spherical nanoparticles to facilitate cellular uptake.[14]
The size of polymersomes depends on the packing of self-assembling amphiphilic polymer chains. The self-assembly is driven primarily by hydrogen bonds between polymers and surrounding water molecules.[15] As a consequence, hydrophobic units aggregate to form a bi-layered structure. In the preparation process, amphiphilic copolymers are first dissolved in a water-miscible organic solvent. Then, water molecules successively added into the polymer solution would form hydrogen bonds with hydrophilic units of polymers, thus driving aggregation between hydrophobic blocks. By doing so, polymers form an ice-like cage structure surrounding the hydrophobic molecules.[15,16]
The total polymer concentration may influence the diameter of polymersomes. For example, the diameter of unilamellar polymersomes increased from 100 to 400 nm when the total polymer concentration was increased by 100-folds.[17] This is likely because the increased number of polymer chains in the same volume resulted in the emergence of polymersomes of larger surface area and, subsequently, the diameter.
The resulting polymersome size can be controlled with the mixing rate (Section 2.1), filtration (Section 2.2), extrusion/sonication (Section 2.3), and polymer chemistry (Section 2.4), as discussed below.
2.1. Role of mixing rate on polymersome size
By altering the mixing rate of water and polymer solution in a micromixer, it is possible to control the size of polymersomes over a broad range. For example, diameters of spherical poly(butadiene)-block-poly(ethylene oxide) polymersomes can be adjusted between 45 nm and 100 nm by varying the polymer concentration and the mixing rate.[18,19] During the mixing of the water and polymer solution, polymer molecules in the organic phase diffuse into the water phase. As such, increasing the mixing rate (total flow rate of 10 mL/min) drives the polymers to diffuse into the water phase quickly and self-assemble into small polymersomes (50 nm in diameter). Conversely, decreasing the mixing rate (total flow rate of 1 mL/min) makes the polymer molecules diffuse into water phase and self-assemble slowly, thus leading to large polymersomes (100 nm in diameter).
However, most approaches used to control the mixing rate macroscopically often result in polymersomes with broad size distribution due to the difficulty in controlling the molecular diffusion rate uniformly. To reduce the polydispersity of particles, purification techniques such as differential centrifugation and size exclusion chromatography are being used to separate polymersomes into distinctive sub-populations of less than 10 nm difference in diameter.[20] Besides, various microfluidic extrusion processes, which have emerged to prepare uniformly sized colloidal particles including liposomes, may be also useful to produce mono-sized polymersomes.[21]
2.2. Filtration for polymersome size control
Polymersome diameters can be controlled with cross-flow filtration followed by differential centrifugation. Filtration separates a mixed population of polymersome by passing the polymersome suspension through a semipermeable membrane with a specific pore size. This excludes polymersomes with larger diameters while allowing those with smaller diameters to pass through the membrane. In the case where the polymersome suspension is passed in the direction through the pores such as in ultrafiltration, the separation is limited by the amount of flow through before the pores are blocked by the larger particles. To minimize the formation of a filter cake that is formed by the accumulation of excluded particles, cross-flow filtration is performed by passing the polymersome suspension flow tangential to the pores.[20]
Differential centrifugation is used to separate a mixed population of polymersomes by applying centrifugal force to sediment larger polymersomes as a pellet. Smaller polymersomes which do not sendiment under the applied centrifugal force are collected in the supernatant. Higher centrifugal force sediments the smaller polymersomes. Thus, different size populations may be obtained by step-wise increments in the applied centrifugal force. However, this method is not effective for separating polymersomes of different shapes.[20]
These techniques were used to prepare poly(2-(methacryloyloxy)ethyl phosphorylcholine)-block-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) polymersomes with six different diameters of 22, 43, 97, 161, 190 and 240 nm.[22] The resulting polymersomes were used to study the effect of polymersome diameter on internalization into neutrophils and further to control inflammation.
According to studies with fluorescently labeled polymersomes, the amount of polymer internalized by neutrophils escalated with increasing polymersome diameters up to 190 nm. The internalization of these PMPC-b-PDPA polymersomes was increased by the class B scavenger receptors, scavenger receptors B1 and B2 and CD36 which have high affinities towards the phosphorylcholine group on the PMPC block.[23] The polymersomes were loaded with (R)-roscovitine and used to treat wounds introduced into zebrafish. These polymersomes reduced the number of neutrophils at the site of injury more effectively than free (R)-roscovitine.[22]
In contrast, effect of particle size on cellular uptake was not marked with a human epithelial cell line due to the low expression of receptors.[22] This selectivity in polymersome internalization is important to prevent the off-target effects of the cargo molecules.
2.3. Extrusion/sonication for polymersome size control
Polymersomes could be resized through shear forces arising from extrusion, where the diameters of poly(ethylene glycol)-block-polystyrene (PEG-b-PS) polymersomes decreased from 400-500 nm to < 100 nm with uniform size distribution. Cryo-transmission electron microscope (TEM) images confirmed no significant changes in the thickness of polymersome membrane before and after extrusion.
Alternatively, polymersome size can be reduced through sonication where sound energy is applied to break the particles into smaller ones. The particle diameters controlled through this process depend on the bilayered membrane rigidity, which is affected by the water content of the PEG-b-PS polymersomes. When the water content was 33.3%, polymersomes with an average diameter of 450 nm were resized to smaller ones (< 100 nm) after 30 s of sonication. On the other hand, when the water content was doubled, even longer sonication time of 15 min did not induce considerable size reduction.[24]
The effect of polymersome diameter on biodistribution was examined by using polymersomes of which diameters were controlled by extrusion or sonication. In this study, polymersomes were prepared with the polybutadiene-block-poly(ethylene glycol) with well-defined diameters between 90 and 250 nm.[11] Polymersomes were formed by slowly diluting the polymer dissolved in tetrahydrofuran with 2-(N-morpholino)ethanesulfonic acid buffer with a pH of 5.5. Then, the samples were extruded through 0.2 μm syringe filters to yield polymersomes of around 250 nm diameter. These polymersomes were extruded through 100 nm filters to yield 120 nm-diameter polymersomes. Alternatively, polymersomes were treated with ultrasonic sound waves at 35 °C to yield 90 nm-diameter polymersomes. Extrusion produces particles with narrow size distribution, while ultrasound waves results in particles with broad size distributions.
These polymersomes with controlled diameters were labeled with surface chelating radioactive 111In to use it as a diagnostic probe for single-photon-emission computed tomography (SPECT) combined with computed tomography (CT). SPECT/CT has been used in clinical diagnosis, specifically in cardiology, neurology and oncology to provide three-dimensional images of radiotracer distribution.[25,26] The 111In-labeled polymersome suspension was then injected via the tail vein of male BALB/c mice to study the biodistribution of the polymersomes after 4 h and 24 h post-injection.[11] Most polymersomes with diameters larger than 120 nm were cleared from the blood within 4 h. In contrast, smaller polymersomes with an average diameter around 90 nm presented the longer circulation time. Also, polymersomes of diameter 90 nm circulated through the liver, lungs, and carotid artery while polymersomes of diameter 20 nm accumulated in the liver and the spleen quickly.[11]
2.4. Chemical modifications of amphiphilic polymers for size control
Alternatively, polymersome size can be controlled by the polymer structure such as the copolymer length and volume fraction of the hydrophilic block. For example, in the case of poly(ethylene glycol)-block-polystyrene (PEG-b-PS), the diameter of the polymersomes was increased from 400 nm to 500 nm as the length of the polystyrene block was increased from 160 to 271 monomer units.[24] The volume fraction of hydrophilic block in the entire copolymer (f) and the molecular weight (Mn) of polymers also modulate the polymersome diameter. For instance, increasing the volume fraction of the poly(ethylene oxide) block and molecular weight of poly(ethylene oxide)-block-polybutadiene led to large polymersomes (Figure 2).[27] As the poly(ethylene oxide) expanded to maximize contact with water, polymersomes with higher poly(ethylene oxide) block volume fraction exhibited larger hydrodynamic diameters. Separately, the longer chains in higher molecular weight polymers formed thicker bilayers that led to an overall increase in diameter.
Figure 2.

Effects of polymer molecular weight (Mn) and volume fraction of hydrophilic block in the entire copolymer (f) on the polymersome diameter (dP). These visualization maps were obtained with the dynamic light scattering (DLS) (a) and freeze-fracture transmission electron microscopy (FF-TEM) (b). Mean polymersome diameter is indicated as full circles (blue and red/orange for DLS and FF-TEM respectively) and the concentric dotted circles indicate mean ± standard deviation. If mean diameter < 400 nm in (a), the circle color is changed to bright blue in and if mean diameter < 400 nm in (b), the circle color is change to bright red. Three regions are indicated: a region with mixed high/medium/low dP values (labeled A), a region with medium/high dP values (labeled B) and a region with medium/low dP values (labeled C). Reproduced with permission.[27]
3. Controlling physicochemical properties of polymersome membrane
The physicochemical properties of the polymersome membrane such as surface charge and permeability influence the rate of cellular internalization and spatial orientation of molecular cargos including magnetic resonance imaging (MRI) contrast agents, fluorescent dyes, and drug molecules. As such, physicochemical properties of the polymersome membrane are engineered to increase the diagnostic and therapeutic efficacy of molecular cargos.
3.1. Controlling surface charge of polymersomes
Surface charge of polymersomes modulates the interaction with the negatively charged mammalian cell membrane (i.e., zeta potential ~ −20 mV).[28] Asymmetrically charged polymersome membranes have been proposed to confer a better control of the surface charge for enhanced endocytosis and endosomal escape.
Asymmetric polymersomes are produced by using triblock polymers consisting of two chemically-different hydrophilic blocks flanking a hydrophobic block. For instance, membrane symmetry of polymersomes prepared from poly(ethylene oxide)-block-poly(caprolactone)-block-poly(acrylic acid) was tailored by varying the volume fraction of poly(acrylic acid) relative to poly(ethylene oxide).[29] Symmetrical polymersomes underwent slow cellular endocytosis due to the repulsive electrostatic interactions. On the other hand, asymmetrical polymersomes demonstrated faster endocytosis than the symmetrical ones. Within 5 minutes, approximately 95 % of HeLa cells, a human cervical cancer cell line, took up asymmetrical polymersomes, while 80 % of cells took up symmetrical polymersomes.
In a similar context, positive charges of polymersomes affect the entry of polymersomes into human dermal fibroblasts, depending on symmetry of block copolymers used to assemble polymersomes (Figure 3).[30] Highly charged polymersomes (e.g., zeta potential ~ 30 mV) were assembled with a block copolymer of poly(ethylene oxide), poly(2-(diisopropylamino)ethyl methacrylate and poly(2-(dimethylamino)ethyl methacrylate). Cells took up these particles rapidly within 1 h. This rapid endocytosis was attributed to the binding of polymersomes to proteoglycan heparin sulfates on the cell membrane. However, this rapid uptake of polymersomes often resulted in undesired cell death within several hours of exposure. However, increasing the volume fraction of poly(ethylene oxide) in the tri-block copolymer made the poly(ethylene oxide) blocks protrude out of the polymersome surface. The resulting polymersomes conferred increased steric repulsive interactions with proteins and cell membranes. As a result, cells took up these polymersomes at a much slower rate, thus reducing toxicity of polymersomes to cells.
Figure 3.

Asymmetrical membranes controlling polymersome surface charge density. TEM images of the polymersomes formed by the following block copolymers: (A) E23-P56-M21; (B) E45-P50-M14; and (C)E113-P84-M13. The white, black and grey chains represent the M block, poly(2-(dimethylamino)ethyl methacrylate); P block, poly(2-(diisopropylamino)ethyl methacrylate); and E block, poly(ethylene oxide), respectively. Reproduced with permission [30].
Also, negatively-charged polymersomes that mimic surface charge of red blood cells were prepared using poly(acrylic acid)-block-poly(1,4-butadiene).[31] The resulting, negatively charged polymersomes displayed significantly different biodistribution from neutral poly(ethylene glycol)-block-poly(1,2-butadiene). Neutral polymersomes accumulated primarily in the liver and spleen while negatively-charged polymersomes accumulated largely in the liver after 24 h post-intravenous injection.
Amphiphilic polymers with positively-charged ammonium moieties have been proposed to be an alternative to antibiotics. The strong electrostatic interaction with the negatively-charged walls leads to the insertion of the hydrophobic part of the polymer into the lipid membranes to lyse the microbes.[32–34] The possibility to develop resistance from this membrane disruption mechanism is lower compared to conventional antibiotics.[33,34] To increase the charge density for more effective interaction with the membranes, polymersomes were made with poly(ethylene oxide)-block-poly(2-diethylaminoethyl methacrylate)-block-poly[(2-tert-butylamino)ethyl methacrylate] triblock copolymer.[35] Polymersomes formed with the polymer with 10 more units of secondary amines per chain were more effective towards killing Escherichia coli.
3.2. Controlling permeability of polymersomes
The polymersome membrane permeability has a large impact on the movement of molecules across the polymersome membrane.[36] The membrane permeability is primarily controlled by the polymer forming the hydrophobic block which affects the fluidity and thickness of the polymersome membrane.
The fluidity of the polymersome membrane may be predicted based on the glass transition temperature of the polymers. For example, the glass transition temperatures for bulk poly(1,2-butadiene) and polystyrene are −20 °C and 100 °C, respectively. At room temperature, the rate of movement of 1-butylimidazole, through glassy membranes made out of polystyrene was three to four times slower than through the rubbery polybutadiene membranes. The rate of movement was not a strong function of temperature for temperatures lower than the glass transition temperature. In contrast, at temperatures above the glass transition temperature of the membrane, the rate of movement increased by about three times.[37]
Relative hydrophilicity of the polymers also affects the ability of molecules to move across the membrane. Polymersome membranes formed with poly(butylene oxide) were almost one order of magnitude more permeable than those formed with phospholipids. This is due to the more hydrophilic nature of the polyether membrane compared to the aliphatic lipid membranes.[38] In contrast, using polymers consisting of 2-methyl-2-oxazoline and dimethylsiloxane to form the membrane, the diffusion coefficients obtained with polymersomes were one order lower than those of liposomes.[39]
The membrane thickness can also affect the rate of movement of molecules. This has been observed in polymersomes made from poly(ethylene oxide) block copolymers with poly(1,2-butadiene),[40] poly(2-vinylpyridine),[41] and poly(butylene oxide) as the hydrophobic block.[38] In each case, the membrane thickness (d) was controlled by increasing the molecular weight of the hydrophobic block (Mhydrophobic) following the scaling of d~Mhydrophobic 0.5.[42] In turn, the membrane permeability is dependent on the membrane thickness according to the Fick’s first law. Using diblock or triblock copolymer of 2-methyl-2-oxazoline and dimethylsiloxane, the diffusion coefficient of polymersome membranes were found to decrease with increasing molecular weight of the copolymer.[39]
Asymmetry in the permeability of the polymersome membrane could be used to enable directionality in the diffusion of solutes out of the polymersome. To form polymersomes with two separate domains of different permeability, two different copolymers were mixed with a molar ratio that controlled the topology of the polymersome membrane.[43] Poly[oligo(ethylene glycol) methyl methacrylate]-block-poly[2-(diisopropylamino)ethyl methacrylate] (POEGMA-b-PDPA) was mixed with poly(ethylene oxide)-block-poly(butylene oxide) (PEO-b-PBO) at an optimal 9:1 molar ratio. This resulted in a spherical POEGMA-b-PDPA polymersome of 100 nm in diameter and membrane thickness of 6.4 nm with a small and more permeable PEO-b-PBO bud of 30 nm in diameter and membrane thickness of 2.4 nm. Enzymes glucose oxidase and catalase were loaded in the polymersome by electroporation to catalyze the sequential production of hydrogen peroxide from glucose oxidation and production of water and oxygen from hydrogen peroxide decomposition, respectively. The reaction in the confined volume produced an overall flux of products that were expelled out of the polymersomes from the more permeable PEO-b-PBO bud. This propelled the polymersomes in the direction of a glucose gradient across the blood brain barrier in rats. The active propulsion delivered 20% of the injected polymersomes into the parenchyma in comparison to only 5% of the injected polymersomes for polymersomes without encapsulated enzymes.
