

Abstract
Purpose
Smoking and the incidence of age-related macular degeneration (AMD) have been linked to an overactive complement system. Here, we examined in a retrospective cohort study whether AMD-associated single nucleotide polymorphisms (SNPs), smoking, ethnicity, and disease status are correlated with blood complement levels.
Methods
Population: The study involved 91 AMD patients and 133 controls, which included 73% Americans of European descent (EUR) and 27% Americans of African descent (AFR) in South Carolina. Readouts: Participants were genotyped for 10 SNPs and systemic levels of complement factor H (CFH) activity, and the complement activation products C3a, C5a, and Bb were assessed. Main Outcome Measures: Univariate and multivariable logistic regression models were used to examine associations between AMD status and distinct readouts.
Results
AMD affects EUR individuals more than AFRs. EUR but not AFR AMD subjects revealed higher levels of Factors C3a and Bb. In all subjects, a 10-unit increase in C3a levels was associated with an approximately 10% increase in the odds of being AMD-positive, and C3a and Bb were associated with smoking. While CFH activity levels were not correlated with AMD, a significant interaction was evident between patient age and CFH activity. Finally, EURs had lower odds of AMD with enhanced copies of rs1536304 (VEGFA) and higher odds with more copy numbers of rs3766404 (CFH).
Conclusions
Our results support previous studies of systemic complement components being potential biomarkers for AMD, but they suggest that smoking and disease do not synergistically affect complement levels. We also suggest a novel susceptibility and protective haplotypes in the South Carolinian AMD population. Our studies indicate that augmented complement activation associated with advanced AMD could be attributed to a decrease in CFH activity in younger patients.
Introduction
Age-related macular degeneration (AMD) is characterized by the progressive loss of central vision resulting from the damage to photoreceptor cells in the central retina and is associated with pathology at the RPE/choroid interface [1,2]. Depending on structural and functional alterations, advanced stages of AMD can be categorized as geographic atrophy (GA; dry) or choroidal neovascularization (CNV; wet) [3]. GA includes the thickening of Bruch’s membrane concomitant with impaired conductivity, the accumulation of large drusen, and RPE atrophy. Meanwhile, CNV is associated with new choroidal blood vessels penetrating through the impaired blood-retina barrier into the subretinal space, with fluid leakage resulting eventually in retinal detachment.
AMD is a multifactorial disease, influenced by both genetic and non-genetic insults [4]. Genetic studies have revealed the association of 52 independent susceptibility variants at 34 loci to advanced AMD [5]. Rare predicted loss-of-function variants in complement pathway genes have been linked to GA and specific endophenotypes [6,7]. The ARMS2/HTRA1 (OMIM 611313/602194; Gene ID 387715/5654) locus and complement factor H (CFH; OMIM 134370 ; Gene ID 3075) exhibit the strongest genetic association, and variants at these loci are linked to subretinal hemorrhage and drusen area phenotypes, respectively [8,9]. Probably the most critical factor linked to AMD is advanced age. The environmental factor unequivocally associated with AMD is smoking [10], which presumably produces oxidative stress.
The activation of a complement pathway has been demonstrated in both early and advanced stages of AMD and is likely activated by oxidative stress-mediated neoepitopes [11,12] or debris accumulating between the RPE and Bruch’s membrane [13], resulting in a chronic inflammatory response [14]. In addition to the association of complement proteins with the pathological features of AMD, such as drusen, basolaminar deposits, GA, CNV, and fibrosis [15-19], elevated levels of complement proteins have been reported in the serum of patients with AMD [20,21]. Cigarette smoking is shown to activate the alternative pathway of complements in vitro [22,23], and serum levels of complement components are correlated with the current cumulative consumption of cigarettes [24].
The complement pathway is an essential part of the evolutionarily ancient innate immune system, responsible for eliminating foreign antigens and pathogens as part of the normal host response [25,26]. However, analyses of autoimmune, inflammatory, and ischemic disease states have suggested the contribution of inappropriate or excessive complement activation to pathology, including AMD [27]. The complement system comprises a cascade of over 30 proteins that can be activated by three distinct pathways, the classical pathway (CP), lectin pathway (LP), and alternative pathway (AP) [28]. Its activation results in the formation of three distinct sets of biologic effector molecules, the anaphylatoxins C3a and C5a; opsonins iC3b, C3d, and C3dg; and the membrane attack complex C5b-9. The risk haplogroups in at least six complement proteins, including complement inhibitors CFH [29] and others [5,30-33], are associated with AMD and predicted to exacerbate complement activation systemically or in affected tissues.
The goal of our study was to further dissect the association between smoking and complement activation products in the serum of AMD patients and normal controls. We tested for levels of the anaphylatoxins C3a and C5a, the catalytic subunit of the alternative pathway serine protease Bb, and the inhibitory activity of CFH. A subset of single nucleotide polymorphisms (SNPs) associated with AMD was included in the analysis to identify SNPs associated with complement dysregulation.
Methods
Subjects
We evaluated 223 subjects, including 90 AMD and 133 control individuals (age 60 or above) from the local South Carolina community, recruited from the ophthalmology clinics at Storm Eye Institute and the Ralph H Johnson Veterans Affairs Medical Center. This study was approved by the local institutional review board (HR 20,083), sponsored by the Veterans Affairs Office of Research and Development as part of an observational trial (NCT01115231), and met the criteria of the Declaration of Helsinki.
Inclusion and exclusion criteria were as follows. The inclusion criteria for AMD patients included age (60 and older); a clear diagnosis of AMD in at least one eye; for controls, less than five small (< 63 μm) hard drusen; and categorized as either non-Hispanic white (henceforth referred to as Americans of European descent [EUR]) or Americans of African descent (AFR). Participants were excluded if they presented themselves with ocular diseases that might simulate AMD or preclude its diagnosis, that exhibit diseases that phenotypically overlap with AMD, or that present themselves with macular dystrophies, toxoplasmosis, histoplasmosis, degenerative myopia, central serous chorioretinopathy, or any disease or treatment that would diminish the ability to recognize drusen, such as laser photocoagulation, prior retinal detachment surgery, posterior uveitis, and trauma. Control subjects were recruited from the General Ophthalmology clinic, and in addition to the exclusion criteria, patients from the experimental group were also required to be free of any ocular complications, including moderate or vision-limiting cataracts in either eye and glaucoma.
Subjects were categorized as either EUR or AFR, and information about gender, ethnicity, and smoking behavior was obtained. No distinction was made between intermediate and advanced AMD or AMD with subfoveal GA or CNV, and no information was obtained on treatment regimens. AMD subjects were recruited in the retina clinics during recurring visits; thus, most patients are presumed to have CNV.
