

The lithiophilicity of carbon materials is explained by the electronegativity, local dipole, charge transfer, and Li bond theory.
Abstract
The uncontrollable growth of lithium (Li) dendrites seriously impedes practical applications of Li metal batteries. Various lithiophilic conductive frameworks, especially carbon hosts, are used to guide uniform Li nucleation and thus deliver a dendrite-free composite anode. However, the lithiophilic nature of these carbon hosts is poorly understood. Herein, the lithiophilicity chemistry of heteroatom-doped carbon is investigated through both first principles calculations and experimental verifications to guide uniform Li nucleation. The electronegativity, local dipole, and charge transfer are proposed to reveal the lithiophilicity of doping sites. Li bond chemistry further deepens the understanding of lithiophilicity. The O-doped and O/B–co-doped carbons exhibit the best lithiophilicity among single-doped and co-doped carbons, respectively. The excellent lithiophilicity achieved by O-doping carbon is further validated by Li nucleation overpotential measurement. This work uncovers the lithiophilicity chemistry of heteroatom-doped carbons and affords a mechanistic guidance to Li metal anode frameworks for safe rechargeable batteries.
INTRODUCTION
With the ever-increasing energy demands from the current world, batteries with high energy densities are highly required. Lithium (Li) metal anodes, with an ultrahigh theoretical specific capacity (3810 mAh g−1) and the lowest negative electrochemical potential (−3.040 V versus the standard hydrogen electrode), are strongly considered as the ultimate anode choice for Li batteries (1–3). However, the practical application of Li metal anodes faces huge challenges, such as the growth of Li dendrites, the low Coulombic efficiency, and the infinite relative volume change during cycling (4). In particular, the uncontrollable growth of Li dendrites, which is considered the most fundamental issue facing Li metal anodes and has attracted much more attention, not only induces the “dead Li” with severe capacity loss but also can penetrate through the separator, inducing an internal short circuit and other potential safety hazards (5, 6).
To address the challenging issue of Li dendrites, several strategies have been proposed, including the construction of three-dimensional (3D) conductive frameworks to guide a uniform Li deposition (7–9), the modification of electrolytes to stabilize the solid electrolyte interphase (SEI) (10–13), and the introduction of high-modulus artificial SEI or solid-state electrolytes to prevent dendrite penetration (14–16). Among them, a desirable conductive framework is supposed to simultaneously resist the growth of Li dendrites and address the infinite relative volume change induced by the “hostless” Li plating/stripping at a practical areal capacity (>4 mAh cm−2). In particular, a variety of carbon materials, including carbon paper (17), carbon nanotubes (18), graphite/graphited carbon (19, 20), graphene (7, 21), and (reduced) graphene oxides (GOs) (22, 23), have been applied as the Li metal anode hosts. For instance, Guo and co-workers demonstrated that a 3D hollow carbon fiber container improves Li deposition behavior (24). To modify the lithiophilicity of routine carbon materials, various doping strategies have been adopted. For instance, nitrogen-doped graphene has been demonstrated to have excellent lithiophilicity to guide uniform Li nucleation, achieving dendrite-free morphology and improved cycling Coulombic efficiency (7). Besides, Cui and co-workers (25) proposed a lithiophilic reduced GO (rGO) with a strong binding interaction toward Li and thus reduced the barrier of heterogenous Li nucleation.
Although a variety of lithiophilic hosts has been proposed and indicate superior performance in suppressing dendritic Li growth, the exact working mechanism between Li and the lithiophilic hosts is tightly sealed and the lithiophilic nature of these carbon hosts is far from clear. If a fundamental understanding of the lithiophilicity chemistry is built, then the rational design of lithiophilic frameworks for dendrite-free Li metal anodes can be achieved.
In this contribution, we probe the lithiophilicity chemistry of heteroatom-doped carbon to guide uniform heterogeneous Li nucleation through both first principles calculations and experimental verifications. Three key factors (electronegativity, local dipole, and charge transfer) are proposed to reveal the lithiophilicity origin of heteroatom-doping sites in carbon materials. O doping exhibits the best performance among all single-doping strategies, which was further validated by Li nucleation overpotential tests and transmission electron microscopy (TEM) characterizations. In addition, O/B co-doping is predicted to achieve better lithiophilicity than any single doping due to its large local dipole. Inspired from the Li bond theory, 7Li nuclear magnetic resonance (NMR) spectroscopy can determine the lithiophilicity of host materials as the linear relationship between Li binding energy and 7Li chemical shift. This work deepens the understanding of lithiophilicity and affords fruitful insights into the rational design of Li metal anode frameworks.
RESULTS AND DISCUSSION
Modeling
In general, a Li ion in electrolyte is driven to a lithiophilic site on the anode framework surface by the electric force, the Li ion concentration gradient, and the electrostatic interaction between the Li ion and the electronegative site in the framework. The third of these factors plays the major role when the Li ion arrives near the host and forms a chemical bond, and it can be precisely regulated by the surface chemistry of anode frameworks. Once adsorbed on the lithiophilic sites, Li ions are reduced to Li metal due to charge transfer (Fig. 1).
Fig. 1. Schematic representation of the Li nucleation on conductive frameworks.
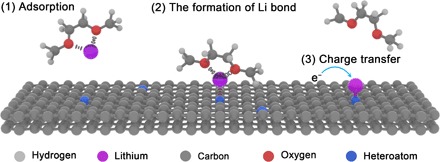
Li nucleation is a very complicated process, controlled by both thermodynamic and kinetic factors. Thermodynamically speaking, nucleation is governed by the decrease in free energy due to phase transition and the increase in surface energy due to the creation of a new interface. The former is the nucleation driving force, while the later is the origin of nucleation barrier. According to the classical heterogeneous nucleation theory, the introduction of the framework will not change the critical nucleation radius (r*) but can lower the nucleation barrier and reduce the nucleation volume needed to reach r* (fig. S1). A larger binding energy can render a lower Li nucleation barrier and, thus, a smaller nucleation overpotential. Therefore, the binding energy between lithium and framework is suggested as the quantitative descriptor of lithiophilicity of anode framework.
