

Abstract
Phosphatases play an important role in cell biology, but there are few suitable probes for selectively imaging phosphatase activity of live cells because the current probes require cell fixation or exhibit considerable cytotoxicity. Here we show that conjugating a D-peptide to a quinazolinone derivative generates cell-compatible, biostable probes for imaging phosphatase activities inside live cells. Moreover, our results show that inhibiting ectophosphatases is a critical factor for imaging intracellular phosphatases. As the first example of using selective inhibitors to ensure intracellular function of molecular probes, this work illustrates a facile approach to design molecular probes for profiling the activities of enzymes in spatial selective manner in a complicated environment.
Keywords: Intracellular phosphatase, ectophosphatase, selectivity, live cells, molecular probe
Graphical Abstract
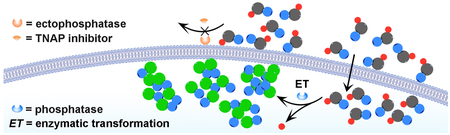
This work reports a conjugate of D-peptide and quinazolinone as cell-compatible, biostable imaging probes for imaging phosphatase activities inside live cells, which illustrates a facile approach to design molecular probes for profiling the activities of enzymes in spatial selective manners.
Being controlled by kinase/phosphatase enzyme switch, protein phosphorylation/dephosphorylation is the most common post-translational mechanism for modulating the functions of proteins in a wide range of cellular processes.[1] Thus, phosphatases play key regulatory roles in many physiological processes, including immune response,[2] pathogen virulence,[3] cancer cell proliferation and metastasis,[4] and host-microbe interaction.[5] More specifically, phosphatases, which catalyze the hydrolysis of phosphorylated substrates, are necessary for embryogenesis,[6] bone formation,[7] and neuron signaling.[8] In addition, certain phosphatases have associated with cancers[4] and have served as tumor markers[4a] for a number of cancers (e.g., pancreatic, ovarian, and testis).[9]
Despite the importance of phosphatases,[1, 10] the activity probes[11] of phosphatase remain less developed.[12] Although there are several agents for detecting phosphatases, they still suffer serious shortcomings. The conventional color assay (p-nitrophenyl phosphate (PNPP)[13]), though being able to detect the presence of phosphatases, is unsuitable for live cell imaging due to the lack of sensitivity. The fluorescent staining agent (ELF®97), which bases on restricting bond rotation to generate fluorescent colloids,[14] still requires cell fixation during the histological sample preparation,[15] and is unsuitable for activity profiling of phosphatases in live cells. The quinazolinone analogs of ELF®97 are unable to achieve satisfactory results on live cells due to its irreversible nucleated crystallization,[16] significant cytotoxicity, or poor spatial resolution.[17] The recently reported D-peptide based phosphatase probes are more suitable to imaging ectophosphatases[18] than intracellular phosphatases of live cells. But many phosphatases associated cellular processes occur intracellularly such as phosphoprotein phosphatase (PPP) family of Ser/Thr protein phosphatases and intracellular pTyr-specific phosphatases (PTPs) including PTP1B, SHP2, and MEG2.[19] Therefore, there still are unmet needs to develop functional probes for imaging the activities of intracellular phosphatases of live cells with high spatiotemporal resolution. To address this previously overlooked problem, we decide to develop a new approach for generating molecular probes that are selective for imaging intracellular phosphatases.
Here we show that the combination of a quinazolinone and a D-tripeptide results a new type of imaging probes that are cell compatible and bio-stable, preserve the sensitivity and excellent imaging contrast of ELF®97, and eliminate the uncontrolled formation of microcrystals without compromising the non-diffusiveness of the probes. Our results show that these probes, working at a wide concentration range, are capable of imaging phosphatase activities inside live cells when ectophosphatases are inhibited (Scheme 1). Illustrating a simple strategy that incorporates D-peptides to generate non-diffusive, amorphous, and biocompatible imaging probes for revealing the activities of intracellular enzymes in complicated cellular environment in the presence of a proper inhibitor, this work contributes to the development of activity probes of important enzymes.
