

Abstract
Activation and transcriptional reprogramming of AR in advanced prostate cancer frequently coincides with the loss of two tumor suppressors, INPP4B and PTEN, which are highly expressed in human and mouse prostate epithelium. While regulation of AR signaling by PTEN has been described by multiple groups, it is not known whether the loss of INPP4B affects AR activity. Using prostate cancer cell lines we showed that INPP4B regulates AR transcriptional activity and the oncogenic signaling pathways Akt and PKC. Analysis of gene expression in prostate cancer patient cohorts showed a positive correlation between INPP4B expression and both AR mRNA levels and AR transcriptional output. Using an Inpp4b-/- mouse model, we demonstrated that INPP4B suppresses Akt and PKC signaling pathways and modulates AR transcriptional activity in normal mouse prostate. Remarkably, PTEN protein levels and phosphorylation of S380 were the same in Inpp4b-/- and WT males, suggesting that the observed changes were due exclusively to the loss of INPP4B. Our data show that INPP4B modulates AR activity in normal prostate and its loss contributes to the AR-dependent transcriptional profile in prostate cancer.
Keywords: INPP4B, androgen receptor, prostate cancer
INTRODUCTION
Inositol polyphosphate 4-phosphatase type II (INPP4B) has been identified as a tumor suppressor in ovarian, breast, thyroid, bladder, and prostate cancers (1–5). INPP4B functions as a dual specificity phosphatase, capable of dephosphorylation of both lipid and protein substrates similar to phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (6). Remarkably, both PTEN and INPP4B are lost in nearly half of all metastatic prostate cancers (7).
Substrates for INPP4B lipid phosphatase activity are PI(3,4)P2, PI(4,5)P2, Ins(1,3,4)P3 (8, 9), and PI(3,4,5)P3 (8). Both PI(3,4,5)P3 and PI(3,4)P2 bind pleckstrin-homology domain of Akt facilitating survival, migration, and invasion of prostate cancer cells (10, 11). We and others have shown that INPP4B suppresses Akt signaling (2, 3, 8). PI(4,5)P2 is hydrolyzed by phospholipase C (PLC) into inositol triphosphate and diacylglycerol (DAG) which activates both conventional (α,β,γ) and novel (ε,δ,η,θ) kinases of the PKC family (12–14). The atypical PKC isoforms (ζ and ι/λ) are activated by lipid membrane components other than DAG (15). Protein levels of PKC α, ε, ζ are increased in prostate and other cancers (16). We have previously shown that INPP4B suppresses PKC signaling in prostate cancer cells (9).
The activation of the androgen receptor (AR) is essential for prostate development and for the initiation and progression of prostate cancer (17, 18). With prostate cancer progression the AR transcriptional profile changes from one of differentiation to proliferation (3). This change is due, in part, to the expression of splice variants of AR, the activation of cell signaling pathways, and posttranslational modifications of AR. AR splice variants lack ligand binding domains and are constitutively active (19) and confer resistance to androgen deprivation therapy (20–22). Activated Akt binds and phosphorylates AR on S213 and S791 (23–25). Phosphorylation of these sites affects AR activity in a cell specific manner (23, 24). In mice, a prostate specific Pten−/− knockout leads to Akt activation and a near complete loss of the AR protein, which is partially reversed by treatment with Akt inhibitors (26). PKC was reported to phosphorylate AR on S578 increasing AR transcriptional activity and regulating its turnover (27). In addition, PKC directly phosphorylates Ack1 and Src which phosphorylate Y267 and Y534 in both full length AR and AR-V7, which stimulates their nuclear translocation and transcriptional activity (28–30). In mouse models, overexpression of PKCε stabilizes the AR protein and leads to prostate hyperplasia and intraepithelial neoplasia (31, 32).
In this report we investigated whether INPP4B modulates transcriptional activity and turnover of full length AR, and the AR splice variant, AR-V7. Unlike full length AR, AR-V7 was unable to induce INPP4B gene expression and did not bind to major regulatory regions in the INPP4B locus that are bound by full length AR. Loss of INPP4B significantly altered a subset of AR, as well as AR-V7 target genes, without altering AR protein levels. We showed that INPP4B suppresses Akt and PKC signaling in independently derived prostate cancer cell lines. Using Akt and PKC specific inhibitors to mimic INPP4B expression we determined that some changes in AR target gene expression are mediated by Akt and PKC. Similar to our observations in human prostate cancer cell lines, induction of prostate specific AR target genes was reduced in Inpp4b−/− mice. In the prostate, Inpp4b knockout mice expressed similar levels of AR, PTEN, and p-PTEN when compared to their wild-type (WT) age-matched controls. Interestingly, the Inpp4b knockout mice displayed increased activity of Akt, PKCζ and PKCβII in the prostate, as indicated by increased phosphorylation levels. Our data indicate that INPP4B suppresses Akt and PKC signaling pathways and modulates AR transcriptional output in human and mouse models of prostate cancer.
RESULTS
Loss of INPP4B changes AR transcriptional output.
INPP4B depletion accelerates LNCaP cell proliferation, an androgen dependent feature of this prostate cancer cell line. To determine whether INPP4B modulates AR transcriptional activity we performed gene expression microarray analysis. Endogenous INPP4B is generally localized to the cell membrane and cytoplasm (Figure 1A), while AR shuttles between the cytoplasm and nucleus (33). Forty eight hours after knockdown INPP4B protein levels were nearly undetectable while AR protein levels were unchanged (Figure 1B). We chose this time point to evaluate changes in transcript levels using Affymetrix microarrays (Supplemental Figure 1A). To determine whether INPP4B regulates AR target genes, we compared AR target genes in LNCaP cells (34) to genes regulated by INPP4B. Expression of a subset of AR target genes was significantly affected by the knockdown of INPP4B (Figure 1C). To confirm INPP4B regulation of AR transcriptional activity we used gene set enrichment analysis (GSEA). An AR signature, created using our previously reported microarrays, GSE60721 (34) (Supplemental Table 2), was significantly represented among INPP4B regulated genes (Normalized Enrichment Score 1.84, p < 0.001) (Figure 1D). We next investigated whether INPP4B expression correlates with AR mRNA levels and the AR signature in prostate cancer patients. In both the Taylor and TCGA cohorts there were statistically significant positive correlations between INPP4B and both AR mRNA and two AR signatures, Nelson and Hieronymus. A weak but significant positive correlation was observed between INPP4B and PTEN mRNA levels in the TCGA and Taylor cohorts (Supplemental Figure 1B). To determine whether INPP4B regulates basal or androgen dependent gene expression, we knocked down INPP4B, treated cells with vehicle or 0.1 nM R1881, and compared the expression of three AR induced genes and three AR repressed genes in LNCaP cells; TARP, TMPRSS2, NNMT, SYTL2, PLA2G2A, and KIAA1324 (Supplemental Figures 2A and 2B). Both induction and repression of these genes were reduced after INPP4B knockdown in medium supplemented with FBS (Supplemental Figure 2C, 2D). Following INPP4B knockdown (Figure 1E), AR dependent repression of KIAA1324 and SYTL2 was abolished (Figures 1F - 1G). While both basal and R1881-dependent induction of TARP, TMPRSS2, and PSA were reduced, fold induction of NNMT was increased by INPP4B loss (Figures 1H – 1K).
Figure 1. Loss of INPP4B changes AR transcriptional activity.
