

Abstract
Endogenous histidyl dipeptides such as carnosine (β-alanine-L-histidine) form conjugates with lipid peroxidation products such as 4-hydroxy-trans-2-nonenal (HNE and acrolein), chelate metals, and protect against myocardial ischemic injury. Nevertheless, it is unclear whether these peptides protect against cardiac injury by directly reacting with lipid peroxidation products. Hence, to examine whether changes in the structure of carnosine could affect its aldehyde reactivity and metal chelating ability, we synthesized methylated analogs of carnosine, balenine (β-alanine-Nτ-methylhistidine) and di-methylbalenine (DMB), and measured their aldehyde reactivity and metal chelating properties. We found that methylation of Nτ residue of imidazole ring (balenine) or trimethylation of carnosine backbone at Nτ residue of imidazole ring and terminal amine group dimethyl balenine (DMB) abolishes the ability of these peptides to react with HNE. Incubation of balenine with acrolein resulted in the formation of single product (m/z 297), whereas DMB did not react with acrolein. In comparison with carnosine, balenine exhibited moderate acrolein quenching capacity. The Fe2+chelating ability of balenine was higher than carnosine, whereas DMB lacked chelating capacity. Pretreatment of cardiac myocytes with carnosine increased the mean lifetime of myocytes superfused with HNE or acrolein compared with balenine or DMB. Collectively, these results suggest that carnosine protects cardiac myocytes against HNE and acrolein toxicity by directly reacting with these aldehydes. This reaction involves both the amino group of β-alanyl residue and the imidazole residue of L-histidine. Methylation of these sites prevents or abolishes the aldehyde reactivity of carnosine, alters its metal-chelating property, and diminishes its ability to prevent electrophilic injury.
Keywords: acrolein, cardiac myocytes, histidyl dipeptides, 4-hydroxy-trans-2-nonenal
Introduction
Extensive evidence suggests that oxidative stress is a significant feature of myocardial injury due to heart failure, ischemia-reperfusion (I/R) injury, pathological hypertrophy and aging (Giordano 2005; Sawyer et al. 2002). However long-term studies with humans that targeted reactive oxygen species with antioxidants such as beta carotene (Hennekens et al. 1996) and vitamin E (Yusuf et al. 2000) failed to show any beneficial effects on cardiovascular disease. The inability of antioxidants to decrease cardiovascular disease risk is likely due to the complex pathophysiological role of ROS, which at low concentrations participate in signaling, whereas at high concentration they cause tissue damage by reacting with proteins, DNA and phospholipids (Halliwell 2015). Oxidation of phospholipids in the fatty acid membranes results in the generation of several highly toxic lipid peroxidation products such as 4-hydroxy-trans-2-nonenal (HNE) and acrolein (Porter et al. 1995; Niki 2009). Increased levels of HNE or HNE-modified protein adducts have been detected in the hearts of dilated cardiomyopathic hamsters (Kato et al. 2010), hypertensive rats (Benderdour et al. 2003; Benderdour et al. 2004), rats infused with isoproterenol (Srivastava et al. 2007), mice subjected to trans aortic constriction (Zhang et al. 2007), heart failure (Liu et al. 2005), rabbits subjected to myocardial infarction-induced heart failure (Qin et al. 2007) and dogs subjected to tachycardia-induced heart failure (Zhang et al. 2009). Lipid peroxidation products such as HNE are metabolically removed by several enzymes including aldose reductase (AR) (Baba et al. 2018; Srivastava et al. 1999; Srivastava et al. 1998), aldehyde dehydrogenase (ALDH2) (Chen et al. 2008; Gomes et al. 2014) and glutathione-S-transferases (GSTs) (Conklin et al. 2015). Although extensive investigations have delineated the pathological role of lipid peroxidation products and identified metabolic pathways that detoxify these toxic aldehydes, non-enzymatic pathways, which can sequester these aldehydes and protect against myocardial injury, have been less well studied.
Most animals and humans synthesize a wide variety of histidyl dipeptides, such as carnosine anserine (β-alanine-Nπ-methylhistidine), balenine (β-alanine-Nτ-methylhistidine) and homocarnosine (γamino-butyryl-histidine) (Boldyrev et al. 2013). Among these dipeptides, carnosine is most abundant in the humans, whereas its methylated analogue - anserine is found mostly in birds (Boldyrev et al. 2013). Balenine is present primarily in whales (Abe 2000). Carnosine is synthesized from β-alanine and histidine by the enzyme carnosine synthase (ATPGD1) (Drozak et al. 2010), which belongs to the ATP grasp family of enzymes (Fawaz et al. 2011). It is accumulated to high abundance in tissues with high metabolic activity, such as the heart, the skeletal muscle, and the brain (Boldyrev et al. 2013). Because these peptides contain the imidazole ring of L-histidine, they can buffer intracellular pH. This buffering activity of histidyl dipeptides is believed to facilitate glycolysis by buffering intracellular pH under anaerobic conditions (Hipkiss 2009). In addition to the imidazole ring, these peptides also contain highly reactive nucleophilic amines, which impart them the ability to bind with a wide variety of lipid peroxidation products such as HNE and acrolein (Aldini et al. 2002; Carini et al. 2003). The reaction products between carnosine HNE and acrolein are extruded out in human urine (Baba et al. 2013). Previous work has shown that in obese diabetic patients, carnosine supplementation increases the extrusion of advanced glycation end products and improves the markers of metabolic syndrome (de Courten et al. 2016).
Recent work from our laboratory has shown that carnosine levels are depleted in hypertrophic and failing hearts (Sansbury et al. 2014). Moreover, we have found that post-ischemic recovery of contractile function after global ischemia is significantly improved in mouse hearts perfused with carnosine (Baba et al. 2013), suggesting that carnosine prevents myocardial injury during conditions of oxidative stress. Nevertheless, the mechanism of this protection remains unclear. During oxidative stress, carnosine can protect the heart either by buffering intracellular pH, chelating metals, quenching singlet oxygen or reacting directly with the electrophilic products of lipid peroxidation. To distinguish between these possibilities, we synthesized methylated analogs of carnosine, balenine and di-methyl balenine (DMB), in which the imidazole ring of L-histidine and the amino group of β-alanine are methylated. We reasoned that if carnosine prevents electrophilic injury due to HNE and acrolein by directly reacting with these aldehydes, then methylation of its reactive groups (the amino group of β-alanine and the imidazole ring of histidyl residue) should diminish its reactivity with HNE and acrolein, and thereby prevent or abolish its protective effects.
Materials and Methods
D-histidine was purchased from TCI America. Hydrochloric acid gas, 1,1’-carbonyldiimidazole, methyl iodide, 3-(dimethylamino) propanoic acid hydrochloride, ethylenediaminetetraacetic acid (EDTA), ferrozine, EDC (N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide), and N-methylmorpholine, carnosine, were purchased from Sigma-Aldrich and tyrosine-histidine (internal standard: IS) was purchased from Bachem. Dry dicholoro methane (DCM) and dry N, N dimethylformamide ( DMF) were purchased from Acros Organics. Adult male C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME).
NMR Spectroscopy and thin layer chromatography
The 1H and 13C NMR spectra were recorded in NMR spectrometers operating at 400.00 MHz for 1H NMR and 100.6 MHz for 13C NMR. The chemical shifts were calibrated from the residual peaks observed for the deuterated solvents such as chloroform (CDCl3), dimethyl sulfoxide (DMSO-d6), and D2O at δ 7.26, 2.50, and 4.79 ppm for 1H NMR, respectively. Chemical shifts were calibrated from the residual peaks observed for the deuterated solvents such as chloroform (CDCl3), and dimethyl sulfoxide (DMSO-d6) at δ 77.0 and 39.5 ppm for 13C NMR, respectively. Multiplicities of 1H NMR spin couplings are: s for singlet, bs for broad singlet, or m for multiplet and overlapping spin systems. Values for the coupling constants (J) are reported in hertz (Hz). Thin layer chromatography (TLC) was performed using pre-coated silica gel plates and visualized by UV light (254 nm).
Synthesis of balenine
(S)-methyl 5-oxo-5,6,7,8-tetrahydroimidazo[1,5-c] pyrimidine-7-carboxylate (2):
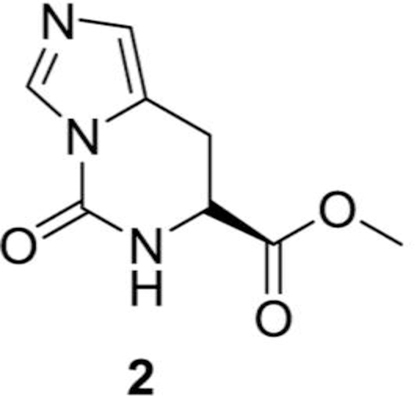
In a round bottomed flask, (S)-methyl 2-amino-3-(1H-imidazol-5-yl) propanoate dihydrochloride (5.0 g, 20.65 mmol), 1,1’-carbonyldiimidazole (3.52 g, 21.7 mmol) were added in DMF (100 mL) at room temperature (RT). The reaction mixture was heated at 60 oC for 6 h and evaporated to take off all the DMF. The crude material was purified with silica gel column chromatography to get the desired compound, (S)-methyl 5-oxo-5,6,7,8-tetrahydroimidazo[1,5-c] pyrimidine-7-carboxylate, (2, 2.9 g, 69%) as a white solid compound.
1H NMR (400 MHz, DMSO d6) δ 8.55–8.53 (m, 1H), 8.10–8.09 (m, 1H), 6.82–6.80 (m, 1H), 4.45–4.42 (m, 1H), 3.62 (s, 3H), 3.22–3.20 (m, 2H) ppm.