To understand the above Li nucleation process and thus unveil the lithiophilicity chemistry, first principles calculations were conducted first to investigate the interaction between Li and conductive carbon frameworks (fig. S2). For modeling, a series of heteroatom-doped graphene nanoribbons (GNRs) were constructed. Specifically, 20 species (pristine or heteroatom doped) were modeled with various dopant forms, including graphitic boron in the bulk phase (bgB), graphitic boron on the edge (egB), B–2C–O–type boron (oB), quaternary nitrogen in the bulk phase (bqN), quaternary nitrogen on the edge (eqN), pyrrolic nitrogen (rN), pyridinic nitrogen (pN), carboxylic group (aO), cyclic oxygen (cO), epoxy group (eO), hydroxyl group (hO), ketone group (kO), fluorine (F), chlorine (Cl), bromine (Br), iodine (I), phosphorus (P), sulfur (S), and sulfonyl group (oS) (Fig. 2, A and B, and figs. S3 to S7) (26–28). GNRs with defects consisting of five-carbon rings (C5), seven-carbon rings (C7), and five-carbon rings adjacent to seven-carbon rings (C5 + C7) on the edge were also considered (fig. S8). 2D single/multilayered graphene and graphite models were also considered.
Fig. 2. Modeling and the summary of Li binding energy.
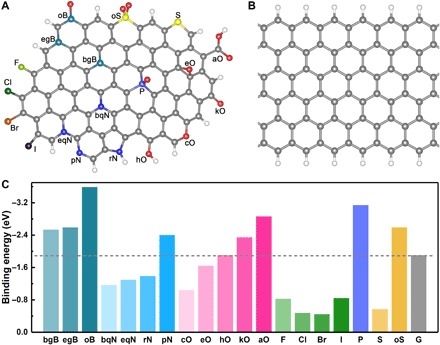
(A) Modeling of heteroatom-doped carbons. (B) Prinstine GNR model. (C) Summary of calculated binding energy between heteroatom-doped carbon and a Li atom. The hydrogen, boron, carbon, nitrogen, oxygen, fluorine, phosphorus, sulfur, chlorine, bromine, and iodine atoms are marked as white, green, gray, blue, red, cyan, wathet, yellow, bottle green, orange, and black, respectively.
Binding energy and charge analyses
The binding energy, geometrical configuration, bond length, and charge density difference were explicitly examined (Figs. 2C, 3, and 4). The binding energies (Eb) are summarized in Fig. 2C and table S1. The adsorption of a Li atom is quite sensitive to dopant elements. GNRs with single doping of bgB, egB, pN, kO, or aO dopant exhibit a larger binding energy (ranging from −2.35 to −2.86 eV) than that of pristine GNR (−1.91 eV). The other O/N-doped GNRs except eO-GNR, such as bqN (Fig. 3E), eqN (Fig. 3F), and cO (Fig. 3J), exhibit a smaller binding energy than pristine GNR, which is induced by the odd electrons in these adsorption models. The eO-GNR interacts with a Li atom from the opposite direction of the functional groups with respect to the carbon plane (Fig. 3K), which is responsible for the small binding energy of eO-GNR toward a Li atom. Therefore, an appropriate introduction of heteroatom into carbon hosts is beneficial for Li heterogeneous nucleation, which is consistent with previous experimental results that N/O-doped carbon can significantly reduce the Li nucleation overpotential and thus render a uniform Li deposition at the lithiophilic sites (7, 19).
Fig. 3. The optimized interaction geometry of a Li atom and heteroatom-doped GNRs.
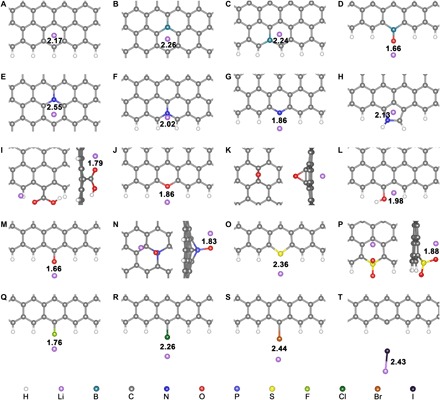
(A) Pristine, (B) bgB-, (C) egB-, (D) oB-, (E) bqN-, (F) eqN-, (G) rN-, (H) pN-, (I) aO-, (J) cO-, (K) eO-, (L) hO-, (M) kO-, (N) P-, (O) S-, (P) oS-, (Q) F-, (R) Cl-, (S) Br-, and (T) I-GNRs. The hydrogen, lithium, boron, carbon, nitrogen, oxygen, fluorine, phosphorus, sulfur, chlorine, bromine, and iodine atoms are marked as white, purple, green, gray, blue, red, cyan, wathet, yellow, bottle green, orange, and black, respectively. The distances between the Li atom and the adjacent doping atom are marked with black numbers (unit: Å).
Fig. 4. The Bader charge and differential charge analyses.
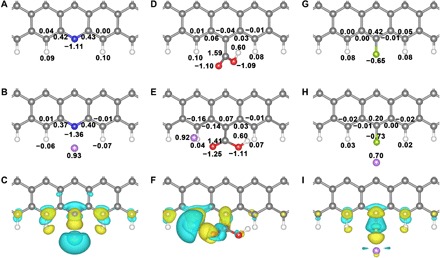
The Bader charge distribution before and after adsorption, and the corresponding differential charge density of Li adsorption site of (A to C) pN-, (D to F) aO-, and (G to I) F-GNRs, respectively. The black numbers are the Bader charge of the nearest atoms. The yellow and cyan surfaces correspond the charge gain and lost regions, respectively (isovalue, 0.0025). The hydrogen, lithium, carbon, nitrogen, oxygen, and fluorine atoms are marked as white, purple, gray, blue, red, and cyan, respectively.