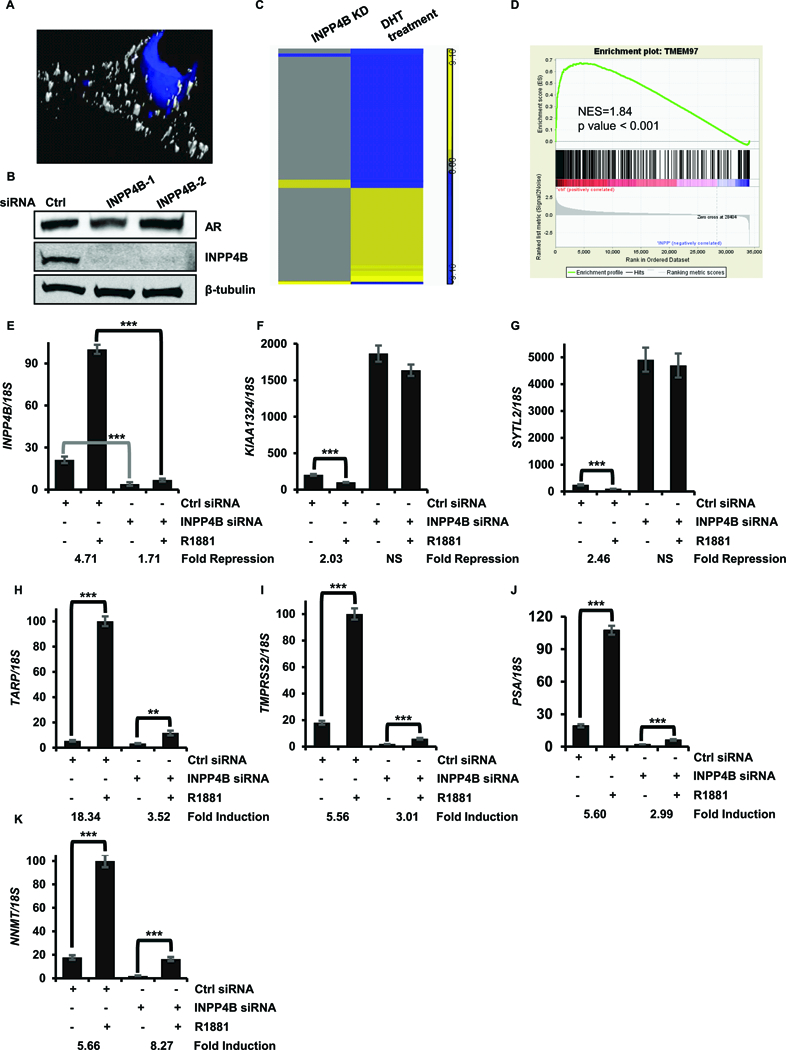
(A) Three dimensional rendering of INPP4B distribution. Cells were fixed and stained for INPP4B (white) and counterstained with DAPI (blue). (B) LNCaP cells were transfected with control (Ctrl) or two independent INPP4B specific siRNAs (INPP4B-1, INPP4B-2) for 48 hours in medium supplemented with FBS. Protein levels of AR, INPP4B, and tubulin were compared by Western blotting. (C) LNCaP cells were transfected as in (B). RNA was extracted and used in microarray analysis. Changes in gene expression were compared to AR regulated genes in LNCaP cells. (D) GSEA of INPP4B regulated genes using an AR transcriptional signature in LNCaP cells (GSE60721). (E-K). LNCaP cells were transfected with either control or INPP4B siRNAs for 48 hour intervals. Forty eight hours later cells were placed in medium supplemented with 10% CSS and treated with either vehicle (ethanol) or 0.1 nM R1881 for 24 hours as indicated. RNA was analyzed for expression of INPP4B (E), KIAA1324 (F), SYTL2 (G), TARP (H), TMPRSS2 (I), PSA (J), and NNMT (K). Expression was normalized to 18S. ** p < 0.01, *** p < 0.001.
Reciprocal regulation of AR-V7 and INPP4B.
We have previously shown that INPP4B expression is stimulated by androgens in LNCaP and VCaP cell lines, and in primary prostate cancer patient derived xenografts (3, 9). While androgen withdrawal reduced INPP4B levels in LNCaP and C4–2 cells, the addition of casodex or enzalutamide (MDV3100) to charcoal stripped serum did not further reduce INPP4B expression in these cell lines (Supplemental Figure 2E). Conversely, androgen independent AR activity in the castration resistant cell line LNCaP-abl did not induce INPP4B expression (GSE39452) (35) suggesting a strict androgen dependence of INPP4B expression. We used the LNCaPAR-V7/pHAGE cell line, which expresses an inducible AR-V7 variant (36), to test whether AR-V7 is able to upregulate expression of endogenous INPP4B. Full length AR induced endogenous INPP4B expressed in an androgen-dependent manner (Figure 2A), while AR-V7 suppressed INPP4B expression on both protein (Figure 2A, lanes 5 and 6) and mRNA levels (Figure 2B, bars 1 and 2). Expression of AR-V7 in LNCaPAR-V7/pHAGE, increased phosphorylation of Akt and PKCβII (Figure 2A). As seen from Figure 2C, INPP4B knockdown significantly decreased the induction of EDN2, an AR-V7 specific target gene (36). These results suggest that INPP4B modulates AR-V7 transcriptional activity and that, unlike full-length AR, AR-V7 is unable to induce INPP4B expression. INPP4B knockdown reduced basal transcription of NNMT and PSA without changing the ability of AR-V7 to induce its expression (Figure 2D-E). Both basal and AR-V7 dependent regulation of TARP, TMPRSS2, KIAA1324, and SYTL2 required INPP4B expression (Figures 2F-I).
Figure 2. Reciprocal regulation of AR-V7 and INPP4B.
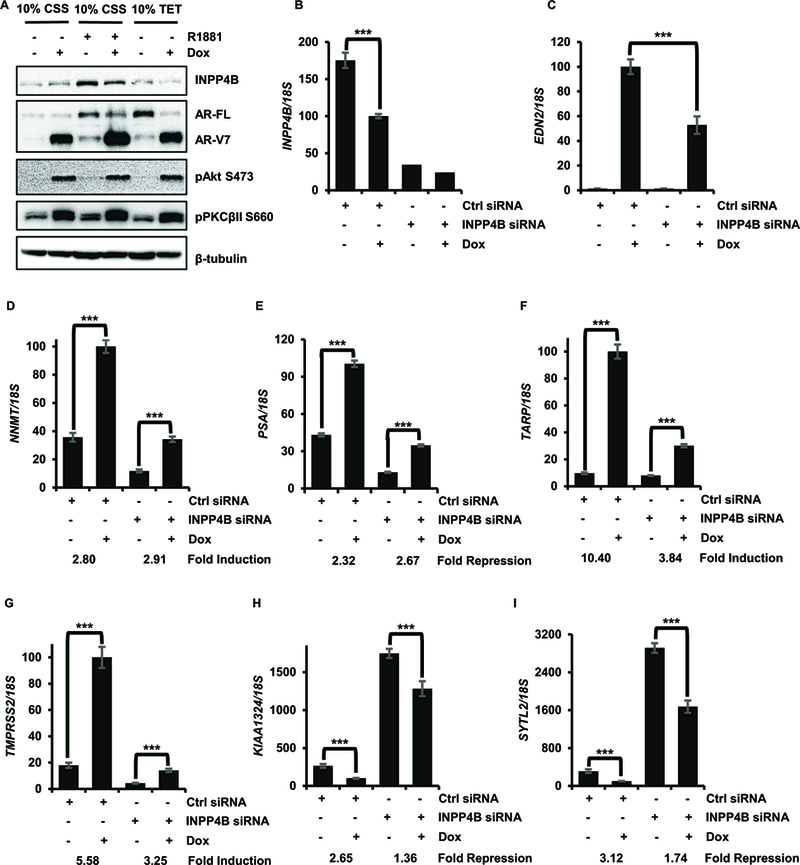
(A) LNCaPAR-V7/pHage cells were placed in medium with 10% CSS or 10% TET FBS serum for 24 hours and treated with 50 ng/ml Dox or 1 nM R1881 as indicated for an additional 36 hours. Protein level of INPP4B, AR-FL, AR-V7, pAkt, pPKCβII, and tubulin were assessed by Western blotting. (B, C) LNCaPAR-V7/pHage cells were transfected with control or INPP4B siRNA and treated with either vehicle or 25 ng/ml Dox for additional 24 hours. Gene expression of INPP4B (B) and EDN2 (C) were analyzed by RT-qPCR using 18S as a control. (D-I) LNCaPAR-V7/pHage cells were transfected and treated as in B. RNA was analyzed for expression of NNMT (D), PSA (E), TARP (F), TMPRSS2 (G), KIAA1324 (H), and SYTL2 (I).*** p < 0.001.
Differential regulation of INPP4B expression by full length AR and AR-V7.