S)-7-(methoxycarbonyl)-2-methyl-5-oxo-5,6,7,8-tetrahydroimidazo[1,5-c] pyrimidin-2-ium iodide (3):
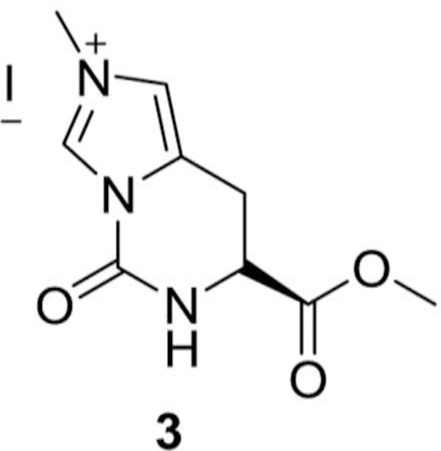
In a round bottomed flask (S)-methyl 5-oxo-5,6,7,8-tetrahydroimidazo[1,5-c] pyrimidine-7-carboxylate (2, 3.00 g, 15.4 mmol) and methyl iodide (3.83 mL, 102.5 mmol) were added in acetonitrile (100 mL) at RT. The reaction mixture was heated at 80 o C for 16 h, and evaporated to dryness to get the desired compound, (S)-7-(methoxycarbonyl)-2-methyl-5-oxo-5,6,7,8-tetrahydroimidazo[1,5-c] pyrimidin-2-ium iodide, (3, 4.9 g, yield = 94 %) as an off-white solid compound.
1H NMR (400 MHz, D2O) δ 9.39 (s, 1H), 7.44 (bs, 1H), 4.77–4.75 (m, 1H), 3.99 (s, 3H), 3.80 (s, 3H), 3.53–3.51 (m, 2H) ppm.
(S)-1-carboxy-2-(1-methyl-1H-imidazol-4-yl) ethanaminium chloride (4):
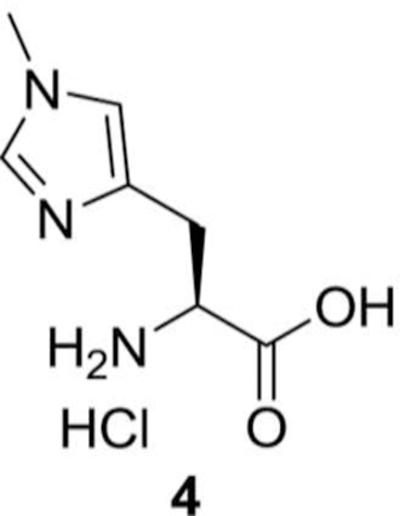
In a round bottom flask (S)-7-(methoxycarbonyl)-2-methyl-5-oxo-5,6,7,8-tetrahydroimidazo[1,5-c] pyrimidin-2-ium iodide (3, 4.00 g, 11.9 mmol) was added to 4N HCl (100 mL). The reaction mixture was refluxed for 6 h, evaporated to dryness, and the solid extract was washed with hexanes and dried to get the desired compound, (S)-1-carboxy-2-(1-methyl-1H-imidazol-4-yl) ethanaminium chloride, (4, 2.10 g, yield = 87%) as yellow solid compound.
1H NMR (400 MHz, D2O) δ 8.69 (bs, 1H), 7.44 (bs, 1H), 4.36 (t, J = 6.60 Hz, 1H), 3.90 (s, 3H), 3.32–3.19 (m, 2H) ppm.
(S)-methyl 2-amino-3-(1-methyl-1H-imidazol-4-yl) propanoate hydrochloride (5):
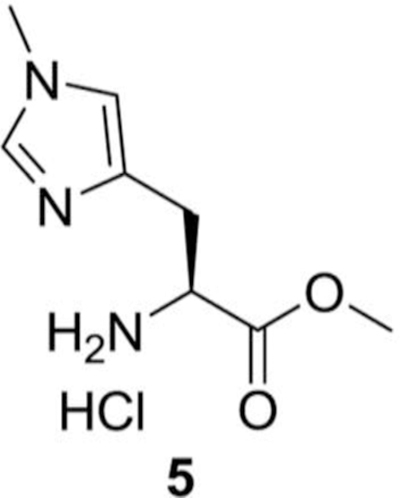
In 300 mL of methanolic hydrochloric acid (S)-2-amino-3-(1-methyl-1H-imidazol-4-yl) propanoic acid hydrochloride (4, 2.60 g, 12.64 mmol) was added at RT. The reaction mixture was then refluxed for 48 h, and then evaporated to dryness to get more than 99% pure of (S)-methyl 2-amino-3-(1-methyl-1Himidazol-4-yl) propanoate hydrochloride, (5, 2.60 g, 94%) as a white solid.
1H NMR (400 MHz, D2O) δ 8.70 (bs, 1H), 7.44 (bs, 1H), 4.51 (t, J = 6.80 Hz, 1H), 3.91 (s, 3H), 3.87 (s, 3H) 3.34–3.20 (m, 2H) ppm.
(S)-methyl 2-(3-(tert-butoxycarbonylamino)propanamido)-3-(1-methyl-1H-imidazol-4-yl)propanoate (6):
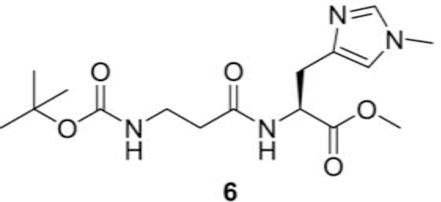
In a reaction flask under nitrogen we sequentially added (S)-methyl 2-amino-3-(1-methyl-1H-imidazol-4yl) propanoate hydrochloride (5, 5.00 g, 22.78 mmol), Boc-b-alanine (4.70 g, 25.04 mmol), EDC (N-(3Dimethylaminopropyl)-N′-ethylcarbodiimide) (4.80 g, 25.04 mmol) followed by DCM (100 mL) and Nmethylmorpholine (10.0 mL, 91.04 mmol via syringe). The contents were stirred at RT under nitrogen and the solids were gradually dissolved. The contents were stirred at RT for 24 h, and slowly diluted into iced water and extracted with DCM. The organic phase was dried evaporated and chromatographed using DCM and methanol as eluents to get (S)-methyl 2-(3-(tert-butoxycarbonylamino) propanamido)-3-(1methyl-1H-imidazol-4-yl) propanoate (6, 3.90 g, yield = 49%) as a white solid compound.
1H NMR (400 MHz, CDCl3) δ 7.46–7.43 (m, 1H), 7.38–7.34 (m, 1H), 6.74 (s, 1H), 5.78 (bs, 1H), 4.91– 4.87 (m, 1H), 3.81 (s, 3H), 3.72 (s, 3H) 3.56–3.51 (m, 2H), 3.20–3.08 (m, 2H), 2.58–2.46 (m. 2H), 1.54 (s, 9H) ppm.
(S)-2-(3-aminopropanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoic acid (7):
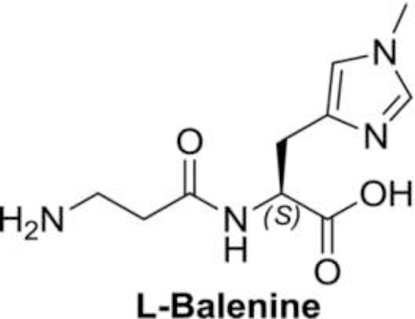
To (S)-methyl 2-(3-(tert-butoxycarbonylamino) propanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoate (6, 4.50 g, 12.7 mmol) was added 2N NaOH (38.1 mL) in THF (70 mL) and methanol (70 mL). Reaction mixture was stirred at RT for 24 h. Solvent was evaporated and reaction mixture was neutralized by 1N HCl to pH 3.0 and stirred for 1h. Reaction was neutralized by saturated sodium bicarbonate. Water was evaporated to dryness and the solid was washed with DCM and dried again. The obtained solid was dissolved in hot ethanol and passed through the celite plug, twice, to get the desired compound, (S)-2-(3aminopropanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoic acid, (7, 2.30 g, yield = 76%) as solid compound.
1H NMR (400 MHz, D2O) δ 7.54 (bs, 1H), 6.94 (bs, 1H), 4.49–4.45 (m, 1H), 3.69 (s, 3H), 3.63–3.57 (m, 1H), 3.14–2.87 (m, 3H), 2.53–2.50 (m, 1H), 2.26–2.23 (m, 1H) ppm.
Synthesis of dimethyl-balenine
Steps 1–5 for dimethyl-balenine synthesis were similar to balenine
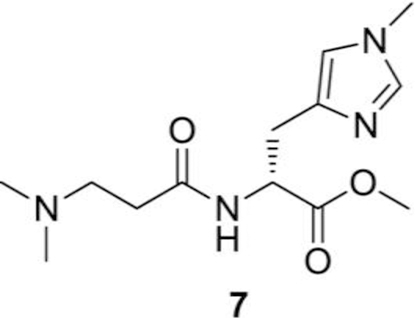
Synthesis of (R)-methyl 2-(3-(dimethylamino)propanamido)-3-(1-methyl-1H-imidazol-4-yl)propanoate (7):
(R)-1-methoxy-3-(1-methyl-1H-imidazol-4-yl)-1-oxopropan-2-aminium chloride (2.5 g, 8.9 mmol), 3-(dimethylamino) propanoic acid hydrochloride (1.5 g, 9.8 mmol), EDC (N-(3-Dimethylaminopropyl)N′-ethylcarbodiimide) (1.9 g, 9.8 mmol) and DCM (100 mL) were added sequentially to a reaction flask under nitrogen. The solution was cooled in an ice water bath under nitrogen. N-methylmorpholine (3.9 mL, 35.8 mmol via syringe) was added slowly. The ice water bath was removed 5–10 min after the completion of addition. The contents were stirred at RT under nitrogen and the solids were gradually dissolved. The reaction mixture was stirred at RT for 48 h. The reaction mixture was dried on reduced pressure and chromatographed using DCM and methanol as eluents to get desired compound, (R)-methyl 2-(3-(dimethylamino) propanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoate (7, 1.1 g, 34%) as a solid compound. 1H NMR (400 MHz, CDCl3) δ 8.89 (bs, 1H), 8.81 (d, J = 7.99 Hz, 1H), 7.13 (bs, 1H), 4.85–4.78 (m, 1H), 3.96 (s, 3H), 3.75 (s, 3H) 3.60–3.52 (m, 1H), 3.37–3.28 (m, 3H), 3.23–3.15 (m. 1H), 2.94– 2.88 (m. 1H), 2.87 (s, 6H) ppm. 13C NMR (100.6 MHz, CDCl3) δ 170.62, 169.36, 135.06, 130.75, 120.49, 53.65, 52.87, 52.30, 50.79, 43.49, 36.23, 30.70, 26.04 ppm.