Among various single-doping GNRs, aO-GNR (Fig. 3I) exhibits the largest binding energy toward a Li atom. Because of the higher electronegativity of the O atom (3.44) against the Li atom (0.98), the interaction between O and Li atoms is very analogous to the “Li bond” that is well explained by the Lewis acid-base theory (27, 29). The aO-GNR with an extra pair of electrons in the carboxylic functional group site is expected as an electron-rich donor that naturally acts as Lewis base sites to strongly absorb Lewis acidic Li ions through acid-base interactions. The length of the as-formed Li–O bond (1.79 Å; Fig. 3I) is even shorter than that in Li2O crystal (2.02 Å; fig. S9), indicating the strong interaction between the Li atom and aO-GNR.
The dopant sites in kO-GNR (Fig. 3M) and pN-GNR (Fig. 3G) are also electronegative and interact with Li ions through a similar mechanism. However, the B atoms in bgB-GNR (Fig. 3B) and egB-GNR (Fig. 3C) are electropositive, and the adjacent carbon atoms are electronegative (fig. S10) due to the smaller electronegativity of the B atom (2.04) against the C atom (2.55). The electronegative carbon sites are preferable to pristine GNR (Fig. 3A), which results in enhanced Li adsorption on bgB-GNR and egB-GNR.
In general, a larger binding energy toward precursor nuclei is advantageous to the heterogeneous nucleation process with a smaller surface tension against the substrate and, thus, a reduced nucleation energy barrier (27). Therefore, carbon hosts with precise O, N, and B functional groups are expected to reduce the Li nucleation overpotential.
On the contrary, other investigated single-doping elements (F, Cl, Br, I, and S) are unsatisfactory to afford adequate interactions toward a Li atom. Although the F atom has the highest electronegativity (3.98), its chemical state in GNR is very different from that of O and N doping sites. All these three atoms form σ-bonds with adjacent carbon and withdraw electrons through inductive effects. However, both O and N atoms participate in the delocalized π system of GNR that additionally contributes to their negative charge states. In the contrast, the filled p orbitals of the F atom form a p–π conjugation with the carbon plane that feedbacks electrons from the F atom to carbon (27). As a result, the F atom is less electronegative than the N and O atoms, which is further validated by the Bader charge analysis (Fig. 4, A, D, and G). Therefore, F-GNR can only afford a weak interaction toward a Li ion (Fig. 3Q). The atomic radii of S, Cl, and Br atoms are much larger than that of a Li ion, which leads to the weak interaction between Li ions and S/Cl/Br-GNR (Fig. 3, O, R, and S). The situation of I-GNR is even worse, as lithium can destroy the structure of I-GNR because the I–C bond is very weak (Fig. 3T). A similar behavior can be found in eO-GNR species (fig. S11).
To investigate the stability of these Li adatoms on doped carbon materials, the adsorption of lithium on Li (100) and (110) surfaces and the interaction between lithium and electrolyte solvents were further probed. The binding energies of lithium on Li (100) and (110) surfaces are −1.33 and −1.41 eV, respectively (table S2). These binding energies are much smaller than that of aO, pN, egB, and bgB doping but larger than that of F, Cl, Br, I, and S doping. Additionally, the binding energies between lithium and a single electrolyte solvent are smaller than −1.0 eV (fig. S12), while the binding energy between lithium and multi-electrolyte solvents can reach to as large as −2.36 eV (fig. S13), which is larger than that of most doping sites. These results additionally enhance the important role of specific doping decoration on carbon materials, especially considering the stability of Li adatom in electrolyte conditions.
The principle of electronegativity and local dipole
Although the elemental electronegativity in combination with the Lewis acid-base theory can well explain why different heteroatoms enjoy varied lithiophilicity, it faces huge challenges when comparing different doping functional groups. This is because the Li affinity is not only determined by the acid-base interaction but also influenced by other factors, such as the local geometrical and electronic structures of the dopant. Herein, the “local dipole,” which is defined as the dipole formed by the doping atom and its adjacent carbon atom in GNR, is further considered. A strong local dipole not only renders a strong ion-dipole force toward Li ions but also delivers an induced dipole to the absorbed Li ions. Therefore, the local dipole can further enhance the anchoring effects. With an overall consideration of elemental electronegativity and the as-defined local dipole (table S3), an implicit correlation between binding energy and “log (0.5 × electronegativity + local dipole)” was proposed (Fig. 5A). A dopant with a large electronegative doping atom and a strong local dipole absolutely delivers a large binding energy.
Fig. 5. The proposed principles for designing highly lithiophilic Li frameworks.
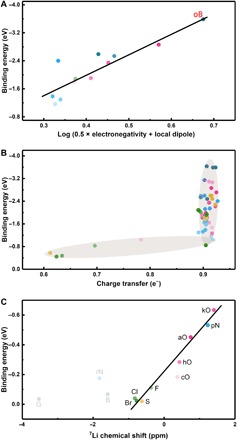
The correlation between Li atom binding energy and (A) log (0.5 × electronegativity + local dipole), (B) charge transfer, and (C) theoretical 7Li chemical shift.
Inspired from the above proposed descriptor, a co-doping strategy is naturally considered as a large local dipole. Elements with a smaller electronegativity against carbon, such as boron, are supposed to induce a stronger local dipole with adjacent oxygen atom. Actually, oB-GNR has a very large local dipole of 3.21 e− Å (table S3) and the largest binding energy (−3.59 eV) among all GNRs herein (Figs. 2C and 3D). Besides, O–S/P co-doped GNRs (Figs. 2C and 3, N and P) also exhibit a large binding energy of −2.60 and −3.14 eV, respectively, which is larger than all single-doping GNR species except aO-GNR. Phosphate-functionalized rGO has been recently demonstrated to strongly bind with lithium and thus achieve low nucleation overpotentials by Archer and co-workers (30), which can validate our proposed P/O the co-doping strategy. Consequently, all these results confirm the reliability of the co-doping strategy and thus validate the proposed log (0.5 × electronegativity + local dipole) principle.