The permeability of the membrane is also dependent on both the solubility and diffusivity of the cargo molecule in the corona formed by the hydrophobic block of the polymer. Similarly sized molecules have the similar diffusivity. However, charged molecules have a higher partitioning difference than neutral molecules because they experience a larger energy barrier to dissolve in the hydrophobic membrane.[44] The rate of movement through the poly(ethylene glycol)-block-poly(1,2-butadiene) membrane for neutral 1-butylimidazole were approximately 10 times faster than that of charged 1-ethyl-3-methylimidazolium.[45]
The dependence of size of the diffusing molecule through poly(ethylene oxide)-block-poly(2-vinylpyridine) was demonstrated with polyethylene oxide molecules of different molecular weights. Increasing the hydrodynamic diameter reduced the rate of movement of polyethylene oxide through the membrane with a linear relationship for molecules with molecular weight less than 3400 g/mol. This suggested that the diffusing polyethylene glycol molecules induced the size-dependent disruption of the intermolecular interactions between the block copolymer molecules. The lower dependence of hydrodynamic diameter on the rate of movement through the membrane suggested that the diffusing polyethylene glycol molecules undergo a conformational change before passing through the membrane.[41]
The polymersome membrane permeability to small molecules may be controlled in situ by installing ionizable functional groups in the hydrophobic block. One example is demonstrated with block copolymers of poly(ethylene glycol) and polymers of piperidine-functionalized methacrylate.[46,47] The pKa of the block copolymer was determined to be 6.95. Thus, when the pH of the media was decreased from 7.4 to 6.8, piperidine groups in the hydrophobic block switched from neutral to positively-charged. The polymersome served to protect the encapsulated glucose oxidase while allowing glucose and oxygen molecules to enter only when the polymersomes reached the acidic tumor microenvironment. Glucose oxidase catalyzes the conversion of glucose and oxygen to hydrogen peroxide. The increase in permeability of polymersomes was demonstrated by detecting the production of hydrogen peroxide when the polymersomes were placed in a glucose solution of pH 6.8.
The increased hydrophilicity which led to increased permeability was also demonstrated in tumor-bearing mice.[46,47] After an intravenous injection of the polymersomes, higher levels of hydrogen peroxide were detected in the tumor where the pH is expected to be about 6.8. In contrast, the hydrogen peroxide levels did not change in the liver and blood where the pH is 7.4. Hydrogen peroxide then cleaved the drugs that were conjugated onto the polymer. This resulted in specific drug release in the tumor and hence lower systemic toxicity.
3.3. Role of polymersome structure on magnetic relaxivity of contrast agents
Magnetic resonance imaging systems are widely being used for clinical diagnosis. Contrasts of magnetic resonance images can be enhanced by contrast agents that serve to increase the proton spin relaxation rates of the water protons in target tissues. For example, gadolinium effectively increases the relaxivity by interacting with water protons through a metal coordination bond. However, the enhancement effect is masked when the gadolinium is unable to interact with water molecules in the tissue environment. In particular, gadolinium loaded into liposomes exhibited 62% lower longitudinal relaxivity per gadolinium than free chelated gadolinium. This is because water diffuses slowly across the lipid bilayers.[48,49]
To overcome the slow water transport across the membrane bilayer, polymersomes with porous membranes were prepared to increase the flux of water across the bilayer.[50][51] The polymersomes were first assembled with poly(ethylene oxide)-block-poly(butadiene) and nonpolymerizable phospholipids (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine.[50] The copolymers were cross-linked by using a chemical initiator and phospholipids were subsequently extracted with a surfactant, thus generating a highly porous outer membrane. The encapsulated gadolinium chelates were attached to dendrimers to prevent their leakage through the pores. The R1 relaxivity per gadolinium for the porous polymersomes was 7.2 mM−1s−1, which is 2.3-fold higher than that of non-porous polymersomes. Due to the large number of gadolinium loaded within a single polymersome core, the paramagnetic porous polymersomes exhibited an amplification of R1 relaxivity by a factor of 100,000 compared to free Gd-DTPA.
Alternatively, porous polymersome membranes were prepared by assembling polymersomes from a mixture of polymersome-forming polymer and an acid degradable polymer, for example, polycaprolactone.[51] The permeability of the membrane was controlled by the number of pores, which could be tuned by controlling the molar ratio of the polycaprolactone in the polymer mixture. Loading 40,000 gadolinium into a single polymersome with increased membrane permeability led to the signal enhancement in the T1-weighted magnetic resonance images of the kidneys and bladder 2 h and 4 h post-injection, respectively (Figure 4).
Figure 4.
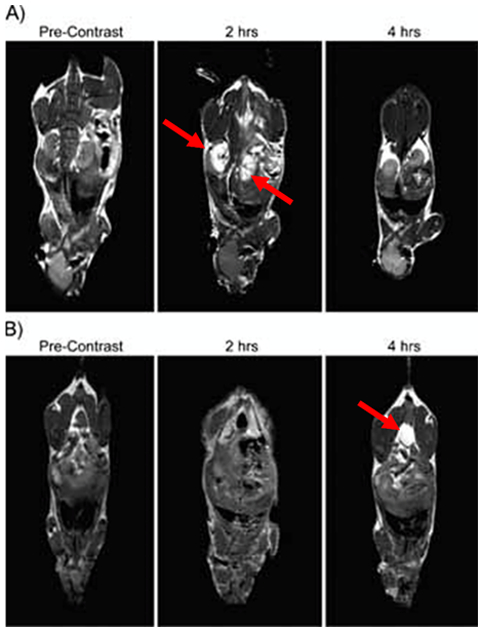
T1 signal magnetic resonance images of C57BL/6 mice at various time points following the intravenous injection of gadolinium-encapsulated porous polymersomes. The local hyperintensity generated by the polymersomes was visualized using a 4.7 T small animal MR. Images of the A) kidney and B) bladder were acquired before injection, and at 2 h and 4 h post-injection, as indicated by the red arrows. Adapted with permission.[51]
The porosity in polymersomes also affected biodistribution of gadolinium.[51] Porous and non-porous polymersomes increased the half-life of gadolinium in circulation to 3.5 h and 15 h, respectively, compared to half-life of free gadolinium chelates which is less than 30 min. The shorter half-life of the porous polymersomes than the non-porous polymersomes were attributed to the different destabilization rate of polymersomes in circulation. This increased circulation time would be beneficial for imaging of targeted organs. However, the slow clearance of gadolinium may result in liver toxicity and, in turn, would be an obstacle towards clinical use.[52]
Another strategy to spatially control the gadolinium in the polymersome is to immobilize the chelated gadolinium on the surface. While this has yet to be demonstrated more systematically, this surface immobilization would decrease the distance between the gadolinium ions and the surrounding water. An additional advantage stemming from the attachment of chelated gadolinium to the polymersome surface is the enhancement in the longitudinal relaxivity per gadolinium by slowing the rotational correlation time, compared with free chelated gadolinium in aqueous media.[53]
The importance of spatial arrangement of contrast agents in the magnetic resonance image contrast was also demonstrated using superparamagnetic iron oxide nanoparticles (SPIONs) loaded in polymersomes.[54,55] Radial arrangement of the SPIONs at the interface between the polymer core and shell provided better water accessibility and hence significantly higher relaxivity rate than when homogenously incorporated into the core of micelles.[54] Poly(acrylic acid)-block-polystyrene polymersomes loaded with 35.8 wt% SPIONs exhibited higher R2 magnetic relaxivity values of 228 ± 4 s−1 mM−1 than the clinically approved formulation Ferucarbotran (Resovist) (R2 186 s−1 mM−1).[56]
The interaction between the polymer chains in the bi-layered polymersome membrane and SPIONs influences the polymersome size and the SPION loading.[55] The formation of polymersomes was favored with the addition SPIONs by reducing the entropy penalty. SPIONs with a diameter of 5.8 nm were dispersed in the bilayer of larger polymersomes with diameters of 513 nm. In contrast, SPIONs with a diameter of 16.3 nm formed a well-ordered superstructure at the interface between the inner and outer layer of the polymersomes bilayer membrane. This spatial organization decreased the polymersome diameter to 257 nm and resulted in the R2 relaxivity of 555 s−1 mM−1.
3.4. Controlling polymersome structure for fluorescence imaging
Fluorescent imaging has been important tools in small animal imaging for cell migration and biodistribution studies. Polymersomes encapsulating near-infrared fluorophores, primarily porphyrin-based fluorophores, seek to overcome limitations such as short half-life in circulation, fast photo-bleaching, broad spectra of emission and poor tissue penetration. Bioresorbable poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) and poly(ethylene oxide)-block-poly(γ-methyl-ε-caprolactone) (PEO-b-PMCL) were used in this study. The intramembraneous polymer-fluorophore physicochemical interactions in these polymersomes profoundly influenced the bulk optical properties of near-infrared-emissive polymersomes (Figure 5A).[57,58]
Figure 5.
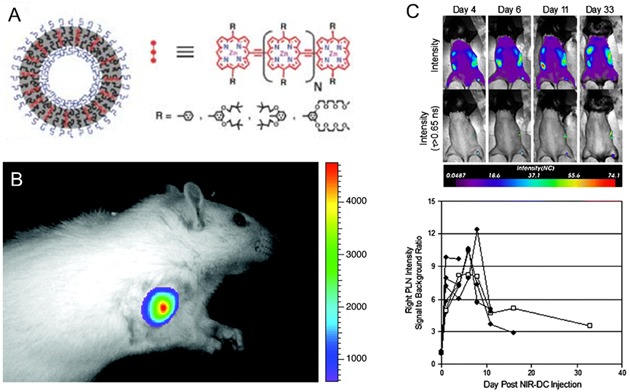
Schematic depiction of the incorporation of various oligo(porphyrin)-based near infrared fluorophores within polymersomes. A. The near infrared fluorophores vary with the number of porphyrin subunits (N), the linkage topology between porphyrin monomers, and the nature and position of ancillary aryl group substituents (R). Reproduced with permission.[58] B. In vivo fluorescence image of 300 nm-sized NIR-emissive polymersomes taken 10 min after direct tumor injection of a 9L glioma-bearing rat. The intensity remained constant between successive images taken during a 20-min interval post-injection. Adapted with permission.[59] Copyright 2005, National Academy of Sciences. C. In vivo longitudinal tracking of NIR-dendritic cells migrating to the popliteal lymph node. Mice were scanned on days 1, 4, 6, 8, 11, 16, and 33 after a single subcutaneous injection of 105 NIR-dendritic cells into the right footpad. Representative intensity maps (top row) and corresponding lifetime-gated intensity maps (bottom row) for a single mouse are presented for days 4, 6, 11, and 33. The measured signal-to-background ratio for right popliteal lymph node intensity is shown versus day. Each trace (n = 6) represents a different mouse and terminates on the day each animal was sacrificed. Trace with open squares is quantification of the images appearing above. Reproduced with permission.[60] Copyright 2009, Springer.
The near infrared-emissive polymersomes were optimized via the collective tuning of several parameters. Specifically, the fluorophore structures need to exhibit large near infrared extinction coefficients and fluorescence quantum yields. The average distance of fluorophores modulates fluorophore emission and loading efficiency of fluorophores, and in turn, signal sensitivity.[57]
As such, polymersomes encapsulating a series of ethynyl-bridged oligo(porphinato)zinc(II)-based supermolecular fluorophores enforced a large barrier to rotational diffusion which otherwise drives polarization loss.[61] The effective dispersion volumes of the fluorophore is controlled by the bilayer thickness which inversely affects the effective fluorophore concentration. Importantly, the fluorescence quantum yield can be modulated through variation of the poly(ethylene oxide)-block-poly(1,2-butadiene) (PEO-b-PBD). The polymer structure influences the hydrophobic bilayer thickness. For instance, polymersomes of PEO30-b-PBD46 and PEO80-b-PBD125 presented a bilayer thickness of 9.6 nm and 14.8 nm, respectively. Loading fluorophores in the thicker hydrophobic bilayer led to the higher excited-state deactivation rate constant for fluorophores.
Also, poly(ethylene oxide)-block-poly(1,2-butadiene) were used to assemble polymersomes that leads to cooperative self-assembly between polymers and multi(porphyrin)-based near infrared fluorophores.[59] The maximal emission intensity was achieved with 100 nm-diameter polymersome that carried 2,500 copies of near infrared-emitting fluorophores and an effective fluorophore concentration of 3 mM. The polymersomes presented a high photobleaching threshold level and, in turn, allowed the in vivo imaging of rat ectopic glioma with tissue penetration up to 1 cm. The high resolution 3D optical scanning also demonstrated a signal-to-background ratio of at least 10:1 (Figure 5B).
These poly(ethylene oxide)-block-poly(1,2-butadiene) polymersomes with porphyrin-based fluorophores were also used to track dendritic cells in vivo.[60] Murine dendritic cells internalized with these polymersomes were delivered either subcutaneously to the right footpad or intravenously via retro-orbital injection to mice. It was possible to track the transport of labeled dendritic cells to the popliteal lymph node for 33 days post-injection (Figure 5C).
4. Controlling polymersome shape
Shape of polymersomes also modulates the transport of polymersomes.[62–65] As individual amphiphilic copolymer chains self-assemble in an aqueous environment, the spatial organization of these molecules between the inner and outer membrane layers contributes to the final shape of the polymersomes. Being the most thermodynamically stable structure owning to the least surface area per unit volume and the lowest surface energy, spherical polymersomes are the most common.[66,67] The transition from spherical to non-spherical polymersomes such as ellipsoidal, tubular and stomatocytes (bowl-shaped) can be induced physically and/or chemically.
4.1. Ellipsoidal polymersomes
A main advantage of using ellipsoidal polymersomes is the enhanced binding efficacy with target cells as compared with their spherical counterparts. Coupling of hydrophilic poly(ethylene glycol) (PEG) chains to alkyl-substituted, poly(2-hydroxyethyl aspartamide) facilitated the formation of ellipsoidal polymersomes.[68] Such shape transition of the polymersomes was due to favorable changes in the bending modulus and spontaneous curvature of the polymeric bilayer. As determined from the autocorrelation function of the dynamic light scattering unit, these ellipsoidal polymersomes exhibited higher diffusivity than spherical vesicles in blood-mimicking plasma solution. A higher diffusivity would facilitate the transport of polymersomes in capillaries where the blood velocity is low.
When the end of PEG grafts was conjugated with cell adhesion peptides containing the integrin-binding Arg-Gly-Asp (RGD) sequence, the ellipsoidal polymersomes exhibited enhanced adhesion to cells overexpressing αvβ3 integrins than spherical ones under flow conditions. Such enhanced adhesion was attributed to a larger surface area and subsequently increased binding affinity when compared with spherical vesicles (Figure 6).
Figure 6.