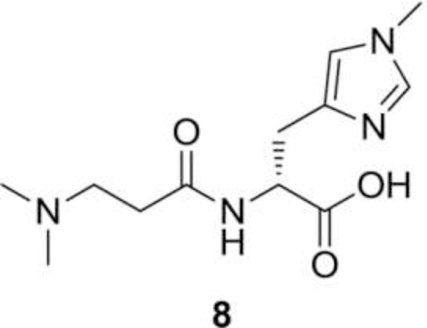
Synthesis of (R)-2-(3-(dimethylamino) propanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoic acid (8):
To (R)-methyl 2-(3-(dimethylamino) propanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoate (0.5 g, 1.8 mmol) was added 2N NaOH (3.54 mL) in THF (10 mL) and methanol (10 mL) and the reaction was stirred for 16 h at RT. Later solvent was evaporated under reduced pressure and pH of reaction mixture was adjusted to pH 7 with 1N HCl solution. Water was evaporated to dryness and the solid product was washed with DCM (2 × 20 mL) and dried again. The obtained solid was dissolved in ethanol (6.0 mL) and passed through the celite plug (5.0 g), twice, to get the desired compound, (R)-2-(3-(dimethylamino) propanamido)-3-(1-methyl-1H-imidazol-4-yl) propanoic acid (8, 360 mg, 76% yield) as an off-white solid compound. 1H NMR (400 MHz, D2O) δ 7.90 (bs, 1H), 6.98 (bs, 1H), 4.40–4.34 (m, 1H), 3.67 (s, 3H), 3.37–3.25 (m, 2H), 3.07–3.00 (m, 1H), 2.94–3.15 (m, 1H), 2.94–2.86 (m, 1H), 2.80 (s, 6H), 2.74–2.63 (m, 2H) ppm.13C NMR (100.6 MHz, D2O) δ 177.25, 170.99, 136.58, 133.92, 119.61, 54.93, 53.63, 42.81, 34.10, 29.73, 28.66 ppm. LC/MS-MS (18C column), tR = 5.4 min, 269.1 ➔ 223.8 m/z.
Rate constants
The rate constants for the reactions of peptides with aldehydes were measured as described previously (Barski et al. 2013). Briefly HNE and acrolein were incubated with carnosine, balenine and DMB (50µM2mM) in 0.15 M potassium phosphate, pH 7.4 at 37°C. Disappearance of acrolein and HNE were monitored by UV-Vis spectrophotometer at approximately 215 nm and 223 nm respectively. A monoexponential equation was used to fit the time course of each reaction and the exponential constant, kobs, was plotted against the histidyl dipeptide concentrations. Plots of kobs vs histidyl dipeptides were straight lines, and the bimolecular rate constants were calculated by their slopes.
Metal chelating capacity
The iron (Fe2+) chelating ability of different histidyl dipeptides was monitored by measuring the formation of ferrous-ferrozine complex as described previously by (Canabady-Rochelle et al. 2015). Briefly, 2 mM ferrous chloride (FeCl2) solution was added to different dipeptide solutions in pure water. After 3 min of incubation at room temperature, the chelation reaction was inhibited by the addition of 5 mM ferrozine solution. The absorbance was measured at 560 nm after 10 min. EDTA was used as a reference chelator and the metal-chelating capacity of dipeptides was calculated in comparison to EDTA. Metal quenching capacity was measured as decrease in the absorbance of ferrous-ferrozine complex and reported as follows:
| Iron chelating capacity (%) = (A0–As/A0) × 100 | (a) |
A0 is the blank absorbance and As is the absorbance of the sample dipeptide. The line slope obtained for each dipeptide was compared with EDTA to determine the EDTA equivalent chelating capacity expressed as µmol EDTA equivalent. EDTA equivalent chelating capacity for each dipeptide was calculated as follows:
| EDTA equivalent chelating capacity= as/ao | (b) |
where as is the line slope of dipeptide for chelating activity, plotted as iron chelating capacity (percent) vs concentration (µM), a0 is the line slope of EDTA for chelating activity, plotted iron chelating capacity (percent) vs concentration (µM).
Synthesis of histidyl-dipeptide-aldehyde conjugates
Histidyl dipeptide-aldehyde conjugates were prepared as described previously (Baba et al. 2013). Briefly, acrolein was synthesized by acid hydrolysis of diethyl acetal acrolein (Sigma) in HCl (0.1 M) at room temperature for 30 min. Carnosine-propanal and balenine-propanal conjugates were synthesized by incubating 100 mM acrolein (20 µL) with 100 mM carnosine (20 µL) or 100 mM balenine (20 µL) in 1mM phosphate buffer pH 7.0 at 37°C for 2 h. Carnosine-HNE conjugate was synthesized by incubating HNE (0.2 mM; Cayman Chemical) with 2 mM carnosine in 1mM phosphate buffer pH 7.0 at 37°C for 2 h at room temperature. For mass spectrometry, the dipeptides and histidyl dipeptide-aldehyde conjugates were individually infused into a stream of 0.55 mL/min 50:50 A: B UPLC solvent going into a Waters Quattro Premier XE triple quadrupole mass spectrometer (Milford, MA). We manually adjusted all voltage and collision energy setting to determine the optimal ionization voltages, collision energies and product ions. Maximum ionization conditions and optimal daughter ions were used for program sensitive MRMs.
Isolation of cardiac myocytes histidyl dipeptide treatment
Cardiac myocytes were isolated from adult C57/BL6 mice by using a modified Langendorff perfusion system with collagenase digestion, as described previously (Keith et al. 2009). Briefly, the mouse hearts were excised, rinsed with physiological saline, and perfused with oxygenated (95% O2–5% CO2), Ca2+free modified Tyrode bicarbonate buffer (buffer A in mM: NaCl 126, KCl 4.4, MgCl2 1.0, NaHCO3 18, glucose 11, HEPES 4, 2,3-butanedione monoxime 10, taurine 30, pH 7.35) at 37°C for 5 min. The extracellular matrix was digested by buffer containing buffer A with Liberase Blendzyme type 1 0.25 mg/mL, 1mg/mL albumin, 0.015 mg/mL DNase, 0.015 mg/mL proteinase in the 50 mL of recirculating digestion buffer (buffer A with 25 µM CaCl2) for 12–15 min. Hearts were separated in mincing buffer (10 mL digestion buffer with 9 mg/mL albumin, and Liberase) and cells were allowed to settle. CaCl2 was reintroduced in a graded fashion at 5-min intervals (five total steps) to sequentially increase the Ca2+ concentration to 500 µM.
Uptake of histidyl dipeptides in myocytes and superfusion with HNE and acrolein
Uptake of histidyl dipeptides in myocytes was measured using Waters Quattro Premier XE mass spectrometer (Blancquaert et al. 2016). Briefly, myocytes isolated from adult mice were pretreated with histidyl dipeptides 1mM: carnosine, balenine and DMB for 12–16 h. Lysates were collected by washing the cells in PBS and collected in 10 mM HCl containing internal standard tyrosine-histidine, sonicated for 10 s, centrifuged at 16000 × g for 10 min and the supernatant was diluted with 3 volumes of ice-cold acetonitrile. The samples were thoroughly vortexed to precipitate proteins and kept on ice for 15 min and centrifuged at 16,000 x g for 10 min at 4˚C. Carnosine, balenine, and DMB were separated by using a Waters Acquity BEH HILIC column (1.7µm, 2.1×50 mm) column and analyzed by mass spectrometry in the positive ion mode. Chromatograms of histidyl dipeptides were acquired using the transitions carnosine: 227→110 m/z, balenine: 241→124 m/z, di-methyl-balenine 269→124 m/z and tyrosinehistidine 319→110 m/z in multiple reaction monitoring (MRM) mode. To examine whether histidyl dipeptides protect against HNE and acrolein induced hypercontracture, myocytes were pretreated with 1mM each of carnosine, balenine and DMB for 12–16 h. The cells were then superfused with either 50–60 μM HNE or 5 μM acrolein for 60 min, and digital images (80–100 cardiac myocytes/field) were acquired at 5 min intervals to quantify numbers of hypercontracted and non-hypercontracted cells.
Statistical analysis
Difference in survival time between the different treatments were analyzed by Student’s t test. Statistical significance was accepted at p<0.05. Group data are mean ± standard deviation (SD). Survival data for cardiac myocytes were modeled using the Weibull survival distribution (Castro and Bhatnagar 1993). In this distribution, the probability density function of time t is:
| f(t)=λγ(λt)γ-t exp[−(λt)γ] | (a) |
γ is the shape parameter, λ is the scale parameter: which are the scale and shape factors respectively. The normalized survival function (at t=0, S=1) is:
| S(t) = exp [(−λt)]γ | (b) |
Mean life times were calculated as: 1/λ Γ (1+1/γ), where Γ represents the Gamma function. Curve fitting was performed using the Proc NLIN procedure in SAS version 9.4 software (SAS Institute, Inc., Cary, North Carolina).
Results
Synthesis of balenine and N, N-dimethyl D-balenine
Histidine has two tautomeric forms N-1 and N-3, which possess both imine and amine character. To selectively alkylate N-3 position of imidazole ring, we temporarily protected the N-1 position of (S)methyl 2-amino-3-(1H-imidazol-5-yl) propanoate dihydrochloride (1) by treating it with 1,1’carbonyldiimidazole in DMF, which afforded the cyclic urea intermediate 2 as a crystalline solid in a good yield. Methylation of urea intermediate 2 with methyl iodide in acetonitrile resulted in the synthesis of N-3 methylated salt 3 in good yield. The cyclic urea 2 was opened with hydrolysis of methyl ester by refluxing in 4N HCl to give the amino acid hydrochloride 4. Acid group of amino acid hydrochloride 4 was protected by refluxing it in methanolic HCl solution. Compound 5 was coupled with Boc-β-alanine using EDC.HCl as the coupling agent and N-methylmorpholine as a base to afford the desired dipeptide 6. Complete deprotection of protecting groups afforded the L-balenine (7) in good yield (Fig. 1).