FOXA1 is a pioneer factor for many AR regulated genes. FOXA1 depletion studies have shown that while some AR regulated genes are insensitive to FOXA1 expression, others require FOXA1 for AR mediated regulation and still others are regulated by AR only when FOXA1 is depleted (37, 38). Many androgen regulated genes in LNCaP cells have overlapping AR and FOXA1 binding sites within their regulatory regions (39). AR binding to FOXA1 requires the complete hinge region of AR that is not present in AR-V7, suggesting that AR-V7 does not have the ability to induce genes requiring direct FOXA1/AR interactions. Consistent with this, a previous study showed that AR-V7 does not induce RASSF3 or EXTL2, two genes that are regulated by AR only in the presence of FOXA1 (36, 40, 41). Analyses of publicly available data sets show that AR binds to several sites in the INPP4B gene where FOXA1 also binds. To test whether FOXA1 is required for INPP4B induction, LNCaPAR-V7/pHAGE cells were treated with control or FOXA1 siRNA followed by treatment with R1881 or doxycycline to induce AR-V7. As shown in Fig 3A, FOXA1 was depleted by the siRNA treatment. Depletion of FOXA1 had no effect on either AR isoform’s ability to induce FKBP5 expression (Figure 3B), but blocked hormone dependent induction of RASSF3 (Figure 3C) as expected. Only the full length AR induced INPP4B expression, and this induction was effectively eliminated with FOXA1 knockdown (Figures 2B and3D). To determine whether AR-V7 could bind to the AR binding sites in INPP4B (Figure 3E), ChIP assays were performed utilizing antibodies against AR and AR-V7. We confirmed the recruitment of both receptors to the PSA enhancer and to the previously reported sites within the INPP4B enhancer and intron 2 (Figures 3F-H) (3, 42). Remarkably, no AR-V7 recruitment was detected at the INPP4B transcription start site or the 5.5 Kb downstream site that feature AR/FOXA1 overlapping binding sites (Figures 3I-J).
Figure 3. INPP4B transcription is regulated by full length AR and FOXA1.
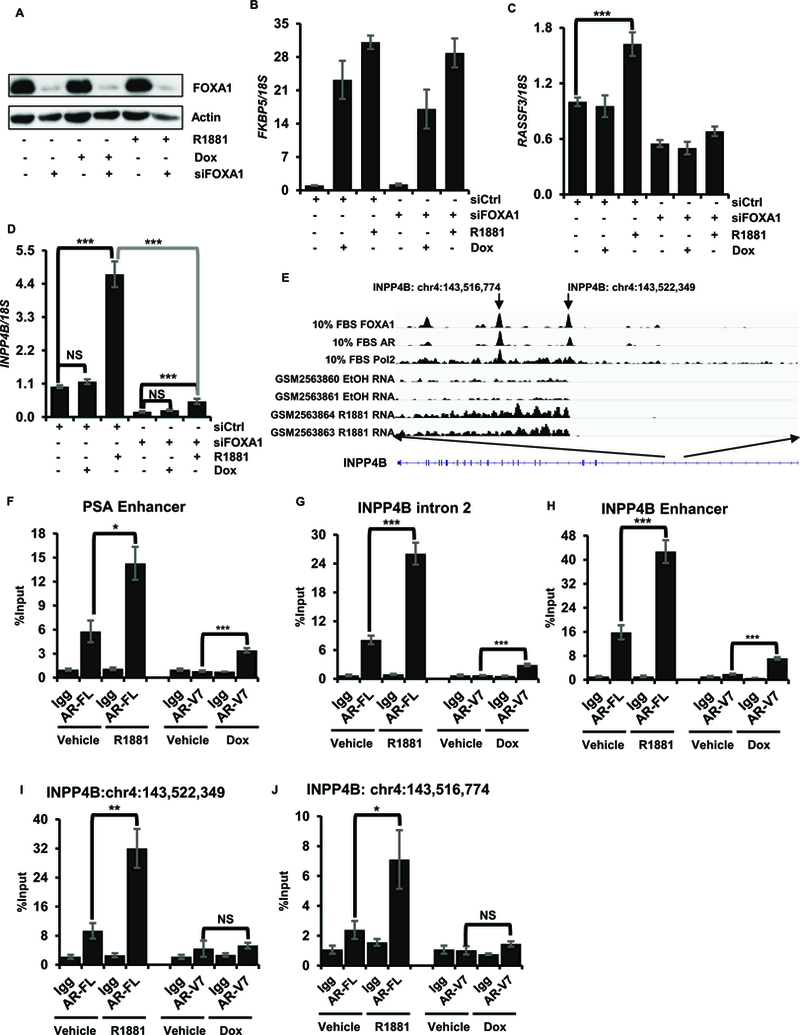
LNCaP cells in 10% CSS were transfected with noncoding control or FOXA1 specific siRNA; cells were treated with R1881 after 24 hours. Cells were harvested after 48 hours and assayed for FOXA1 expression by western blotting (A), RNA isolated and assayed for FKBP5 (B), RASSF3 (C), and INPP4B (D). (E) A diagram of AR, FOXA1, and Pol2 recruitment and RNA-seq in LNCaP cells. (F-J) LNCaPAR-V7/pHage cells were placed in medium with 10% CSS for 24 hours and treated with 50 ng/ml Dox or 10 nM R1881 overnight as indicated. Recruitment of AR-FL and AR-V7 to PSA enhancer (F), INPP4B intron 2 (G), INPP4B enhancer region (H), INPP4B transcription start site chr4:143522349 (I), and chr4:143514774 (J) were measured by ChIP-qPCR using Igg as control. * p < 0.05, ** p < 0.01, *** p < 0.001.
INPP4B regulates the AR transcriptome via inhibition of PI3K/Akt and PKC signaling pathways.
Based on the disparate spatial distribution of INPP4B and AR, we tested whether INPP4B regulation of the AR transcriptome includes intermediate signaling pathways. We and others have reported that INPP4B suppresses phosphorylation of Akt (1–3, 43, 44) and PKC (9). As seen in Figure 4A, knockdown of INPP4B but not PTEN, increased phosphorylation of PKCζ on T410 by 1.84 ± 0.19 fold and PKCβII on S660 by 1.70 ± 0.04 fold in the androgen sensitive VCaP cell line, which expresses INPP4B and PTEN. In LNCaP cells, pPKCζ and pPKCβII were increased 2.52 ± 0.77 fold and 2.23 ± 0.09 fold, respectively (Figure 4B). In C4–2 cells, INPP4B knockdown upregulated phosphorylation levels of PKCζ by 1.89 ± 0.30 fold (Figure 4C). pAkt levels were increased 48 hours after INPP4B knockdown in LNCaP and C4–2 cell lines (Figure 4B-C). We next tested whether Akt and PKC specific inhibitors would phenocopy the effects of INPP4B transcriptional regulation (Supplemental Figure 2C-D). AZD5363 and bisindolymaleimide I (BIM-I) were used to test whether INPP4B regulates AR transcriptional activity through the Akt and PKC pathways, respectively. The inhibitor activity was confirmed by decreased phosphorylation of the downstream target S6 protein (Figure 4D). PLA2G2A mRNA was reduced following BIM-I or AZD5363 treatment while expression of KIAA1324 was decreased by the PKC inhibitor (Figures 4E-F). AZD5363 increased expression of TARP and NNMT, as did INPP4B (Figures 4G-H). However, the lack of concordant regulation of TMPRSS2 and SYTL2 (Figures 4I-J) suggests additional Akt and PKC independent modes for INPP4B regulation of AR activity.
Figure 4. Suppression of Akt and PKC signaling contributes to INPP4B regulation of AR transcriptional activity.