Scheme 1.

Schematic illustration of conjugating a D-tripeptide to the motif of a phosphatase probe (i.e., a quinazolinone derivative) that generates a cell-compatible, biostable, and non-diffusive functional probe for selectively imaging intracellular phosphatases.
Scheme 1 shows the design of the D-peptide conjugated molecular probes containing a quinazolinone derivative that is based on ELF®97. As a good substrate for phosphatases, ELF®97 has several unique merits, including significant signal changes, excellent photo stability, and prompt responses. Upon enzymatic cleavage, ELF®97, from being soluble and weakly blue-fluorescent, yields a bright green fluorescent precipitate.[14] However, the use of ELF®97 requires the fixation of cells and is unsuitable for live cells. Moreover, the dephosphorylation of ELF®97 results in uncontrolled crystallization that is detrimental for achieving high spatial resolution within a single cell. To achieve live cell imaging, to target exclusively intracellular phosphatases, and to eliminate uncontrolled nucleation of the crystallization, we modify ELF®97 with a small D-peptide sequence (DPhe-DPhe-DTyr) to yield precursor 1. In addition, we choose to use neutral small functional group for replacing the carboxylic acid group at the C-terminus of the peptides for enhancing their self-assembling ability.[20] Such a design not only preserves the excellent fluorescence of ELF®97, but also efficiently changes the crystallization to a self-assembly process since the sequence of DPhe-DPhe-DTyr have been proven to be an excellent motif for self-assembly.[20–21]
Scheme S1 shows the synthetic route based on the design. We use a multi-step synthesis to generate the phosphorylated quinazolinone derivative (3), then use solid phase peptide synthesis (SPPS)[22] to obtain intermediates (4) that contain carboxylic acid group at C-terminus, and finally select methylacetate (-COOMe) or N-methylacetamide (-CONHMe) group to replace the carboxylic acid terminus of 4 to result in D-peptide conjugated quinazolinone derivatives, 1a or 1b, respectively. The difference of 1a and 1b is that 1a is a substrate of carboxylesterases, but 1b is not.
As shown in the insets of Figure 1a, 1a (or 1b) forms a transparent solution. Transmission electron microscopy (TEM) images show that the solution of 1a contains sparse nanofibers (8 nm ± 2 nm) while the solution of 1b produces aggregates (with the particle sizes of 8–19 nm) upon drying. After the addition of alkaline phosphatase (ALP), the solution of 1a becomes a stable hydrogel, which exhibits bright green fluorescence under UV irradiation, and the solution of 1b transforms to a viscous liquid that shows yellow fluorescence. As revealed by TEM, the hydrogel contains dense nanofibers (diameter of ~8 nm), and the viscous liquid consists of the mixture of nanofibers (diameter of ~8 nm) and aggregates (particle sizes of 8–19 nm). Before the addition of ALP, static light scattering (SLS) (Figure 1b) of the solutions of 1a and 1b exhibits signals slightly higher than and almost the same as that of PBS buffer, respectively. The intensities of the SLS signals increase drastically after the addition of ALP to the solutions. These results confirm that soluble precursors 1a and 1b become fluorescent and insoluble nanofibers or aggregates after the dephosphorylation catalyzed by the phosphatases. The precursor 1a undergoes ALP catalyzed dephosphorylation, but only reaches 50% conversion after 30 h when the concentration of ALP is 0.1 U/mL (Figure 1c and S1). The incomplete dephosphorylation likely is resulted from the incorporation of the precursors into the nanofibers. So the nanofibers consist of 1a and 2a. The precursor 1b also dephosphorylated with the addition of ALP but only achieves 31% conversion after 30 h (Figure S2), corresponding with the TEM that exhibits a mixture of nanofibers and aggregates.