Fig. 1. Preparation of balenine.
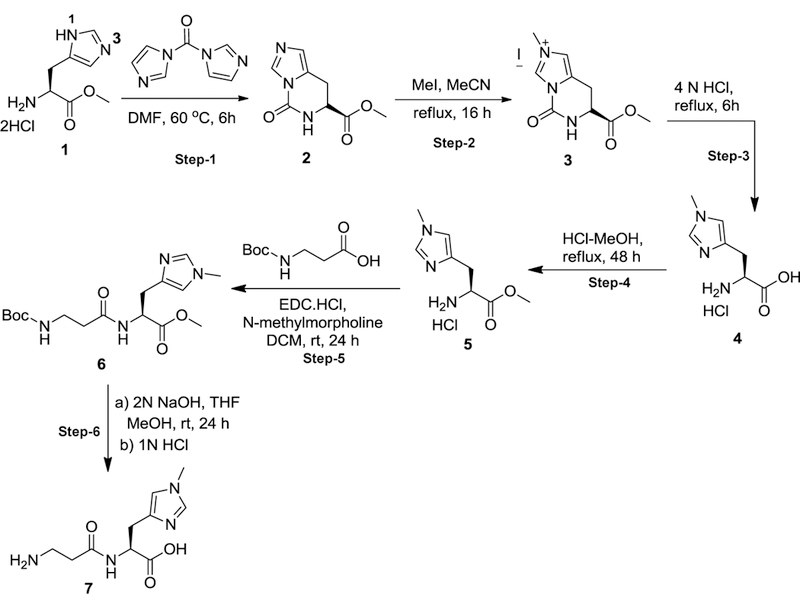
Reagents and conditions for synthesis of natural occurring histidyl dipeptide balenine steps 1–6.
The N, N-dimethyl-D-Balenine (DMB) was synthesized following the steps 1–5 described for balenine. The acid group of amino acid hydrochloride 5 was protected by refluxing it in methanolic HCl solution. Compound 6 was coupled with 3-(dimethylamino) propanoic acid using EDC.HCl as the coupling agent and N-methylmorpholine as a base to afford the desired dipeptide 7. Complete deprotection of protecting groups afforded the N, N-dimethyl-D-Balenine (8) in good yield (Fig. 2).
Fig. 2. Preparation of di-methyl-balenine (DMB).
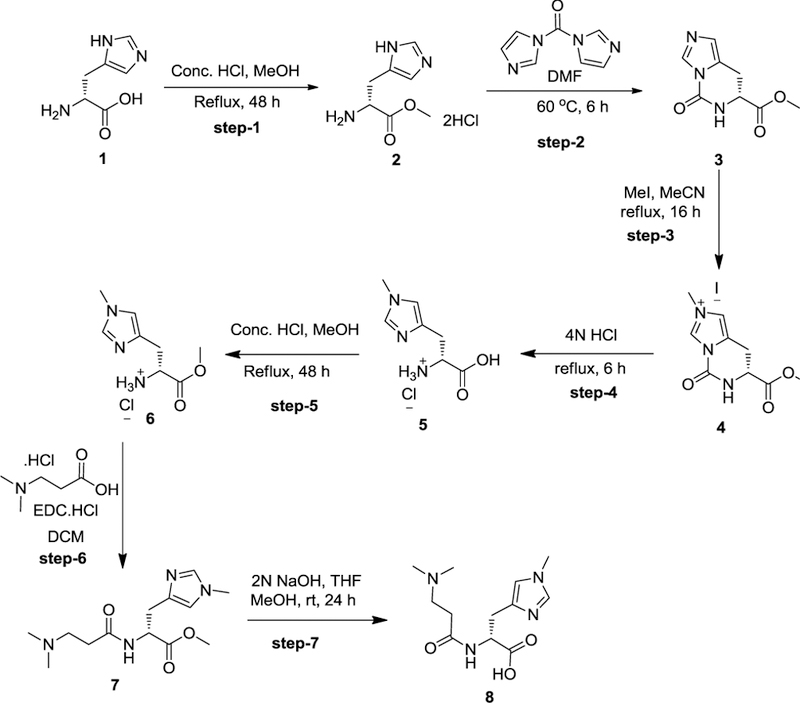
Reagents and conditions for synthesis of synthetic carnosine analogue DMB steps 1–7
Methylation of carnosine diminishes aldehyde binding capacity
Because Nτ atom of imidazole ring and the tertiary amino group of β-alanine are requisite for binding with reactive aldehydes (Carini et al. 2003), we evaluated the effects of methylation on these residues on aldehyde binding. To optimize the conditions for characterization of aldehyde-histidyl dipeptide conjugate formation, carnosine was used as a reference dipeptide. As reported previously, incubation of carnosine with HNE (10:1 molar ratio) led to the formation of one reaction product (m/z 383), which could be ascribed to a Michael adduct between the C3 of HNE and Nτ atom of imidazole ring (Fig.3A) (Baba et al. 2013; Aldini et al. 2002). Incubation of acrolein with carnosine (10:1 molar ratio) resulted in the formation of multiple ionic species (m/z 265, 283, 303). Both balenine, which is methylated at Nτ atom of imidazole ring (Fig. 3C), and DMB, in which three CH3 groups are attached on the carnosine backbone (Fig. 3E), were unable to form conjugates with HNE (Results not shown). Incubation of balenine with acrolein (10:1 molar ratio) led to the formation of one major ionic species with m/z of 297 (Fig. 3D), which is likely a Michael adduct between the carbonyl atom of acrolein and the amino-terminal group of β-alanine or Nτ of histidine.
Fig. 3. LC/MS spectra of different histidyl dipeptides with lipid peroxidation products.
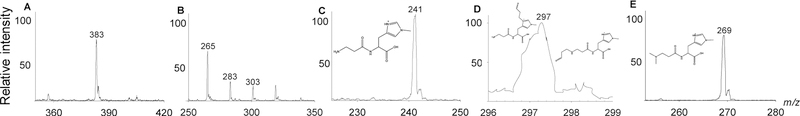
Histidyl dipeptides: carnosine, balenine, and di-methyl-balenine (DMB) were incubated with HNE or acrolein (10:1 molar ratio) in 1mM phosphate buffer (pH 7.4). Ionic species were monitored by ESI/MS - (A) carnosine-HNE (m/z 383), (B) carnosine-propanal (m/z 265, 283, 303), and (C) balenine (m/z 241). Inset shows the chemical structure of (D) balenine-propanal (m/z 297); the Michael adduct of balenine with acrolein; and (E) di-methyl-balenine (m/z 269).
To examine relative reactivity of the peptides, we compared the second order rate constants for the reaction of HNE and acrolein with carnosine, balenine, and DMB. In agreement with our previous observations, incubation of carnosine with acrolein led to a time-dependent decrease in the acrolein concentration (Fig. 4 A) (Barski et al. 2013), suggesting rapid reactivity between acrolein and carnosine. In contrast, acrolein concentration was only slightly decreased in the balenine-acrolein mixture compared with acrolein alone after 5 h of incubation (Fig. 4 C). No change in HNE or acrolein concentrations were observed when the aldehydes were incubated with DMB, compared with the HNE and acrolein incubated alone for 5 h (Fig. 4 E, F). The rate of decrease in HNE and acrolein concentration could be described by a single exponential: At=A0e−kobs.t, where At is the concentration of aldehyde at time t, A0 is the initial aldehyde concentration, and kobs is the apparent rate constant of the reaction. The rate constants were calculated using this equation at several different concentrations of dipeptides. As shown in Fig. 4 B, D and Table I the values of kobs showed a linear dependence on carnosine and balenine concentration with a slope equal to the second order rate constant, which were used to calculate the rate of reaction for acrolein and HNE with these dipeptides. Among different histidyl dipeptides, carnosine exhibited highest reactivity with HNE and acrolein (Table I). The second order rate constant for the reaction between acrolein and balenine was 19% of the rate constant of the reaction between carnosine and acrolein. These observations suggest that methylation of Nτ atom of imidazole ring or terminal amino group decreases or diminishes the aldehyde binding ability of carnosine.
Fig. 4. Methylation of carnosine backbone diminishes its aldehyde quenching potential.
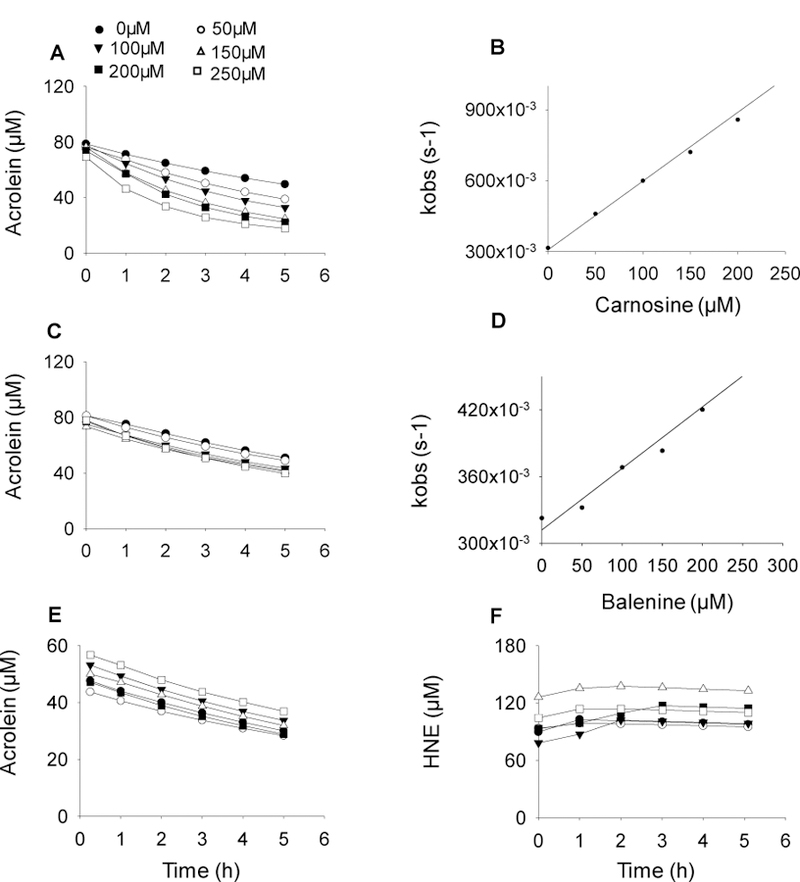
Rate of disappearance of acrolein (100 μM) in a reaction mixture containing different concentrations of (A) carnosine, (C) balenine, and (E) dimethyl balenine (50–250 µM) in 0.15M potassium phosphate buffer, pH 7.4. (F) Rate of disappearance of HNE incubated with DMB. The reaction mixtures were incubated at 37°C and the decrease in absorbance of acrolein and HNE were monitored at 215 nm and 223 nm respectively. (B, D) Data are shown as discrete points and curves were best fit of a single exponential equation to the data [y=Ae−kobs.t]. Concentration dependence of kobs. Second order rate constants were calculated from the best fits of the linear dependence and are shown in Table I.