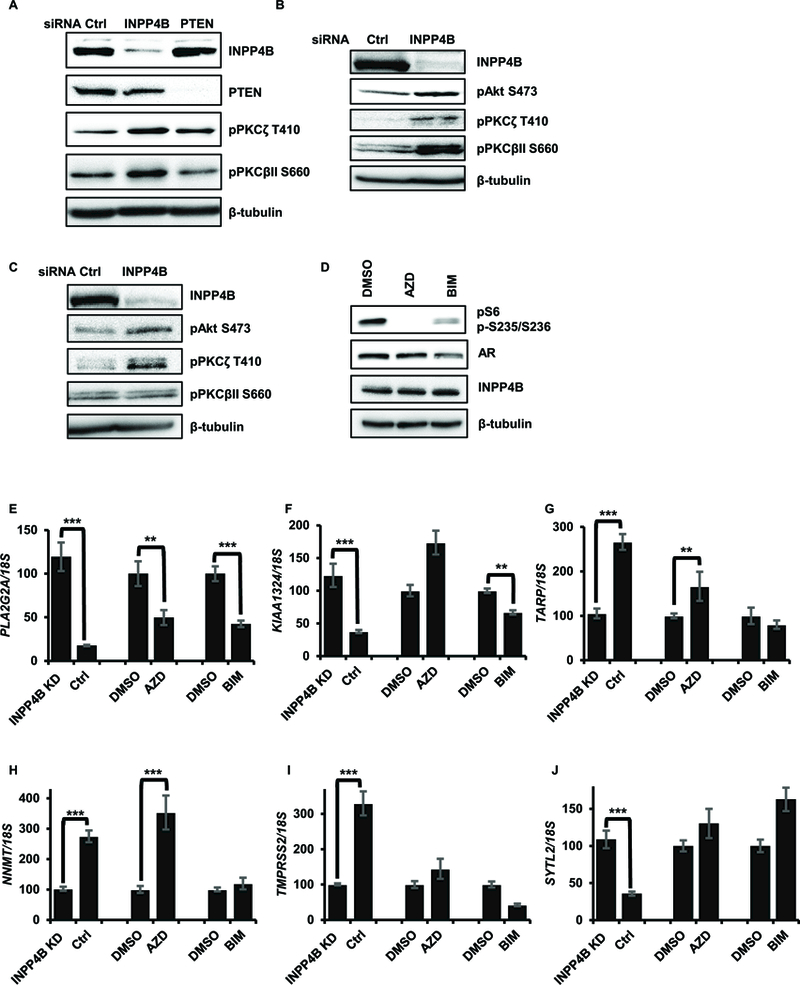
(A) VCaP cells were transfected with control, INPP4B, or PTEN specific siRNAs. Expression levels of INPP4B, PTEN, pPKCζ, pPKCβII and β-tubulin were measured by Western blotting. (B) LNCaP cells were transfected with control or INPP4B siRNAs for 48 hours in medium supplemented with 10% FBS. Cellular lysates were assayed for INPP4B, pAkt, pPKCζ, pPKCβII and β-tubulin by Western blotting. (C) C4–2 cells were transfected and assayed in parallel with B. (D) LNCaP cells were plated in complete medium and treated with indicated inhibitors for 8 hours. Protein extracts were assayed for AR, INPP4B, pS6, and tubulin levels by Western blotting. (E-J) LNCaP cells were plated in medium supplemented with 10% CSS with either vehicle or 1 nM R1881. Twenty four hours later cells were treated with vehicle (DMSO), 5μM AZD5363, or 2 μM BIM-I for an additional 8 hours. In parallel, LNCaP cells were transfected with control or INPP4B siRNA for 48 hours in 10% CSS medium supplemented with 1 nM R1881. RNA was purified, reverse transcribed, and expression levels of AR regulated genes PLA2G2A (E), KIAA1324 (F), TARP (G), NNMT (H), TMPRSS2 (I) and SYTL2 (J) compared by RT-qPCR. ** represents p < 0.01; *** represents p < 0.001.
INPP4B signaling in the normal mouse prostate.
To investigate the function of INPP4B in the normal prostate we used an Inpp4b−/− mouse model with deletion of exon 11 (45) which results in a frame shift at codon 213 and the generation of a stop signal at codon 217 (Supplemental Figure 3E). As expected, no expression of INPP4B was detected in the prostates of Inpp4b−/− mice and immunohistochemical analysis confirmed the lack of INPP4B protein in prostate epithelial cells (Figures 5A–5B). Mutant male mice were fertile and displayed no difference in body, prostate, and testis weights (Supplemental Figures 3A-3C). The ratio of seminal vesicles versus body weight showed a modest but statistically significant increase in Inpp4b−/− mice compared to age-matched WT males (Supplemental Figure 3D).
Figure 5. Loss of INPP4B in mouse prostate activates PKC and Akt signaling without changing AR levels.
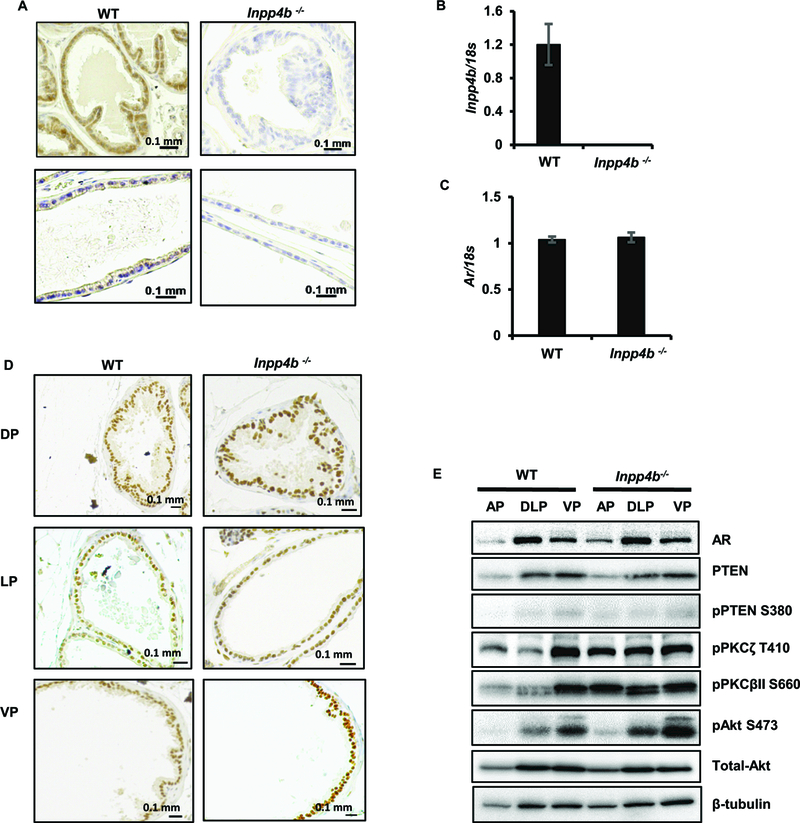
(A) Prostates from 2 month old WT or Inpp4b−/− males were stained with INPP4B antibodies and counterstained with hematoxylin. (B-C) RNA was extracted from prostates of WT (N=11) or Inpp4b−/− (N=11) males and analyzed for expression of Inpp4b (B) and Ar (C) by RT-qPCR. (D) Slides from A were stained with AR antibody and counterstained with hematoxylin. (E) Anterior (AP), dorsolateral (DLP), and ventral (VP) prostate lobes from WT or Inpp4b−/− males were analyzed for AR, total PTEN, pPTEN, pPKCζ, pPKCβII, pAkt, total Akt, and tubulin levels by Western blotting.
Consistent with our observations in prostate cancer cell lines, AR mRNA and protein levels did not change in Inpp4b−/− mouse prostates compared to those of the WT mice (Figures 5C-E). Anterior (AP), dorsolateral (DLP), and ventral (VP) prostate lobes of WT and Inpp4b−/− mice had similar expression of AR protein (Figures 5D-E). We observed an increase of pPKCβII (3.98 fold ± 1.01) and pPKCζ (2.83 fold ± 0.39) in the AP and an increase of pPKCβII (2.11 fold ± 0.24) and pPKCζ (2.28 fold ± 0.26) in the DLP of Inpp4b−/− mice compared to the WT controls (Figure 5E). An increase in pAkt levels in the DLP (1.52 fold ± 0.08) and VP (1.51 fold ± 0.20) occurred without changes in PTEN protein amounts or phosphorylation levels (Figure 5E).
INPP4B regulates AR transcriptional activity in normal mouse prostate.