In vitro analysis of the binding affinity of ellipsoidal polymersomes to the model target tissue in a flow chamber. a) The experimental setup of a flow chamber designed to evaluate binding affinity of polymersomes to bone marrow stromal cells sheets. The polymersomes encapsulated with fluorophores were added to media that flowed at a rate of 200 ml/h. b) Confocal microphotographs of bone marrow stromal cells exposed to fluorescent polymersomes modified with varying degree of substitution for poly(ethylene glycol) (DSPEG) and degree of substation for RGD peptides (DSRGD). (c) Quantitative analysis of the effects of DSPEG and DSRGD on the number of polymersomes adhered to bone marrow stromal cell sheets. Reproduced with permission.[68]
4.2. Tubular polymersomes
Similar to ellipsoidal polymersomes, tubular polymersomes offer a higher adhesion affinity than spherical counterparts due to increased surface area. The larger surface area is presents a more peptides or antibodies on the surfaces than the spherical ones.[69] As such, the tubular shaped polymersomes conjugated with cell adhesion peptides exhibit the increased adhesion to cell surfaces. Tubular polymersomes can be obtained using the film rehydration method where a thin film of amphiphilic copolymer is solvent-casted and placed in contact with an aqueous solution under stirring. For example, film rehydration of the poly(2-(methacryloyloxy)ethylphosphorylcholine)-block-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) resulted in the production of a mixture of spherical and tubular polymersomes.[65] The tubular polymersomes can be purified through centrifugation. Furthermore, the yield of tubular polymersomes can be modified with cholesterol and phosphocholine. For instance, phosphocholine promoted the formation of spherical polymersomes, while addition of cholesterol favored the formation of tubular polymersomes over time.
Separately, polymersomes can undergo a sphere-to-tubule transition through chemical modification of the polymeric bilayer. For example, poly(ethylene glycol)-block-poly(styrene-co-4-vinylbenzyl azide) polymersomes was modified through a strain-promoted alkyne-azide cycloaddition reaction between azide handles and a bicyclo[6.1.0]nonyne-cross-linker.[70] This process created a gradient of the bicyclo[6.1.0]nonyne over the polymersomes. The number of azides in proximity to the hydrophilic outer shell was higher than those attached to the inner leaflet of the polymersomes. Subsequently, a curvature of the bilayer was spontaneously driven by the asymmetry in the cross-link density of the membrane. With an increase in the total mass of the vesicle membrane and tension between the polymers, the spherical polymersomes were stretched in one dimension to form tubular polymersomes. This elongation process can be reversible by introducing a reducible disulfide bridge in the cross-linker. Interestingly, the tubular polymersomes reverted back to their original spherical shape after incubating them in the media dissolved with a reducing agent, tris(2-carboxyethyl)phosphine hydrochloride.
Tubular polymersomes were also prepared from the elongation of spherical polymersomes comprising biodegradable copolymer poly(ethylene glycol)-block-poly(D,L-lactide).[71] This shape transformation was controlled osmotically using dialysis against aqueous solutions with varying concentrations of sodium chloride. At salt concentrations of 10 and 50 mM, elongated nanotubes were mainly observed. Further increase of salt concentration to 100 mM led to the formation of elongated ribbons (i.e., collapsed nanotubes). Under hypotonic conditions, spherical polymersomes deflated into nanotubes and elongated ribbons with increasing salt concentration.
Dialysis temperature also influences the extent of shape transformation.[71] When the polymersomes were dialyzed at 4°C, shape transformation from spherical to tubular polymersomes was evident, and the tubular shape was maintained. In contrast, the polymersomes remained spherical at 25 °C. Elongated ribbons were observed at 30 °C. These polymersomes were functionalized with surface-bound biomolecules for biological applications. For instance, green fluorescent protein was tethered on the nanotubes through the clickable unnatural amino acid bicyclo[6.1.0]nonyne-L-lysine.
Despite possessing larger luminal volume, which increases the loading content of drug loads, tubular polymersomes display different internalization kinetics from spherical ones. One of the characteristics found in fibroblasts derived from patients with Parkinson’s disease is the presence of mutations in the parkin gene which results in mitochondrial dysfunction. Hydrophobic steroid-like compounds ursolic acid and ursocholanic acid are candidate drugs found to restore mitochondrial function in mitochondrial dysfunctional-fibroblasts derived from patients with mutations in the parkin gene. Therefore, in an effort to increase their transport to the brain, ursolic acid or ursocholanic acid were loaded into poly(2-(methacryloyloxy)ethyl phosphorylcholine)-block-poly(2-di-isopropylamino)ethyl methacrylate) (PMPC-b-PDPA) polymersomes.[72] Both spherical and tubular polymersomes were prepared. Although the tubular polymersomes contained more ursolic acid or ursocholanic acid, the parkin-mutant fibroblast took up more spherical polymersomes than tubular polymersomes. As a result, the tubular polymersomes were less efficacious than spherical polymersomes in increasing the cellular ATP levels following 24 h incubation with parkin-mutant fibroblast.
Similarly, according to the study with rhodamine-conjugated PMPC-b-PDPA polymersomes, neutrophils and FaDu human epithelial cells took up spherical polymersomes rapidly.[65] In contrast, the tubular-shaped polymersomes exhibited a two-phase internalization kinetics: an initial quick binding followed by a slow internalization. The slow internalization of tubular polymersomes was attributed to the increased energy requirement for endocytosis of particles.
4.3. Stomatocytes
Stomatocytes are polymersomes with a bowl-shaped structure. These polymersomes have been successfully transformed from spherical polymersomes comprising poly(ethylene glycol)-block-polystyrene.[73–75] The key factor to induce such transformation was the ratio between water and organic solvent used during preparation of the polymersomes. The copolymer was first dissolved in a mixture of tetrahydrofuran and dioxane. Water was then slowly introduced to the solution to form spherical polymersomes. Subsequent dialysis of the resulting polymersomes in an equimolar mixture of water and organic solvents induced the formation of stomatocytes. The entry of the organic solvent into the polymersome membrane increased the membrane permeability, which facilitated the outflow of water from the polymersomes. This water transport reduced the volume of the inner compartment, and in turn, led to inward folding of the membrane (Figure 7). In situ morphological transition from spherical to stomatocytes can be monitored using magnetic bifringence with diamagnetic polymersomes.[74]
Figure 7.
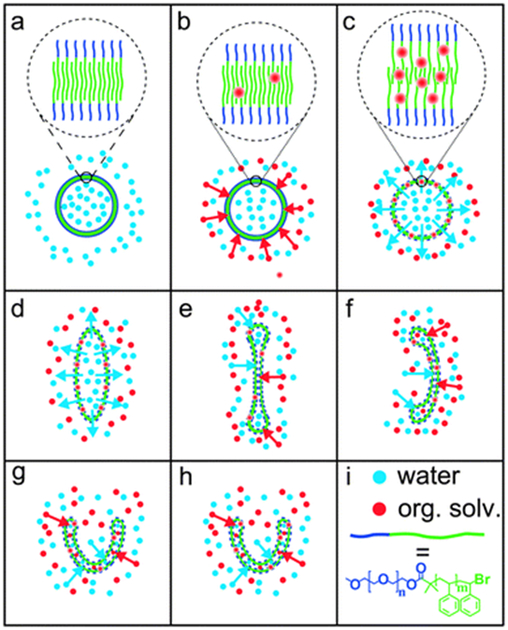
(a–h) Proposed mechanism of the dialysis of polymersomes against 50% water, 40% tetrahydrofuran and 10% 1,4-dioxane. In (a–c), the polymersome membrane is shown enlarged. The organic solvent acts as a plasticizer, which swells the membrane. Swollen membranes are indicated by dashed lines in figures (c–h) compared to the solid lines in figures (a) and (b). (i) Red and blue dots represent organic solvent and water, respectively. The block copolymer is drawn schematically as a short blue line (poly(ethylene glycol)) connected to a longer green line (polystyrene). Reproduced with permission.[74] Copyright 2014, The Royal Society of Chemistry.
The inner cavity of the poly(ethylene glycol)-block-polystyrene stomatocytes was utilized as a nanocontainer of platinum nanoparticles, which could be used as catalysts for the decomposition of hydrogen peroxide into oxygen and water.[76] The discharge of decomposed products induced thrust and, in turn, enabled autonomous movement of the polymersomes. Such design may be useful to deliver drug molecules through cell and tissue barriers.[77,78]
Biodegradable stomatocytes were also fabricated from a mixture of poly(ethylene glycol)-block-poly(ε-caprolactone) and poly(ethylene glycol)-block-polystyrene.[79] Under the presence of hydrogen peroxide, the introduction of poly(ε-caprolactone) in the system did not affect the velocity of the stomatocytes (i.e., 39 μm/s). Water-soluble doxorubicin was loaded into the lumen of the resulting polymersomes while the platinum nanoparticles were encapsulated in the inner cavity. A sustained release of doxorubicin at acidic pH 5.0 was observed because of the pores created in the stomatocytes due to acidic degradation of poly(ε-caprolactone) segments.
Overall, depending on the application, it is important to optimize the shape of the polymersomes in use. While spherical polymersomes are the most thermodynamically stable structure, their capability to cellular or tissue adhesion may not be as superior as non-spherical polymersomes. On the other hand, if a high cellular uptake is desired, spherical polymersomes, which require the least energy for endocytosis as compared to non-spherical ones of the same volume, may be more desirable.[80]
5. Surface activities of polymersomes
Capability of polymersomes to reach and adhere to target cells and tissue is an important parameter for an effective delivery of therapeutic and/or imaging probes. It is common to engineer the surface of polymersomes with homing ligands including metabolites, peptides, aptamers, and antibodies. These bioactive moieties should associate with their corresponding receptors that are overexpressed in the diseased organs, tissues or cells. Table 1 summarizes the ligands that have been used to functionalize polymersomes for targeted delivery. In this section, we will discuss the extent to which these ligands control delivery efficiency of polymersomes, and conversely, the extent that the polymersomes mediate bioactivity of these molecules.
Table 1.
Summary of ligands used to functionalize polymersomes for targeted delivery.
| Model | Target receptor | Ligand | Chemistry used for conjugation | Enhancement | Reference |
|---|---|---|---|---|---|
| Small molecule metabolites | |||||
| Breast cancer | Folate receptor | Folic acid | Coupling of aminated folic acid and PLGA-NHS | 1.7-fold higher uptake | [81] |
| Liver cancer | Folate receptor | Folic acid | Physical conjugation | 3.5-fold lower IC50 | [82] |
| Cervical cancer | Folate receptor | Folic acid | Amino group of copolymer and folic acid | Halved tumor volume | [83] |
| Bacteria | Concanavalin A | Glucose | Displacement of pentafluorophenol by β-D-glucosyloxyethylamine | 8-fold higher binding | [84] |
| Bacteria | Concanavalin A | Glucose | Atom transfer radical polymerization of methacrylated-glucose monomers | 60% increased concanavalin A binding | [85] |
| Peptides | |||||
| Blood brain barrier | Low-density protein receptor-related protein 1 | Angiopep-2 | Maleimide terminated polymer with cysteine-terminated peptide | 3 to 4-fold higher uptake in the brain and spinal cord | [86] |
| Blood brain barrier | GM1 and GT1b receptors | G23 | Maleimide functionalized polymer with cysteine bearing peptide | 4-fold increment in transcytosed polymersomes | [87] |
| Lung cancer | α3β1 integrins | cyclic peptide cNGQGEQc | N-hydroxysuccinimide-functionalized polymer with amino-functionalized peptide | 3-fold lower IC50 | [88] |
| Lung cancer | α3β1 integrins | cyclic peptide cNGQGEQc | N-hydroxysuccinimide-functionalized polymer with amino-functionalized peptide | Increased median survival time from 30 to 54 days | [89] |
| Dendritic cells | Cellular membrane | Tat | Succinimide-terminated polymer with amino-functionalized peptide | 2 h reduction in time to obtain half-maximal fluorescent intensity; 7-fold increase maximal fluorescent intensity |
[90] |
| Aptamers | |||||
| Lung cancer | Epithelial cell-adhesion molecule | 19-mer EpCAM RNA aptamer | Carboxylic acid-functionalized polymer with amine-terminated aptamer | Halved tumor volume | [91] |
| Protein ligands | |||||
| Blood brain barrier | Low-density lipoprotein receptor family | Lactoferrin | Maleimide-functionalized polymers with sulfhydrated-lactoferrin | 2-fold increase in brain permeability surface area product | [92] |
| Blood brain barrier | Transferrin receptor | Transferrin | Maleimide-functionalized polymers with sulfhydrated-transferrin | 2.3-fold increase in brain uptake | [93] |
| Antibodies | |||||
| Breast cancer | Human Epidermal Growth Factor Receptor-2 | Trastuzumab | Maleimide-functionalized polymer with thiol-derivatized trastuzumab | 2-fold increase in signal-to-noise ratio | [94] |
| Inflamed epithelial | Endothelial cell adhesion molecules ICAM-1 | Anti-ICAM1 | Neutravidin-coated polymersomes with biotinylated antibodies | 7.5-fold increase in binding rate to inflamed versus uninflamed cells | [95] |
| Blood brain barrier | Transferrin receptor | Transferrin receptor monoclonal antibodies | Maleimide-functionalized polymer with thiol-derivatized antibodies | 2.6-fold increase in accumulation in the brain | [96] |
5.1. Functionalization with small molecule-based metabolites
5.1.1. Folic acid-conjugated polymersomes
Folate receptor is commonly up-regulated in more than 40% of human cancers, including breast, and liver.[97–99] The expression of folate receptor is also related to the progress of cancer. To this end, dextran-block-poly(lactide-co-glycolide) was conjugated with folic acid. The resulting molecule was used to build polymersomes encapsulated with anticancer drug for breast cancer chemotherapy.[81] For instance, docetaxel was loaded in folic acid-conjugated polymersomes via the nanoprecipitation method. A mixture of docetaxel and the copolymer dissolved in an organic solvent was added drop-wise to deionized water to form the polymersomes. High resolution transmission electron microscopy demonstrated that these particles existed as spherical vesicles with a bilayer in which the hydrophobic docetaxel molecules were loaded.
In comparison to polymersomes without folic acid, folic acid-conjugated polymersomes achieved approximately 1.7-fold higher accumulation in breast cancer 4T1 and MCF-7 cell lines due to increased adhesion, cellular uptake, and reduced clearance from the tumor tissue. In particular, the higher cellular internalization was attributed to the folate receptor-mediated endocytosis. Using allograft models of 4T1 mice breast cancer adenocarcinoma, tumor volume of mice treated with a single intravenous injection of folic acid-conjugated polymersomes was half of that of mice with treated with folic acid-free polymersomes.
Targeted co-delivery of anticancer drugs has also been explored by using polymersomes. Triblock copolymer, poly(ε-caprolactone)-block-poly(ethylene glycol)-block-poly(ε-caprolactone), was used to encapsulate paclitaxel via the thin film rehydration method followed by loading of doxorubicin.[82] Folic acid was conjugated to the polymersomes for targeted delivery capability. The presence of folic acid did not affect the loading content of doxorubicin (~9.0 wt%) and paclitaxel (~3.5 wt%), and the drug release profile was comparable to that without folic acid. According to the study of intracellular distribution of polymersomes, a larger number of folic acid-conjugated polymersomes was found in the nuclei and cytoplasm of cells than polymersomes free of folic acids due to the increased receptor-mediated endocytosis. As such, the folic acid-conjugated polymersomes increased anti-tumor activity of loaded drug molecules, as confirmed with the decreased tumor volume in vivo.
Folic acid-conjugated polymersomes were also used to improve the capability to detect and image tumor.[83] For instance, poly (L-glutamic acid)-block-poly(ε-caprolactone) conjugated with folic acid self-assembled into polymersomes in aqueous solution. The negatively charged interior of coronas of the polymersomes were loaded with positively charged superparamagnetic iron oxide nanoparticles (SPIONs), a magnetic resonance imaging contrast agent. The hydrophilic form of doxorubicin was encapsulated into the core of the polymersomes. The resulting polymersomes demonstrated the negative contrast enhancement within the tumor of nude mice bearing the HeLa tumor. The polymersomes also resulted in a two-fold decrease in the tumor volume.