Table I.
Bimolecular rate constant of histidyl dipeptides with HNE and acrolein. Not detected (n.d).
| Aldehydes | Carnosine (M−1 s−1) |
Balenine (M−1 s−1) |
Dimethyl balenine (M−1 s−1) |
|---|---|---|---|
| 4HNE | 0.035±0.003 | n.d | n.d |
| Acrolein | 0.288±0.013 | 0.055±0.005 | n.d |
Metal quenching capacity of histidyl dipeptides
Given that carnosine chelates numerous first transition metals (Baran 2000), we next determined whether methylation at Nτ atom of imidazole ring (balenine) or trimethylation (DMB) could affect its metal quenching capacity. Using EDTA as a reference chemical chelator, we compared the Fe2+ chelating ability of these dipeptides and calculated the EDTA equivalent chelating capacity. EDTA as a reference chelator showed a linearization of metal quenching in the range 0 to 50 µM (Fig. 5 A, D), carnosine and balenine showed linearization of metal quenching in the range between 0 to 500 µM (Fig. 5 B, D) and 0 to 250 µM (Fig. 5 C, D) respectively. In reference to EDTA, balenine exhibited the highest chelating capacity (0.179±0.002 µmol of EDTA equivalent) compared with carnosine (0.087±0.005 µmol of EDTA equivalent; p<0.05), whereas trimethylation of carnosine backbone abolished the metal chelating property (Fig. 1 D). Taken together, these results suggest that single methylation enhances and trimethylation diminished metal chelating property of carnosine.
Fig.5. Metal chelating capacity of different histidyl dipeptides.
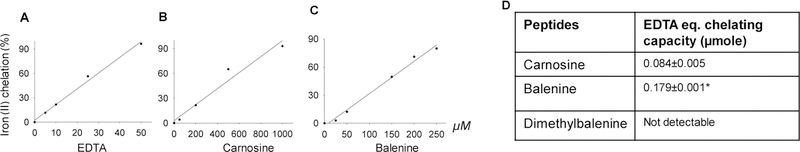
Iron (Fe2+) chelating capacity of (A) EDTA (B) carnosine and (C) balenine. (D) EDTA equivalent chelating activity of different histidyl dipeptides.
Carnosine protects against HNE-induced hypercontracture in myocytes
Because carnosine binds with reactive aldehydes (Aldini et al. 2002) and excessive HNE generation is a hallmark of cardiac dysfunction (Baba et al. 2018), we next tested whether enhancing carnosine levels in the isolated adult mouse cardiac myocytes would prevent against HNE-induced myocyte injury. For this, we first measured carnosine levels in the isolated adult cardiac myocytes using LC/MS/MS and detected ~0.249±0.035 nmole/mg protein of carnosine in these cells. To ensure that the dipeptides are taken up by the cardiac myocytes, we incubated these cells with 1mM of each carnosine, balenine and DMB for 12–16 h and dipeptide concentration was measured by LC/MS/MS. Our results showed that under similar incubation conditions, the intracellular concentrations of the dipeptides in cardiac myocytes were comparable, indicating that all three dipeptides are taken up by isolated myocytes (carnosine: 2.77±1.032 nmole/mg protein; balenine: 3.759±1.388 nmole/mg protein; and DMB: 6.243±1.482 nmole/mg protein; Fig. 6A). To test whether these dipeptides protect against HNE-induced hypercontracture, myocytes were pretreated with 1mM each of carnosine, balenine, and DMB for 12–16 h and superfused with HNE (50–60 µM) for 60 min. The cells were then continuously monitored under the light microscope and survival was computed by comparing their survival using the Weibull distribution. We found that during the initial phase of superfusion, there was a progressive increase in the hypercontracture; however, after 40–45 min of superfusion, the number of hypercontracted cells in the presence of HNE increased abruptly (Fig. 6B). The mean lifetime of the myocytes superfused with HNE was 42±6 min, which was significantly increased to 53±8 min (p<0.05) in myocytes preloaded with carnosine. In contrast, balenine and DMB pretreatments were unable to diminish HNE toxicity and thus affect the mean lifetime of superfused myocytes (balenine: 41±9 min; DMB: 45±8 min; Fig. 6C).
Fig. 6. Carnosine pretreatment prevents HNE-induced hypercontracture.
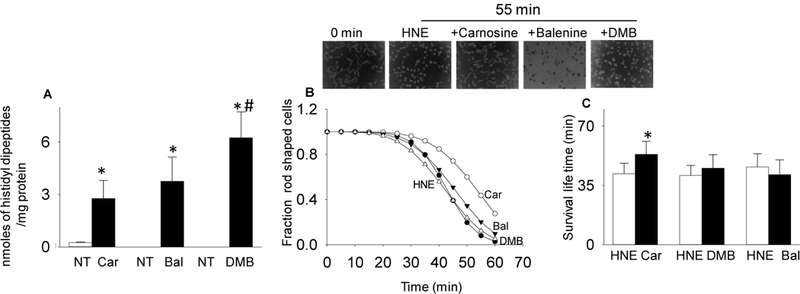
(A) Levels of histidyl dipeptides in myocytes non-treated (NT) or treated with 1mM each of carnosine (car), balenine (bal), and di-methyl-balenine (DMB). Inset shows representative images of myocytes at 0 min and after 55 min of superfusion with HNE (50–60 μM) with and without pretreatment with the indicated histidyl dipeptides. Hypercontracture was monitored at 5 min intervals for 60 min. (B) Mean lifetime of myocytes was calculated using the Weibull Survival distribution function (S(t)=exp-(λt)γ). (C) Data for mean life time are presented as mean ± SD. *p<0.05 vs HNE treated myocytes, # p<0.05 vs carnosine and balenine treated myocytes n= 3–5 mice in each group.
Carnosine pretreatment prevents acrolein-induced hypercontracture in cardiomyocyte
Because a wide variety of reactive aldehydes are generated in diseased myocardium (Baba et al. 2018) and carnosine has the ability to bind several different aldehydes (Carini et al. 2003), we further tested whether carnosine could also prevent acrolein-induced hypercontracture. As before, adult cardiac myocytes were pretreated with 1mM each of carnosine, DMB and balenine for 12–16 h and then superfused with 5 µM acrolein. Hypercontracture in the superfused myocytes was continuously monitored for 60 min. As shown in Fig. 7, the mean lifetime of myocytes superfused with acrolein was 41±3 min which was significantly increased to 50±4 min in myocyte pretreated with carnosine. In contrast, balenine and DMB pretreatments did not reduce acrolein-induced toxicity or affect myocyte survival (balenine: 43±3 min; DMB: 41±3 min). Taken together, these results suggest that methylation of carnosine either at the amino group of β-alanine or the imidazole ring of L-histidine abolishes its ability to prevent electrophilic injury to cardiac myocytes. This observation is consistent with the notion that carnosine prevents HNE and acrolein-induced injury by forming conjugates with these aldehydes.
Fig. 7. Acrolein-induced hypercontracture is attenuated by carnosine pretreatment.
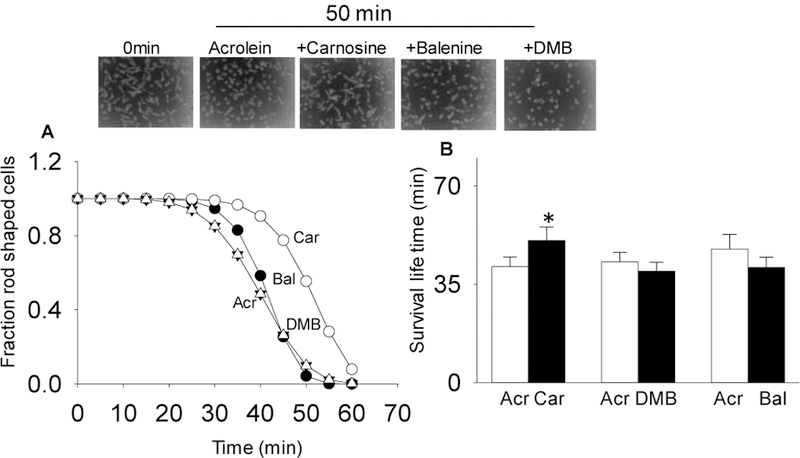
(A) Hypercontracture was monitored at 5 min interval for 60 min. Mean-lifetime was calculated using the Weibull distribution function. Inset shows the representative images of myocytes at 0 min and after 50 min of acrolein (5 μM) superfusion. (B) Data for mean lifetime are presented as mean ± SD. *p<0.05 vs acrolein treated myocytes, n=4–5 mice in each group.