The principle of charge transfer
On the basis of a fundamental understanding on the Li adsorption process, the charge transfer is further investigated through Bader charge and charge density difference analyses. As illustrated in Fig. 4 (B, E, and H), there is an evident charge transfer above 0.9 e− from Li to pN/aO-GNR, which is around 0.2 e− larger than that of F-GNR. The transferred charge is delocalized in the conjugation structure. The charge density difference analysis further confirms that the charge transfer between Li and N/O atoms is obviously increased, indicating the formation of a strong Li-N/O interaction (Fig. 4, C and F). However, the charge gain between Li and F atoms is less palpable, further confirming the lessened charge transfer between Li and F-GNR (Fig. 4I). Therefore, an efficient charge transfer is necessary for a large binding energy.
To determine the critical value of an efficient charge transfer, the correlation between binding energy and charge transfer is further probed. As indicated in Fig. 5B, when charge transfer increases up to 0.9 e−, a surge of the binding energy is sparked. Therefore, a critical charge transfer around 0.9 e− is determined. When charge transfer is below this critical value, the binding energy is absolutely small, indicating the critical role of charge transfer during heterogeneous Li nucleation.
Li bond and NMR
According to lithium bond chemistry, the chemical shift of 7Li NMR spectroscopy is suggested to serve as a quantitative descriptor of Li bond strength in Li-S batteries. Therefore, NMR spectroscopy calculation was conducted to probe the Li nucleation at the anode side by characterizing the formed Li bond when the solvated Li ions interact with lithiophilic sites during the initial plating process. To calculate the 7Li chemical shift, benzene, borabenzene, pyridine, pyrrole, benzoic acid, furan, phenol, quinomethan, thiophene, fluorobenzene, chlorobenzene, and bromobenzene molecules were selected as modeling molecules of the corresponding pristine, egB-, pN-, rN-, aO-, cO-, hO-, kO-, S-, F-, Cl-, and Br-GNRs, respectively (figs. S14 and S15). Although the calculated binding energies between these small molecules and the Li+–1,2-dimethoxyethane complex are different from that calculated on the basis of the GNR models due to change of calculation models and methods, the results mostly agree with each other.
In the cluster calculations, the aO, pN, and kO exhibited the largest binding energy, followed by hO, cO, and rN. The F, Cl, Br, and S can only afford a very weak binding toward the Li ion, similar to G (benzene) (Fig. 5C). The only point where the cluster and periodic model diverge was the borabenzene- and boron-doped GNR species. The divergence is induced by the very different local chemical environments around B atoms in these two models (figs. S3 and S14A). Despite this disparity, when we correlated the binding energy and 7Li chemical shift collected from the cluster models, an excellent linear relationship was obtained (Fig. 5C). The 7Li chemical shift is a quantitative descriptor of the electron shielding effects on the 7Li nucleus. In detail, when the Li ions in G, B, and rN clusters interact with the overall quintuple/hexatomic ring, the electron shielding effects are thus enhanced, resulting in a more negative 7Li chemical shift. Consequently, as demonstrated by the cluster model calculations, 7Li chemical shift can be used to evaluate the interaction strength between Li ions and lithiophilic sites, affording fruitful insights into the Li nucleation process.
Theoretically predicted rules for designing highly lithiophilic frameworks
According to the insights gained from Fig. 5 (A to C), we can propose several rules for creating a strong anchoring effect toward Li ions for doped carbons:
(1) The doping atom should form an electronegative site (the doping atom or the adjacent carbon atom) as a Lewis base to interact with the Lewis acidic Li ions. A more negative charge state site is thus supposed to induce a stronger interaction toward Li ions.
(2) The doping atom should form a strong local dipole to induce a strong ion-dipole force and generate an induced dipole around the absorbed Li ions to further enlarge the binding energy. A co-doping strategy (e.g., O-B/S/P co-doping) is inferred to deliver a larger binding energy than single-doping of carbons.
(3) An efficient charge transfer above 0.9 e− is necessary to achieve a strong binding interaction. An electron-deficient site or a doping atom with large electronegativity is thus preferred.
(4) The 7Li chemical shift can be used as another quantitative descriptor to judge the interaction strength between Li ions and carbon hosts. The NMR technique affords emerging chance for bridging the gap between theory and experiment to screen Li metal anode frameworks.
On the basis of these considerations, O is better than B, N, and F as a single dopant to guide the uniform Li nucleation. The O–B/S/P co-doping can even achieve a stronger interaction toward Li ions than any single doping. These principles acquired from single-layered GNR models are also applicable to 2D multilayered graphene and graphite models (figs. S16 and 17).
Experimental verifications
To validate the reliability of the above predicted principles from first principles calculations, Li nucleation overpotential tests were used to evaluate the lithiophilicity on three kinds of graphene materials: pristine graphene (G), nitrogen-doped graphene (NG), and oxygen-containing graphene (OG). According to the classical nucleation and growth theory, the driving force for the electrocrystallization process can be divided into the reaction potential, charge transfer overpotential, diffusion overpotential, and crystallization overpotential (31–33). Under a small nucleation current, the variation of charge transfer overpotential and diffusion overpotential could be ignored. Therefore, the as-obtained nucleation overpotential mainly changes with the crystallization overpotential, which is determined by the heterogeneous nucleation energy barrier (lithiophilicity) of the electrode surface. As shown in Fig. 6A, the G, NG, and OG samples have a nucleation overpotential of 22.5, 19.4, and 15.0 mV, respectively, illustrating that OG affords the lowest nucleation energy barrier and enjoys the best lithiophilicity. Consequently, the lithiophilicity of O-doped carbons predicted by the above-mentioned principles is validated.
Fig. 6. Li nucleation characterizations.