5.1.2. Glycosylated polymersomes
Many biological processes, including cellular adhesion, migration, inflammation, and virus docking, and cancer metastasis, are heavily dependent on carbohydrate-lectin interactions.[100–102] As one of the most common carbohydrates, glucose exhibits weak individual interactions with its lectin receptor, concanavalin A. However, polymers conjugated with multiple glucose units display strong binding through multi-valency.[103] As such, controlled numbers of glucose molecules were coupled to the surface of polymersomes.
In one example, polymers with activated ester monomer, pentafluorophenyl acrylate, were polymerized followed by chain extension with n-butyl acrylate via reversible addition-fragmentation chain-transfer polymerization (Figure 8).[84] The pentafluoroester was later replaced with the amine-functionalized glucose [p(NβGluEAM-b-BA)]. The polymersomes were formed using the electrofomation method where a film of the resulting glycosylated polymer was deposited onto indium tin oxide coated electrodes, followed by budding off from the film in a chamber of sucrose solution under an AC field. The extent of interactions between the Rhodamine B-labeled glycosylated polymersomes and fluorescein-labeled concanavalin A-functionalized polystyrene beads was investigated using confocal microscopy. These giant polymersomes demonstrated eight times higher binding affinity with concanavalin A-functionalized polystyrene beads than non-functionalized ones.
Figure 8.
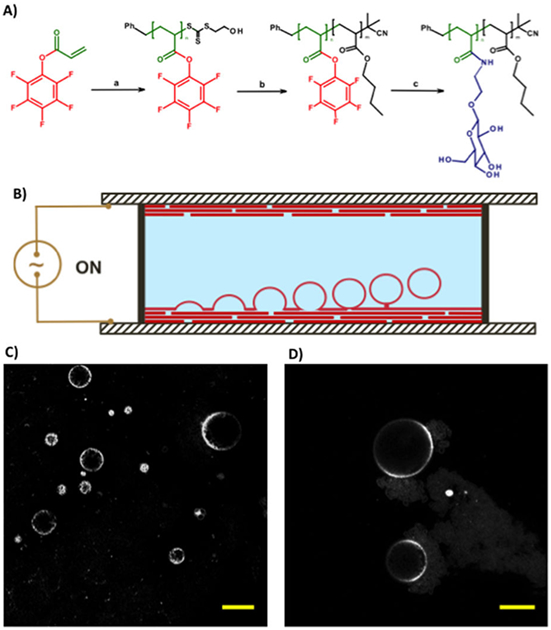
(A) Synthesis of amphiphilic glycopolymers by (a) reversible addition-fragmentation chain-transfer polymerization of pentafluorophenyl acrylate; (b) chain extension with n-butyl acrylate; and (c) displacement of pentafluorophenol by β-D-glucosyloxyethylamine. (B) Schematic of electroformation apparatus for the construction of the giant polymersomes. A polymer film is deposited onto indium tin oxide-coated glass slides, which are separated by a rubber O-ring. The chamber is filled with sucrose solution. A sinusoidal electric field is applied to form giant polymersomes form by budding off from the film on the conductive substrate. (C, D) Fluorescence microscopy images of glycosylated giant polymersomes stained with rhodamine B octadecyl ester perchlorate (scale bar: 20 μm). Reproduced with permission.[84] Copyright 2016, Nature Publishing Group.
Separately, block copolymers were synthesized by polymerizing methacrylated-glucose monomer followed by a sparingly water-soluble diethylene glycol methacrylate, via reversible addition-fragmentation chain-transfer (RAFT) polymerization of pentafluorophenyl acrylate (Figure 9).[85] The resulting polymersomes displayed more efficient surface binding with concanavalin A than a linear homopolymer of the methacrylated-glucose monomers. The increased binding affinity was likely due to the spatial accumulation of glucose molecules on the vesicular exterior, which increased multivalent capacity.
Figure 9.
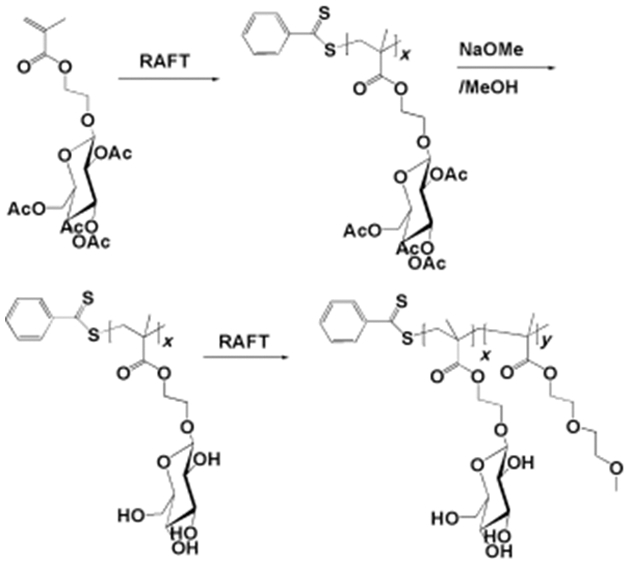
Synthesis of block copolymers of poly(2-glucosyloxyethyl methacrylate) and poly(diethyleneglycol methacrylate). x=28; y=36. Adapted with permission.[85]
Molecular transport by the polymersomes was investigated with bacteria expressing the fim H protein, which binds specifically to glucose and mannose. Within 30 minutes, ethidium bromide, a dye with an orange-red color, was detected in the bacteria, because cells attached with the polymersomes had taken up the dye (Figure 10).[85]
Figure 10.

Interaction of glycosylated polymersomes with E.coli bacterial cells. (a) polymersomes and cells in the phase contrast mode, (b) same cells in the fluorescence mode. Green and orange red color represent bacterial cells and polymersomes, respectively. Insets in (b) show vesicles at higher image contrast and ×2 magnification. Scale bars: 1 μm. Adapted with permission.[85]
5.2. Peptide-conjugated polymersomes
The central nervous system (CNS) houses a tight network of membrane barriers such as the blood brain barrier (BBB) to regulate specific molecular transport. Given the highly selective and specific regulation of the BBB, delivery of molecules of interests to target pathologic brain tissue is often challenging. To this end, enhanced delivery of therapeutic agents into brain tissues has been achieved with peptide-conjugated polymersomes via transcytosis across the BBB.
Several receptors involved in transcytosis are expressed on the endothelial cells of the BBB. These receptors include the low-density lipoprotein receptor-related protein 1, monosialotetrahexosylganglioside (GM1) and trisialganglioside (GT1b) receptors.[104,105] To target the lipoprotein receptor-related protein 1, polymers of poly[2-(methacryloyloxy)ethyl phosophcholine] were synthesized.[86] One end of polymer was capped with maleimide group, which was later reacted with cysteine-terminated Angiopep-2 peptides. This 19-amino acid long peptide binds with the lipoprotein receptor-related protein 1 and in turn, has a high brain penetration capability. Following intravenous injection of the resulting polymersomes in mice, pharmacokinetics analysis of the brain and spinal cord revealed a 3 to 4-fold increment in the concentration of peptide-functionalized polymersomes than non-functionalized ones.
Separately, polymersomes prepared with block copolymers of polybutadiene and poly(ethylene glycol) were coupled with G23 dodecamer peptides that recognized gangliosides GM1 and GT1b receptors in BBB.[87] According to the transwell assay conducted with human cerebral microvascular endothelial cells, polymersomes tagged with G23 peptides exhibited a 4-fold increment in the amount of transcytosed polymersomes as compared with unmodified counterparts. Following injection into the intracarotid artery of mice, the polymersomes conjugated with G23 peptides accumulated in the cortex, forebrain, midbrain, pons, and cerebellum. In contrast, unmodified polymersomes were only found in the leaky vessels of the fourth ventricle, ependymal cells of the aqueduct, and only occasionally in brain parenchyma.
In the case of human lung cancer, α3β1 integrins are up-regulated in A549 cancer cells. Cyclic peptides including NGQ sequence, denoted as NGQ peptides, are highly specific to these integrins.[106] Poly(ethylene glycol)-block-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) were functionalized with NGQ peptides via ring-opening copolymerization.[88] Doxorubicin was loaded into polymersomes by a pH-gradient method, where the polymer was first dissolved in citric acid of pH 4.0 followed by adjustment to pH 8.0 before adding doxorubicin.
Interestingly, dithiolane rings in the polymer backbone cross-linked spontaneously, which in turn, exhibited excellent colloidal stability in blood-mimicking cell culture media. Glutathione in the cytoplasm subsequently would de-crosslink thiol linkages to release molecular cargos such as doxorubicin. These polymersomes inhibited the growth of A549 lung cancer cells overexpressing α3β1 integrin by 3-fold higher than that of polymersomes without the NGQ peptides. In addition, the inhibitory effect of polymersomes with NGQ peptides was demonstrated in nude mice bearing subcutaneous A549 xenografts. According to histological analyses, tumor tissue of mice treated with polymersomes conjugated with NGQ peptides and loaded with doxorubicin became necrotic while insignificant damage was observed in liver, kidney and heart.
These NGQ peptides-conjugated polymersomes also mediated the delivery of Polo-like kinase1 specific silencing RNA (siRNA).[89] These polymersomes were formed from the co-self-assembly of asymmetric poly(ethylene glycol)-block-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate)-block-polyethylenimine tiblock copolymer. siRNA-loaded polymersomes were prepared via the solvent exchange method where dimethyl sulfoxide dissolved copolymer with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer containing siRNA were dialyzed HEPES. Since the polymersomes displayed a close to neutral surface charge, the siRNA was loaded into the aqueous lumen of the polymersomes. The peptide-functionalized polymersomes displayed better silencing activity than polymersomes without the peptides with 73.2% and 52.7% silencing, respectively. The in vivo studies using nude mice bearing A549 lung tumors revealed a long circulation duration, a high tumor accumulation and selectivity, and an effective suppression of tumor growth with the polymersomes loaded with siPLK1 and functionalized with NGQ peptides.
Polymersomes functionalized with cell permeable peptide, denoted as Tat, were also used to track dendritic cells in cancer therapy.[107] Derived from the trans-activating transcriptional activator (TAT) of human immunodeficiency virus, the Tat peptides are known for their efficacious internalization by numerous cell types in culture.[108] Polymersomes consisting of poly(ethylene glycol)-block-poly(1,4-butadiene) were fabricated via self-assembly with porphyrin-based near-infrared fluorophores.[90] A significant uptake of these polymersomes in dendritic cells was observed when Tat peptide was conjugated to the polymersomes.
In the absence of conjugated Tat peptides, the extent of uptake by dendritic cells varied among the cell populations. Consequently, polymersomes conjugated with Tat peptides displayed the half-maximal fluorescent intensity at 5h. In contrast, polymersomes without Tat peptides resulted in half-maximal fluorescent intensity at 7h. The fluorescent intensity was also decreased. The number of Tat-conjugated polymersomes taken up by a cell was estimated as 70,000, equivalent to 0.7 fmol of near infrared fluorophore.[90]
5.3. Aptamer-conjugated polymersomes
Epithelial cell-adhesion molecule (EpCAM) is highly expressed in non-small cell lung cancer (NSLCL).[109,110] The 19-mer EpCAM RNA aptamer was demonstrated to specifically bind to the extracellular domain of the epithelial cell adhesion molecule. As such, polymersomes composed of poly(ethylene glycol)-block-poly(lactide-co-glycolide) was used to load doxorubicin via thin film rehydration of the polymer followed by loading of the drugs.[91] The polymersomes, which contained carboxylic acid functional groups, were covalently linked to the EpCAM aptamer with terminal amine functional group via aqueous carbodiimide chemistry. According to electron microscopy images, EpCAM aptamers coupled to the polymersomes caused a 10 nm increase in the particle diameter. The presence of EpCAM aptamers increased the cellular uptake of doxorubicin-loaded polymersomes in SK-MES-1 and A549 lung cancer cells. As reflected in mice bearing SK-MES-1 xenografts, the tumor volume was almost halved within 15 days for mice treated with doxorubicin loaded aptamer-conjugated polymersomes.
Section 5.4. Protein ligand-conjugated polymersomes
Specialized brain capillary endothelial cells restricts the free exchange of most solutes between the plasma and the extracellular fluid in the brain. Only specific ligands such as lactoferrin and transferrin are able to cross the blood brain barrier via the low-density lipoprotein receptor family and transferrin receptor-mediated transcytosis, respectively.[111] Therefore, more efficient transport of drug molecules across the brain endothelium could be achieved with polymersomes displaying transferrin and lactoferrin proteins.
Humanin, a 24-amino acid neuroprotective peptide inhibits neuronal death by specifically-blocking Alzheimer’s disease-related apoptosis induced by amyloid-β. S14G-humanin is a more efficacious variant of humanin. However, its therapeutic application is restricted by its instability and poor blood brain barrier penetration. To form the polymersome carrier, poly(ethylene glycol)-block-poly(D,L-lactic-co-glycolic acid) with maleimide end group was conjugated with sulfhydrated-lactoferrin.[92] The conjugation of 101 lactoferrin molecules per polymersome increased the brain permeability surface area product of the polymersome by 2-folds. When the S14G-humanin-loaded polymersomes, polymersomes modified with lactoferrin improved the impaired learning and spatial memory in a dose-dependent pattern on rats impaired by amyloid-β25–35.
Besides lactoferrin, transferrin has also been conjugated to polymersomes to enhance the transport of biomolecules. Using Traut’s reagent, the transferrin protein was sulfhydrated and then conjugated onto maleimide-functionalized poly(ethylene glycol)-block-poly(caprolactone) polymersomes.[93] With 35 transferrin molecules per polymersome, the brain uptake for transferrin-conjugated polymersomes at 2 h increased by 2.3-fold when compared with polymersomes without transferrin-conjugated.
Polymersome-mediated delivery may be improved by conjugating more than one targeting ligand. Poly(ethylene glycol)-block-poly(D,L-lactic-co-glycolic acid) polymersomes were decorated with transferrin and Tet-1 peptides.[112] Tet-1 is a 12-amino acid peptide which has high affinity for GT1B receptors on neurons. These polymersomes were loaded with curcumin, a polyphenol which is found to promote the degradation of amyloid-β proteins in Alzheimer’s disease but is poorly soluble in water and poorly absorbed in the brain. The bioconjugation of both transferrin and Tet-1 improved the transport of the curcumin-loaded polymersomes by 2-3 times than polymersomes conjugated with only either one of them. Upon their release in the brain, curcumin molecules bound with amyloid-β proteins and prevent their pathological aggregation in intrahippocampal amyloid-β1–42-injected mice.
5.5. Antibody-conjugated polymersomes
Antibodies have been used as targeting ligands extensively. For instance, approximately 20% of breast cancer overexpresses the membrane tyrosine kinase human epidermal growth factor receptor-2 (HER2). This finding prompted the development of Trastuzumab, a FDA-approved monoclonal antibody targeting HER2.[113] To this end, polymersomes comprising a mixture of amphiphilic block copolymers: poly(trimethylene carbonate)-block-poly(glutamic acid) and the same copolymer end-capped with a maleimide group for conjugation with thiol-derivatized Trastuzumab were prepared. For imaging analysis, polymersomes were also coated with superparamagnetic iron oxide nanoparticles (SPIONs).[94] Following incubation of BT-474 breast cancer cells with the resulting polymersomes for 3.5 h, polymersomes functionalized with Trastuzumab demonstrated a clear targeting capability. The number of polymersomes internalized into the HER2-positive cancer cells was significantly higher than those without Trastuzumab. Concurrently, these polymersomes marked the cancer cells with negative contrast in the magnetic resonance image.