Discussion
The major finding of this study is that carnosine protects against aldehyde-induced hypercontracture in isolated cardiac myocytes and that this protection is related to the ability of this dipeptide to directly scavenge electrophilic aldehydes by forming covalent adducts. This notion is supported by the observation showing that the decrease in aldehyde binding capacity by methylation of Nτ of imidazole ring, but preserved metal chelating property and trimethylation of histidyl dipeptides was accompanied by decreased ability of the dipeptide to protect against aldehyde induced hypercontracture. Collectively, these results add a new facet to the biology of carnosine and corroborate the critical role of its direct reactive ability in preventing electrophilic injury.
Previous work has shown that carnosine forms a conjugate with HNE through a mechanism that involves the formation of reversible α, β unsaturated imine between the amino group on the β-alanyl residue and the aldehyde carbonyl of HNE. This intermediate imine acts as a catalyst for the Michael addition between the C-3 of HNE and the Nτ atom of imidazole ring (Aldini et al. 2002). It has been shown that HNE also binds with anserine, which has a methyl residue attached to Nτ atom of imidazole (Aldini et al. 2002) indicating that Nτ atom of imidazole is necessary for HNE binding. However, less is known about the aldehyde binding ability of balenine, which is methylated at Nτ atom of imidazole ring and is largely present in whales (Abe 2000). Our LC/MS results showed that balenine was unable to react with HNE, which further highlights the role of Nτ atom in imidazole ring for HNE binding. Although it was expected that an intermediate imine derivative between the amino group of β-alanine and the aldehyde carbonyl of HNE might be formed, however our results suggest that the suppression of Michael type addition between the C-3 of HNE and the Nτ atom of imidazole ring in balenine prevents the formation of this imine derivative.
Incubation of carnosine with acrolein yielded several ionic species (m/z 265, 283, 321, 339 ) which are formed due to the binding of either one, two or three acrolein molecules with Nτ, Nπ atom of the imidazole ring or terminal amino group of β-alanine (Carini et al. 2003). Our results showing that incubation of balenine with acrolein furnishes only one main products at m/z 297, suggest-that only one mole of acrolein binds to one mole of balenine through Michael addition to either Nτ atom of imidazole ring or to the amino group of β-alanine. The inability of DMB to react with HNE or acrolein further confirms the role of Nτ of imidazolic ring and terminal amine group in aldehyde conjugation. Although it was expected that acrolein may form a conjugate with the Nπ atom of imidazole ring in DMB, however our ESI/MS did not detect any ionic species conjugated with acrolein, suggesting that methylation at three residue of carnosine blocks its accessibility to small, unsaturated aldehydes. Furthermore, the second order rate constants for the reaction of HNE and acrolein with different histidyl dipeptides showed that carnosine reacts faster with acrolein than balenine or DMB. Previously, we had reported that anserine had either equal or high, acrolein and HNE binding rate (Barski et al. 2013), which further strengthens the rationale that Nτ of imidazole moiety is essential for aldehyde binding. Collectively these results suggest that carnosine displays the most efficient reaction with reactive aldehydes than balenine and that Nτ of imidazole ring of histidyl dipeptides is essential for conjugation with the bulkier aldehydes such as HNE.
Several analytical studies have suggested that carnosine forms complexes with the transition metals such as Cu2+, Co2+, Ni2+ (Baran 2000). Among these complexes, Cu2+-carnosine metal complex is the most well studied model for metal binding (Dobbie and Kermack 1955) (Lenz and Martell 1964). It was first proposed by Dobbie and Kermack that the binding of carnosine with Cu2+ occurs as a monomer through the Nτ of imidazole group, terminal NH2 and the deprotonated peptide nitrogen of carnosine. Later structural studies using NMR, X-ray crystallographic studies revealed that Cu2+-carnosine complex exists as a dimer, in which Cu2+ ion binds with the terminal amino group, amide nitrogen, and the carboxylate oxygen of one dipeptide molecule and the Nπ of imidazole group of the other dipeptide (Freeman and Szymanski 1967). However, subsequent studies at low or room temperature revealed that the Cu2+-carnosine complexes could exist as a dimer and monomer in equilibrium (Brown 1980 ) (Brown 1982). In addition to copper, carnosine may also chelate iron. Carnosine is capable of inhibiting ironcatalyzed oxidation of lipid phosphohatidycholine liposomes and lipid peroxides, however the inability of carnosine to form complex with Fe2+ has also been reported (Eric A Decker 1992). Recent study by Rochelle et. al., (Canabady-Rochelle et al. 2015), using EDTA as a reference metal chelator reported that carnosine chelates Fe2+, equivalent to 0.2 μmole of EDTA equivalent chelating capacity. In line with these observations we found that carnosine has 0.08 µmole of EDTA equivalent chelating capacity and the methylation of carnosine at Nτ of histidine enhanced it to 0.2 µmole of EDTA equivalent chelating capacity, whereas trimethylation completely abolished this property. The reason, why methylation of Nτenhanced the metal chelating ability is not clear. Previous studies suggest that the metal chelating capacity of carnosine is dependent on pH (Dobbie and Kermack 1955) (Lenz and Martell 1964) and carnosine has a pKa of 7.1, whereas balenine has a pKa of 6.9 (Okuma 1991). It seems likely that these differences in pKa might regulate the formation of metal-carnosine complexes. However, future studies are needed to delineate the mechanism by which methylation at Nτ enhances the metal chelating capacity and whether this methylation prefers the monomeric or the dimeric complexes. Our results showing that DMB was unable to chelate metals confirms earlier findings that NH2 terminal groups of the carnosine backbone are essential for metal binding (Dobbie and Kermack 1955). Although we did not test the metal quenching ability of anserine, which has a pKa 7.15 (Okuma 1991), future studies are needed to compare the metal quenching ability of this dipeptide and determine how methylation at Nπ of histidine effects metal chelating capacity. Taken together, these findings suggest that the single methylation of carnosine on Nτ of imidazole group does not abolish its metal chelating property, but diminishes its aldehyde quenching ability, whereas trimethylation of carnosine abolishes both the metal chelating and the aldehyde binding ability of peptide.
Of the several lipid peroxidation products, HNE and acrolein are some of the most toxic species that have been implicated as significant mediators of cardiac injury under a variety of conditions, including heart failure (Ismahil et al. 2011), pathological hypertrophy (Baba et al. 2018), I/R injury (Conklin et al. 2015) and aging (Moreau et al. 2003). Normal cellular levels of HNE range from 0.8 to 2.8 µM (Esterbauer et al. 1991) although up to 10–100 mM HNE has been measured in oxidizing microsomes (Benedetti et al. 1984; Benedetti et al. 1982). Exposure of isolated myocytes to HNE and acrolein causes prolongation of the action potential, increases arrhythmogenesis, elevation in intracellular sodium, and cell death (Conklin et al. 2015; Bhatnagar 1995). Recent reports from our laboratory demonstrated that superfusion with HNE increases autophagy in cardiac myocytes (Baba et al. 2018). Perfusion of isolated hearts with HNE leads to coronary vasodilation (van der Kraaij et al. 1990) and progressive decrease in the peak systolic pressure (Ishikawa et al. 1986). Similarly, it has been shown that exposure to acrolein increases, heart rate variability, left ventricular developed pressure and arrhythmia (Kurhanewicz et al. 2017). In agreement with these observations, we found that exposure of isolated myocytes to HNE or acrolein causes a time-dependent hypercontracture and cell death, indicating that both these aldehydes are highly toxic to cardiac myocytes. Previous studies have shown that overexpression or activation of enzymes that detoxify aldehydes protect against cardiac injury mediated by reactive aldehydes. Overexpression of ALDH2 in the cardiac tissue diminishes the aldehydic load in cardiac tissues and ameliorates doxorubicin-induced myocardial dysfunction (Sun et al. 2014a), improves contractile function in heart failure mice (Sun et al. 2014b) and protects the heart against I/R injury (Endo et al. 2009). Activation of ALDH2 also reduces ischemic damage to the heart (Chen et al. 2008) and improves ventricular remodeling through detoxification of HNE (Gomes et al. 2014). Similarly, activation of AR during late phase of ischemic preconditioning diminishes the myocardial injury by metabolizing toxic aldehydes (Shinmura et al. 2002). Reports from our laboratory and others have shown that the deletion of enzymes that detoxify reactive aldehydes exacerbates cardiac injury via excessive accumulation of lipid peroxidation products. Genetic deletion of GSTP increases the levels of acrolein and acrolein-protein adducts in the ischemic hearts and sensitizes the hearts to I/R injury (Conklin et al. 2015). Similarly, deletion of AR or ALDH2 enhances accumulation of HNE protein adducts in the cardiac tissue, promotes pathological remodeling (Baba et al. 2018) and aggravates doxorubicin-induced myocardial dysfunction respectively (Sun et al. 2014a). However, even though the pathological role of reactive aldehydes and the cardioprotective role of aldehyde-metabolizing enzymes has been established, gene therapy with these enzymes to protect myocardium against aldehyde induced injury is complicated and not a viable therapeutic option, and therapy with small cardioprotective peptides may be more feasible. Indeed, our previous work has shown that perfusion of isolated hearts with carnosine promotes post-ischemic recovery (Baba et al. 2013), suggesting that carnosine could be a therapeutically viable option for preventing myocardial injury.
Carnosine is a multifaceted molecule that could scavenge ROS, quench singlet oxygen, sequester reactive aldehydes, chelate metals and buffer intracellular pH(Boldyrev et al. 2013). Thus, during ischemia, it could preserve myocardial integrity by targeting multiple mediators of cardiac injury. However, our results showing that carnosine increased the survival of cardiac myocytes superfused with reactive aldehydes in comparison with histidyl dipeptides that have diminished ability to bind these aldehydes, suggest that direct sequestering of aldehydes by carnosine might be one of the underlying mechanisms to protect against myocardial injury.