Complement activation
Venous blood was obtained at the University phlebotomy laboratory for measurement of the complement components, processed for both plasma (CFH activity) and serum (C3a, C5a, and Bb) collection within 30 min of collection and stored at −80 °C until use. A complement component analysis was performed as a fee for service at the National Jewish Health Advanced Diagnostic Laboratories (Denver, CO).
Bb and C3a was measured by Quidel ELISA (Quidel, San Diego, CA) using microtiter plates precoated with a specific monoclonal antibody against Bb [34] or C3a [35], respectively. The standards, controls, and test specimens were diluted and placed in duplicate into the wells and incubated to allow binding of the split product to the antibodies in the well. After washing away unbound proteins, a second anti-split antibody, conjugated to an enzyme (horseradish peroxidase) was allowed to react with the Bb (or C3a) bound to the first antibody on the plate. After an appropriate incubation time and washing, a chromogenic substrate for the enzyme was added to the wells and the amount of color that developed was determined spectrophotometrically. All assays were performed with two quality control (QC) specimens supplied by the manufacturer, along with an additional in-laboratory characterized QC specimen.
C5a was measured by PharMingen (San Diego, CA) OptEIATM ELISA tests and solid-phase sandwich enzyme-linked immunosorbent assays (ELISAs) using monoclonal antibodies specific to human C5a-desArg coated on 96-well microtiter plates [36,37]. Standards and specimens were added to the wells, and any available C5a-desArg would bind to the appropriate immobilized antibody. The wells were washed, and a mixture of biotinylated polyclonal anti-human C5a antibody and avidin-horseradish peroxidase was added, producing an antibody-antigen-antibody “sandwich.” The wells were again washed and a substrate solution was added, producing a blue color in direct proportion to the amount of the anaphylatoxin present in the initial specimen. The Stop Solution changed color from blue to yellow, and the samples were read at 450 nm.
The method for testing CFH function was based on published work [38], with modifications. Uncoated sheep cells were the target of lysis, and serial dilutions of the test specimen were mixed with equal volumes of the cells. All components except CFH were added in excess, and the patient samples supplied the factor H. If the patient’s CFH were fully functional, there would be no lysis, as the alternative pathway would be controlled. The results were based on the dilution necessary to achieve 50% lysis of the cells, but as the function of CFH would be inversely proportional to the amount of lysis, the results are expressed as percent lysis.
Genotyping
Genomic DNA was extracted from lymphocytes per the manufacturer’s instructions (QIAmp® DNA Mini kit; Qiagen, Germantown, MD). Representative SNPs were selected by their reported association in AMD consortium studies [5,39]. At the CFH locus, we incorporated two additional SNPs spanning CFHR5 [40]. Thus, a total of ten SNPs were genotyped at the ARMS2 (rs10490924, rs3793917), CFH (rs3766404, rs393955), VEGFA (Gene ID 7422; OMIM 601398; rs1536304), CETP (rs1864163; Gene ID 1071; OMIM 118470), C3 (OMIM 120700; Gene ID 718; rs2230199), CFHR5 (OMIM 608593; Gene ID 81494; rs6667243, rs10922153), and COL8A1 (OMIM 12051; Gene ID 1295; rs1308155) loci using PCR-based assays (TaqMan assays, Applied Biosystems, Foster City, CA), per manufacturer’s instructions.
The genotypic data was subject to the following QC filters: markers that did not statistically conform to the Hardy–Weinberg equilibrium (HWE; at a p < 0.001) in controls, markers with >20% missing data, and markers with a minor allele frequency (MAF) of p < 0.05 were excluded from the analysis. The SNPs meeting these QC thresholds included seven of the original ten SNPs.
Statistical analysis
Descriptive statistics by AMD status were estimated for all variables in the data, with continuous variables being reported as the mean (±standard deviation) and categorical variables reported as n (%).
Associations with AMD
Associations between AMD status and complement levels, ethnicity, gender, and smoking status were examined using a series of univariate and multivariable logistic regression models in the combined data across all ethnicities and stratified by ethnicity. Smoking was considered as ever versus never, as no significant differences between current versus former were noted. Variables with p values of ≤ 0.2 were considered in a multivariable logistic regression model. The final model was selected using backwards selection based on the model with the smallest Akaike’s information criterion (AIC).
Associations between AMD and patient genotype were examined using a logistic regression approach stratified on ethnicity. Multiple imputation was conducted to impute all missing SNP values to generate ten datasets with complete SNP information using the haplo.stats library in R (R v 3.2.5). The haplo.stats library employs an expectation-maximization algorithm for the imputation of missing SNP values and uses the estimated linkage disequilibrium between SNPs during imputation to account for the linkage between SNPs when imputing missing values. For tests of associations between the SNPs and AMD status, we considered three different genetic models: additive, dominant, and recessive. The SNPs for which no subjects were homozygous for the minor allele, we only examined the dominant model. For SNPs with fewer than three subjects homozygous for the minor allele, only the dominant and additive models were considered. We also evaluated multivariable logistic regression models including smoking status, genotype, and the interaction between smoking status and genotype to examine the joint impact of smoking status and each SNP on AMD. As these analyses are exploratory, the p values given were not adjusted for multiple testing. Therefore, these findings will require further verification in additional studies.
Associations with complement levels
Complement levels or activity was assessed in two shipments, which necessitated data normalization within the two groups to remove batch effects. Associations between complement levels and smoking status were examined using a series of linear regression models. Levels of complement components were log-transformed to meet linear model assumptions, and complement levels are therefore reported as geometric means. As a secondary analysis, differences in complement levels were examined by race and between races by AMD status. The association between race and complement levels was evaluated using a two-sample t test or Wilcoxon rank sum test where appropriate. The association between race by AMD status and complement levels was evaluated using an ANOVA or Kruskal–Wallis test approach. Pair-wise comparisons between groups were examined for significant associations between complement levels and race by AMD based on Tukey’s honestly significant difference (HSD) test to adjust for multiple comparisons. P values of <0.05 are accepted as significant in all analyses.
Results
Ninety AMD patients and 133 controls were included in this study, with a mean age of 73.5±8.0 years. Most of the study participants were EUR (73.5%) and female (60.1%). Approximately 40% of the study participants have a positive diagnosis of AMD. Twenty-nine percent of subjects were current or former smokers at the time the data were collected. AFRs in the study constituted about 26% of the population and were significantly younger than the EURs (p = 0.007). Characteristics of the patient population are reported in Table 1.