These polymersomes were further tested in a bone metastasis model where BT-474 breast cancer cells were injected directly in the femoral bone of mice. When polymersomes loaded with Trastuzumab and SPIONs were administered, tumor growth was observable inside the bone lumen, and outside of the bone at a later stage. Polymersomes without Trastuzumab, on the contrary, did not give a good contrast in the tumor area of either inside or outside the one. Such observation was attributed to the enhanced capability of polymersomes to reach and retain at the tumor. This, in turn, gave rise to a more persistent T2* contrast effect at the tumor site.
The inflammatory environment is characterized by the up-regulation of intracellular adhesion molecule-1 (ICAM-1) on the vascular endothelium, and hence represents a key therapeutic target in ischemia and inflammation.[114] As such, leukocytes in circulation are able to adhere to the inflamed endothelium because they present surface receptors binding ICAM-1. To impart the adhesive properties of leukocytes on polymersomes, the surfaces of polymersomes were coated with anti-ICAM-1 molecules.[95,115] Biocytin-coated polymersomes were first made by esterification of the hydroxyl terminal end of poly(ethylene glycol)-block-poly(1,2-butadiene) and 4-fluoro-3-nitrobenozic acid, followed by a nucleophilic substitution with biocytin (Figure 11).[95] The biocytin-polymersomes were coated with neutravidin, which binds to biotinylated anti-ICAM-1. With the ICAM-1-coated microspheres, studies reported that anti-ICAM-1-conjugated polymersomes present a larger binding affinity than the antibody-free polymersomes. The adhesion strength increased in proportion to the surface density of anti-ICAM-1 molecules.
Figure 11.

An esterification between the hydroxyl-terminated polymer poly(ethylene glycol)-block-poly(1,2-butadiene) (OB29-OH) and 4-fluoro-3-nitrobenzoic acid linker (A) followed by a nucleophilic aromatic substitution with biocytin to obtain the biotin-coated polymersomes (B). Reproduced with permission.[95]
Additionally, the molecular topology of the targeting ligands is an important feature affecting the adhesion of polymersomes. A diblock copolymer, poly(ethylene oxide)-block-poly(1,4-butadiene) modified with biotin yielded a membrane with a surface brush topology.[116] Biotin molecules impart specific adhesiveness while poly(ethylene oxide) imparts stability by prohibiting the approach of opsonins, lipoproteins and other polymersomes. The biotin, which protruded out of the poly(ethylene glycol) layer, facilitated the binding with the avidin. The extent of adhesiveness of polymersomes could be tuned by mixing polymers of different backbone chain lengths.[117]
Polymersomes were also used to regulate degranulation of leukocytes. Neutrophils activated in response to infection degranulate and expel destructive contents to surrounding area for sterilization and prevention of microorganism escape. Unfortunately, this degranulation often damages tissues. Thus, stimulating apoptosis of neutrophils is a potential therapeutic strategy to prevent excessive degranulation. Very few vectors have been found to enable the efficient intracellular delivery of cargos into neutrophils to kill these cells.
However, rapid internalization of poly(2-(methacryloyloxy)ethyl phosphorylcholine)-block-poly(2-(diisopropylamino)ethyl methacrylate) was observed with neutrophils which also express class B scavenger receptors and CD36.[22] These polymersomes were then used to target and reduce the number of neutrophils in an in vivo zebrafish wound injury model. The polymersomes encapsulating (R)-roscovitine reduced the number of neutrophils at the site of injury more effectively than free (R)-roscovitine by 1.3-fold.
Peptide-based treatments for neurological disorders are limited by their poor transport across the blood brain barrier and susceptibility to endopeptidase. To aid the transcytosis across the brain endothelium, poly(ethylene glycol)-block-polycaprolactone, were conjugated with transferrin receptor monoclonal antibodies by the maleimide-thiol chemistry.[96] With 34 antibodies per polymersome, 2.6-fold higher dose of polymersomes were accumulated in the brain. These polymersomes protected NC-1900, a peptide candidate for the treatment of Alzheimer’s disease. An improvement in scopolamine-induced learning and memory impairments in a water maze task when mice were administered intravenously with the polymersomes loaded with peptide at a much lower dose than when the peptide alone was injected intravenously.
As exemplified by the numerous abovementioned examples, functionalization of polymersomes with ligands is an effective strategy to improve target specificity, cellular adhesion and uptake. The choice of ligands depends on the expression level of the corresponding receptor in the target, diseased sites and the simplicity of ligand conjugation to the copolymers of interest.
6. Stimuli-responsive polymersomes
Intravenous injection is one of the most preferred routes of administration due to its minimal invasiveness. Following intravenous injection, particles are exposed to extensive dilution or opsonization by proteins/serum factors in the blood stream.[118–120] Polymersomes, which are formed through physical self-assembly, have a higher tendency to either dissociate or aggregate in physiological media than nano- or microparticles formed from covalent crosslinks.[121] This, in turn, could lead to premature release of the cargo, thus reducing the bioavailability of drug cargos in target diseased tissue.
To prolong the lifetime of polymersomes in circulation, hydrophilic poly(ethylene glycol) was presented at the surface of polymersomes. Polymersomes with poly(ethylene glycol) displayed a two-fold longer circulating half-time in rats (20-30 hours) than liposomes of similar poly(ethylene glycol) molecular weight (10-15 hours).[122] This enhanced stability was attributed to the delayed opsonization due to the denser brush-like layer of poly(ethylene glycol) on the polymersome surface which reduced the adsorption of plasma proteins. Consequently, phagocytosis of the polymersomes occurred after the half-time. On the other hand, the liposomes contained only less than 10% of the poly(ethylene glycol)-lipid, resulting in poor distribution of the hydrophilic molecules.
Separately, covalent crosslinking between polymeric bilayer has been widely employed to prevent dilution-induced destabilization of polymersomes. For example, poly(ethylene glycol) was modified by linking the terminal hydroxyl groups on poly(ethylene glycol) and hydrophobic 2,4,6-trimethoxybezylidene-1,1,1-tris(hydroxymethyle) ethane using fumaryl chloride.[123] After their self-assembly to form polymersomes, the fumarate segments cross-linked the polymer chains via radical polymerization. When the polymersomes with the cross-linked bilayer were exposed to serum, surfactants or saline solution, polymersomes displayed a minimal change of the hydrodynamic size as determined with the dynamic light scattering.
Cross-linking within the polymersome bilayer using epoxy-amine chemistry improved the stability of polymersomes even in the presence of surfactant. Epoxy-functional block copolymers of poly(glycidyl methacrylate) and poly(2-hydroxypropyl methacrylate) were cross-linked in the aqueous solution by diamine cross-linkers.[124] The size of the resulting polymersomes remained stable in the presence of nonionic surfactant. However, exposure to ionic surfactants resulted in a drastic size change from 218 nm to 10-800 nm. Destabilization of polymersomes due to ionic surfactants was likely caused by dissociation of their delicate vesicular nanostructures in the presence of charge.
Besides epoxy-amine cross-linking reactions, cross-linking between methacrylate groups in the bilayer has shown to enhance stability of polymersomes in physiological media. Methacrylate groups in poly(2-hydroxyethyl-co-2-methacryloxyethyl-co-octadecyl aspartamide) polymersomes were cross-linked upon exposure to ultraviolet light.[118] When challenged with physiological media, polymersomes with the cross-linked bilayer reduced the extent of size increment by ~1.2-fold over a period of 12 days. Accordingly, the ability to withstand destabilization during blood circulation resulted in a significantly higher accumulation in the tumor sites of squamous cell carcinoma bearing mice.
Cross-linking within the bilayer using disulfide groups also increased structural stability of polymersomes. In this study, poly(ethylene glycol)-block-poly(acrylic acid)-block-poly(2-diethyl amino)ethyl methacrylate) triblock copolymer was modified with cysteamine to yield thiol-containing copolymer.[125] When the resulting polymersomes were exposed to oxygen, the thiol groups underwent cross-linking through oxidative reaction, forming disulfide crosslinked polymersomes. The hydrodynamic diameter of these polymersomes was minimally changed against 100-fold dilution with the phosphate buffer saline and against 2 M NaCl solution. The cross-linked bilayer of the polymersome did not impose detrimental effects on the release of cargo.
Higher stability of polymersomes minimizes premature release of the cargo during circulation. On the other hand, overly stable polymersomes may encounter the problem of lethargic release at the target site. Given that the disease environments are biochemically different from a normal physiological state, there are emerging efforts to utilize these differences to increase the efficacy of diagnostic probes and drug carriers. As such, polymersomes are designed to undertake structural changes in response to biochemicals up-regulated in the pathologic tissues and, in turn, release molecular cargos and switch on imaging probes. These internal stimuli include low pH, changes in redox signaling molecules (i.e., glutathione), and elevated enzymes such as hyaluronidase (Figure 12). Alternatively, polymersomes are engineered to respond to external energies in the form of ultrasound, magnetic field and light. We will discuss various approaches to engineer polymersomes responsive to internal (Section 6.1) and external stimuli (Section 6.2).
Figure 12.

Summary of the various internal stimuli for triggered release of cargos.
6.1. Polymersomes responsive to internal stimuli
6.1.1. pH-responsive polymersomes
The pH of the extracellular matrix and blood under normal metabolic conditions is maintained at 7.4. In contrast, in a tumor microenvironment, the extracellular pH goes down to 6.5-6.9. This is due to the combination of poor perfusion and increased glucose metabolism by the tumor cells that produce more H+ ions than normal cells.[126] To respond to the different pH, polymersomes are tagged with pH-responsive Nile Blue-based molecules for their uses as a pH-responsive colorimetric/fluorescent biosensor during the far-red and near-infrared imaging of live cells.[127] These polymersomes are able to detect interstitial hypoxic/acidic regions in a tumor model with a pH-dependent colorimetric shift.
Highly metabolizing bacteria also decrease pH levels in the surrounding media. To target bacteria-causing periodontitis, metronidazole was encapsulated in poly(2-(methacryloyloxy)ethyl phosphorylcholine-block-poly(2-(diisopropylamino)ethyl methacrylate) (PMPB-b-PDPA) polymersomes.[128] The polymersomes released metronidazole only when polymersomes were incubated in media with pH 6.0. Efficacy of pH-responsive polymersomes to inhibit growth of intracellular P. gingivalis was evaluated by using a tissue-engineered human oral mucosal model. This model presented an epithelium of the human buccal mucosa TR146 cells. Polymersomes were accumulated in the upper layer of the epithelium. Some of polymersomes also penetrated approximately two-thirds of the thickness of the epithelium of the mucosal model. Then, these polymersomes entered the cytoplasm and peri-nuclear regions of infected TR146 cells. As a result, the polymersome-mediated delivery reduced the number of intracellular P. gingivalis by 80% when compared with the condition with free metronidazole.
Separately, the pH of endosomes and lysosomes are 5.0-6.0 and 4.5-5.0, respectively. These acidic conditions can catalyze hydrolytic degradation of polymersomes to release cargo molecules. This strategy was demonstrated with polymersomes formed from poly(D,L-lactide)-block-poly(2-methacryloyloxyethyl phosphorylcholine) (PLA-b-PMPC).[129] Hydrophilic doxorubicin-hydrochloric acid and hydrophobic doxorubicin were encapsulated in the core and membrane of polymersomes, respectively. At endosomal pH of 5.0, ester hydrolysis of the poly(D,L-lactide) block was accelerated by 2-fold than at pH 7.0. Therefore, the drug-loaded polymersomes entered hepatocarcinoma cell line, escaped from endosomes, and released the drug into the cytosol within 3 h.
Besides esters, acetals are also degradable in an acidic environment. Diblock copolymer of poly(ethylene glycol) and polycarbonate containing trimethoxybenzylidene acetals were prepared for the loading of doxorubicin hydrochloride and paclitaxel (Figure 13).[130] Hydrophobic paclitaxel and hydrophilic doxorubicin hydrochloride were encapsulated in the bilayer and core of polymersomes, respectively. By varying the hydrophobic block volume fraction, both micelles and polymersomes could be formed. At pH 4.0, polycarbonates containing trimethoxybenzylidene acetals were hydrolyzed into non-cytotoxic 2,4,6-trimethoxybenzaldehye, and released the drugs. Within 24 hours, the release rate of paclitaxel and doxorubicin hydrochloride from the polymersomes was approximately 3-fold and 2-fold, respectively, faster in acidic pH 4.0 than in neutral pH. Degradation of polycarbonate containing trimethoxybenzylidene acetals resulted in a significant increase in the diameter of polymersomes from 100-200 nm to over 1,000 nm within 24 h.
Figure 13.
Illustration of pH-sensitive degradable polymersomes based on poly(ethylene glycol)-block-poly(2,4,6-trimethoxybenzylidenepentaerythritol carbonate) (PEG-b-PTMBPEC) diblock copolymer for triggered release of both hydrophilic and hydrophobic anticancer drugs. Acid-labile acetals in the PTMBPEC block, indicated in red, hydrolyze at pH 5.0 and the encapsulated drugs are released from the polymersomes. Adapted with permission.[130] Copyright 2010, Elsevier.
Another strategy to form pH-sensitive polymersomes is to prepare polymers with ionizable amine groups. Tertiary amines diisopropylamine and diethylamines in poly(ethylene glycol)-block-polypeptide, were functionalized via the copper-catalyzed azide-alkyne cycloaddition (Figure 14).[131] The pKa of the copolymers was between pH 5.5 and 7.7. As demonstrated with fluorescence resonance energy transfer experiment, the polymersomes in acidic pH were destabilized due to the increased repulsion between positively charged copolymers.
Figure 14.

Synthetic route to the pH-responsive copolymers. Poly(ethylene glycol)-block-poly(γ-propargyl l-glutamate) (PEG-b-PPLG) copolymers were first synthesized (first row). Then, the PPLG block, indicated in red, was modified with azido-functionalized tertiary amines, diisopropylamine or diethylamine, by copper catalyzed azide–alkyne cycloaddition to form copolymers 1 to 4. Adapted with permission.[131] Copyright 2014, American Chemical Society.
Similar observations were noted in copolymers containing hydrophobic pH-sensitive poly(2-(diisopropylamino)ethyl methacrylate) (pKa of 6.4).[132] In the endosomes and lysosomes with pH below 6.4, the tertiary amine groups on the poly(2-(diisopropylamino)ethyl methacrylate) chains became extensively protonated, rendering the polymer chains to be hydrophilic. As a consequence, polymersomes were disintegrated owing to the charge repulsion and increased water solubility. The dramatic increase in the number of polymer chains resulting from the polymersome dissolution, in turn, triggered a rise in osmotic pressure that temporarily lysed the endosomal membrane. As a consequence, an increased mass of cargo molecules was released into the cell cytosol (Figure 15a). In contrast, a minimal amount of cargo was released from polymersomes formed from pH-insensitive polymers (Figure 15b).
Figure 15.

Confocal laser scanning microscope (CLSM) images and colocalization study examining endosomal escape by the rhodamine‐labeled pH-sensitive poly(2-(methacryloyloxy)ethyl phosphorylcholine)-block-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) chains. (a) CLSM z‐stack micrographs showing live human dermal fibroblast(HDF) cells containing PMPC-b-PDPA polymersomes loaded with rhodamine octadecyl ester perchlorate B (red color). Polymersomes were then treated with lysotracker (yellow color) and DNA staining SYTO9 (green color). b) CLSM z‐stack micrographs showing live HDF cells incubated with pH-insensitive poly(ethylene glycol)-block-poly(butylene glycol) polymersomes. Polymersomes were loaded with rhodamine octadecyl ester perchlorate B (red color) and DNA staining SYTO9 (green color). Reproduced with permission.[132]
The efficient cytosolic delivery by the pH-responsive poly(2-(methacryloyloxy)ethyl phosphorylcholine)-block-poly(2-(diisopropylamino)ethyl methacrylate) (PMPC-b-PDPA) polymersomes were also used to address challenges in transport of fluorogenic cell stains[133] and antibodies[134] across the membrane of live cells. Polymersomes delivered fluorophores into different types of animal and human cells, including both primary cells and cell lines.[133] Polymersomes were dissociated immediately within the endolysosomal compartments. As a consequence, fluorophores were released from polymersomes.