In our experiment, we used isolated adult cardiac myocytes, which enabled us to individually test the efficacy of different histidyl dipeptides against HNE and acrolein. Although, several assays have been used to measure cytotoxicity in cardiac myocytes, our previous work has shown that measurements of hypercontracture are reflective of calcium overload secondary to sodium influx (Conklin et al. 2015). This assay is therefore a robust and sensitive indicator of myocyte injury induced by oxidants (Castro and Bhatnagar 1993). Before inducing hypercontracture, aldehydes such as HNE alter the inactivation properties of the sodium channel (Bhatnagar Circ Res 1993), which leads to sodium influx and a resultant increase in intracellular calcium via the sodium-calcium exchanger (Conklin et al. 2015). Although the specific molecular events of HNE and acrolein toxicity were not reexamined in this study, we believe that carnosine prevents HNE and acrolein-induced hypercontracture, not by interfering with molecular events of injury, but by directly quenching these aldehydes, and thereby diminishing their effective cytotoxic concentrations. Our results showed that the cardiac myocytes pretreated with carnosine imparted protection, whereas pretreatment with balenine, which had preserved metal chelating property, is taken up by cardiomyocytes to the similar extent as carnosine, but had diminished aldehyde binding capacity, lacked the ability to protect cardiac myocytes from aldehyde induced toxicity. These observations define more clearly, the role of carnosine as an effective aldehyde sequestering agent. Furthermore, we observed that the D-analogue of trimethylated carnosine, which are suggested to be resistant to hydrolysis (Orioli et al. 2011), enhanced bioavailability, gives the leverage to test the aldehyde quenching ability of these dipeptides, lacked the ability to scavenge reactive aldehydes, was unable to protect the aldehyde induced cell death. These results further validate our hypothesis, that aldehyde sequestering by these dipeptides could be one of the major routes for cardio-protection. Nevertheless, in future, both in-vivo and ex-vivo experiments are required to determine the relative efficacy of carnosine and its methylated analogs to determine whether the protective effects of carnosine are due to direct binding to lipid peroxidation products.
In summary, this is the first report to compare the aldehyde-quenching and metal chelating efficacy of naturally occurring histidyl dipeptides balenine and synthetic dipeptide DMB with carnosine. By synthesizing and studying the analogues in which the Nτ of histidine was methylated (balenine) or the Nτ of histidine and amino-group of β-alanine were methylated (DMB), we could examine, for the firsttime, the role of Nτ of imidazole ring in aldehyde binding and metal chelating, and further define the adduction chemistry of carnosine. Our results showing that balenine, which was unable to form a conjugate with HNE or react avidly with acrolein suggest that Nτ of histidine is necessary to complete the conjugation reaction with bulkier aldehydes and to maintain high reactivity with small unsaturated aldehydes. Thus, both balenine and DMB represent a new series of natural and synthetic carnosine analogues that could be used to investigate the biological relevance of carnosine, especially under conditions where reactive aldehydes or metal toxicity is suspected to be an important mediator of pathological stress. Our data showing that the quenching of reactive aldehydes by carnosine in myocytes lay down the foundation for investigating the role of enzyme ATPGD1 or carnosine in clinical outcomes such as acute myocardial infarction or heart failure.
Acknowledgements
We would like to thank Bioanalytical Core of the Diabetes and Obesity Center for biochemical analysis.
Funding Sources
This work was supported by grants from the National Institutes of Health, R01HL122581-01 (SPB), R01HL55477 and GM103492 (AB).
Footnotes
Notes
Conflict of Interest
All authors declare that no competing financial interest exists.
Ethical approval
All treatments and protocols were approved by the University of Louisville, Institutional Animal Care and Use Committee. The ethical approval number is 15387.
References
- Abe H (2000) Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochemistry (Mosc) 65 (7):757–765 [PubMed] [Google Scholar]
- Aldini G, Carini M, Beretta G, Bradamante S, Facino RM (2002) Carnosine is a quencher of 4hydroxy-nonenal: through what mechanism of reaction? Biochem Biophys Res Commun 298 (5):699–706 [DOI] [PubMed] [Google Scholar]
- Baba SP, Hoetker JD, Merchant M, Klein JB, Cai J, Barski OA, Conklin DJ, Bhatnagar A (2013) Role of aldose reductase in the metabolism and detoxification of carnosine-acrolein conjugates. J Biol Chem 288 (39):28163–28179. doi: 10.1074/jbc.M113.504753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba SP, Zhang D, Singh M, Dassanayaka S, Xie Z, Jagatheesan G, Zhao J, Schmidtke VK, Brittian KR, Merchant ML, Conklin DJ, Jones SP, Bhatnagar A (2018) Deficiency of aldose reductase exacerbates early pressure overload-induced cardiac dysfunction and autophagy in mice. J Mol Cell Cardiol 118:183–192. doi: 10.1016/j.yjmcc.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran EJ (2000) Metal complexes of carnosine. Biochemistry (Mosc) 65 (7):789–797 [PubMed] [Google Scholar]
- Barski OA, Xie Z, Baba SP, Sithu SD, Agarwal A, Cai J, Bhatnagar A, Srivastava S (2013) Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 33 (6):1162–1170. doi: 10.1161/ATVBAHA.112.300572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benderdour M, Charron G, Comte B, Ayoub R, Beaudry D, Foisy S, Deblois D, Des Rosiers C (2004) Decreased cardiac mitochondrial NADP+-isocitrate dehydrogenase activity and expression: a marker of oxidative stress in hypertrophy development. American journal of physiology Heart and circulatory physiology 287 (5):H2122–2131. doi: 10.1152/ajpheart.00378.2004 [DOI] [PubMed] [Google Scholar]
- Benderdour M, Charron G, DeBlois D, Comte B, Des Rosiers C (2003) Cardiac mitochondrial NADP+-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: an event that precedes hypertrophy development. J Biol Chem 278 (46):45154–45159. doi: 10.1074/jbc.M306285200 [DOI] [PubMed] [Google Scholar]
- Benedetti A, Comporti M, Fulceri R, Esterbauer H (1984) Cytotoxic aldehydes originating from the peroxidation of liver microsomal lipids. Identification of 4,5-dihydroxydecenal. Biochim Biophys Acta 792 (2):172–181 [DOI] [PubMed] [Google Scholar]
- Benedetti A, Fulceri R, Ferrali M, Ciccoli L, Esterbauer H, Comporti M (1982) Detection of carbonyl functions in phospholipids of liver microsomes in CCl4-and BrCCl3-poisoned rats. Biochim Biophys Acta 712 (3):628–638 [DOI] [PubMed] [Google Scholar]
- Bhatnagar A (1995) Electrophysiological effects of 4-hydroxynonenal, an aldehydic product of lipid peroxidation, on isolated rat ventricular myocytes. Circ Res 76 (2):293–304 [DOI] [PubMed] [Google Scholar]
- Blancquaert L, Baba SP, Kwiatkowski S, Stautemas J, Stegen S, Barbaresi S, Chung W, Boakye AA, Hoetker JD, Bhatnagar A, Delanghe J, Vanheel B, Veiga-da-Cunha M, Derave W, Everaert I (2016) Carnosine and anserine homeostasis in skeletal muscle and heart is controlled by beta-alanine transamination. J Physiol doi: 10.1113/JP272050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldyrev AA, Aldini G, Derave W (2013) Physiology and pathophysiology of carnosine. Physiol Rev 93 (4):1803–1845. doi: 10.1152/physrev.00039.2012 [DOI] [PubMed] [Google Scholar]
- Brown CE, Vidrine DW, Julian RL, and Froncisz W (1982) Copper (II) dimers in solution:evidence for motional averaging of coupling tensors without chemical dissociation. J CehmSocDalton Transact (12):2371–2377 [Google Scholar]
- Brown CEA, W. E, and Froncisz W (1980. ) Multiple forms of the copper(II)-carnosine complex. JChemSoc,Dalton Trans (4):590–596 [Google Scholar]
- Canabady-Rochelle LL, Harscoat-Schiavo C, Kessler V, Aymes A, Fournier F, Girardet JM (2015) Determination of reducing power and metal chelating ability of antioxidant peptides: revisited methods. Food Chem 183:129–135. doi: 10.1016/j.foodchem.2015.02.147 [DOI] [PubMed] [Google Scholar]
- Carini M, Aldini G, Beretta G, Arlandini E, Facino RM (2003) Acrolein-sequestering ability of endogenous dipeptides: characterization of carnosine and homocarnosine/acrolein adducts by electrospray ionization tandem mass spectrometry. J Mass Spectrom 38 (9):996–1006. doi: 10.1002/jms.517 [DOI] [PubMed] [Google Scholar]
- Castro GJ, Bhatnagar A (1993) Effect of extracellular ions and modulators of calcium transport on survival of tert-butyl hydroperoxide exposed cardiac myocytes. Cardiovasc Res 27 (10):1873–1881 [DOI] [PubMed] [Google Scholar]
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321 (5895):1493–1495. doi: 10.1126/science.1158554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin DJ, Guo Y, Jagatheesan G, Kilfoil PJ, Haberzettl P, Hill BG, Baba SP, Guo L, Wetzelberger K, Obal D, Rokosh DG, Prough RA, Prabhu SD, Velayutham M, Zweier JL, Hoetker JD, Riggs DW, Srivastava S, Bolli R, Bhatnagar A (2015) Genetic Deficiency of Glutathione STransferase P Increases Myocardial Sensitivity to Ischemia-Reperfusion Injury. Circ Res 117 (5):437–449. doi: 10.1161/CIRCRESAHA.114.305518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Courten B, Jakubova M, de Courten MP, Kukurova IJ, Vallova S, Krumpolec P, Valkovic L, Kurdiova T, Garzon D, Barbaresi S, Teede HJ, Derave W, Krssak M, Aldini G, Ukropec J, Ukropcova B (2016) Effects of carnosine supplementation on glucose metabolism: Pilot clinical trial. Obesity (Silver Spring) 24 (5):1027–1034. doi: 10.1002/oby.21434 [DOI] [PubMed] [Google Scholar]