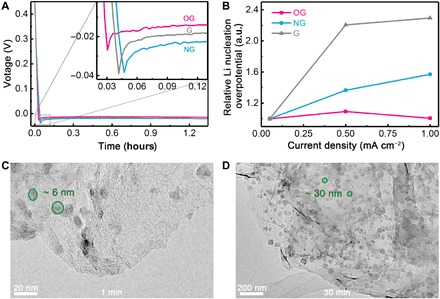
(A) Voltage-time curves during Li nucleation at 0.50 mA cm−2 on G, NG, and OG electrodes. (B) Relative Li nucleation overpotentials on G, NG, and OG electrodes at different current densities (0.05, 0.50, and 1.00 mA cm−2). The nucleation overpotential at 0.05 mA cm−2 is set to be 1. TEM images of Li deposits at the current density of 0.5 mA cm−2 at (C) 1 and (D) 30 min, respectively. a.u., arbitrary units.
When comparing the Li deposition at different current densities ranging from 0.05 to 1.00 mA cm−2, the nucleation overpotential of NG and G enlarges 1.57 and 2.29 times, respectively, showing the significant influence of current density (Fig. 6B and fig. S18). However, OG exhibits a very steady nucleation overpotential due to its super lithiophilicity though the current density is increased by 20 times. Besides, the specific surface areas of OG, NG, and G are 758, 1166, and 604 m2 g−1, respectively (figs. S19 and S20), indicating that the low overpotential of OG does not originate from a large specific surface area to reduce local current density but its excellent lithiophilicity. To exclude the influence of different amounts of defects in G, NG, and OG (fig. S21), GNRs with C5, C7, and C5 + C7 defects, which can afford binding energies of −2.65, −1.89, and −1.96 eV, respectively, were also considered during first principles calculations (figs. S22 and S23). The binding energy of C5 is larger than that of pristine graphene (−1.91 eV) but smaller than that of aO (−2.85). Edge also has a certain impact on binding energy as lithium prefers to be adsorbed on the edge (−1.91 eV) of GNR rather than the bulk region (−1.66 eV). Besides, the binding energy of the pristine GNR (−1.91 eV) is larger than that of pristine 2D periodic graphene (−1.15 eV). These results agree with recent report that edge-rich graphene nanosheets are beneficial for enhancing the surface electronegativity, attract Li ions, and promote the Li nucleation (34). However, all these binding energies are smaller than that of aO (−2.86 eV), indicating O doping but not edge or defect plays the major role in reducing nucleation overpotential. Therefore, the lithiophilicity of the Li metal host is the most important factor in determining the Li nucleation overpotential, and designing a highly lithiophilic conductive framework is thus of great significance to practical Li metal anodes at large current densities.
The Li nucleating and plating processes induced by the OG electrode were further illustrated by TEM characterizations (Fig. 6, C and D). The metallic Li deposited at a current density of 0.50 mA cm−2. After only 1.0 min of plating, uniform Li nucleation sites emerge on the surface of OG due to its excellent lithiophilicity. After 30 min of plating, the nucleation sites grow from 6 to 30 nm but keep a uniform morphology, further confirming the important role of lithiophilicity in the Li nucleation process.
CONCLUSIONS
The lithiophilicity chemistry of heteroatom-doped carbons as heterogeneous Li deposition substrates was probed through first principles calculations and experimental characterizations. Three key factors account for the excellent lithiophilicity achieved by B, N, and O single-doping and O-B/S/P co-doping strategies, including the electronegativity, the local dipole, and the charge transfer. Inspired from lithium bond chemistry, the 7Li chemical shift is further validated as a quantitative descriptor to determine binding strength between solvated Li ions and carbons. On the basis of these principles, O doping and O/B co-doping are predicted to produce the best lithiophilicity among single-doped and co-doped carbons, respectively. The Li nucleation overpotential tests further demonstrate the excellent lithiophilicity induced by O doping carbon frameworks, which can induce a uniform Li deposition and thus dendrite-free metal anodes, confirmed by TEM images. This work reveals the lithiophilic nature of heteroatom-doped carbons and renders a rational strategy of designing lithiophilic frameworks for Li metal anodes with a low Li nucleation overpotential, dendrite-free morphology, and thus safe batteries with Li metal anodes.
MATERIALS AND METHODS
Computational details
The periodic density functional theory calculation was conducted in the Vienna Ab initio Simulation package (35), and the results were visualized in the Visualization for Electronic and Structural Analysis (36) program. The projector augmented wave (37) pseudopotentials and the Perdew-Burk-Ernzerhof generalized gradient approximation functional (38) were adopted. The energy cutoff was set to 520 eV (fig. S2A). The self-consistent field and geometry convergence tolerance were set to 1 × 10−5 and 1 × 10−4 eV, respectively.
Considering the electrical conductivity and stability, zigzag rather than armchair GNR with a 20-Å vacuum layer in both the slip direction and normal direction was chosen as the basic model. The decorations with B, N, O, F, P, S, Cl, Br, and I dopants were further systematically investigated. A G-centered k-point grid through the Monkhorst-Pack method was set to 6 × 1 × 1 for GNR models (fig. S2B). The binding energy between GNRs and a Li atom was defined as following
where Etotal, EG, and ELi are the total energy of GNR bound with a Li atom, the pristine GNR, and a single Li atom, respectively.
Cluster-based calculations were conducted using the Gaussian (G09) (39) suite of programs. Becke’s three-parameter hybrid method using the Lee-Yang-Parr correlation functional (B3LYP) was chosen in this study. Geometries were optimized and vibrational modes were calculated in G09 at a B3LYP/6-311++G(2d,p) level of theory. The 7Li chemical shifts were calculated using fully optimized geometry with the integral equation formalism variant of the Polarizable Continuum Model as implemented with parameters of dielectric constant ε = 7.2 and solvent radius of 4.1 Å, and referenced with respect to hydrated Li+, i.e., [Li (3H2O)]+, the same method adopted in our previous studies (29, 40).