To investigate whether INPP4B modulates AR target gene expression, we selected three genes strongly induced by AR and three genes regulated by PTEN in the mouse prostate (26, 46). In previous reports, DHT supplementation of castrated mice significantly induced Msmb, Apof, and Nkx3.1 (46). We show here that INPP4B is required for their optimal expression (Figures 6A-C). Expression of Pten, and the AR and PTEN-regulated genes Pbsn and Clu, was similar in the prostates of WT and Inpp4b−/− males (Figures 6D-E). The lack of INPP4B led to histological abnormalities in the prostates of aged mice. Compared to the one year old WT males (Figure 6G), age-matched Inpp4b−/− mice displayed significant alterations in prostate gland histology including tufting of the secretory epithelial layer (Figure 6H), micropapillary structures lacking fibrovascular core (Figure 6I), and cribriform formations (Figure 6J). Interestingly, Pten knockout in the mouse prostate did not alter INPP4B mRNA levels in two independent microarray profiles (Supplemental Figure 3F). This data suggests that the observed INPP4B-dependent changes are due specifically to the loss of INPP4B, as the levels of total and phosphorylated PTEN did not change.
Figure 6. INPP4B is required for optimal expression of a subset of AR target genes in mouse prostate.
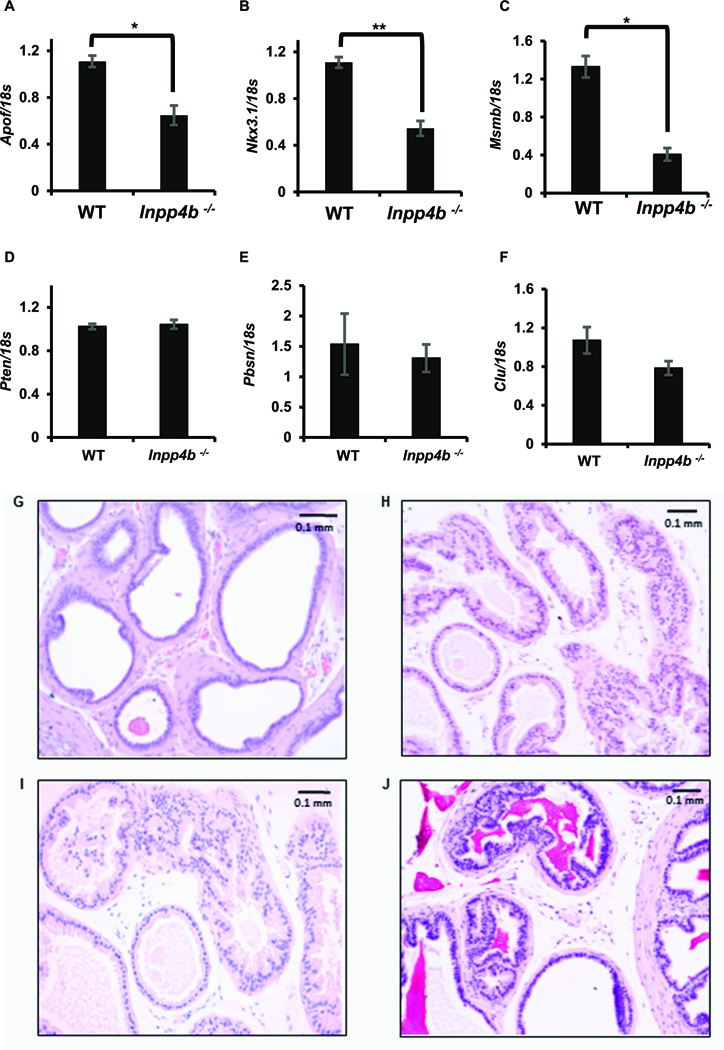
Whole prostates were dissected from 2 month old WT (N=11) and Inpp4b−/− (N=11) males and expression levels of Apof (A), Nkx 3.1 (B), Msmb (C), Pten (D), Pbsn (E) and Clu (F) were compared by RT-qPCR. (G) Histology of a 1 year old WT mouse prostate. (H-J) Examples of histology of 1 year old Inpp4b−/− mouse prostate. Examples of major HG PIN architectural patterns are shown: tufting (H), micropapillary structures lacking fibrovascular core (I), and cribriform formations (J). * p < 0.05, ** p < 0.01
Discussion:
We have previously reported that a decline in INPP4B protein level occurs in primary prostate cancer, and the progressive loss of INPP4B is correlated with biochemical recurrence of the disease (3). Thus, we investigated how the loss of INPP4B affects AR signaling in prostate cancer cell lines, mouse models, and patient gene expression analysis.
In LNCaP cells, INPP4B knockdown modulated the ability of AR to activate and repress target genes (Figures 1F – 1K). Supporting this observation, the AR transcriptional signature is significantly represented among genes regulated by INPP4B in the LNCaP cell line (Figure 1D). Importantly, statistically significant correlations between INPP4B expression and AR mRNA levels and transcriptional output were observed in both the TCGA and Taylor cohorts. While it is well established that PTEN regulates AR transcriptional output in prostate cancer, INPP4B correlations with AR signatures were stronger than those of PTEN (Supplemental Figure 1B) in both cohorts.
The loss of INPP4B and expression of AR splice variants occurs most frequently in the advanced stages of prostate cancer (7, 47). We used a well characterized LNCaPAR-V7/pHAGE cell line which retains endogenous full length AR and INPP4B expression and signaling to investigate the reciprocal regulation of INPP4B and AR-V7 (36, 48). Consistent with its oncogenic function, AR-V7 is unable to induce expression of the tumor suppressor INPP4B and may in fact reduce INPP4B expression (Figures 2A and2B). This could be explained by concordant regulation of INPP4B expression by AR and FOXA1 (Figure 3D). Similar to a previous report (36), AR-V7 failed to bind overlapping AR/FOXA1 sites at the INPP4B transcription start site or at intragenic regulatory regions that activate INPP4B transcription (Figures 3E, 3I – 3J). INPP4B however, retains its ability to regulate AR-V7 transcriptional activity (Figures 2C – 2H).
While AR shuttles between the nucleus and the cytoplasm in both a ligand dependent and independent manner (33, 49), INPP4B localizes mostly to cytoplasmic and membrane compartments (1, 9). This localization could be due in part to the INPP4B C2 domain that binds phosphatidylinositol polyphosphates, a lipid component of cellular membranes (50). Similar to observations in mammary epithelium and PC3 cells (1, 9), INPP4B shows focal cytoplasmic localization in LNCaP cells (Figure 1A). Membranes contain the INPP4B lipid substrates PI(3,4)P2 and PI(4,5)P2 which activate Akt and PKC respectively. Activation of PKC has been linked to AR phosphorylation and prostate cancer progression (16, 51, 52) and was shown to regulate angiogenesis, apoptosis, cell proliferation, invasion, and metastasis (51, 53–55). The PI3K/Akt signaling pathway is activated in nearly half of all primary prostate tumors and in all metastatic prostate tumors (7). Akt and PKC play essential roles in the regulation of activities, stabilities, and cellular localization of both full-length (24, 26, 28, 56) and splice variants of AR (30, 57, 58), as well as AR coregulators (59–61). Importantly, increased levels of Y267, Y534, and S578 phosphorylation correlate with resistance to castration therapies in prostate cancer patients (27, 29, 52, 62, 63). Since INPP4B suppresses Akt and PKC phosphorylation in multiple cell lines (Figures 4A – 4C), we reasoned that if treatment with an inhibitor mimicked the effect of INPP4B on gene expression, the corresponding kinase might contribute to the INPP4B regulation of AR transcriptional activity. As seen from Figures 4E – 4F, expression of PLA2G2A was lower in the presence of INPP4B. BIM-I and AZD5363 decreased expression of PLA2G2A implicating both PKC and Akt in the regulation of this gene. Only PKC contributed to the repression of KIAA1324. Both INPP4B and AZD5363 treatment increased expression of TARP and NNMT (Figures 4G-H) suggesting that Akt mediates INPP4B-dependent induction of these genes. However, INPP4B regulation of SYTL2 and TMPRSS2 were due to mechanisms other than inhibition of PKC and PI3K/Akt signaling. This could be attributed to INPP4B protein phosphatase (43, 44) or other activities. We have previously shown that INPP4B can dephosphorylate Akt on Y residues (44) two of which,Y176 and Y284, positively correlate with prostate cancer progression and inversely correlate with patient survival (63), suggesting a role for INPP4B protein phosphatase activity in prostate cancer progression.