- Dobbie H, Kermack WO (1955) Complex-formation between polypeptides and metals. 2. The reaction between cupric ions and some dipeptides. Biochem J 59 (2):246–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozak J, Veiga-da-Cunha M, Vertommen D, Stroobant V, Van Schaftingen E (2010) Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J Biol Chem 285 (13):9346–9356. doi: 10.1074/jbc.M109.095505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo J, Sano M, Katayama T, Hishiki T, Shinmura K, Morizane S, Matsuhashi T, Katsumata Y, Zhang Y, Ito H, Nagahata Y, Marchitti S, Nishimaki K, Wolf AM, Nakanishi H, Hattori F, Vasiliou V, Adachi T, Ohsawa I, Taguchi R, Hirabayashi Y, Ohta S, Suematsu M, Ogawa S, Fukuda K (2009) Metabolic remodeling induced by mitochondrial aldehyde stress stimulates tolerance to oxidative stress in the heart. Circ Res 105 (11):1118–1127. doi: 10.1161/CIRCRESAHA.109.206607 [DOI] [PubMed] [Google Scholar]
- Eric A Decker ADC, and John T.Calvert (1992) Differnces in teh antioxidant mechanism of carnosine in the presence of copper and iron. J Agric Food Chem 40 (5):756–759 [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11 (1):81–128 [DOI] [PubMed] [Google Scholar]
- Fawaz MV, Topper ME, Firestine SM (2011) The ATP-grasp enzymes. Bioorg Chem 39 (5–6):185191. doi: 10.1016/j.bioorg.2011.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman HC, Szymanski JT (1967) Crystallographic studies of metal-peptide complexes. V. (Beta-alanyl-L-histidinato)copper(II)dihydrate. Acta Crystallogr 22 (3):406–417 [DOI] [PubMed] [Google Scholar]
- Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115 (3):500508. doi: 10.1172/JCI24408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH, Magno P, Chen CH, Brum PC, Kowaltowski AJ, Mochly-Rosen D, Ferreira JC (2014) Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res 103 (4):498–508. doi: 10.1093/cvr/cvu125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell BGJ (2015) Free radical in biology and medicine Oxford University Press, Oxfore [Google Scholar]
- Hennekens CH, Buring JE, Manson JE, Stampfer M, Rosner B, Cook NR, Belanger C, LaMotte F, Gaziano JM, Ridker PM, Willett W, Peto R (1996) Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N Engl J Med 334 (18):1145–1149. doi: 10.1056/NEJM199605023341801 [DOI] [PubMed] [Google Scholar]
- Hipkiss AR (2009) Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res 57:87–154. doi: 10.1016/S1043-4526(09)57003-9 [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Esterbauer H, Sies H (1986) Role of cardiac glutathione transferase and of the glutathione S-conjugate export system in biotransformation of 4-hydroxynonenal in the heart. J Biol Chem 261 (4):1576–1581 [PubMed] [Google Scholar]
- Ismahil MA, Hamid T, Haberzettl P, Gu Y, Chandrasekar B, Srivastava S, Bhatnagar A, Prabhu SD (2011) Chronic oral exposure to the aldehyde pollutant acrolein induces dilated cardiomyopathy. Am J Physiol Heart Circ Physiol 301 (5):H2050–2060. doi: 10.1152/ajpheart.00120.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Iwase M, Ichihara S, Kanazawa H, Hashimoto K, Noda A, Nagata K, Koike Y, Yokota M (2010) Beneficial effects of growth hormone-releasing peptide on myocardial oxidative stress and left ventricular dysfunction in dilated cardiomyopathic hamsters. Circ J 74 (1):163–170 [DOI] [PubMed] [Google Scholar]
- Keith RJ, Haberzettl P, Vladykovskaya E, Hill BG, Kaiserova K, Srivastava S, Barski O, Bhatnagar A (2009) Aldose reductase decreases endoplasmic reticulum stress in ischemic hearts. Chem Biol Interact 178 (1–3):242–249. doi: 10.1016/j.cbi.2008.10.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurhanewicz N, McIntosh-Kastrinsky R, Tong H, Ledbetter A, Walsh L, Farraj A, Hazari M (2017) TRPA1 mediates changes in heart rate variability and cardiac mechanical function in mice exposed to acrolein. Toxicol Appl Pharmacol 324:51–60. doi: 10.1016/j.taap.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz GR, Martell AE (1964) Metal Complexes of Carnosine. Biochemistry 3:750–753 [DOI] [PubMed] [Google Scholar]
- Liu YH, Carretero OA, Cingolani OH, Liao TD, Sun Y, Xu J, Li LY, Pagano PJ, Yang JJ, Yang XP (2005) Role of inducible nitric oxide synthase in cardiac function and remodeling in mice with heart failure due to myocardial infarction. American journal of physiology Heart and circulatory physiology 289 (6):H2616–2623. doi: 10.1152/ajpheart.00546.2005 [DOI] [PubMed] [Google Scholar]
- Moreau R, Heath SH, Doneanu CE, Lindsay JG, Hagen TM (2003) Age-related increase in 4hydroxynonenal adduction to rat heart alpha-ketoglutarate dehydrogenase does not cause loss of its catalytic activity. Antioxid Redox Signal 5 (5):517–527. doi: 10.1089/152308603770310167 [DOI] [PubMed] [Google Scholar]
- Niki E (2009) Lipid peroxidation: physiological levels and dual biological effects. Free Radic Biol Med 47 (5):469–484. doi: 10.1016/j.freeradbiomed.2009.05.032 [DOI] [PubMed] [Google Scholar]
- Okuma HAaE (1991) Effect of Temperature on the buffering capacities of histidine-related compounds and fish skeletal muscle. Nippon Suisan Gakkaishi 57 (11):2101–2107 [Google Scholar]
- Orioli M, Vistoli G, Regazzoni L, Pedretti A, Lapolla A, Rossoni G, Canevotti R, Gamberoni L, Previtali M, Carini M, Aldini G (2011) Design, synthesis, ADME properties, and pharmacological activities of beta-alanyl-D-histidine (D-carnosine) prodrugs with improved bioavailability. ChemMedChem 6 (7):1269–1282. doi: 10.1002/cmdc.201100042 [DOI] [PubMed] [Google Scholar]
- Porter NA, Caldwell SE, Mills KA (1995) Mechanisms of free radical oxidation of unsaturated lipids. Lipids 30 (4):277–290 [DOI] [PubMed] [Google Scholar]
- Qin F, Simeone M, Patel R (2007) Inhibition of NADPH oxidase reduces myocardial oxidative stress and apoptosis and improves cardiac function in heart failure after myocardial infarction. Free Radic Biol Med 43 (2):271–281. doi: 10.1016/j.freeradbiomed.2007.04.021 [DOI] [PubMed] [Google Scholar]
- Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP, Hill BG (2014) Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail 7 (4):634–642. doi: 10.1161/CIRCHEARTFAILURE.114.001151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS (2002) Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol 34 (4):379–388. doi: 10.1006/jmcc.2002.1526 [DOI] [PubMed] [Google Scholar]
- Shinmura K, Bolli R, Liu SQ, Tang XL, Kodani E, Xuan YT, Srivastava S, Bhatnagar A (2002) Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circ Res 91 (3):240–246 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Chandrasekar B, Gu Y, Luo J, Hamid T, Hill BG, Prabhu SD (2007) Downregulation of CuZn-superoxide dismutase contributes to beta-adrenergic receptor-mediated oxidative stress in the heart. Cardiovasc Res 74 (3):445–455. doi: 10.1016/j.cardiores.2007.02.016 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Harter TM, Chandra A, Bhatnagar A, Srivastava SK, Petrash JM (1998) Kinetic studies of FR-1, a growth factor-inducible aldo-keto reductase. Biochemistry 37 (37):12909–12917. doi: 10.1021/bi9804333 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Watowich SJ, Petrash JM, Srivastava SK, Bhatnagar A (1999) Structural and kinetic determinants of aldehyde reduction by aldose reductase. Biochemistry 38 (1):42–54. doi: 10.1021/bi981794l [DOI] [PubMed] [Google Scholar]
- Sun A, Cheng Y, Zhang Y, Zhang Q, Wang S, Tian S, Zou Y, Hu K, Ren J, Ge J (2014a) Aldehyde dehydrogenase 2 ameliorates doxorubicin-induced myocardial dysfunction through detoxification of 4-HNE and suppression of autophagy. J Mol Cell Cardiol 71:92–104. doi: 10.1016/j.yjmcc.2014.01.002 [DOI] [PubMed] [Google Scholar]
- Sun A, Zou Y, Wang P, Xu D, Gong H, Wang S, Qin Y, Zhang P, Chen Y, Harada M, Isse T, Kawamoto T, Fan H, Yang P, Akazawa H, Nagai T, Takano H, Ping P, Komuro I, Ge J (2014b) Mitochondrial aldehyde dehydrogenase 2 plays protective roles in heart failure after myocardial infarction via suppression of the cytosolic JNK/p53 pathway in mice. J Am Heart Assoc 3 (5):e000779. doi: 10.1161/JAHA.113.000779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kraaij AM, de Jonge HR, Esterbauer H, de Vente J, Steinbusch HW, Koster JF (1990) Cumene hydroperoxide, an agent inducing lipid peroxidation, and 4-hydroxy-2,3nonenal, a peroxidation product, cause coronary vasodilatation in perfused rat hearts by a cyclic nucleotide independent mechanism. Cardiovasc Res 24 (2):144–150 [DOI] [PubMed] [Google Scholar]
- Yusuf S, Dagenais G, Pogue J, Bosch J, Sleight P (2000) Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med 342 (3):154–160. doi: 10.1056/NEJM200001203420302 [DOI] [PubMed] [Google Scholar]
- Zhang P, Hou M, Li Y, Xu X, Barsoum M, Chen Y, Bache RJ (2009) NADPH oxidase contributes to coronary endothelial dysfunction in the failing heart. American journal of physiology Heart and circulatory physiology 296 (3):H840–846. doi: 10.1152/ajpheart.00519.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y (2007) Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res 100 (7):1089–1098. doi: 10.1161/01.RES.0000264081.78659.45 [DOI] [PMC free article] [PubMed] [Google Scholar]