Figure 1.

(a) TEM images of the solutions of 1a and 1b (0.5 wt%) without or with the addition of ALP at physiological pH (pH = 7.4) in PBS buffer (insert: corresponding optical images; [ALP] = 1 U/mL; bar = 100 nm). (b) Static light scattering (SLS) signals of the solutions of 1a and 1b before and after the treatment of ALP at the concentration of 100 μM ([ALP] = 1 U/mL). (c) Time-dependent dephosphorylation process of the precursor 1a with the addition of ALP at 37 °C in PBS buffer ([1a]0 = 200 μM, [ALP] = 0.1 U/mL). (d) Fluorescent confocal microscopy images show the fluorescence emission in Saos-2 cells with the treatment of 1a at the concentration of 250 μM for 0 min, 15 min and 30 min, respectively. [TNAP inhibitor] = 5 μM; scale bar = 20 μm. (e) Fluorescent confocal microscopy images show the fluorescence emission in HeLa cells with the treatment of 1a at different concentrations (50 μM and 100 μM) for 4 h, respectively. (f) Fluorescent confocal microscopy images of HeLa cells treated with 1a at the concentration of 50 μM for 4 h and then stained with endoplasmic reticulum (ER) tracker. Pearson’s R value is 0.725 from 30 cells. Scale bar = 50 μm. (g) Fluorescent confocal microscopy images show the fluorescence in different cell lines (HepG2 and VCaP) with the treatment of 1a at the concentration of 50 μM for 4 h, respectively. Scale bar = 50 μm. (h) 3D construction of HeLa and Saos-2 cells with the treatment of 1a at the concentration of 50 μM for 4 h, respectively.
After confirming that dephosphorylation of 1a results in a fluorescent gel of 2a, we incubate Saos-2 cells with 1a and monitor the change of the fluorescence in the cell culture. We choose Saos-2 cells because they, overexpressing tissue non-specific alkaline phosphatase (TNAP,[23] an ectophosphatase), offer a fine case to test the probes in a complicated environment (i.e., extracellular and intracellular phosphatases co-existing). The time-dependent fluorescence of 2a (Videos S1 and S2 in SI) reveals the extracellular and intracellular distribution of phosphatases without and with a TNAP inhibitor (2,5-dimethoxy-N-(quinolin-3-yl) benzenesulfon-amide (DQB)[24]). As shown in the fluorescent images of live Saos-2 cells at different time points (0 min, 15 min, and 30 min (Figure 1d)), the fluorescence of 2a appears in the cytoplasm, suggesting that the cells uptake 1a and the intracellular phosphatases turn 1a to 2a to result in the fluorescence. Without the addition of any phosphatase inhibitor, beside the bright fluorescence in the cytoplasm, considerable amount of fluorescent spots exist outside cells. While the intracellular fluorescence increases moderately from 15 to 30 min, the amount of extracellular fluorescent particles increases considerably. This result, besides agreeing with that dephosphorylating 1a generates insoluble, fluorescent 2a, indicates that ectophosphatases (e.g., TNAP) or the phosphatases in the culture medium exhibit different dynamics from intracellular phosphatases. Considering that TNAP is overexpressed on Saos-2 cells, we treat the cells with DQB, the TNAP inhibitor, 2 h before imaging, then co-culture the cells with 1a and DQB together during imaging. As shown in Figure 1d, after the addition of DQB, extracellular fluorescence reduces significantly, indicating that inhibiting ectophosphatases reduces the dephosphorylation of 1a outside cells effectively. On the other hand, the intracellular fluorescent increases over time, indicating that the inhibition of ectophosphatases improves the accuracy of imaging the activity of intracellular phosphatase. Although the fibers and aggregates in TEM images of dried samples indicate that the probes might self-assemble before activation, the probe, in fact, dissolves well in water and culture medium at the concentration used. From both of time-dependent and dephosphorylation process of 1a with the addition of ALP (Figure 1c) and the time-dependent fluorescence of 1a in live Saos-2 cells within 30 min (Figure 1d), one can conclude that the activation of the probe is fast. In addition, the fluorescence appears after dephosphorylation rather self-assembly. Because the aggregation rate is relevant to phosphatase imaging in live cells, it is important to choose the concentrations of the probes according to cell types or treat the cells with the probes of a series of concentrations for rate determination.