Table 1. Characteristics of the study population.
| Characteristic | # subjects | All subjects |
|---|---|---|
| AMD (Yes) |
223 |
90 (40.4) |
| Age (years) |
223 |
73.2 (8.00) |
| Sex (Male) |
223 |
89 (39.9) |
| Race (EUR) |
221 |
164 (73.5) |
| Smoking status |
223 |
|
| Never |
158 (70.9) |
|
| Former |
42 (18.8) |
|
| Current | 23 (10.3) |
Continuous variables are reported as mean (SD) and categorical variables are reported as n (%).
Association of AMD with demographic variables and complement factors
AMD is reported to be associated with patient age and race [41]. Likewise, across all subjects, individuals with AMD had higher levels of C3a (p = 0.032) and Bb (p = 0.007); however, surprisingly, levels of C5a were lower (p = 0.002), and CFH activity levels were unaffected (p = 0.864; Table 2; all subjects).
Table 2. Univariate associations between AMD status across race and within race.
| Characteristic | No AMD | AMD | p |
|---|---|---|---|
|
All |
No AMD (n=133) |
AMD (n=90)* |
p |
| Age |
70.6 (6.30) |
77.8 (8.33) |
<0.001 |
| Sex (Male) |
51 (38.4) |
38 (42.2) |
0.562 |
| Race (EUR) |
81 (60.9) |
83 (92.2) |
<0.001 |
| Smoking (Ever) |
34 (25.6) |
31 (34.4) |
0.152 |
| C3a |
95.3 (34.3) |
113.1 (58.8) |
0.032 |
| C5a |
103.7 (23.9) |
93.1 (24.4) |
0.002 |
| Bb |
94.7 (42.5) |
116.6 (71.5) |
0.007 |
| Factor H |
98.3 (52.1) |
104.8 (63.2) |
0.864 |
|
AFR |
No AMD (n=52) |
AMD (n=7) |
p |
| Age |
70.9 (6.95) |
70.9 (10.4) |
0.993 |
| Sex (Male) |
20 (38.5) |
2 (28.6) |
0.702 |
| Smoking (Ever) |
9 (17.3) |
3 (42.9) |
0.141 |
| C3a |
96.1 (29.3) |
115.7 (55.0) |
0.371 |
| C5a |
112.3 (24.6) |
92.3 (26.1) |
0.045 |
| Bb |
93.5 (35.7) |
94.1 (49.1) |
0.592 |
| Factor H |
95.9 (58.9) |
135.9 (73.1) |
0.103 |
|
EUR |
No AMD (n=81) |
AMD (n=83)* |
p |
| Age |
70.6 (6.03) |
78.3 (7.94) |
<0.001 |
| Sex (Male) |
31 (38.3) |
36 (43.4) |
0.506 |
| Smoking (Ever) |
25 (30.9) |
28 (33.7) |
0.694 |
| C3a |
94.8 (37.3) |
112.9 (59.5) |
0.038 |
| C5a |
98.1 (21.8) |
93.1 (24.5) |
0.17 |
| Bb |
95.4 (46.6) |
118.5 (73.0) |
0.004 |
| Factor H | 99.9 (47.5) | 102.1 (62.0) | 0.946 |
Continuous variables are reported as mean (SD) and categorical variables are reported as n (%).*One patient missing all complement data.
As race and genetics are predicted to affect the readouts examined here (C3a, C5a, and Bb complements and CFH activity levels), additional univariate analyses were stratified on race. Among EURs (Table 2; EUR), the occurrence of AMD was associated with patient age (p<0.001) and C3a (p = .038) and Bb levels (p = 0.004). However, AMD status was not associated with C5a levels (p = 0.12) or CFH activity (p = 0.946). Among AFRs, the occurrence of AMD was only associated with C5a levels (p = 0.045; Table 2; AFR).
To test whether race and AMD are both associated with complement components, we examined differences in complement activity levels by both ethnicity and disease status (Table 3). C5a levels were higher in AFRs than in EURs (p < 0.001), whereas C3a, Bb, and CFH activity levels were similar. When analyzed by ethnicity and disease, significant differences were identified for C5a and Bb levels (global p = 0.003 and 0.020). Specifically, AFR control subjects had higher levels of C5a relative to EUR controls and EUR patients (p = 0.005 and p < 0.001, respectively; adjusted using Tukey’s HSD). EUR AMD patients also had higher levels of Bb relative to EUR controls and near-significant differences relative to AFR controls (p = 0.027 and 0.058, respectively).
Table 3. Associations between race by AMD status and complement levels.
| Complement | Healthy controls |
AMD |
p | ||
|---|---|---|---|---|---|
| AFR (n=52) | EUR (n=81) | AFR (n=7) | EUR (n=82)* | ||
| C3a |
96.1 (29.3) |
94.8 (37.3) |
115.7 (55.0) |
113.6 (60.0) |
0.282 |
| C5a |
112.3 (24.6) |
98.1 (21.8) |
92.3 (26.1) |
92.4 (24.3) |
0.003 |
| Bb |
93.5 (35.7) |
95.4 (46.6) |
94.1 (49.1) |
119.8 (73.4) |
0.02 |
| Factor H | 95.9 (58.9) | 99.9 (47.5) | 135.9 (73.1) | 102.6 (62.5) | 0.422 |
*One of the Caucasian patients is missing all complement information. p values are reported for the global test that at least one of the groups is different.
A multivariable logistic regression model of AMD was developed for all participants (Table 4). All collected subject variables were considered in the final model, including age, race, C3a levels, C5a levels, CFH activity levels, and the interaction between CFH activity and age. EURs have 5.5 times the odds of developing AMD relative to AFRs after controlling for age and complement levels (p < 0.001, odds ratio [OR]: 5.52, 95% confidence interval [CI]: 2.16–14.0). A ten-unit increase in the level of C3a is associated with a 10% increase in the odds of AMD after controlling for other factors (p = 0.019, OR: 1.10, 95% CI: 1.02–1.19). Meanwhile, a ten-unit increase in C5a levels is associated with a 17% decrease in the odds of being AMD positive after controlling for other factors (p = 0.019, OR: 0.83, 95% CI: 0.72–0.97). A significant interaction was evident between patient age and levels of CFH activity. The model indicates that as age increases, the impact of the level of CFH activity on the odds of having AMD decreases, such that there is only a significant impact in younger individuals (Figure 1). Similarly, the higher the level of CFH activity, the lesser the effect age has on the odds of developing AMD. However, increasing age has a strong impact on the odds of having AMD in the observed range of CFH activity.