In order to validate the INPP4B regulation of AR activity in vivo we used an Inpp4b knockout mouse (45). Deletion of exon 11 in this model causes a frame shift leading to the addition of four amino acids after Ala 212 and protein truncation (Supplemental Figure 3E). Male Inpp4b−/− mice are fertile and their prostates are histologically normal at 11 weeks of age (Figure 5A and5D). Similar to our observations in prostate cancer cell lines, the loss of INPP4B changed neither the mRNA or protein levels of AR (Figures 5C–5E). However, INPP4B regulates AR transcriptional activity in mouse prostates. Similar to PTEN (26, 64), Inpp4b knockout reduced expression of Apof, Nkx3.1, and Msmb mRNAs (Figure 6A-C). In contrast to the prostate specific Pten knockouts (65, 66), we observed no decline in Pbsn or increase in Clu mRNA levels in Inpp4b−/− prostates (Figure 6E-F). A significant increase of pAkt in the dorsolateral and ventral prostate lobes, and increased pPKCζ and PKCβII in the anterior and dorsolateral prostate lobes of Inpp4b−/−, compared to age-matched WT males was consistently observed (Figure 5E). Of note, the dorsolateral lobe of the mouse prostate is considered a functional model for the peripheral zone of the human prostate, the site of origin for most prostate cancers. The cumulative changes in AR activity and cell signaling pathways lead to histological abnormalities in aged mice (Figures 6G – 6J). Since levels of total and phospho-Pten S380 remained the same, PTEN was unlikely to have contributed to these changes. Thus, differential regulation of AR target genes and AR protein levels signifies divergent functions for INPP4B and PTEN in the prostate epithelium.
Using the observations from both the human and mouse models we propose that castration therapy first initiates a decline in INPP4B expression. The loss of INPP4B protein activates the Akt and PKC signaling pathways to induce the reprogramming of full length AR and AR splice variants, which ultimately leads to the resistance of castration therapies (Figure 7). INPP4B is lost at a higher rate than PTEN during prostate cancer progression (7) and its loss is a strong predictor of disease recurrence (3). Our data indicates that tumors lacking INPP4B use different mechanisms of progression than tumors featuring PTEN loss and that these patients may require a different course of treatment.
Figure 7. INPP4B loss activates Akt and PKC signaling and leads to resistance of castration therapies.
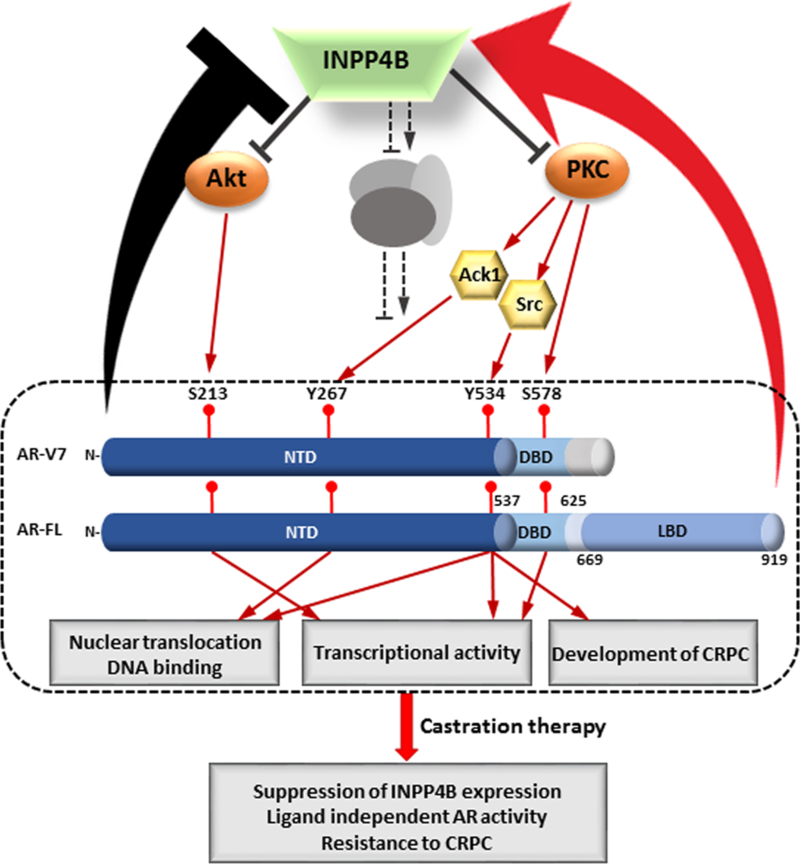
Loss of INPP4B activity in primary prostate cancer (3) leads to accumulation of its substrates PI(3,4)P2 and PI(4,5)P2 which activate Akt and PKC respectively. Activation of Akt and PKC kinases increase phosphorylation of both full length AR and AR splice variants stimulating their nuclear translocation, transcriptional activation and reprogramming, and ligand-independent transcriptional activity. Castration elevates levels of AR splice variants (73) and decreases androgen-dependent INPP4B expression (74), further activating Akt and PKC signaling pathways and leading to resistance to castration therapies. Unlike full length AR, AR-V7 is unable to induce INPP4B expression and may interfere with androgen dependent induction of INPP4B expression
Materials and Methods
Cell culture and reagents
The human prostatic carcinoma cell lines, LNCaP and VCaP, were purchased from ATCC (Manassas, VA) and maintained under ATCC-recommended conditions. C4–2 cells were obtained from UroCor (Oklahoma City, OK) and maintained in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS). All experiments were performed during first 6 passages after purchasing cell lines from ATCC. FBS and charcoal stripped serum (CSS) were purchased from Sigma-Aldrich (St. Louis, MO). LNCaPAR-V7/pHAGE maintenance was described previously (36). Tetracycline-screened FBS (TET FBS) was purchased from GE Healthcare (Chicago, IL) and geneticin from Thermo Fisher Scientific. R1881 was purchased from Perkin Elmer (Waltham, MA), AZD5363 from Selleckchem (Houston, TX), and BIM-I from AdipoGen (San Diego, CA).
Immunofluorescence
LNCaP cells were grown on etched coverslips and fixed and processed as previously described (9). Fixed cells were incubated with INPP4B primary antibody (Cell Signaling, Danvers, MA) at 1:1000 dilution in 4°C overnight and Alexa Fluor conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific) at 1:1000 dilution at 4°C for 1 hour. Cells were counterstained with DAPI (Sigma) and visualized using Axio Imager 2 (Zeiss, ST Petersburg, FL). Three dimensional rendering was performed using AxioVision LE software (Zeiss).
siRNA transfections
Noncoding control, INPP4B, PTEN, and FOXA1 siRNAs were transfected at 50–100 pmol siRNA per well in 6 well cell culture plate using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific) as recommended by the manufacturer. All siRNAs used in this manuscript are listed in Supplemental Table 3.
Microarray Analysis
LNCaP cells were transfected with noncoding control (n=4) or INPP4B specific (n=4) siRNAs. Forty eight hours later, RNA was extracted using Tri reagent (Molecular Research Center, Cincinnati, OH),and analyzed using the Illumina Human HT-12v4 expression Beadchip platform exactly as described in our previous publication (9). Gene expression data were deposited into GEO repository, series number GSE111725.
Gene Set Enrichment Analysis (GSEA)
GSEA analysis was performed using JAVA program http://software.broadinstitute.org/gsea as previously described (34, 67). AR signature (Supplemental Table 2) was generated from LNCaP which includes 422 genes significantly changed (p<0.01) after 48 hours of DHT treatment (GSE60721) (34).
Correlation with published AR signatures and key genes in prostate cancer cohorts
Previously published signatures of prostate cancer cells transcriptional response to androgen were derived from the Gene Expression Omnibus (GEO), termed Hyeronimus and Nelson AR signatures (68, 69). We downloaded gene expression datasets from multiple previously reported human prostate cancer specimen cohorts: Taylor et al. (7), and The Cancer Genome Atlas (TCGA) (70). Within each dataset, we utilized the expression of each gene to calculate its respective z-score for each sample, relative to the normal prostate gland specimens available in that cohort. For each of two published androgen-induced signatures, Nelson and Hieronymus, we computed the sum z-score for each sample (the z-scores of downregulated genes were subtracted from the z-scores of upregulated genes), as described previously (7). Finally, for each pair of genes or signatures we computed the Pearson Correlation Coefficient R and associated p-value using the R statistical system.