In order to investigate the intracellular uptake of the imaging probe more accurately, we select HeLa cells as the model cells to avoid the influence of too much extracellular fluorescence since it is known that expression of ectophosphatases on HeLa cells is less than on Saos-2 cells.[18b] We incubate HeLa cells with 1a at different concentration (i.e., 5, 10, 50, and 100 μM) for 4 h, and then switch to fresh live cell imaging solution. When the incubating concentration is 5 or 10 μM, we can hardly observe any fluorescence, suggesting the cellular uptake is less pronounced at such low concentration (Figure S3). It appears that 50 μM of 1a is sufficient to reveal the distribution of intracellular phosphatase because the fluorescence increases only slightly with 1a at 100 μM (Figure 1e). We also measured the photostability of 1a by irradiating the cells for 10 minutes and found that the fluorescence of the probe hardly reduced. This observation (Figure S9) indicates the excellent photostability of the assemblies of the probes. In the optimal condition, we next use confocal microscopy to examine the intracellular localization of 2a in HeLa cells. As shown in Figure 1f, most of the green fluorescence in the HeLa cells co-localize well with the fluorescence from endoplasmic reticulum (ER) tracker, suggesting that intracellular phosphatases such as PTP1B likely dephosphorylate 1a to form 2a. This result agrees with our previous report.[25] As PTP1B is a phosphatase known to localize at the cytoplasmic face of the ER, we used an inhibitor of PTP1B (CinnGEL) to co-incubate with the HeLa cells and 1a (Figure S10). Increasing the concentration of CinnGEL results in weak fluorescence, confirming that PTP1B is likely the major phosphatase for catalyzing the dephosphorylation of 1a and activating the probes.
We also apply 1a on other human cell lines, including four cancer cell lines (HepG2, MCF7, VCaP, and PC-3 cells) and one normal cell line (HS-5), to compare the activities of intracellular phosphatases (Figure 1g and S4). After 4 h of incubation with 1a, HepG2, MCF7, or PC-3 cells only exhibit weak fluorescence, suggesting the lower activity of their intracellular phosphatases than those of intracellular phosphatases of HeLa cells. Compared with these cells lines, VCaP cells exhibit a little brighter fluorescence but still much weaker than that on Saos-2 and HeLa cells. As a normal cell line, HS-5 cell hardly exhibits fluorescence, indicating low activity of phosphatases inside the cells, consistent with the results using commercially available ELF®97 (Figure S6). This result agrees with dysregulation of phosphorylation and dephosphorylation in malignant cells.[26] To further examine the distribution of the probes when they are incubated with Saos-2 and HeLa cells, we use confocal microscopy to generate 3D constructions of the fluorescence. Figure 1h shows that most of fluorescent puncta distribute through the cytoplasms, with a small amount of extracellular fluorescence remains. This result further proves 1a is sensitive for imaging phosphatases and selective to reveal the activities intracellular phosphatases. Our study also shows that the probes exhibit slightly toxicity to HeLa and Saos-2 cells, but are largely innocuous to HepG2 and HS-5 cells (Figure S11). This result suggests that overexpressed ALP on HeLa and Saos-2 cells, indeed, is responsible for the conversion of 1a to 2a and the subsequent inhibition of these two types of cancer cells.