Table 4. Multivariable logistic regression model of AMD status across all participants in the study.
| Variable | Odds Ratio (95% CI) | p |
|---|---|---|
| Race (EUR versus AFR) |
5.52 (2.16, 14.01) |
<0.001 |
| C3a (10 units increase) |
1.10 (1.02, 1.19) |
0.019 |
| C5a (10 units increase) |
0.83 (0.72, 0.97) |
0.016 |
| Age (years) |
See Below |
<0.001 |
| Age x CFH activity |
See Below |
0.058 |
| Age (10 year increase) |
||
| If CFH activity is 60 |
3.71 (2.06, 6.69) |
|
| If CFH activity is 90 |
2.90 (1.80, 4.65) |
|
| If CFH activity is 120 | 2.26 (1.45, 3.53) |
Results for race are reported as odds ratio of AMD for EUR participants relative to AFR participants. Results for C3a and C5a that are not in an interaction are reported as odds ratios estimated for a specific increase in complement levels. Results for CFH activity are reported as odds ratios for a specific increase in CFH activity at specific patient ages. Similarly, results for age are reported as odds ratios for a specific increase in Age at specific levels of CFH activity to account for the interaction between age and CFH activity observed in the model. P values reported in the table are for the estimated regression coefficient from the model.
Figure 1.
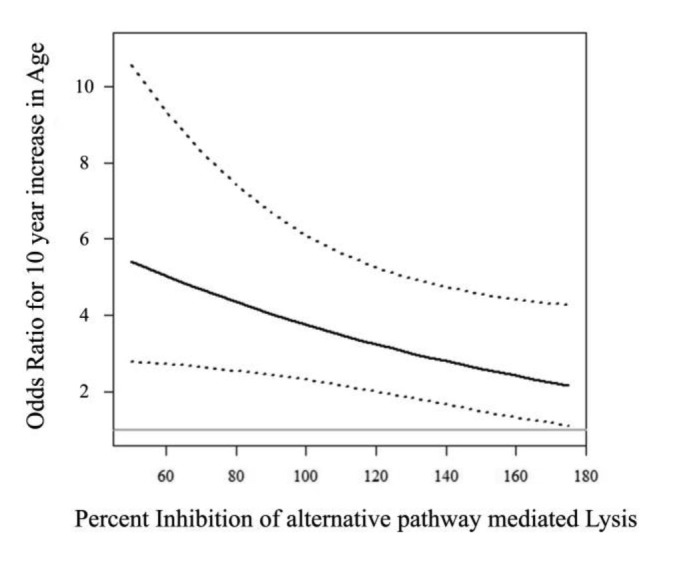
Plot of the OR for a ten-year increase in age with increasing levels of the inhibition of alternative pathway-mediated lysis by factor H. Percent inhibition of lysis has been normalized to 100 as described in the methods. The solid line is the OR from the multiple logistic regression model of AMD presented in Table 3. The dashed lines represent the 95% confidence interval for the OR at specific values of the inhibition of lysis. The solid gray line represents the null OR of 1. Values of the inhibition of lysis range from 50 to 175, which are the 10th and 90th percentiles of observed values in our study population. The plot indicates that as the inhibition of lysis increases, the impact of increasing age on the probability of developing AMD decreases.
Associations between Smoking and Complement Factors
Associations between complement levels with smoking status were examined using a linear regression analysis. Given that the dichotomous smoking variable exhibited stronger associations, we only report on the association between smoking as ever versus never. The complement factors C3a (p < 0.001) and Bb (p = 0.002) were associated with smoking, such that individuals across ethnicities that reported ever smoking had higher levels of these complement factors (Table 5, all subjects). In a stratified analysis, this association held for EURs (p = 0.001 and 0.006, respectively; Table 5, EUR), but there was no apparent difference among AFRs (p = 0.132 and 0.252, respectively; Table 5, AFR).
Table 5. Associations between smoking status and complement levels.
| Complement factor | Never | Ever | p |
|---|---|---|---|
|
All |
Never (n=158) |
Ever (n=64)* |
p |
| C3a |
97.2 (46.1) |
115.4 (45.2) |
<0.001 |
| C5a |
100.1 (25.4) |
97.8 (22.7) |
0.396 |
| Bb |
97.8 (52.5) |
117.5 (64.6) |
0.002 |
| Factor H |
101.1 (58.8) |
100.4 (51.6) |
0.279 |
|
EUR |
Never (n=111) |
Ever (n=52)* |
p |
| C3a |
97.8 (50.3) |
116.8 (48.6) |
0.001 |
| C5a |
95.1 (23.8) |
96.9 (22.1) |
0.864 |
| Bb |
99.9 (57.3) |
122.2 (69.8) |
0.006 |
| Factor H |
101.3 (57.5) |
100.4 (50.3) |
0.204 |
|
AFR |
Never (n=47) |
Ever (n=12) |
p |
| C3a |
95.6 (34.5) |
109.3 (26.7) |
0.132 |
| C5a |
112.1 (25.2) |
101.7 (25.7) |
0.152 |
| Bb |
92.6 (39.1) |
97.4 (28.2) |
0.252 |
| Factor H | 100.6 (62.7) | 100.8 (59.1) | 0.992 |
*One of the EUR ever smokers is missing all complement information
Associations between AMD with single nucleotide polymorphisms
Data were stratified based on ethnicity to examine the association between patient genotype and AMD status. Representative AMD-associated SNPs [5,39,40] were included in this analysis. Estimable ORs and 95% CIs for SNPs considered in the analysis are shown in Figure 2A (EURs) and Figure 2B (AFRs).
Figure 2.

Association between advanced AMD and common SNPs in EURs and AFRs. A forest plot of the univariate association between advanced AMD and common SNPs in A: EURs and B: AFRs. The table describes the SNP being considered, the model type, the estimated OR for AMD, and the associated p value. The plot shows the estimated OR with 95% CIs for the respective SNP and model. The gray vertical line represents the null OR of 1. Recessive SNP models missing values in the table and on the plot indicate that there were fewer than three subjects in the study population with two copies of the minor allele for that SNP. Additive models with missing values indicate that no subjects were recessive for the minor allele.
In EURs, the occurrence of AMD was associated with the additive effect of rs1536304 VEGFA (p = 0.043); marginal associations were also noted with the additive effect of the rs3766404 CFH (p = 0.075). Specifically, EUR participants had a 39% decrease in the odds of AMD with increasing copies of rs1536304 (OR = 0.61, 95% CI 0.38–0.98) and higher odds of having AMD with increasing copy numbers of rs3766404, though the association was not significant (OR = 0.20, 95% CI 0.93–4.42).