Experimental details
The graphene (G) and oxygen-containing graphene (OG) were obtained by thermal reduction of GO, which was prepared by a modified Hummers method at 1000° and 250°C, respectively. The N-doped graphene (NG) here was obtained by treating the as-obtained graphene (G) with NH3 flowing in a quartz tube.
Two-electrode cells (Li versus G/NG/OG) were used with standard CR2025 coin-type cells (Shenzhen Kejingstar Technology Ltd.), which were assembled in an Ar-filled glove box with O2 and H2O content below 1.0 ppm. The G/NG/OG powder and polyvinylidene fluoride binder (PVDF) (mass ratio of G/NG/OG:PVDF = 5:1) were stirred into a slurry in N-methylpyrrolidone for 24 hours. The slurry was coated onto a Cu foil and dried in a vacuum drying oven at 60°C for 6.0 hours. The as-obtained G/NG/OG-coated Cu foil was punched into disks with a diameter of 13.0 mm as the working electrode. A 0.5-mm-thick Li metal foil with a diameter of 16.0 mm (China Energy Lithium Co. Ltd.) was used as the counter electrode. A 1,3-dioxolane/1,2-dimethoxyethane (v/v = 1:1) (Beijing Chemical Industry Group Co. Ltd.) electrolyte within 1.0 M LiTFSI salt was used in each coin cell.
The coin cells were monitored in galvanostatic mode in Land CT2001 multichannel battery tester. In each galvanostatic cycle, the discharge time was fixed in the lithium plating process, and the charge time in the lithium stripping process was controlled by a cutoff voltage. The nucleation tests with a low current density of 0.5 mA cm−2 were performed on Solartron 1470E electrochemical workstation (Solartron Analytical, UK) for 2 hours.
The morphology was characterized by a JEM 2010 (JEOL Ltd., Tokyo, Japan) TEM operated at 120.0 kV. N2 isotherms of OG, NG, and G were obtained at −196°C (77 K) with an Autosorb-IQ2-MPC system (Quantachrome, USA). The specific surface area was determined by the Brunauer-Emmett-Teller method.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Key Research and Development Program (2016YFA0202500 and 2016YFA0200102) and the National Natural Scientific Foundation of China (21676160, 21825501, and 21808121), the China Postdoctoral Science Foundation (2018M631480 and BX201700125), and the Tsinghua University Initiative Scientific Research Program. The authors acknowledged the support from Tsinghua National Laboratory for Information Science and Technology for theoretical simulations. We thank J. Q. Huang and H. J. Peng for the helpful discussions. Author contributions: Q.Z. conceived the study and supervised this work. X.C. and Q.Z. proposed the concept. X.C. carried out the theoretical calculation, while T.-Z.H. was involved into Bader charge distribution analysis. X.-R.C. synthesized and characterized the host materials. X.-R.C., B.-Q.L., X.-B.C., and R.Z. evaluated the electrochemical performance. X.C. and Q.Z. wrote the paper. All authors contributed to the discussion. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaau7728/DC1
Fig. S1. Schematic representation of the Li nucleation on conductive frameworks based on the classical heterogeneous nucleation model.
Fig. S2. Periodic calculation method test.
Fig. S3. The optimized configuration of carbons with bgB, egB, and oB dopant.
Fig. S4. The optimized configuration of carbons with bqN, eqN, pN, and rN dopant.
Fig. S5. The optimized configuration of carbons with aO, cO, eO, hO, and kO dopant.
Fig. S6. The optimized configuration of carbons with F, Cl, Br, and I dopant.
Fig. S7. The optimized configuration of carbons with P, oS, and S dopant.
Fig. S8. The optimized configuration of carbons with C5, C7, and C5+C7 defects.
Fig. S9. The optimized crystal structures of Li2O and Li3N.
Fig. S10. The Bader charge analyses of bgB- and egB-GNRs.
Fig. S11. The optimized interaction geometry of a Li atom interacts with eO-GNR.
Fig. S12. The optimized structures and binding energies between a Li atom and a solvent molecule.
Fig. S13. The optimized structures and binding energies between a Li atom and a multisolvent molecule.
Fig. S14. The optimized structures of borabenzene, pyridine, pyrrole, benzoic acid, furan, phenol, quinomethan, thiophene, fluorobenzene, chlorobenzene, and bromobenzene molecules.
Fig. S15. The optimized interaction geometries of Li+–DME complex and borabenzene, pyridine, pyrrole, benzoic acid, furan, phenol, quinomethan, thiophene, fluorobenzene, chlorobenzene, and bromobenzene molecules.
Fig. S16. The optimized interaction geometry of a Li atom and single-/multilayered graphene and graphite.
Fig. S17. A summary of calculated binding energy between a Li atom and single/multilayered graphene and graphite.
Fig. S18. The Li nucleation overpotentials on G, NG, and OG electrodes at different current densities (0.05, 0.50, and 1.00 mA cm−2).
Fig. S19. The N2 isotherms of OG, NG, and G materials.
Fig. S20. The pore size distribution derived from the N2 isothermal adsorption plot of OG, NG, and G materials.
Fig. S21. The Raman spectra of OG, NG, and G materials.
Fig. S22. The optimized interaction geometry of Li atom on GNR with C5, C7, and C5+C7 defects.
Fig. S23. The summary of binding energy of a Li atom adsorbed on GNR with C5, C7, and C5 + C7 defects.
Table S1. A summary of binding energy between nanoribbons and a Li atom.
Table S2. A summary of Li binding energy on Li (100) and (110) surfaces and total energy per bulk lithium.
Table S3. A summary of local dipole in heteroatom-doped nanoribbons.