Considering that the culture media contain soluble esterases,[27] we incubate HeLa, Saos-2, HepG2, or HS-5 cells with the molecular probe 1b, to avoid the interference of esterase. Being treated by 1b, HeLa or Saos-2 cells exhibit bright fluorescence, which is comparable to the fluorescence from the same cells treated 1a. There is hardly any fluorescence in HepG2 and HS-5 cells after being treated by 1b (Figure 2a). This result suggests negligible amount of esterases in culture media has minimal impact on 1a. However, the addition of exogenous carboxylesterase (CES) (e.g., 1 U/mL) significantly decreased the fluorescence when we incubate Saos-2 cells with the probe 1a, and the fluorescence become weaker gradually with the amount of CES increasing (Figure S5). These observations further support our design that using small neutral group to capping the C-terminals of the peptides enhances their self-assembling ability.
Figure 2.

(a) Fluorescent confocal microscopy images show the fluorescence emission in different cell lines (HeLa, Saos-2, HepG2 and HS-5) with the treatment of 1b at the concentration of 50 μM for 4 h, respectively. Scale bar = 50 μm. Fluorescent confocal microscopy images show the fluorescence emission in (b) HeLa and (c) Saos-2 cells with the treatment of 1a at 37 °C and 4 °C, or with the treatment of 1a plus Mβ-CD, CPZ, EIPA and filipin III at 37 °C for 4 h, respectively. [1a] = 50 μM, [Mβ-CD] = 5 mM, [CPZ] = 30 μM, [EIPA] = 50 μM, [filipin III] = 5 μg/mL. Scale bar = 50 μm.
We carry out a preliminary study to understand the mechanism of the uptake of the probes. As shown in Figure 2b, we first incubate HeLa cells with 1a at 4 and 37 °C, respectively. The fluorescence in cell milieu reduces significantly at 4 °C, suggesting that the uptake is an energy-dependent process. Then we use inhibitors of different endocytotic processes to determine the possible pathways of cellular uptake. We use methyl-β-cyclodextrin (Mβ-CD) for inhibiting lipid raft-dependent endocytosis, chlorpromazine (CPZ) for clathrin-mediated endocytosis, ethylisopropylamiloride (EIPA) for macropinocytosis, and filipin III for caveolae-mediated endocytosis. We find that only the addition of Mβ-CD results in significance decrease of fluorescence, suggesting that the probes likely mainly enter the cells via lipid raft-dependent endocytosis. We also carry out the same experiments using Saos-2 cells and find that the addition of Mβ-CD results in moderate decrease of fluorescence (Figure 2c). These results indicate that the lipid raft-dependent endocytosis of HeLa cells differs from that of Saos-2, though the exact origin of such a difference remains to be elucidated.
In conclusion, the D-peptide conjugated molecular probes containing a quinazolinone derivative are suitable for selectively imaging intracellular phosphatases of live cells. Moreover, our results confirm the designed molecular probes not only preserving the sensitivity and excellent imaging contrast of commercially available ELF®97, also eliminating the uncontrolled formation of microcrystals without compromising the non-diffusiveness of the probes. Because the molecular probes are suitable for activity profiling of phosphatases in live cells (i.e., it omits cell fixation), and yield reproducible results, they will provide a facile means to target intracellular phosphatases for understanding the characteristics of the intracellular phosphatases, especially their activities. It should provide useful insights for developing applications based on enzymatic reactions, such as instructed-assemblies[11c, 25, 27-28] for molecular imaging and cancer therapy, screening intracellular phosphatases inhibitors using cell assays, and understanding of the biochemical underpinnings of intracellular phosphatases (e.g., PTPs) regulation in both health and disease tissues. Future work will extend to use other sequences conjugated probes to increase the specificity of intracellular phosphatase probes.
Supplementary Material
Acknowledgements
This work was partially supported by NIH (R01CA142746, R21 AI130560). JZ is an HHMI international student fellow.