In AFRs, the occurrence of AMD was not significantly associated with any of the SNPs under consideration. Nevertheless, there was a marginal association between AMD status with the rs10490924 ARMS2 in the additive and dominant models (p = 0.060 and 0.062, respectively). Participants with at least one copy of the minor allele for rs10490924 had 5.2 times the odds of AMD relative to participants with no copies, although this association was not significant (p = 0.062, OR: 5.24, 95% CI 0.92–29.9).
Associations between SNPs and Complement Factors
Data were stratified based on ethnicity and SNPs to examine the association between genotype and complement activation status (Figure 3).In EURs, there was a significant association between rs2230199 (C3) and C3a levels (p = 0.05) in the recessive model, indicating that two copies of the allele are required for an increase in complement activation in the common terminal pathway. An association was also detected between rs2230199 and Bb levels (p = 0.049) in the dominant model, indicating that at least one copy of the minor allele resulted in decreased complement activation. Increased activity was not due to a decreased level of CFH activity or associated with higher levels of C5a.
Figure 3.

Association between complement levels and SNPs across all subjects. A forest plot of the association between complement levels and SNPs by race (EUR or AFR). The table describes the SNP and complement factor being considered, the SNP model type, the estimated difference in mean complement levels by SNP status, and the associated p value. The plot shows the estimated mean difference in complement levels with 95% CIs for the respective SNP and model. The gray vertical line represents a null mean difference of 0. Recessive SNP models missing values in the table and on the plot indicate that there were fewer than three subjects in the study population with two copies of the minor allele for that SNP. Additive models with missing values indicate that no subjects were recessive for the minor allele.
In AFRs, there was an association with the dominant effect of rs6667243 (CFHR5) on C3a levels (p = 0.044) and additive and dominant effects on C5a (p = 0.041 and 0.027, respectively). Participants with at least one copy of the minor allele for rs6667243 had higher levels of anaphylatoxins relative to participants with no copies of the minor allele. In addition, marginal associations were noted for the dominant effect of rs1864163 (CETP; p = 0.064) and rs1308155 (COL8A1; p = 0.092) on C3a levels. Participants with at least one copy of the minor allele for rs1864163 had lower levels of C3a, whereas those with at least one copy of the minor allele for rs1308155 had higher C3a levels relative to participants with no copies.
Discussion
AMD is a multifactorial disease caused by the combined effects of lifestyle, environmental factors, race, and variants at multiple loci. Here, we explored the association between complement activation and AMD, focusing on genetic risk factors, race, smoking, and complement dysregulation in a South Carolinian population. Specifically, in the context of systemic complement components in serum as potential biomarkers for AMD, we asked whether complement components in serum are affected by race, whether their levels are affected in a synergistic way between smoking and disease, and whether they correlate with particular SNPs. In addition, and novel to this study, we examined whether CFH activity rather than level differed in AMD and control subjects and how those activity levels change with age.
The original reports on the Y402H polymorphism in CFH as a major risk factor for AMD suggested that an overactive complement system is associated with AMD disease and risk; thus, elevated complement components and their breakdown products have been examined as potential biomarkers. While the best combination of markers still needs to be confirmed due to differences in analyses, the subjects included in the studies (all cases, GA or CNV only) had elevated plasma markers of chronic activation (Ba and C3d) [20,42] [43]; an increased C3d/C3 ratio [21,43]; elevated anaphylatoxins C3a, C4a, and C5a [43,44]; and altered levels of factor B, factor D, and factor I [20,42,43,45]. A few studies have indicated impairment of CFH-mediated complement inhibition, based on reduced CFH protein levels in an Indian AMD cohort [46]. In addition, it has been reported that AMD patients might have higher concentrations of nitrated CFH, a form that exhibits impaired GAG and C3b binding, as well as cofactor activity [47]. Here, we confirmed that AMD is associated with elevated levels of the anaphylatoxin C3a, and we showed that a 10-unit/mL increase in the level of C3a is associated with a 10% increase in the odds of AMD. Most of the biologic effectors of complement activation appear to be generated by the AP, a prediction that is supported by the elevated levels of the active AP serine protease Bb identified in AMD patients. The lack of increased levels of C5a in American AMD subjects contradicts some previous findings; in two European studies of either neovascular AMD [43] or predominantly neovascular AMD [48], elevated levels were reported, whereas in two studies with US participants, levels of C5a were only associated with GA but not CNV [49], or no association was found [20].
Surprisingly, in our cohort, AMD was significantly associated with reduced levels of the anaphylatoxin C5a, an association that was driven by ethnicity, as there was no association between C5a levels and AMD in EURs, but there was in AFRs. Based on this observation, we reexamined all our complement data between EURs and AFRs and suggested that complement levels can only be used as biomarkers in EURs, but not in AFRs, as elevated levels in C3a and Bb are only present in EURs.
Smoking is a major modifiable risk factor for AMD, and the association between risk for AMD and smoking has been documented in many epidemiological studies. Smoking not only increases the overall risk of developing AMD [50], but it also promotes the progression of AMD from the atrophic to neovascular form [51], and it does so significantly earlier in smokers compared to non-smokers [52]. In addition, the additive effects of smoking and genetic risk factors in the development of AMD have been shown [53]. Three pathogenic mechanisms have been examined extensively, including the role of cigarette smoke and nicotine on angiogenesis and neovascularization, the generation of oxidative stress due to the presence of pro-oxidant compounds in smoke, and the direct toxic effects of compounds, such as polycyclic aromatic hydrocarbons, on ocular cells (reviewed in [10]). An observation that we have discussed previously [54] is that compounds in cigarette smoke can directly activate the AP in vitro [22], which might explain why serum levels of complements are associated with current or cumulative cigarette consumption [24]. Here, we report that ever smokers had higher levels of C3a and Bb. However, despite the known risk of smoking and AMD, in our cohort, an association between smoking and AMD was not found in either the EUR or the AFR group. This lack of association might be due to the small sample size and lack of information on the extent of smoking (e.g., pack years for ever smokers or years since quitting for former smokers). The lack of synergism between smoking and higher levels of complement activation might suggest that not all changes in complement levels with smoking are associated with disease.