As such, the polymersome-mediated staining combined with confocal laser scanning microscopy allowed the study of cells in complex 3D environments.[133] For instance, to study the migration of cells into a tissue-engineered oral mucosa, primary human oral fibroblast and human oral keratinocytes were labeled with polymersomes loaded with red Rhodamine B octadecyl ester and far-red, 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate, respectively. This study revealed that fibroblasts migrate continuously during mucosa formation, while keratinocytes prefer to remain on the surface and organize into a thick epithelium.
Antibody delivery across the membrane of live cells has been one of the biggest challenge in the biotechnology field. Approaches to bind antibodies to membrane-permeable peptide carriers often ended up with the loss of antibody efficacy and the concern with cytotoxicity. To resolve these challenges, antibodies were physically-encapsulated into polymersomes prepared with biocompatible PMPC-b-PDPA.[134] Polymersomes escaped from the endosomes and lysosomes transported the antibodies into the cytosol. The retention of antibody structure and function by the polymersomes allowed for the real-time imaging of the NF-κB translocation upon proinflammatory stress stimulation in human dermal fibroblasts. Additionally, this route of transport through the polymersome carrier complements the need to deliver functional antibodies to intracellular targets for antibody-mediated therapy. For instance, the expression of NF-κB inducible cytokines by lipopolysaccharides were effectively inhibited by delivering antibodies towards the p65 subunit of NF-κB.
6.1.2. Redox-responsive polymersomes
Besides H+ ions, concentration of the biological reducing agent, glutathione, is also found to vary with intracellular compartments. The glutathione concentration in the cytosol ranges between 1-10 mM while plasma glutathione concentration is in the micromolar range.[135] With exceptions such as the brain and gastrointestinal cancers, the glutathione level in tumor is recorded to be higher than that of disease-free tissues.[136] Therefore, stimulus-responsive release of cargo molecules could be achieved by incorporating linkages cleaved by glutathione into polymersome-forming polymers. By doing so, polymersomes are destabilized exclusively under reducing conditions.[125,137,138]
For instance, a reduction-sensitive disulfide bond is introduced between the hydrophilic and hydrophobic blocks of diblock copolymers.[137] To probe the release triggered by the high glutathione concentration, the polymersomes were loaded with calcein, a hydrophilic fluorescent molecule. When calcein was concentrated within the polymersomes, no fluorescence was detected due to the auto-quenching of the fluorescent molecule. Upon the addition of glutathione, a rapid increase in fluorescence emission was observed. Calcein was released rapidly upon cellular uptake by mouse macrophage J774A-1 cells. After 10 min, most of the cells exhibited punctate intracellular fluorescence, which suggested intraendosomal release but not endosomal rupture. After 2 h, the intracellular fluorescence was diffused into cell cytosol, which indicated the endosomal disruption by the reduction-sensitive block copolymer.
Another example to form reduction-sensitive polymersomes is the insertion of a disulfide linkage between the hydrophobic cucurbit[6]uril and hydrophilic hexaethylene glycol blocks (Figure 16A).[138] In the presence of dithiothreitol, a reducing agent, polymersomes precipitated due to the loss of amphiphilicity following the cleavage of disulfide bonds. These polymersomes influenced the intracellular transport of doxorubicin. More doxorubicin was detected in the nuclei of HeLa cells treated with polymersomes containing the disulfide linkage. In contrast, doxorubicin was found to accumulate in the cytoplasm when treated with polymersomes without the disulfide linkage. This result indicated that the reduction-sensitivity accelerated the release of doxorubicin and allowed doxorubicin to reach nucleic acids in the cell nuclei (Figure 16B).
Figure 16.
(A) Chemical structure of polymersomes with disulfide linkage. (B) Confocal laser scanning microscopy images of HeLa cells incubated for 2 h with (a & b) doxorubicin-loaded polymersomes without disulfide linkage and (c & d) doxorubicin-loaded polymersomes with disulfide linkage [red: doxorubicin (DOX), blue: 4’,6-diamidino-2-phenylindole (DAPI), pink: overlay of DOX and DAPI, scale bar: 20 μm]. Adapted with permission.[138]
6.1.3. Enzyme-responsive polymersomes
Polymersomes are degraded by a relatively slow hydrolytic degradation of amphiphilic polymers over time. Therefore, the drug release rate is limited under normal physiological conditions. Efforts were made to accelerate the hydrolytic degradation rate by making the polymersomes sensitive to enzymes such as lysosomal enzymes and microorganism-secreting enzymes. The resulting polymersomes were triggered to release their cargo only in the presence of these enzymes.
However, in comparison to H+ and glutathione-responsive polymersomes, the enzyme-triggered destabilization of polymersomes may be still slower due to the steric-hindrance of bulky enzymes. According to the kinetic study of Proteinase K-mediated hydrolysis, the polymersome degradation process was initiated by enzyme diffusion to the hydrophobic poly(D,L-lactide) block and cleavage of the ester bonds.[139] As such, the degradation rate was dependent of the enzyme concentration. Thereafter, the poly(D,L-lactide) was hydrolyzed into smaller oligomers.
Separately, peptides including the sequence of Gly-Phe-Leu-Gly-Phe, which is cleavable by lysozyme, was introduced between poly(ethylene glycol) and poly(D,L-lactide) blocks to increase the degradation rate of polymersomes in the lysosome.[140] Following the incubation with lysosomal enzyme cathepsin B, the poly(ethylene glycol) block was separated from the hydrophobic poly(D,L-lactide). These polymersomes exhibited larger intracellular release of fluorescent cargos than those without the peptide linker, as observed with the breast cancer cell line SKBR3.
Polysaccharides can also be a target for the enzyme degradation. One example is hyaluronic acid, which is naturally hydrolyzed by glucosidases such as hyaluronidase. Hyaluronidase is secreted by bacteria such as Staphylococcus aureus, Clostridium and Streptococcus spp. so as to use hyaluronan as a carbon source. Thus, in the preparation a bacteria-detecting polymersomes, hyaluronic acid was covalently linked to poly(ɛ-caprolactone) through a 1,3-Huisgen dipolar cyclo-addition.[141] According to the analysis conducted with the dynamic light scattering, the mean diameters of polymersomes quickly decreased within 30 min of contact with hyaluronidase.
Polymersomes responsive to the enzymatic degradation by penicillin G amidase and β-lactamase were formed from the self-assembly of diblock copolymers consisting of hydrophilic poly(ethylene glycol) and hydrophobic block containing side groups with specific enzyme-cleavable linkages.[142] Penicillin G amidase and β-lactamase were chosen as they are enzymes expressed by bacterial strains that have developed antibiotic resistance but not by beneficial gut bacteria. Thus, the loaded vancomycin only inhibited bacterial growth when the polymersomes were incubated with methicillin-resistant Staphylococcus aureus but not when they were incubated with Lactobacillus acidophilus.
6.2. External stimuli-controlled molecular release
6.2.1. Focused ultrasound-responsive polymersomes
One of the greatest challenges of cancer therapy is the deep penetration of therapeutics into the tumor tissue. Specifically, these are solid tumors that reside far away from the vasculature and are often deprived of oxygen. The hypoxic condition contributes to resistance to chemotherapy due to the poor diffusion of drugs in the avascular area with high interstitial fluid pressure.
To resolve this challenge, monocytes have been employed as cellular carrier of therapeutic polymersomes and echogenic polymer bubbles. Poly(acrylic acid)-block-poly(distearin acrylate) of two different length of hydrophobic distearin acrylate blocks were used to prepare doxorubicin-loaded polymersomes and perfluoropentane-loaded polymer bubbles. To prepare the cellular carrier, monocytes were incubated with the polymersomes and polymer bubbles. Monocytes engulfed both the polymersomes and polymer bubbles with minimal loss in viability. The application of focused ultrasound caused the polymer bubble collapse. Subsequent shear force disrupted the vesicle structure and in turn, released doxorubicin into the extracellular media. The intercellular doxorubicin transport from the monocytes to Tramp-C1 prostate cancer cells led to a 2-fold reduction in cancer cell viability. Also, monocytes that engulfed polymersomes and polymer bubbles were injected into mice pretreated with γ-ray irradiation to induce hypoxia conditions in the tumor. The subsequent application of focused ultrasound increased doxorubicin levels in tumor sections. Importantly, the combination of monocyte homing and focused ultrasound led to the deep tumor penetration up to 150 μm from the nearest blood vessel.[143]
6.2.2. Magnetic field-responsive polymersomes
External high frequency magnetic field was also used to induce the release of oxygen-loaded polymer bubbles.[144] This is mediated by superparamagnetic iron oxide nanoparticles (SPIONs), which create local heating in response to the high frequency magnetic field and in turn, releases oxygen from the core. Only in the presence of high frequency magnetic field, the oxygen dissolved in the media increased from 3.6 to 8.5 ppm. In this example, the released oxygen is coupled to antitumor photodynamic therapy by co-encapsulation of photosensitizer chlorin e6 into the polymersome bilayer. Upon laser irradiation at 660 nm, chlorin e6 converts oxygen to cytotoxic reactive oxygen species.
Taking advantage of the innate tropism to hypoxic regions, bone marrow-derived monocytes/macrophages were used to deliver the polymer bubbles to the tumor site. The high frequency magnetic field was then applied to induce the death of monocytes by hyperthermia from the SPIONs, and also to trigger the release of oxygen from the polymer bubbles.
When Tramp-C1 prostate cancer cells were treated with monocytes in the absence of high frequency magnetic field or photodynamic therapy, cancer cell viability remained high. In contrast, the combined procedure of high frequency magnetic field and photodynamic therapy led to a significant drop of cell viability. This result was attributed to the hyperthermia-triggered, rapid oxygen release from monocytes and subsequent entry of reactive oxygen species into cancer cells via molecular diffusion. This combined high frequency magnetic field and photodynamic therapy significantly increased therapeutic efficacy of the monocyte to inhibiting the tumor growth by 50% in mice bearing Tramp-C1 xenografts.
6.2.3. Light-responsive polymersomes
Alternatively, efforts were made to assemble the light-sensitive polymersomes. To do so, photosensitizers may be encapsulated within the membrane or core of the polymersomes. Then, in the presence of external irradiation, the photosensitizers would induce a structural change of the polymers and subsequently deform the membrane. Finally, this structural deformation would release of molecular cargo driven by diffusion.
Photo-responsive polymersomes were prepared from self-assembly of poly(ethylene oxide)-block-poly(1,2-butadiene). A meso-to-meso ethyne-bridged bis[(porphinato)zinc] (PZn2) fluorophore was encapsulated within the 10 nm-thick hydrophobic polymersome membrane.[145] Dextran was added into the core to increase the rate of deformation by decreasing the elastic modulus of the polymersome membrane. Excitation with visible or near infrared light resulted in the relaxation of the electronically excited PZn2 and the generation of local heat. At the same time, the interaction of dextran added in the aqueous core with the inner leaflet of the polymersome membrane reduced the membrane elasticity. The combined effect led to a local asymmetric membrane stretching; irreversible membrane deformation; and finally rupture (Figure 17). The frequency of the membrane rupture events was controlled by the molecular weight of dextran. The hydrophobicity of dextran and hence rate of aggregation led to increased destabilization of the polymersome membrane.
Figure 17.
(A) Types of membrane deformation of polymersomes observed following optical excitation. (B) Membrane rupture of a dextran-encapsulated polymersome following optical excitation (λex = 458, 488, 515, 543, and 633 nm; laser power = 33 mW). Top panels represent phase contrast images and bottom panels represent emitted PZn2 fluorescence images, before and after optical excitation. (C) Frequency of deformation events for different molecular weights of dextran (MW). Reproduced with permission.[145]
Similar polymersome rupture was observed in zebrafish embryos, which were injected with the nano-sized polymersomes containing PZn2 in the hydrophobic membrane and dextran in the aqueous core.[146] Compared to micron-sized polymersomes, the polymersomes with diameters of 100 – 200 nm were ruptured by the PZn2-induced thermal expansion because of the increased curvature. After irradiation, many non-vesicular structures including worm-like micelles and “dumbbell”-shaped vesicles were found. This result suggested that the membrane buckled and folded over itself. Or, the membrane was disrupted and later reassembled.
Polymersomes of poly(2-vinylpyridine) with an amphiphilic sulfonic acid, 4’-[3,5-di(trideca-2,4-diynyloxyl)]azobenzene-4-sulfonic acid, contained an azo group that rendered the polymersomes to lose the smectic layer that made up the membrane in response to light.[147] Small-angle X-ray scattering data showed that polymer chains in this system were mainly parallel to the vesicle surface, contributing to their mechanical stability. A collapse of the vesicle could be induced by UV irradiation at 350 nm. This result is related to the pi-pi* transition of the azo group. The cis isomer had a bent shape, which could perturb the ordered structure of the membrane (Figure 18A). Thus, upon irradiation, the trans–cis transition of the azo groups led to the isotropization of the layered structure. This mechanism served to form polymersomes with locally disrupted regions that could form outlets for cargo release (Figure 18B).
Figure 18.
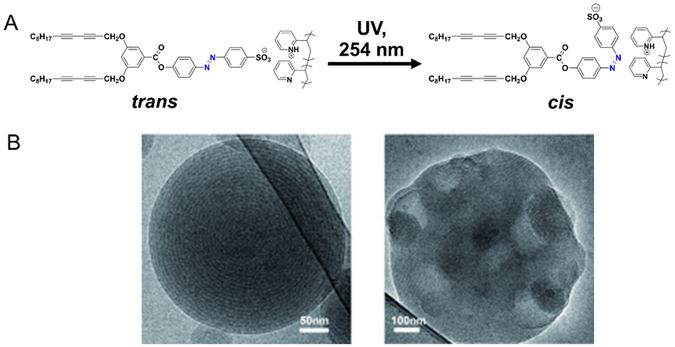
(A) Chemical structures of complexes of poly(2-vinylpyridine) and 4’-[3,5-di(trideca-2,4-diynyloxyl)]azobenzene-4-sulfonic acid. Before UV irradiation at 254 nm, the azo group exists as a trans isomer. Upon irradiation, the cis isomer has a bent shape that perturbs the ordered structure of the membrane. (B) Cryo-TEM micrographs of the multilayer polymersomes before (left) and after (right) UV irradiation for 45 min. Adapted with permission.[147]
Photochromic polymersomes exhibiting photo-switchable and reversible bilayer permeability were prepared from poly(ethylene glycol)-block-PSPA, where SPA is spiropyran-based monomer containing carbamate linkage.[148] DAPI, a cell nuclei DNA-intercalating dye with blue emission, was encapsulated into the aqueous interior of polymersomes. Upon intracellular release from polymersomes, DAPI spontaneously entered the cell nuclei and reported the extent of release through enhanced fluorescence emission. After endocytosis of polymersomes loaded with DAPI, the intracellular, photo-switching process was investigated by the confocal laser scanning microscopy. Laser irradiation at 405 nm triggered the isomer transition between hydrophobic spiropyran and zwitterionic merocyanine states (Figure 19). The increased permeability of polymersome bilayer due to the increased hydrophilicity in the bilayer led to efficient DAPI release and binding to the nuclear DNA.
Figure 19.

Amphiphilic diblock copolymers consisting poly(ethylene oxide) and spiropyran-based monomers containing the carbamate linkage self-assemble into polymersomes with hydrophobic bilayers. Spiropyran moieties within polymersome bilayers undergo reversible isomerization between hydrophobic spiropyran (SP, λ2 > 450 nm irradiation) and zwitterionic merocyanine (MC, λ1 < 420 nm irradiation). Reproduced with permission.[148] Copyright 2015, American Chemical Society.