References
- [1].Lodish H, Berk A, Kaiser CA, Krieger M, Bretscher A, Ploegh H, Amon A, Scott MP, Mol. Cell Biol. 7th ed., W. H. Freeman, 2012. [Google Scholar]
- [2].a) Khalil AM, Cambier JC, Shlomchik MJ, Science 2012, 336, 1178–1181; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vivier E, Nunes JA, Vely F, Science 2004, 306, 1517–1519. [DOI] [PubMed] [Google Scholar]
- [3].Broberg CA, Zhang LL, Gonzalez H, Laskowski-Arce MA, Orth K, Science 2010, 329, 1660–1662. [DOI] [PubMed] [Google Scholar]
- [4].a) Fishman WH, Inglis NR, Green S, Anstiss CL, Gosh NK, Reif AE, Rustigia R, Krant MJ, Stolbach LL, Nature 1968, 219, 697–699; [DOI] [PubMed] [Google Scholar]; b) Pospisil P, Iyer LK, Adelstein SJ, Kassis AI, BMC Bioinform. 2006, 7, 354–364; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, Rodrigues DN, Renwick A, Seal S, Ramsay E, Duarte SD, Rivas MA, Warren-Perry M, Zachariou A, Campion-Flora A, Hanks S, Murray A, Pour NA, Douglas J, Gregory L, Rimmer A, Walker NM, Yang TP, Adlard JW, Barwell J, Berg J, Brady AF, Brewer C, Brice G, Chapman C, Cook J, Davidson R, Donaldson A, Douglas F, Eccles D, Evans DG, Greenhalgh L, Henderson A, Izatt L, Kumar A, Lalloo F, Miedzybrodzka Z, Morrison PJ, Paterson J, Porteous M, Rogers MT, Shanley S, Walker L, Gore M, Houlston R, Brown MA, Caufield MJ, Deloukas P, McCarthy MI, Todd JA, Turnbull C, Reis JS, Ashworth A, Antoniou AC, Lord CJ, Donnelly P, Rahman N, Nature 2013, 493, 406–410; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Saha S, Bardelli A, Buckhaults P, Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C, Kinzler KW, Vogelstein B, Science 2001, 294, 1343–1346. [DOI] [PubMed] [Google Scholar]
- [5].Bates JM, Akerlund J, Mittge E, Guillemin K, Cell Host & Microbe 2007, 2, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, Alfonso ZC, Fraser JK, Benhaim P, Hedrick MH, Mol. Biol. Cell 2002, 13, 4279–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawasehara A, Suda T, J. Cell Biol. 1994, 127, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fonta C, Négyessy L, Subcell. Biochem. 2015, 76, 85–106. [DOI] [PubMed] [Google Scholar]
- [9].a) Uhlén M, Pontén F, Lindskog C, Science 2015, 347, 1274–1274; [Google Scholar]; b) Millán JL, Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology, John Wiley & Sons, 2006. [Google Scholar]
- [10].Liberti S, Sacco F, Calderone A, Perfetto L, Iannuccelli M, Panni S, Santonico E, Palma A, Nardozza AP, Castagnoli L, Cesareni G, Febs J 2013, 280, 379–387. [DOI] [PubMed] [Google Scholar]
- [11].a) Ye FM, Wu CF, Jin YH, Chan YH, Zhang XJ, Chiu DT, J. Am. Chem. Soc. 2011, 133, 8146–8149; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mutch SA, Gadd JC, Fujimoto BS, Kensel-Hammes P, Schiro PG, Bajjalieh SM, Chiu DT, Nat. Protoc. 2011, 6, 1953–1968; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ye DJ, Shuhendler AJ, Cui LN, Tong L, Tee SS, Tikhomirov G, Felsher DW, Rao JH, Nat. Chem. 2014, 6, 519–526; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liang G, Ren H, Rao J, Nat Chem 2010, 2, 54–60; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shuhendler AJ, Pu KY, Cui L, Uetrecht JP, Rao JH, Nat. Biotechnol. 2014, 32, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sacco F, Perfetto L, Castagnoli L, Cesareni G, FEBS Lett. 2012, 586, 2732–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lorenz U, Curr. Protoc. Immunol. 2011, Chapter 11, Unit 11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Larison KD, Bremiller R, Wells KS, Clements I, Haugland RP, J. Histochem. Cytochem. 1995, 43, 77–83. [DOI] [PubMed] [Google Scholar]