Variants at multiple genetic loci are independently associated with AMD [5,39], and a risk calculator has been developed based on ten variants [55]. Here, we tested two SNPs that are part of the risk calculator, ARMS2 (rs10490924) and C3 (rs2230199), as well as eight additional SNPs: ARMS2 (rs3793917), CFH (rs3766404, rs393955), VEGFA (rs1536304), CETP (rs1864163), C3 (rs2230199), CFHR5 (rs6667243, rs10922153), and COL8A1 (rs1308155), for their association with AMD. In EUR participants, the occurrence of AMD was associated with the additive effect of rs1536304 VEGFA (significant) and rs3766404 CFH (marginal). While the rs3766404 CFH locus has been shown previously to predispose individuals to AMD [56], no reports are currently available for rs1536304 VEGFA. In AFRs, marginal associations between AMD status with the known ARMS2 SNP (rs10490924) [57] in the additive and dominant models were identified.
Interactions between risk factors are expected in AMD. To date, the additive effects of smoking and genetic risk factors in the development of AMD have been shown for HTRA1 and CFH [53,58]. The association between genotype and levels of complement activation products has been examined in the literature. The CFH [43] and ARMS2 [21,43] risk genotypes have been shown to be independently associated with an elevated C3d/C3 ratio, and carriers of the CFB (OMIM 138470, Gene ID 629) protective allele had lower serum levels of CFB [43]. Here, we added to this pool of information that in EURs, rs2230199 C3 was associated with an increased complement activation, as demonstrated by elevated levels of C3a and C5a; whereas in AFRs, complement activation (C3a and C5a) was associated with a dominant effect of rs1536304 VEGFA and potentially with a dominant effect of rs1864163 CETP and rs1308155 COL8A1.
Finally, because CFH plasma concentrations range considerably between individuals (250 to 564 µg/ml) [59], rather than examining levels of CFH, we used a modified erythrocyte lysis assay to determine the inhibitory capacity of CFH present in the serum. CFH activity was not correlated with AMD or ethnicity. However, a significant interaction between patient age and levels of CFH activity was observed. Importantly, the model indicated that there is only a significant impact in younger individuals. In other words, the model indicates that as age increases, the impact of the level of CFH activity on the odds of having AMD decreases, and alternatively, the higher the levels of CFH activity, the less impact age has on the odds of having AMD. This observation might imply that CFH-based complement inhibition should become less efficacious with advanced age. A marginal correlation was identified between age and CFH activity (p = .0965), which is significant if levels in younger (below 70 years-of-age) subjects are compared to those in older subjects (above 70 years of age; p<0.05). We suggest that augmented complement activation in advanced AMD might be attributed to a decrease in CFH activity in younger patients, a hypothesis that needs further investigation. Importantly, this observation should be considered in the context of data obtained in young healthy CFH homozygous CC haplotype subjects who exhibit an abnormal choroidal blood flow regulation decades before potentially developing the disease. The authors have speculated that this might lead to ischemia or hypoxia [60]. These data in the context of our results suggest that AMD in CFH-risk subjects might be a disease that needs to be prevented early in life rather than treated when fully established.
We note several limitations to the study. As indicated previously, a relatively small sample size does not allow us to examine the association between AMD and smoking, and it might also responsible for the lack of association between AMD and C5a levels in EURs or the apparent association between AMD and low C5a levels in AFR subjects. The small sample size prohibits the detection of moderate to weak associations between genetic factors with complement activation products and AMD status. This is even more relevant when stratifying on race, as the study includes only 59 AFRs, among whom only seven had AMD. A posteriori power calculation shows that for common SNPs occurring in 50% of AFRs, our study was powered to detect a larger OR of 5.8. For rarer SNPs (10% of AFRs have the minor allele), the study was powered to detect only much larger ORs (>11). As we had a limited sample size, we could not adjust for multiple comparisons when examining associations between SNPs with complement components or with AMD status. However, given the exploratory nature of this study for evaluating genetic factors, the goal was to identify those SNPs that are likely associated with AMD and complement activation and that are worth further examination. In addition, race was self-reported. Thus, AFR cases and controls might be genetically dissimilar, but the insufficient sample size and SNPs did not permit the examination of genetic admixture. All these results require further validation in a larger cohort of AFRs that such a study accounts for genetic admixture. Nevertheless, our findings are intriguing and indicate that complement activation may differ in patients with and without AMD, and this association may be tempered by ethnicity and genetic factors. Within the context of race, we note that non-Whites do not respond as well to intravitreal ranibizumab treatment for wet AMD as compared to Whites, suggesting differences in disease etiology across ethnic groups [61].
In summary, our results first validate and further extend reports of systemic complement components, such as C3a and Bb, in serum as potential biomarkers for AMD. However, the lack of association between AMD and complement components in AFR subjects suggests that complement dysregulation might not underlie AMD pathology in all races. Second, they suggest that smoking and disease might not synergistically affect complement levels. Third, our results suggest that augmented complement activation in advanced AMD might be attributed to a decrease in CFH activity in younger patients. Though these findings are intriguing, they need verification in a larger study.
Acknowledgments
The authors thank Drs. E Bowie, J Gross, N Patel and C Grice and the student volunteers, J Pire, G Beinbrink and L Crawford for help with identifying and recruiting patients and the patients who participated in this research making it possible. This work was sponsored in part by a Department of Veterans Affairs merit award RX000444 and BX003050 (B.R.), a National Institutes of Health grant R01EY019320 (B.R.), the SmartState Endowment of the State of South Carolina, the Intramural Research Program of the National Eye Institute (ZIAEY000546; A.S.), and the South Carolina Clinical & Translational Research Institute, Medical University of South Carolina’s CTSA, NIH/NCRR Grant Number UL1RR029882.