REFERENCES AND NOTES
- 1.Tarascon J.-M., Armand M., Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Chu S., Majumdar A., Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Cheng X.-B., Zhang R., Zhao C.-Z., Zhang Q., Toward safe lithium metal anode in rechargeable batteries: A review. Chem. Rev. 117, 10403–10473 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Albertus P., Babinec S., Litzelman S., Newman A., Status and challenges in enabling the lithium metal electrode for high-energy and low-cost rechargeable batteries. Nat. Energy 3, 16–21 (2018). [Google Scholar]
- 5.Xu W., Wang J., Ding F., Chen X., Nasybulin E., Zhang Y., Zhang J.-G., Lithium metal anodes for rechargeable batteries. Energy Environ. Sci. 7, 513–537 (2014). [Google Scholar]
- 6.Chen X., Hou T.-Z., Li B., Yan C., Zhu L., Guan C., Cheng X.-B., Peng H.-J., Huang J.-Q., Zhang Q., Towards stable lithium–sulfur batteries: Mechanistic insights into electrolyte decomposition on lithium metal anode. Energy Storage Mater. 8, 194–201 (2017). [Google Scholar]
- 7.Zhang R., Chen X.-R., Chen X., Cheng X.-B., Zhang X.-Q., Yan C., Zhang Q., Lithiophilic sites in doped graphene guide uniform lithium nucleation for dendrite-free lithium metal anodes. Angew. Chem. Int. Ed. 56, 7764–7768 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Yang C.-P., Yin Y.-X., Zhang S.-F., Li N.-W., Guo Y.-G., Accommodating lithium into 3D current collectors with a submicron skeleton towards long-life lithium metal anodes. Nat. Commun. 6, 8058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang R., Chen X., Shen X., Zhang X.-Q., Chen X.-R., Cheng X.-B., Yan C., Zhao C.-Z., Zhang Q., Coralloid carbon fiber-based composite lithium anode for robust lithium metal batteries. Joule 2, 764–777 (2018). [Google Scholar]
- 10.Zhang X. Q., Chen X., Cheng X.-B., Li B.-Q., Shen X., Yan C., Huang J.-Q., Zhang Q., Highly stable lithium metal batteries enabled by regulating the solvation of lithium ions in nonaqueous electrolytes. Angew. Chem. Int. Ed. 57, 5301–5305 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Zheng J. M., Yan P., Mei D., Engelhard M. H., Cartmell S. S., Polzin B. J., Wang C., Zhang J.-G., Xu W., Highly stable operation of lithium metal batteries enabled by the formation of a transient high-concentration electrolyte layer. Adv. Energy Mater. 6, 1502151 (2016). [Google Scholar]
- 12.Chen X., Shen X., Li B., Peng H.-J., Cheng X.-B., Li B.-Q., Zhang X.-Q., Huang J.-Q., Zhang Q., Ion–solvent complexes promote gas evolution from electrolytes on a sodium metal anode. Angew. Chem. Int. Ed. 57, 734–737 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Zhang X.-Q., Cheng X.-B., Chen X., Yan C., Zhang Q., Fluoroethylene carbonate additives to render uniform Li deposits in lithium metal batteries. Adv. Funct. Mater. 27, 1605989 (2017). [Google Scholar]
- 14.Tu Z. Y., Choudhury S., Zachman M. J., Wei S., Zhang K., Kourkoutis L. F., Archer L. A., Designing artificial solid-electrolyte interphases for single-ion and high-efficiency transport in batteries. Joule 1, 394–406 (2017). [Google Scholar]
- 15.Zhao C.-Z., Zhang X.-Q., Cheng X.-B., Zhang R., Xu R., Chen P.-Y., Peng H.-J., Huang J.-Q., Zhang Q., An anion-immobilized composite electrolyte for dendrite-free lithium metal anodes. Proc. Natl. Acad. Sci. U.S.A. 114, 11069–11074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li N. W., Shi Y., Yin Y.-X., Zeng X.-X., Li J.-Y., Li C.-J., Wan L.-J., Wen R., Guo Y.-G., A flexible solid electrolyte interphase layer for long-life lithium metal anodes. Angew. Chem. Int. Ed. 57, 1505–1509 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Lu Z., Zhang Z., Chen X., Chen Q., Ren F., Wang M., Wu S., Peng Z., Wang D., Ye J., Improving Li anode performance by a porous 3D carbon paper host with plasma assisted sponge carbon coating. Energy Storage Mater. 11, 47–56 (2018). [Google Scholar]
- 18.Wang Y., Shen Y., Du Z., Zhang X., Wang K., Zhang H., Kang T., Guo F., Liu C., Wu X., Lu W., Chen L., A lithium–carbon nanotube composite for stable lithium anodes. J. Mater. Chem. A 5, 23434–23439 (2017). [Google Scholar]
- 19.Liu L., Yin Y.-X., Li J.-Y., Wang S.-H., Guo Y.-G., Wan L.-J., Uniform lithium nucleation/growth Induced by lightweight nitrogen-doped graphitic carbon foams for high-performance lithium metal anodes. Adv. Mater. 30, 1706216 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Zuo T.-T., Wu X.-W., Yang C.-P., Yin Y.-X., Ye H., Li N.-W., Guo Y.-G., Graphitized carbon fibers as multifunctional 3D current collectors for high areal capacity Li anodes. Adv. Mater. 29, 1700389 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Liu S., Wang A., Li Q., Wu J., Chiou K., Huang J., Luo J., Crumpled graphene balls stabilized dendrite-free lithium metal anodes. Joule 2, 184–193 (2018). [Google Scholar]
- 22.Wang A., Tang S., Kong D., Liu S., Chiou K., Zhi L., Huang J., Xia Y.-Y., Luo J., Bending-tolerant anodes for lithium-metal batteries. Adv. Mater. 30, 1703891 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Liu Y., Lin D., Jin Y., Liu K., Tao X., Zhang Q., Zhang X., Cui Y., Transforming from planar to three-dimensional lithium with flowable interphase for solid lithium metal batteries. Sci. Adv. 3, eaao0713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L., Yin Y.-X., Li J.-Y., Li N.-W., Zeng X.-X., Ye H., Guo Y.-G., Wan L.-J., Free-standing hollow carbon fibers as high-capacity containers for stable lithium metal anodes. Joule 1, 563–575 (2017). [Google Scholar]