- [15].Cox WG, Singer VL, J. Histochem. Cytochem. 1999, 47, 1443–1455. [DOI] [PubMed] [Google Scholar]
- [16].Wang KT, Kirichian AM, Al Aowad AF, Adelstein SJ, Kassis AI, Bioconjug. Chem. 2007, 18, 754–764. [DOI] [PubMed] [Google Scholar]
- [17].Kim TI, Kim H, Choi Y, Kim Y, Chem. Commun. 2011, 47, 9825–9827. [DOI] [PubMed] [Google Scholar]
- [18].a) Wang H, Feng Z, Del Signore SJ, Rodal AA, Xu B, J. Am. Chem. Soc. 2018, 140, 3505–3509; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, Xu B, Chem 2016, 1, 246–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yu Z-H, Zhang Z-Y, Chem. Rev. 2018, 118, 1069–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Feng Z, Wang H, Du X, Shi J, Li J, Xu B, Chem. Comm. 2016, 52, 6332–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Du X, Zhou J, Shi J, Xu B, Chem. Rev. 2015, 115, 13165–13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Weng C, Peter D, Fmoc Slild Phase Peptide Synthesis: A Practical Approach, Oxford: Oxford University Press, 2000. [Google Scholar]
- [23].Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, Pashkuleva I, J. Am. Chem. Soc. 2015, 137, 576–579. [DOI] [PubMed] [Google Scholar]
- [24].Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, Smith LH, O’Neill WC, Millan JL, Cosford NDP, J. Med. Chem. 2009, 52, 6919–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gao Y, Shi J, Yuan D, Xu B, Nat. Comm. 2012, 3, 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Hunter T, Cold Spring Harbor Perspect. Biol 2014, 6, a020644; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sawyers C, Nature 2004, 432, 294–297. [DOI] [PubMed] [Google Scholar]
- [27].Li J, Bullara D, Du X, He H, Sofou S, Kevrekidis IG, Epstein IR, Xu B, ACS nano 2018, 12, 3804–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Zhou J, Du X, Li J, Yamagata N, Xu B, J. Am. Chem. Soc. 2015, 137, 10040–10043; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Feng Z, Wang H, Chen X, Xu B, J. Am. Chem. Soc. 2017, 139, 15377–15384; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kuang Y, Xu B, Angew. Chem. Int. Ed. 2013, 52, 6944–6948; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhan J, Cai Y, He S, Wang L, Yang Z, Angew. Chem. Int. Ed. 2018, 57, 1813–1816; [DOI] [PubMed] [Google Scholar]; e) Hai Z, Li J, Wu J, Xu J, Liang G, J. Am. Chem. Soc. 2017, 139, 1041–1044; [DOI] [PubMed] [Google Scholar]; f) Kumar M, Ing NL, Narang V, Wijerathne NK, Hochbaum AI, Ulijn RV, Nat. Chem. 2018, 10, 696–703; [DOI] [PubMed] [Google Scholar]; g) Yu G, Zhang M, Saha ML, Mao Z, Chen J, Yao Y, Zhou Z, Liu Y, Gao C, Huang F, Chen X, Stang PJ, J. Am. Chem. Soc. 2017, 139, 15940–15949; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Aggarwal BB, Harikumar KB, Int. J. Biochem. Cell Biol. 2009, 41, 40–59; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Li J, Xie C, Huang J, Jiang Y, Miao Q, Pu K, Angew. Chem. Int. Ed. 2018, 57, 3995–3998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.