References
- 1.Xu X, Liu X, Wang X, Clark ME, McGwin G, Jr, Owsley C, Curcio CA, Zhang Y. Retinal pigment epithelium degeneration associated with subretinal drusenoid deposits in age-related macular degeneration. Am J Ophthalmol. 2017;175:87–98. doi: 10.1016/j.ajo.2016.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33:295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudnicka AR, Kapetanakis VV, Jarrar Z, Wathern AK, Wormald R, Fletcher AE, Cook DG, Owen CG. Incidence of Late-Stage Age-Related Macular Degeneration in American Whites: Systematic Review and Meta-analysis. Am J Ophthalmol. 2015;160:85–93. doi: 10.1016/j.ajo.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu Rev Genomics Hum Genet. 2014;15:151–71. doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, Burdon KP, Hebbring SJ, Wen C, Gorski M, Kim IK, Cho D, Zack D, Souied E, Scholl HP, Bala E, Lee KE, Hunter DJ, Sardell RJ, Mitchell P, Merriam JE, Cipriani V, Hoffman JD, Schick T, Lechanteur YT, Guymer RH, Johnson MP, Jiang Y, Stanton CM, Buitendijk GH, Zhan X, Kwong AM, Boleda A, Brooks M, Gieser L, Ratnapriya R, Branham KE, Foerster JR, Heckenlively JR, Othman MI, Vote BJ, Liang HH, Souzeau E, McAllister IL, Isaacs T, Hall J, Lake S, Mackey DA, Constable IJ, Craig JE, Kitchner TE, Yang Z, Su Z, Luo H, Chen D, Ouyang H, Flagg K, Lin D, Mao G, Ferreyra H, Stark K, von Strachwitz CN, Wolf A, Brandl C, Rudolph G, Olden M, Morrison MA, Morgan DJ, Schu M, Ahn J, Silvestri G, Tsironi EE, Park KH, Farrer LA, Orlin A, Brucker A, Li M, Curcio CA, Mohand-Said S, Sahel JA, Audo I, Benchaboune M, Cree AJ, Rennie CA, Goverdhan SV, Grunin M, Hagbi-Levi S, Campochiaro P, Katsanis N, Holz FG, Blond F, Blanche H, Deleuze JF, Igo RP, Jr, Truitt B, Peachey NS, Meuer SM, Myers CE, Moore EL, Klein R, Hauser MA, Postel EA, Courtenay MD, Schwartz SG, Kovach JL, Scott WK, Liew G, Tan AG, Gopinath B, Merriam JC, Smith RT, Khan JC, Shahid H, Moore AT, McGrath JA, Laux R, Brantley MA, Jr, Agarwal A, Ersoy L, Caramoy A, Langmann T, Saksens NT, de Jong EK, Hoyng CB, Cain MS, Richardson AJ, Martin TM, Blangero J, Weeks DE, Dhillon B, van Duijn CM, Doheny KF, Romm J, Klaver CC, Hayward C, Gorin MB, Klein ML, Baird PN, den Hollander AI, Fauser S, Yates JR, Allikmets R, Wang JJ, Schaumberg DA, Klein BE, Hagstrom SA, Chowers I, Lotery AJ, Leveillard T, Zhang K, Brilliant MH, Hewitt AW, Swaroop A, Chew EY, Pericak-Vance MA, DeAngelis M, Stambolian D, Haines JL, Iyengar SK, Weber BH, Abecasis GR, Heid IM. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134–43. doi: 10.1038/ng.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pietraszkiewicz A, van Asten F, Kwong A, Ratnapriya R, Abecasis G, Swaroop A, Chew EY. Association of Rare Predicted Loss-of-Function Variants in Cellular Pathways with Sub-Phenotypes in Age-Related Macular Degeneration. Ophthalmology. 2018;125:398–406. doi: 10.1016/j.ophtha.2017.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorin MB, Weeks DE, Baron RV, Conley YP, Ortube MC, Nusinowitz S. Endophenotypes for Age-Related Macular Degeneration: Extending Our Reach into the Preclinical Stages of Disease. J Clin Med. 2014;3:1335–56. doi: 10.3390/jcm3041335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Asten F, Simmons M, Singhal A, Keenan TD, Ratnapriya R, Agron E, Clemons TE, Swaroop A, Lu Z, Chew EY. A Deep Phenotype Association Study Reveals Specific Phenotype Associations with Genetic Variants in Age-related Macular Degeneration: Age-Related Eye Disease Study 2 (AREDS2) Report No. 14. Ophthalmology. 2018;125:559–68. doi: 10.1016/j.ophtha.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kavanagh D, Yu Y, Schramm EC, Triebwasser M, Wagner EK, Raychaudhuri S, Daly MJ, Atkinson JP, Seddon JM. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum Mol Genet. 2015;24:3861–70. doi: 10.1093/hmg/ddv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Velilla S, Garcia-Medina JJ, Garcia-Layana A, Dolz-Marco R, Pons-Vazquez S, Pinazo-Duran MD, Gomez-Ulla F, Arevalo JF, Diaz-Llopis M, Gallego-Pinazo R. Smoking and age-related macular degeneration: review and update. J Ophthalmol. 2013;2013:895147. doi: 10.1155/2013/895147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joseph K, Kulik L, Coughlin B, Kunchithapautham K, Bandyopadhyay M, Thiel S, Thielens NM, Holers VM, Rohrer B. Oxidative Stress Sensitizes RPE Cells to Complement-Mediated Injury in a Natural Antibody-, Lectin Pathway- and Phospholipid Epitope-Dependent Manner. J Biol Chem. 2013;288:12753–65. doi: 10.1074/jbc.M112.421891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zipfel PF, Lauer N, Skerka C. The role of complement in AMD. Adv Exp Med Biol. 2010;703:9–24. doi: 10.1007/978-1-4419-5635-4_2. [DOI] [PubMed] [Google Scholar]
- 14.Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina. 2010;30:1350–67. doi: 10.1097/IAE.0b013e3181f57e30. [DOI] [PubMed] [Google Scholar]
- 15.Kijlstra A, La Heij E, Hendrikse F. Immunological factors in the pathogenesis and treatment of age-related macular degeneration. Ocul Immunol Inflamm. 2005;13:3–11. doi: 10.1080/09273940590909185. [DOI] [PubMed] [Google Scholar]
- 16.Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–96. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 17.Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 18.Lommatzsch A, Hermans P, Muller KD, Bornfeld N, Bird AC, Pauleikhoff D. Are low inflammatory reactions involved in exudative age-related macular degeneration? Morphological and immunhistochemical analysis of AMD associated with basal deposits. Graefes Arch Clin Exp Ophthalmol. 2008;246:803–10. doi: 10.1007/s00417-007-0749-4. [DOI] [PubMed] [Google Scholar]