- 25.Lin D., Liu Y., Liang Z., Lee H.-W., Sun J., Wang H., Yan K., Xie J., Cui Y., Layered reduced graphene oxide with nanoscale interlayer gaps as a stable host for lithium metal anodes. Nat. Nanotechnol. 11, 626–632 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Jiao Y., Zheng Y., Jaroniec M., Qiao S. Z., Origin of the electrocatalytic oxygen reduction activity of graphene-based catalysts: A roadmap to achieve the best performance. J. Am. Chem. Soc. 136, 4394–4403 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou T.-Z., Chen X., Peng H.-J., Huang J.-Q., Li B.-Q., Zhang Q., Li B., Design principles for heteroatom-doped nanocarbon to achieve strong anchoring of polysulfides for lithium–sulfur batteries. Small 12, 3283–3291 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Tang C., Wang H.-F., Chen X., Li B.-Q., Hou T.-Z., Zhang B., Zhang Q., Titirici M.-M., Wei F., Topological defects in metal-free nanocarbon for oxygen electrocatalysis. Adv. Mater. 28, 6845–6851 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Hou T. Z., Xu W.-T., Chen X., Peng H.-J., Huang J.-Q., Zhang Q., Lithium bond chemistry in lithium–sulfur batteries. Angew. Chem. Int. Ed. 56, 8178–8182 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Kim M. S., Ryu J.-H., Deepika, Lim Y. R., Nah I. W., Lee K.-R., Archer L. A., Cho W. I., Langmuir–Blodgett artificial solid-electrolyte interphases for practical lithium metal batteries. Nat. Energy 3, 889–898 (2018). [Google Scholar]
- 31.W. Plieth, Electrochemistry for Materials Science (Elsevier Science, 2008). [Google Scholar]
- 32.Winand R., Electrocrystallization: Fundamental considerations and application to high current density continuous steel sheet plating. J. Appl. Electrochem. 21, 377–385 (1991). [Google Scholar]
- 33.Pei A., Zheng G., Shi F., Li Y., Cui Y., Nanoscale nucleation and growth of electrodeposited lithium metal. Nano Lett. 17, 1132–1139 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Song Q., Yan H., Liu K., Xie K., Li W., Gai W., Chen G., Li H., Shen C., Fu Q., Zhang S., Zhang L., Wei B., Vertically grown edge-rich graphene nanosheets for spatial control of Li nucleation. Adv. Energy Mater. 8, 1800564 (2018). [Google Scholar]
- 35.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Momma K., Izumi F., VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011). [Google Scholar]
- 37.Blöchl P. E., Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 39.M. J. Frisch, G. W. Trucks, H. B. Schlegel, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, Gaussian 09, Revision A.02 (Gaussian, Inc., 2009). [Google Scholar]
- 40.Chen X., Li H.-R., Shen X., Zhang Q., The origin of reduced reductive stability of ion–solvent complexes on alkali and alkaline earth metal anodes. Angew. Chem. Int. Ed. 57, 16643–16647 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaau7728/DC1
Fig. S1. Schematic representation of the Li nucleation on conductive frameworks based on the classical heterogeneous nucleation model.
Fig. S2. Periodic calculation method test.
Fig. S3. The optimized configuration of carbons with bgB, egB, and oB dopant.
Fig. S4. The optimized configuration of carbons with bqN, eqN, pN, and rN dopant.
Fig. S5. The optimized configuration of carbons with aO, cO, eO, hO, and kO dopant.
Fig. S6. The optimized configuration of carbons with F, Cl, Br, and I dopant.
Fig. S7. The optimized configuration of carbons with P, oS, and S dopant.
Fig. S8. The optimized configuration of carbons with C5, C7, and C5+C7 defects.
Fig. S9. The optimized crystal structures of Li2O and Li3N.
Fig. S10. The Bader charge analyses of bgB- and egB-GNRs.
Fig. S11. The optimized interaction geometry of a Li atom interacts with eO-GNR.
Fig. S12. The optimized structures and binding energies between a Li atom and a solvent molecule.
Fig. S13. The optimized structures and binding energies between a Li atom and a multisolvent molecule.
Fig. S14. The optimized structures of borabenzene, pyridine, pyrrole, benzoic acid, furan, phenol, quinomethan, thiophene, fluorobenzene, chlorobenzene, and bromobenzene molecules.
Fig. S15. The optimized interaction geometries of Li+–DME complex and borabenzene, pyridine, pyrrole, benzoic acid, furan, phenol, quinomethan, thiophene, fluorobenzene, chlorobenzene, and bromobenzene molecules.
Fig. S16. The optimized interaction geometry of a Li atom and single-/multilayered graphene and graphite.
Fig. S17. A summary of calculated binding energy between a Li atom and single/multilayered graphene and graphite.
Fig. S18. The Li nucleation overpotentials on G, NG, and OG electrodes at different current densities (0.05, 0.50, and 1.00 mA cm−2).
Fig. S19. The N2 isotherms of OG, NG, and G materials.
Fig. S20. The pore size distribution derived from the N2 isothermal adsorption plot of OG, NG, and G materials.
Fig. S21. The Raman spectra of OG, NG, and G materials.
Fig. S22. The optimized interaction geometry of Li atom on GNR with C5, C7, and C5+C7 defects.
Fig. S23. The summary of binding energy of a Li atom adsorbed on GNR with C5, C7, and C5 + C7 defects.
Table S1. A summary of binding energy between nanoribbons and a Li atom.
Table S2. A summary of Li binding energy on Li (100) and (110) surfaces and total energy per bulk lithium.
Table S3. A summary of local dipole in heteroatom-doped nanoribbons.