Western blotting
Proteins were extracted from cultured cells as previously described (9). Mouse prostate tissues from 3 animals were pooled and homogenized in ice-cold RIPA buffer with protease and phosphatase inhibitors. Cleared lysates were diluted to 4μg/μl and 10–25 μg of protein was resolved on SDS-PAGE. Primary antibodies against INPP4B (Cell Signaling, #8450), AR (Santa Cruz Biotechnology, Dallas, TX, sc-816), pAkt S473 (Cell Signaling, #4051), pPKCζ T410 (Cell Signaling, #2060), pPKCβII S660 (Cell Signaling, #9371, 1:1000), PTEN (Cell Signaling, #9188), total Akt (Cell Signaling, #4691), pS6 S235/236 (Cell Signaling, #2211), p44/42 MAPK (Cell Signaling, #4370) and β-tubulin (Millipore Sigma, #05–661) were used to compare protein levels. Signal was captured by Gel Logic 2000 imaging system with Carestream Molecular Imaging software (Carestream, Rochester, NY).
Gene expression analysis.
RNA was prepared from prostate cancer cells or mouse prostates using Tri Reagent (Molecular Research Center) and reverse transcribed using Verso cDNA synthesis Kit (Thermo Fisher Scientific) or AmfiRivert cDNA Synthesis kit (GenDEPOT, Katy, TX). cDNA was used as a template for Real Time PCR using primers and probes from Universal ProbeLibrary (Supplemental Table 1) (Roche, Basel, Switzerland) and Roche 480 LightCycler (Roche) or StepOnePlus™ Real-Time PCR System (ThermoFisher). For cell lines, three or more independent experiments were performed and representative experiment is shown. In each experimental point an average of 3 biological replicates, standard error of the mean, and p value calculated by T test are shown. For animal tissues, WT group contained 11 animals and Inpp4b−/− group contained 12 animals.
Chromatin immunoprecipitation assay.
ChIP assays were performed exactly as previously described (71). Cell lysates were incubated with 5 μg AR-FL antibody (Millipore #06–680), 5 μg AR-V7 antibody (Cell Signaling #68492) or 5 μg rabbit anti-mouse Igg (Santa Cruz sc2027) respectively. The AR and AR-V7 recruitment were measured by qPCR using the primers and probes listed in Supplemental Table 1.
Animal Studies
Animals were housed at AAALAC certified barrier facility at Florida International University. All procedures were reviewed and approved by the Institutional Animal Care and Use Committee at FIU and conducted in accordance with the National Academy of Science Guide for Care and Use of Laboratory Animals. C57BL/6 Inpp4b−/− mice was kindly provided by Dr. Vacher (Institut De Recherches Cliniques De Montreal, Canada), and bred with FVB for at least three generations. Genotyping procedure was described in original publication (45). Two month old male mice were euthanized, weighed, their urogenital organs dissected and weighed, prior to RNA and protein extraction or formaldehyde fixation and paraffin embedding. In addition prostates of 374 days old males were fixed and used for histological analysis.
H&E and Immunohistochemistry (IHC)
Whole prostates were fixed in 4% formaldehyde (Electron Microscopy Sciences, Hatfield, PA) and embedded in paraffin. H&E staining was performed as previously described (72). Antigen was retrieved by heating slides in 0.01 M sodium citrate buffer (pH 6.0). Primary antibodies for INPP4B (Santa Cruz Biotechnology) or AR (Santa Cruz Biotechnology) were used at 1:800 dilution and sections were counterstained with hematoxylin (EMD Millipore). All images were acquired by AxioCam camera and were processed by AxioVision LE software (Zeiss).
Statistical analysis
Comparisons of mean levels of expression of specific mRNAs were done as previously described (44). Student t-tests were used to test for inequality of means from two independent samples sets, unless the normality assumptions about the data are violated, in which case the Welch’s t test was used. P-values less than 0.05 were considered statistically significant. Values are presented as mean ± SEM. Gene expression analysis was done using Prism 7.0 software. Biological triplicates were used for every point in individual experiments for evaluating changes in gene expression in cell lines. All experiments were repeated at least three times, unless stated otherwise.
Supplementary Material
Acknowledgments
Financial support: This work was supported by NCI grant R15 CA179287–01A1 (IUA), NCI grant P30CA125123 (CC), CPRIT grant RP150648 (NLW, CC), and CPRIT grant RP170005 (CC).
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest.
Reference:
- 1.Fedele CG, Ooms LM, Ho M, Vieusseux J, O’Toole SA, Millar EK, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(51):22231–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16(2):115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodgson MC, Shao LJ, Frolov A, Li R, Peterson LE, Ayala G, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71(2):572–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu I, Yeh CR, Slavin S, Miyamoto H, Netto GJ, Tsai YC, et al. Estrogen receptor alpha prevents bladder cancer via INPP4B inhibited akt pathway in vitro and in vivo. Oncotarget. 2014;5(17):7917–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Chew C, Lunardi A, Gulluni F, Ruan DT, Chen M, Salmena L, et al. In Vivo Role of INPP4B in Tumor and Metastasis Suppression through Regulation of PI3K-AKT Signaling at Endosomes. Cancer Discov. 2015;5(7):740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget. 2011;2(4):321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kofuji S, Kimura H, Nakanishi H, Nanjo H, Takasuga S, Liu H, et al. INPP4B Is a PtdIns(3,4,5)P3 Phosphatase That Can Act as a Tumor Suppressor. Cancer Discov. 2015;5(7):730–9. [DOI] [PubMed] [Google Scholar]
- 9.Hodgson MC, Deryugina EI, Suarez E, Lopez SM, Lin D, Xue H, et al. INPP4B suppresses prostate cancer cell invasion. Cell Commun Signal. 2014;12(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Nowak DG, Trotman LC. Molecular pathways: PI3K pathway phosphatases as biomarkers for cancer prognosis and therapy. Clin Cancer Res. 2014;20(12):3057–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton AC. Diacylglycerol’s affair with protein kinase C turns 25. Trends Pharmacol Sci. 2004;25(4):175–7. [DOI] [PubMed] [Google Scholar]
- 13.Newton AC. Regulation of the ABC kinases by phosphorylation : protein kinase C as a paradigm. Biochemical Journal. 2003;370(8):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creba JA, Downes CP, Hawkins PT, Brewster G, Michell RH, Kirk CJ. Rapid breakdown of phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate in rat hepatocytes stimulated by vasopressin and other Ca2+-mobilizing hormones. Biochemical Journal. 1983;212(3):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishizuka Y The Protein Kinase C Family and Lipid Mediators for Transmembrane Signaling and Cell Regulation. Alcohol Clin Exp Res. 2001;25. [DOI] [PubMed] [Google Scholar]
- 16.Cornford P, Evans J, Dodson A, Parsons K, Woolfenden A, Neoptolemos J, et al. Protein Kinase C Isoenzyme Patterns Characteristically Modulated in Early Prostate Cancer. The American Journal of Pathology. 1999;154(1):137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balk SP. Androgen receptor as a target in androgen-independent prostate cancer. Urology. 2002;60(3 Suppl 1):132–8; discussion 8–9. [DOI] [PubMed] [Google Scholar]
- 18.Yeh S, Tsai M-Y, Xu Q, Mu X-M, Lardy H, Huang K-E, et al. Generation and characterization of androgen receptor knockout (ARKO) mice: An in vivo model for the study of androgen functions in selective tissues. PNAS, Proceedings of the National Academy of Sciences. 2002;99(21):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakazawa M, Antonarakis ES, Luo J. Androgen receptor splice variants in the era of enzalutamide and abiraterone. Horm Cancer. 2014;5(5):265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69(6):2305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72(14):3457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin HK, Hu YC, Yang L, Altuwaijri S, Chen YT, Kang HY, et al. Suppression versus induction of androgen receptor functions by the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer LNCaP cells with different passage numbers. J Biol Chem. 2003;278(51):50902–7. [DOI] [PubMed] [Google Scholar]
- 24.Lin HK, Yeh S, Kang HY, Chang C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(13):7200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taneja SS, Ha S, Swenson NK, Huang HY, Lee P, Melamed J, et al. Cell-specific regulation of androgen receptor phosphorylation in vivo. J Biol Chem. 2005;280(49):40916–24. [DOI] [PubMed] [Google Scholar]