An alternative to chemically incorporating photosensitive groups into the polymersomes, photo-fragmenting dyes such as calcein or methylene blue were incorporated into poly(ethylene oxide)-block-poly(1,2-butadiene) polymersomes. Upon irradiation in the visible light region, calcein or methylene blue generated reactive oxygen species. As a consequence, the increased osmotic pressure within the lumen of the polymersomes led to vesicle bursting.[149]
The main limitation of using light as an external trigger is the poor tissue penetration. Therefore, the clinical procedure may require an endoscope combined with the light-sensitive polymersomes. It may also be desirable to develop polymersomes that are responsive to light in the near-infrared range, which penetrates skin and blood more efficiently than visible light.
7. Conclusions and future directions
Given the numerous advantages, polymersomes are well-received as the next generation carrier for delivery of diagnostic and therapeutic molecules. The ability to customize polymer functionality has made it amenable to impose design specifics such as optimal particle size, structure and shape for transport of imaging probes and therapeutic drugs. Furthermore, enhancement in cargo delivery have been demonstrated by tethering targeting moieties onto the surface of polymersomes and installing stimuli-responsive chemistries within the particle. However, there are still needs to improve the controllability of properties, functionality, and reproducibility of polymersomes before they can be used in biomedical diagnostics and therapy.
To achieve this goal, molecular simulations can be employed as tools for the design of polymers. By doing so, it will be possible to optimize the size, shape and surface charge of polymersomes while minimizing experimental trial-and-error. Simulations of the polymer interactions with imaging probes and drug candidates may guide the polymer synthesis to attain desired morphological and chemical properties of polymersomes. Simulations of functionalized polymersomes in the complex biological fluids with confounding protein and salt interactions will also improve the stability of polymersomes under physiological conditions, thus extending the lifetime of molecular cargos.
Another need lies in better understanding of the interactions between molecular cargos and physiological environments. In particular, diagnostic probes such as magnetic resonance imaging contrast agents display varying levels of contrast signals depending on the interaction efficiency with aqueous media because of the differed relaxation rate of water protons. For stimuli-responsive polymersomes to release molecular cargos at the target site, careful design of the polymer is required to prevent premature release but to accelerate the release in response to pathological signals. The sensitivity of the stimuli-responsive groups is affected by both accessibility of the signals and the perturbations at the molecular level to facilitate the outward diffusion of molecular cargos.
Moreover, there should be efforts to study and improve the encapsulation efficiency of molecular cargos. Improving the encapsulation efficiency of molecular cargos is essential to reduce the amount of cargo feeds required during fabrication of the polymersomes. This is especially important for large-scale manufacturing of the drug-loaded polymersomes, which in turn, imply lower costs of production. Polymer design could be centered on physical and chemical properties of the desired molecular cargos. The cargo of interests may possess inherent moieties which can facilitate attractive interactions (e.g. electrostatics, hydrogen bonding, van der Waals) with the polymersomes and the cooperative stacking of molecules within the confined space. Thus, higher encapsulation efficiency is achievable with greater affinity between polymers and cargos molecules.
Finally, manufacturability of polymersomes is another challenge to resolve. Current methods of polymersome assembly are limited to a batch process, which often results in particles with a broad size distribution, and necessitates subsequent filtration steps. Uniform-sized polymersomes can be obtained with the help of technologies, and in turn, minimize the need for filtration. This increases the yield of polymersomes needed for clinical applications. While technologies offer the benefits of automation with precise control of physiochemical properties of polymersomes, the ability to manufacture homogenous particles in a scalable manner remains a challenge. Accordingly, collaborations between the scientists, engineers, and clinicians must be established and be supported so as to accelerate the translation of these polymersomes into clinical use.
Acknowledgements
This work was supported by the National Institutes of Health (Grant 1R01 HL109192 to H. K.) and National Institutes of Health/National Eye Institute Award (K08EY024339 to V. K. A.). This work was also supported by the Department of Defense Vision Research Program under Award W81XWH-17-1-022. J. L. and J. Y. T. acknowledge the A*STAR Graduate Scholarship (Overseas) from the Agency for Science, Technology and Research (A*STAR). Y. Y. Y. acknowledges the support from the Institute of Bioengineering and Nanotechnology (A*STAR), Singapore.
Biography

Jiayu Leong is a graduate student in the Department of Chemical and Biomolecular Engineering at the University of Illinois, Urbana-Champaign and at the A*STAR Institute of Bioengineering and Nanotechnology, Singapore. She received her B.Eng. in Chemical and Biomolecular Engineering from the National University of Singapore in 2013. Her research interests are in designing polymers for drug and stem cell delivery for treating cancer, inflammation and bacterial infections.

Jye Yng Teo is currently undertaking her Ph.D. under the guidance of Dr. Yi Yan Yang at the A*STAR Institute of Bioengineering and Nanotechnology, Singapore, and Professor Hyunjoon Kong in the Department of Chemical and Biomolecular Engineering at the University of Illinois at Urbana-Champaign, U.S.A. She graduated from Nanyang Technological University, Singapore, in 2012 with a B.Eng. (Honors) in Bioengineering. Her current research interests include drug delivery using stimuli responsive polymeric nanoparticles for cancer therapy, and surface engineering of therapeutic stem cells with nanomaterials for the treatment of vascular diseases.

Dr. Hyunjoon Kong is a professor in the Department of Chemical and Biomolecular Engineering at the University of Illinois at Urbana-Champaign. He also holds affiliation with the Department of Bioengineering, Department of Pathobiology, Center for Biophysics and Computational Biology, and Neuroscience Program. He received his engineering education from the University of Michigan at Ann Arbor (Ph. D. 2001) and performed post-doctoral research at the University of Michigan and Harvard University. He joined the University of Illinois in 2007. He has been developing a series of soft matters that can regulate molecular and cell transports for diagnosis and treatments of vascular and pro-inflammatory diseases.
Contributor Information
Jiayu Leong, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA, Institute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore 138669, Singapore.
Jye Yng Teo, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA, Institute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore 138669, Singapore.
Prof. Dr. Vinay K. Aakalu, Illinois Eye and Ear Infirmary, University of Illinois at Chicago, Department of Ophthalmology and Visual Sciences, Chicago, IL 60612, USA
Dr. Yi Yan Yang, Institute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore 138669, Singapore
Prof. Dr. Hyunjoon Kong, Department of Chemical and Biomolecular Engineering, Department of Bioengineering, Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA, hjkong06@illinois.edu
References
- [1].Wu NZ, Da D, Rudoll TL, Needham D, Whorton AR, Dewhirst MW, Cancer Res. 1993, 53, 3765. [PubMed] [Google Scholar]
- [2].Barenholz Y, J. Control. Release 2012, 160, 117. [DOI] [PubMed] [Google Scholar]
- [3].Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S, Front. Pharmacol 2015, 6, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Uziely B, Jeffers S, Isacson R, Kutsch K, Wei-Tsao D, Yehoshua Z, Libson E, Muggia FM, Gabizon A, J. Clin. Oncol 1995, 13, 1777. [DOI] [PubMed] [Google Scholar]
- [5].Lee H, Larson RG, Biomacromolecules 2016, 17, 1757. [DOI] [PubMed] [Google Scholar]
- [6].Sarin H, J. Angiogenes. Res 2010, 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK, Proc. Natl. Acad. Sci. U. S. A 1998, 95, 4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM, Am. J. Pathol 2000, 156, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yhee JY, Son S, Son S, Joo MK, Kwon IC, in Cancer Target. Drug Deliv, Springer New York, New York, NY, 2013, pp. 621–632. [Google Scholar]
- [10].Liu Y, Yuan H, Fales AM, Register JK, Vo-Dinh T, Front. Chem 2015, 3, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brinkhuis RP, Stojanov K, Laverman P, Eilander J, Zuhorn IS, Rutjes FPJT, van Hest JCM, Bioconjug. Chem 2012, 23, 958. [DOI] [PubMed] [Google Scholar]
- [12].Yang NJ, Hinner MJ, in Site-Specific Protein Labeling Methods Protoc, NIH Public Access, 2015, pp. 29–53. [Google Scholar]
- [13].Akinc A, Battaglia G, Cold Spring Harb. Perspect. Biol 2013, 5, a016980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao H, Shi W, Freund LB, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bowron DT, Filipponi A, Roberts MA, Finney JL, Phys. Rev. Lett 1998, 81, 4164. [Google Scholar]
- [16].Cheng Y-K, Rossky PJ, Nature 1998, 392, 696. [DOI] [PubMed] [Google Scholar]
- [17].Anraku Y, Kishimura A, Oba M, Yamasaki Y, Kataoka K, J. Am. Chem. Soc 2010, 132, 1631. [DOI] [PubMed] [Google Scholar]
- [18].Thiermann R, Mueller W, Montesinos-Castellanos A, Metzke D, Löb P, Hessel V, Maskos M, Polymer (Guildf). 2012, 53, 2205. [Google Scholar]
- [19].Bleul R, Thiermann R, Maskos M, Macromolecules 2015, 48, 7396. [Google Scholar]
- [20].Robertson JD, Rizzello L, Avila-Olias M, Gaitzsch J, Contini C, Magoń MS, Renshaw SA, Battaglia G, Sci. Rep 2016, 6, 27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clay NE, Whittenberg JJ, Leong J, Kumar V, Chen J, Choi I, Liamas E, Schieferstein JM, Jeong JH, Kim DH, Zhang ZJ, Kenis PJA, Kim IW, Kong H, Nanoscale 2017, 9, 5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Robertson JD, Ward JR, Avila-Olias M, Battaglia G, Renshaw SA, J. Immunol 2017, 1601901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Colley HE, Hearnden V, Avila-Olias M, Cecchin D, Canton I, Madsen J, Macneil S, Warren N, Hu K, McKeating JA, Armes SP, Murdoch C, Thornhill MH, Battaglia G, Mol. Pharm 2014, 11, 1176. [DOI] [PubMed] [Google Scholar]
- [24].Men Y, Peng F, Tu Y, van Hest JCM, Wilson DA, Polym. Chem 2016, 7, 3977. [Google Scholar]
- [25].Franc BL, Acton PD, Mari C, Hasegawa BH, J. Nucl. Med 2008, 49, 1651. [DOI] [PubMed] [Google Scholar]
- [26].Madsen MT, J Nucl Med 2007, 48, 661. [DOI] [PubMed] [Google Scholar]
- [27].Habel J, Ogbonna A, Larsen N, Schulte L, Almdal K, Hélix-Nielsen C, J. Polym. Sci. Part B Polym. Phys 2016, 54, 699. [Google Scholar]
- [28].V Bondar O, V Saifullina D, Shakhmaeva II, Mavlyutova II, Abdullin TI, Acta Naturae 2012, 4, 78. [PMC free article] [PubMed] [Google Scholar]
- [29].Liu Q, Chen J, Du J, Biomacromolecules 2014, 15, 3072. [DOI] [PubMed] [Google Scholar]
- [30].Blanazs A, Massignani M, Battaglia G, Armes SP, Ryan AJ, Adv. Funct. Mater 2009, 19, 2906. [Google Scholar]
- [31].Christian DA, Garbuzenko OB, Minko T, Discher DE, Macromol. Rapid Commun. 2010, 31, 135. [DOI] [PubMed] [Google Scholar]
- [32].Yang C, Krishnamurthy S, Liu J, Liu S, Lu X, Coady DJ, Cheng W, De Libero G, Singhal A, Hedrick JL, Yang YY, Adv. Healthc. Mater 2016, 5, 1272. [DOI] [PubMed] [Google Scholar]
- [33].Cheng J, Chin W, Dong H, Xu L, Zhong G, Huang Y, Li L, Xu K, Wu M, Hedrick JL, Yang YY, Fan W, Adv. Healthc. Mater 2015, 4, 2128. [DOI] [PubMed] [Google Scholar]
- [34].Zhong G, Cheng J, Liang ZC, Xu L, Lou W, Bao C, Ong ZY, Dong H, Yang YY, Fan W, Adv. Healthc. Mater 2017, 6, 1601134. [DOI] [PubMed] [Google Scholar]
- [35].Wang T, Jiang J, Xiao Y, Zou Y, Gao J, Du J, RSC Adv. 2015, 5, 55602. [Google Scholar]
- [36].Wu J, Eisenberg A, J. Am. Chem. Soc 2006, 128, 2880. [DOI] [PubMed] [Google Scholar]
- [37].So S, Yao LJ, Lodge TP, J. Phys. Chem. B 2015, 119, 15054. [DOI] [PubMed] [Google Scholar]
- [38].Battaglia G, Ryan AJ, Tomas S, Langmuir 2006, 22, 4910. [DOI] [PubMed] [Google Scholar]
- [39].Itel F, Chami M, Najer A, Lörcher S, Wu D, Dinu IA, Meier W, Macromolecules 2014, 47, 7588. [Google Scholar]
- [40].Bai Z, Zhao B, Lodge TP, J. Phys. Chem. B 2012, 116, 8282. [DOI] [PubMed] [Google Scholar]
- [41].Leson A, Hauschild S, Rank A, Neub A, Schubert R, Förster S, Mayer C, Small 2007, 3, 1074. [DOI] [PubMed] [Google Scholar]
- [42].Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE, Macromolecules 2002, 35, 8203. [Google Scholar]
- [43].Joseph A, Contini C, Cecchin D, Nyberg S, Ruiz-Perez L, Gaitzsch J, Fullstone G, Tian X, Azizi J, Preston J, Volpe G, Battaglia G, Sci. Adv 2017, 3, e1700362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Paula S, Volkov AG, Deamer DW, Biophys. J 1998, 74, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].So S, Lodge TP, J. Phys. Chem. C 2014, 118, 21140. [Google Scholar]