- 26.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponguta LA, Gregory CW, French FS, Wilson EM. Site-specific androgen receptor serine phosphorylation linked to epidermal growth factor-dependent growth of castration-recurrent prostate cancer. J Biol Chem. 2008;283(30):20989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatin F, Varon C, Genot E, Moreau V. A signalling cascade involving PKC, Src and Cdc42 regulates podosome assembly in cultured endothelial cells in response to phorbol ester. J Cell Sci. 2006;119(Pt 4):769–81. [DOI] [PubMed] [Google Scholar]
- 29.Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell. 2006;10(4):309–19. [DOI] [PubMed] [Google Scholar]
- 30.Karaca M, Liu Y, Zhang Z, De Silva D, Parker JS, Earp HS, et al. Mutation of androgen receptor N-terminal phosphorylation site Tyr-267 leads to inhibition of nuclear translocation and DNA binding. PloS one. 2015;10(5):e0126270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benavides F, Blando J, Perez CJ, Garg R, Conti CJ, DiGiovanni J, et al. Transgenic overexpression of PKCepsilon in the mouse prostate induces preneoplastic lesions. Cell Cycle. 2011;10(2):268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hafeez BB, Zhong W, Weichert J, Dreckschmidt NE, Jamal MS, Verma AK. Genetic ablation of PKC epsilon inhibits prostate cancer development and metastasis in transgenic mouse model of prostate adenocarcinoma. Cancer Res. 2011;71(6):2318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saporita AJ, Zhang Q, Navai N, Dincer Z, Hahn J, Cai X, et al. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J Biol Chem. 2003;278(43):41998–2005. [DOI] [PubMed] [Google Scholar]
- 34.Lopez SM, Agoulnik AI, Zhang M, Peterson LE, Suarez E, Gandarillas GA, et al. Nuclear Receptor Corepressor 1 expression and output declines with prostate cancer progression. Clin Cancer Res. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338(6113):1465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krause WC, Shafi AA, Nakka M, Weigel NL. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol. 2014;54:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J. 2011;30(19):3962–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474(7351):390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132(6):958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao N, Zhang J, Rao MA, Case TC, Mirosevich J, Wang Y, et al. The role of hepatocyte nuclear factor-3 alpha (Forkhead Box A1) and androgen receptor in transcriptional regulation of prostatic genes. Mol Endocrinol. 2003;17(8):1484–507. [DOI] [PubMed] [Google Scholar]
- 41.Gao N, Ishii K, Mirosevich J, Kuwajima S, Oppenheimer SR, Roberts RL, et al. Forkhead box A1 regulates prostate ductal morphogenesis and promotes epithelial cell maturation. Development. 2005;132(15):3431–43. [DOI] [PubMed] [Google Scholar]
- 42.He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(51):18261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo ST, Chi MN, Yang RH, Guo XY, Zan LK, Wang CY, et al. INPP4B is an oncogenic regulator in human colon cancer. Oncogene. 2016;35(23):3049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez SM, Hodgson MC, Packianathan C, Bingol-Ozakpinar O, Uras F, Rosen BP, et al. Determinants of the tumor suppressor INPP4B protein and lipid phosphatase activities. Biochem Biophys Res Commun. 2013;440(2):277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferron M, Boudiffa M, Arsenault M, Rached M, Pata M, Giroux S, et al. Inositol polyphosphate 4-phosphatase B as a regulator of bone mass in mice and humans. Cell Metab. 2011;14(4):466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang XD, Wang BE, Soriano R, Zha J, Zhang Z, Modrusan Z, et al. Expression profiling of the mouse prostate after castration and hormone replacement: implication of H-cadherin in prostate tumorigenesis. Differentiation. 2007;75(3):219–34. [DOI] [PubMed] [Google Scholar]
- 47.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120(8):2715–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget. 2015;6(31):31997–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jenster G, Trapman J, Brinkmann AO. Nuclear import of the human androgen receptor. Biochem J. 1993;293 ( Pt 3):761–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kooijman EE, King KE, Gangoda M, Gericke A. Ionization properties of phosphatidylinositol polyphosphates in mixed model membranes. Biochemistry. 2009;48(40):9360–71. [DOI] [PubMed] [Google Scholar]
- 51.Kim J, Choi YL, Vallentin A, Hunrichs BS, Hellerstein MK, Peehl DM, et al. Centrosomal PKCbetaII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res. 2008;68(16):6831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patek S, Willder J, Heng J, Taylor B, Horgan P, Leung H, et al. Androgen receptor phosphorylation status at serine 578 predicts poor outcome in prostate cancer patients. Oncotarget. 2017;8(3):4875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gavrielides MV, Gonzalez-Guerrico AM, Riobo NA, Kazanietz MG. Androgens regulate protein kinase Cdelta transcription and modulate its apoptotic function in prostate cancer cells. Cancer Res. 2006;66(24):11792–801. [DOI] [PubMed] [Google Scholar]
- 54.Henttu P, Vihko aP. The Protein Kinase C Activator, Phorbol Ester, Elicits Disparate Functional Responses in Androgen-Sensitive and Androgen-Independent Human Prostatic Cancer Cells Biochemical and Biophysical Research Communications. 1998;224(1):5. [DOI] [PubMed] [Google Scholar]
- 55.Wu D, Foreman TL, Gregory CW, McJilton MA, Wescott GG, Ford OH, et al. Protein Kinase Cε Has the Potential to Advance the Recurrence of Human Prostate Cancer. Cancer Research. 2002;62(8):7. [PubMed] [Google Scholar]
- 56.Darne C, Veyssiere G, Jean C. Phorbol ester causes ligand-independent activation of the androgen receptor. Eur J Biochem. 1998;256(3):9. [DOI] [PubMed] [Google Scholar]
- 57.Mediwala SN, Sun H, Szafran AT, Hartig SM, Sonpavde G, Hayes TG, et al. The activity of the androgen receptor variant AR-V7 is regulated by FOXO1 in a PTEN-PI3K-AKT-dependent way. Prostate. 2013;73(3):267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, Xie N, Gleave ME, Rennie PS, Dong X. AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget. 2015;6(32):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garabedian MJ, Logan SK. Atypical regulation of SRC-3. Trends Biochem Sci. 2008;33(7):301–4. [DOI] [PubMed] [Google Scholar]
- 60.Wu RC, Feng Q, Lonard DM, O’Malley BW. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129(6):1125–40. [DOI] [PubMed] [Google Scholar]
- 61.Jo YS, Ryu D, Maida A, Wang X, Evans RM, Schoonjans K, et al. Phosphorylation of the nuclear receptor corepressor 1 by protein kinase B switches its corepressor targets in the liver in mice. Hepatology. 2015;62(5):1606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahajan K, Challa S, Coppola D, Lawrence H, Luo Y, Gevariya H, et al. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate. 2010;70(12):1274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PloS one. 2010;5(3):e9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19(6):792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Thomas JJGV, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. CANCER CELL. 2003;4. [DOI] [PubMed] [Google Scholar]
- 66.Lokody IB, Francis JC, Gardiner JR, Erler JT, Swain A. Pten Regulates Epithelial Cytodifferentiation during Prostate Development. PloS one. 2015;10(6):e0129470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10(4):321–30. [DOI] [PubMed] [Google Scholar]
- 69.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(18):11890–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang M, Krause WC, Agoulnik IU. Techniques for Evaluation of AR Transcriptional Output and Recruitment to DNA. Methods Mol Biol. 2018;1786:219–36. [DOI] [PubMed] [Google Scholar]
- 72.Hodgson MC, Vanostran G, Alghamdi S, Poppiti RJ, Agoulnik AI, Agoulnik IU. Reduced androgen receptor expression accelerates the onset of ERBB2 induced breast tumors in female mice. PloS one. 2013;8(4):e60455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guedes LB, Morais CL, Almutairi F, Haffner MC, Zheng Q, Isaacs JT, et al. Analytic Validation of RNA In Situ Hybridization (RISH) for AR and AR-V7 Expression in Human Prostate Cancer. Clin Cancer Res. 2016;22(18):4651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hodgson MC, Deryugina EI, Suarez E, Lopez SM, Lin D, Xue H, et al. INPP4B suppresses prostate cancer cell invasion. Cell Commun Signal. 2014;12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.