- [46].Li J, Dirisala A, Ge Z, Wang Y, Yin W, Ke W, Toh K, Xie J, Matsumoto Y, Anraku Y, Osada K, Kataoka K, Angew. Chemie 2017, 129, 14213. [DOI] [PubMed] [Google Scholar]
- [47].Li J, Li Y, Wang Y, Ke W, Chen W, Wang W, Ge Z, Nano Lett. 2017, 17, 6983. [DOI] [PubMed] [Google Scholar]
- [48].Fossheim SL, Fahlvik AK, Klaveness J, Muller RN, Magn. Reson. Imaging 1999, 17, 83. [DOI] [PubMed] [Google Scholar]
- [49].Smith CE, Shkumatov A, Withers SG, Yang B, Glockner JF, Misra S, Roy EJ, Wong CH, Zimmerman SC, Kong H, ACS Nano 2013, 7, 9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cheng Z, Tsourkas A, Langmuir 2008, 24, 8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cheng Z, Thorek DLJ, Tsourkas A, Adv. Funct. Mater 2009, 19, 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Khawaja AZ, Cassidy DB, Al Shakarchi J, McGrogan DG, Inston NG, Jones RG, Insights Imaging 2015, 6, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Turner JL, Pan D, Plummer R, Chen Z, Whittaker AK, Wooley KL, Adv. Funct. Mater 2005, 15, 1248. [Google Scholar]
- [54].Hickey RJ, Haynes AS, Kikkawa JM, Park SJ, J. Am. Chem. Soc 2011, 133, 1517. [DOI] [PubMed] [Google Scholar]
- [55].Hickey RJ, Koski J, Meng X, Riggleman RA, Zhang P, Park SJ, ACS Nano 2014, 8, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bin Na H, Song IC, Hyeon T, Adv. Mater 2009, 21, 2133. [Google Scholar]
- [57].Ghoroghchian PP, Frail PR, Li G, Zupancich JA, Bates FS, Hammer DA, Therien MJ, Chem. Mater 2007, 19, 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ghoroghchian PP, Therien MJ, Hammer DA, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology 2009, 1, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ghoroghchian PP, Frail PR, Susumu K, Blessington D, Brannan AK, Bates FS, Chance B, Hammer DA, Therien MJ, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Christian NA, Benencia F, Milone MC, Li G, Frail PR, Therien MJ, Coukos G, Hammer DA, Mol. Imaging Biol 2009, 11, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Duncan TV, Ghoroghchian PP, Rubtsov IV, Hammer DA, Therien MJ, J. Am. Chem. Soc 2008, 130, 9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Champion JA, Mitragotri S, Proc. Natl. Acad. Sci. U. S. A 2006, 103, 4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE, Nat. Nanotechnol 2007, 2, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Robertson JD, Yealland G, Avila-Olias M, Chierico L, Bandmann O, Renshaw SA, Battaglia G, ACS Nano 2014, 8, 4650. [DOI] [PubMed] [Google Scholar]
- [66].Banerjee A, Qi J, Gogoi R, Wong J, Mitragotri S, J. Control. Release 2016, 238, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ponchel G, Cauchois O, in Polym. Nanoparticles Nanomedicines, Springer International Publishing, Cham, 2016, pp. 159–184. [Google Scholar]
- [68].Lai MH, Jeong JH, Devolder RJ, Brockman C, Schroeder C, Kong H, Adv. Funct. Mater 2012, 22, 3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Jeong JH, Schmidt JJ, Kohman RE, Zill AT, Devolder RJ, Smith CE, Lai MH, Shkumatov A, Jensen TW, Schook LG, Zimmerman SC, Kong H, J. Am. Chem. Soc 2013, 135, 8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Van Oers MCM, Rutjes FPJT, Van Hest JCM, J. Am. Chem. Soc 2013, 135, 16308. [DOI] [PubMed] [Google Scholar]
- [71].Abdelmohsen LKEA, Williams DS, Pille J, Ozel SG, Rikken RSM, Wilson DA, van Hest JCM, J. Am. Chem. Soc 2016, 138, 9353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yealland G, Battaglia G, Bandmann O, Mortiboys H, Neurosci. Lett 2016, 630, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rikken RSM, Engelkamp H, Nolte RJM, Maan JC, van Hest JCM, Wilson DA, Christianen PCM, Nat. Commun 2016, 7, 12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Rikken RSM, Kerkenaar HHM, Nolte RJM, Maan JC, van Hest JCM, Christianen PCM, Wilson DA, Chem. Commun 2014, 50, 5394. [DOI] [PubMed] [Google Scholar]
- [75].Meeuwissen SA, Kim KT, Chen Y, Pochan DJ, Van Hest JCM, Angew. Chemie - Int. Ed 2011, 50, 7070. [DOI] [PubMed] [Google Scholar]
- [76].Wilson DA, Nolte RJM, van Hest JCM, Nat. Chem 2012, 4, 268. [DOI] [PubMed] [Google Scholar]
- [77].Gao W, Wang J, Nanoscale 2014, 6, 10486. [DOI] [PubMed] [Google Scholar]
- [78].Peng F, Tu Y, Adhikari A, Hintzen JCJ, Löwik DWPM, Wilson DA, Chem. Commun 2017, 53, 1088. [DOI] [PubMed] [Google Scholar]
- [79].Tu Y, Peng F, André AAM, Men Y, Srinivas M, Wilson DA, ACS Nano 2017, 11, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Chen L, Xiao S, Zhu H, Wang L, Liang H, Soft Matter 2016, 12, 2632. [DOI] [PubMed] [Google Scholar]
- [81].Alibolandi M, Abnous K, Hadizadeh F, Taghdisi SM, Alabdollah F, Mohammadi M, Nassirli H, Ramezani M, J. Control. Release 2016, 241, 45. [DOI] [PubMed] [Google Scholar]
- [82].Zhu D, Wu S, Hu C, Chen Z, Wang H, Fan F, Qin Y, Wang C, Sun H, Leng X, Kong D, Zhang L, Acta Biomater. 2017, 58, 399. [DOI] [PubMed] [Google Scholar]
- [83].Liu Q, Song L, Chen S, Gao J, Zhao P, Du J, Biomaterials 2017, 114, 23. [DOI] [PubMed] [Google Scholar]
- [84].Kubilis A, Abdulkarim A, Eissa AM, Cameron NR, Sci. Rep 2016, 6, 32414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pasparakis G, Alexander C, Angew. Chemie - Int. Ed 2008, 47, 4847. [DOI] [PubMed] [Google Scholar]
- [86].Tian X, Nyberg S, Sharp PS, Madsen J, Daneshpour N, Armes SP, Berwick J, Azzouz M, Shaw P, Abbott NJ, Battaglia G, Sci. Rep 2015, 5, 11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Georgieva JV, Brinkhuis RP, Stojanov K, Weijers CAGM, Zuilhof H, Rutjes FPJT, Hoekstra D, Van Hest JCM, Zuhorn IS, Angew. Chemie - Int. Ed 2012, 51, 8339. [DOI] [PubMed] [Google Scholar]
- [88].Zou Y, Meng F, Deng C, Zhong Z, J. Control. Release 2016, 239, 149. [DOI] [PubMed] [Google Scholar]
- [89].Zou Y, Zheng M, Yang W, Meng F, Miyata K, Kim HJ, Kataoka K, Zhong Z, Adv. Mater 2017, 1703285. [DOI] [PubMed] [Google Scholar]
- [90].Christian NA, Milone MC, Ranka SS, Li G, Frail PR, Davis KP, Bates FS, Therien MJ, Ghoroghchian PP, June CH, Hammer DA, Bioconjug. Chem 2007, 18, 31. [DOI] [PubMed] [Google Scholar]
- [91].Alibolandi M, Ramezani M, Abnous K, Sadeghi F, Atyabi F, Asouri M, Ahmadi AA, Hadizadeh F, J. Control. Release 2015, 209, 88. [DOI] [PubMed] [Google Scholar]
- [92].Yu Y, Jiang X, Gong S, Feng L, Zhong Y, Pang Z, Nanoscale 2014, 6, 3250. [DOI] [PubMed] [Google Scholar]
- [93].Pang Z, Gao H, Yu Y, Chen J, Guo L, Ren J, Wen Z, Su J, Jiang X, Int. J. Pharm 2011, 415, 284. [DOI] [PubMed] [Google Scholar]
- [94].Pourtau L, Oliveira H, Thevenot J, Wan Y, Brisson AR, Sandre O, Miraux S, Thiaudiere E, Lecommandoux S, Adv. Healthc. Mater 2013, 2, 1420. [DOI] [PubMed] [Google Scholar]
- [95].Robbins GP, Saunders RL, Haun JB, Rawson J, Therien MJ, Hammer DA, Langmuir 2010, 26, 14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Pang Z, Lu W, Gao H, Hu K, Chen J, Zhang C, Gao X, Jiang X, Zhu C, J. Control. Release 2008, 128, 120. [DOI] [PubMed] [Google Scholar]
- [97].Sega EI, Low PS, Cancer Metastasis Rev. 2008, 27, 655. [DOI] [PubMed] [Google Scholar]
- [98].Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP, Anal. Biochem 2005, 338, 284. [DOI] [PubMed] [Google Scholar]
- [99].Zwicke GL, Ali Mansoori G, Jeffery CJ, Nano Rev. 2012, 3, 18496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Varki A, Glycobiology 1993, 3, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sharon N, Lis H, Science 1989, 246, 227. [DOI] [PubMed] [Google Scholar]
- [102].Dwek RA, Chem. Rev 1996, 96, 683. [DOI] [PubMed] [Google Scholar]
- [103].Eissa AM, Cameron NR, Adv. Polym. Sci 2013, 253, 71. [Google Scholar]
- [104].Jones AR, V Shusta E, Pharm. Res 2007, 24, 1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].V Georgieva J, Hoekstra D, Zuhorn IS, Pharmaceutics 2014, 6, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lau D, Guo L, Liu R, Marik J, Lam K, Lung Cancer 2006, 52, 291. [DOI] [PubMed] [Google Scholar]
- [107].Anguille S, Smits EL, Lion E, Van Tendeloo VF, Berneman ZN, Lancet Oncol. 2014, 15, e257. [DOI] [PubMed] [Google Scholar]
- [108].Wagstaff K, Jans D, Curr. Med. Chem 2006, 13, 1371. [DOI] [PubMed] [Google Scholar]
- [109].Kim Y, Hyo SK, Zheng YC, Lee HS, Jin SA, Chan KP, Park K, Ahn MJ, Anticancer Res. 2009, 29, 1817. [PubMed] [Google Scholar]
- [110].Litvinov SV, Balzar M, Winter MJ, Bakker HAM, Briaire-De Bruijn IH, Prins F, Fleuren GJ, Warnaar SO, J. Cell Biol 1997, 139, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Fillebeen C, Descamps L, Dehouck MP, Fenart L, Benaïssa M, Spik G, Cecchelli R, Pierce A, J. Biol. Chem 1999, 274, 7011. [DOI] [PubMed] [Google Scholar]
- [112].Jia TT, Sun ZG, Lu Y, Gao J, Zou H, Xie FY, Zhang GQ, Xu H, Sun DX, Yu Y, Zhong YQ, Int. J. Nanomedicine 2016, 11, 3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Burstein HJ, Engl N. J. Med 2005, 353, 1652. [DOI] [PubMed] [Google Scholar]
- [114].Dustin ML, Springer TA, J. Cell Biol 1988, 107, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lin JJ, Ghoroghchian PP, Zhang Y, Hammer DA, Langmuir 2006, 22, 3975. [DOI] [PubMed] [Google Scholar]
- [116].Kim DH, Klibanov AL, Needham D, Langmuir 2000, 16, 2808. [Google Scholar]
- [117].Lin JJ, Silas JA, Bermudez H, Milam VT, Bates FS, Hammer DA, Langmuir 2004, 20, 5493. [DOI] [PubMed] [Google Scholar]
- [118].Lai MH, Lee S, Smith CE, Kim K, Kong H, ACS Appl. Mater. Interfaces 2014, 6, 10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H, Nano Today 2015, 10, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Chen F, Wang G, Griffin JI, Brenneman B, Banda NK, Holers VM, Backos DS, Wu L, Moghimi SM, Simberg D, Nat. Nanotechnol 2017, 12, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Liu Y, Palmans ARA, in Single-Chain Polym. Nanoparticles, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, pp. 183–216. [Google Scholar]
- [122].Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE, J. Control. Release 2003, 90, 323. [DOI] [PubMed] [Google Scholar]
- [123].Liu X, Yaszemski MJ, Lu L, Biomater. Sci 2016, 4, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Chambon P, Blanazs A, Battaglia G, Armes SP, Langmuir 2012, 28, 1196. [DOI] [PubMed] [Google Scholar]
- [125].Sun H, Meng F, Cheng R, Deng C, Zhong Z, Acta Biomater. 2014, 10, 2159. [DOI] [PubMed] [Google Scholar]
- [126].Stubbs M, McSheehy PMJ, Griffiths JR, Bashford CL, Mol. Med. Today 2000, 6, 15. [DOI] [PubMed] [Google Scholar]
- [127].Madsen J, Canton I, Warren NJ, Themistou E, Blanazs A, Ustbas B, Tian X, Pearson R, Battaglia G, Lewis AL, Armes SP, J. Am. Chem. Soc 2013, 135, 14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wayakanon K, Thornhill MH, Douglas CWI, Lewis AL, Warren NJ, Pinnock A, Armes SP, Battaglia G, Murdoch C, FASEB J. 2013, 27, 4455. [DOI] [PubMed] [Google Scholar]
- [129].Liu G-Y, Lv L-P, Chen C-J, Liu X-S, Hu X-F, Ji J, Soft Matter 2011, 7, 6629. [Google Scholar]
- [130].Chen W, Meng F, Cheng R, Zhong Z, J. Control. Release 2010, 142, 40. [DOI] [PubMed] [Google Scholar]
- [131].Quadir MA, Morton SW, Deng ZJ, Shopsowitz KE, Murphy RP, Epps TH, Hammond PT, Mol. Pharm 2014, 11, 2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Massignani M, Lopresti C, Blanazs A, Madsen J, Armes SP, Lewis AL, Battaglia G, Small 2009, 5, 2424. [DOI] [PubMed] [Google Scholar]
- [133].Massignani M, Canton I, Sun T, Hearnden V, MacNeil S, Blanazs A, Armes SP, Lewis A, Battaglia G, PLoS One 2010, 5, e10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Canton I, Massignani M, Patikarnmonthon N, Chierico L, Robertson J, Renshaw SA, Warren NJ, Madsen JP, Armes SP, Lewis AL, Battaglia G, FASEB J. 2013, 27, 98. [DOI] [PubMed] [Google Scholar]
- [135].Forman HJ, Zhang H, Rinna A, Mol. Aspects Med 2009, 30, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Gamcsik M, Kasibhatla M, Teeter S, Colvin O, Biomarkers 2012, 17, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Cerritelli S, Velluto D, Hubbell JA, Biomacromolecules 2007, 8, 1966. [DOI] [PubMed] [Google Scholar]
- [138].Park KM, Lee DW, Sarkar B, Jung H, Kim J, Ko YH, Lee KE, Jeon H, Kim K, Small 2010, 6, 1430. [DOI] [PubMed] [Google Scholar]
- [139].Wang S, Chen Z-R, Photochem. Photobiol. Sci 2017, 16, 155. [DOI] [PubMed] [Google Scholar]
- [140].Lee JS, Groothuis T, Cusan C, Mink D, Feijen J, Biomaterials 2011, 32, 9144. [DOI] [PubMed] [Google Scholar]
- [141].Haas S, Hain N, Raoufi M, Handschuh-Wang S, Wang T, Jiang X, Schönherr H, Biomacromolecules 2015, 16, 832. [DOI] [PubMed] [Google Scholar]
- [142].Li Y, Liu G, Wang X, Hu J, Liu S, Angew. Chemie - Int. Ed 2016, 55, 1760. [DOI] [PubMed] [Google Scholar]
- [143].Huang WC, Chiang WH, Cheng YH, Lin WC, Yu CF, Yen CY, Yeh CK, Chern CS, Chiang CS, Chiu HC, Biomaterials 2015, 71, 71. [DOI] [PubMed] [Google Scholar]
- [144].Huang WC, Shen MY, Chen HH, Lin SC, Chiang WH, Wu PH, Chang CW, Chiang CS, Chiu HC, J. Control. Release 2015, 220, 738. [DOI] [PubMed] [Google Scholar]
- [145].Kamat NP, Robbins GP, Rawson J, Therien MJ, Dmochowski IJ, Hammer DA, Adv. Funct. Mater 2010, 20, 2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Griepenburg JC, Sood N, Vargo KB, Williams D, Rawson J, Therien MJ, Hammer DA, Dmochowski IJ, Langmuir 2015, 31, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Li L, Rosenthal M, Zhang H, Hernandez JJ, Drechsler M, Phan KH, Rütten S, Zhu X, Ivanov DA, Möller M, Angew. Chemie 2012, 124, 11784. [DOI] [PubMed] [Google Scholar]
- [148].Wang X, Hu J, Liu G, Tian J, Wang H, Gong M, Liu S, J. Am. Chem. Soc 2015, 137, 15262. [DOI] [PubMed] [Google Scholar]
- [149].Peyret A, Ibarboure E, Tron A, Beauté L, Rust R, Sandre O, McClenaghan ND, Lecommandoux S, Angew. Chemie - Int. Ed 2017, 56, 1566. [DOI] [PubMed] [Google Scholar]