- 19.Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, Wang K, Tucker BA, Stone EM. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. 2014;184:3142–53. doi: 10.1016/j.ajpath.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hecker LA, Edwards AO, Ryu E, Tosakulwong N, Baratz KH, Brown WL, Issa PC, Scholl HP, Pollok-Kopp B, Schmid-Kubista KE, Bailey KR, Oppermann M. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet. 2010;19:209–15. doi: 10.1093/hmg/ddp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saksens NT, Lechanteur YT, Verbakel SK, Groenewoud JM, Daha MR, Schick T, Fauser S, Boon CJ, Hoyng CB, den Hollander AI. Analysis of Risk Alleles and Complement Activation Levels in Familial and Non-Familial Age-Related Macular Degeneration. PLoS One. 2016;11:e0144367. doi: 10.1371/journal.pone.0144367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kew RR, Ghebrehiwet B, Janoff A. Cigarette smoke can activate the alternative pathway of complement in vitro by modifying the third component of complement. J Clin Invest. 1985;75:1000–7. doi: 10.1172/JCI111760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunchithapautham K, Atkinson C, Rohrer B. Smoke Exposure Causes Endoplasmic Reticulum Stress and Lipid Accumulation in Retinal Pigment Epithelium through Oxidative Stress and Complement Activation. J Biol Chem. 2014;289:14534–46. doi: 10.1074/jbc.M114.564674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyatt RJ, Bridges RB, Halatek DG. Complement levels in cigarette smokers: elevation of serum concentrations of C5, C9, and C1-inhibitor. J Clin Lab Immunol. 1981;6:131–5. [PubMed] [Google Scholar]
- 25.Fearon DT. The complement system and adaptive immunity. Semin Immunol. 1998;10:355–61. doi: 10.1006/smim.1998.0137. [DOI] [PubMed] [Google Scholar]
- 26.Holers VM. Phenotypes of complement knockouts. Immunopharmacology. 2000;49:125–31. doi: 10.1016/s0162-3109(00)80298-2. [DOI] [PubMed] [Google Scholar]
- 27.Holers VM. The complement system as a therapeutic target in autoimmunity. Clin Immunol. 2003;107:140–51. doi: 10.1016/s1521-6616(03)00034-2. [DOI] [PubMed] [Google Scholar]
- 28.Muller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57:321–47. doi: 10.1146/annurev.bi.57.070188.001541. [DOI] [PubMed] [Google Scholar]
- 29.Tan PL, Bowes Rickman C, Katsanis N. AMD and the alternative complement pathway: genetics and functional implications. Hum Genomics. 2016;10:23. doi: 10.1186/s40246-016-0079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100–4. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Group AMDGCS, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–61. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 33.Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, Goldstein JI, Triebwasser M, Anderson HE, Zerbib J, Kavanagh D, Souied E, Katsanis N, Daly MJ, Atkinson JP, Raychaudhuri S. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013;45:1366–70. doi: 10.1038/ng.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pavlov IY, De Forest N, Delgado JC. Specificity of EIA immunoassay for complement factor Bb testing. Clin Lab. 2011;57:225–8. [PubMed] [Google Scholar]
- 35.Magen E, Feldman A, Cohen Z, Alon DB, Linov L, Mishal J, Schlezinger M. Potential link between C3a, C3b and endothelial progenitor cells in resistant hypertension. Am J Med Sci. 2010;339:415–9. doi: 10.1097/MAJ.0b013e3181d7d496. [DOI] [PubMed] [Google Scholar]
- 36.Volokhina EB, Bergseth G, van de Kar NC, van den Heuvel LP, Mollnes TE. Eculizumab treatment efficiently prevents C5 cleavage without C5a generation in vivo. Blood. 2015;126:278–9. doi: 10.1182/blood-2015-03-637645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehling C, Amon O, Bommer M, Hoppe B, Kentouche K, Schalk G, Weimer R, Wiesener M, Hohenstein B, Tonshoff B, Buscher R, Fehrenbach H, Gok ON, Kirschfink M. Monitoring of complement activation biomarkers and eculizumab in complement-mediated renal disorders. Clin Exp Immunol. 2017;187:304–15. doi: 10.1111/cei.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, Lopez-Trascasa M. Functional analysis in serum from atypical Hemolytic Uremic Syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol. 2004;41:81–4. doi: 10.1016/j.molimm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, Zack DJ, Arakawa S, Cipriani V, Ripke S, Igo RP, Jr, Buitendijk GH, Sim X, Weeks DE, Guymer RH, Merriam JE, Francis PJ, Hannum G, Agarwal A, Armbrecht AM, Audo I, Aung T, Barile GR, Benchaboune M, Bird AC, Bishop PN, Branham KE, Brooks M, Brucker AJ, Cade WH, Cain MS, Campochiaro PA, Chan CC, Cheng CY, Chew EY, Chin KA, Chowers I, Clayton DG, Cojocaru R, Conley YP, Cornes BK, Daly MJ, Dhillon B, Edwards AO, Evangelou E, Fagerness J, Ferreyra HA, Friedman JS, Geirsdottir A, George RJ, Gieger C, Gupta N, Hagstrom SA, Harding SP, Haritoglou C, Heckenlively JR, Holz FG, Hughes G, Ioannidis JP, Ishibashi T, Joseph P, Jun G, Kamatani Y, Katsanis N. C NK, Khan JC, Kim IK, Kiyohara Y, Klein BE, Klein R, Kovach JL, Kozak I, Lee CJ, Lee KE, Lichtner P, Lotery AJ, Meitinger T, Mitchell P, Mohand-Said S, Moore AT, Morgan DJ, Morrison MA, Myers CE, Naj AC, Nakamura Y, Okada Y, Orlin A, Ortube MC, Othman MI, Pappas C, Park KH, Pauer GJ, Peachey NS, Poch O, Priya RR, Reynolds R, Richardson AJ, Ripp R, Rudolph G, Ryu E, Sahel JA, Schaumberg DA, Scholl HP, Schwartz SG, Scott WK, Shahid H, Sigurdsson H, Silvestri G, Sivakumaran TA, Smith RT, Sobrin L, Souied EH, Stambolian DE, Stefansson H, Sturgill-Short GM, Takahashi A, Tosakulwong N, Truitt BJ, Tsironi EE, Uitterlinden AG, van Duijn CM, Vijaya L, Vingerling JR, Vithana EN, Webster AR, Wichmann HE, Winkler TW, Wong TY, Wright AF, Zelenika D, Zhang M, Zhao L, Zhang K, Klein ML, Hageman GS, Lathrop GM, Stefansson K, Allikmets R, Baird PN, Gorin MB, Wang JJ, Klaver CC, Seddon JM, Pericak-Vance MA, Iyengar SK, Yates JR, Swaroop A, Weber BH, Kubo M, Deangelis MM, Leveillard T, Thorsteinsdottir U, Haines JL, Farrer LA, Heid IM, Abecasis GR, Consortium AMDG. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45:433–9. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–7. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 41.Vanderbeek BL, Zacks DN, Talwar N, Nan B, Musch DC, Stein JD. Racial differences in age-related macular degeneration rates in the United States: a longitudinal analysis of a managed care network. Am J Ophthalmol. 2011;152:273–82. doi: 10.1016/j.ajo.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hecker LA, Edwards AO. Genetic control of complement activation in humans and age related macular degeneration. Adv Exp Med Biol. 2010;703:49–62. doi: 10.1007/978-1-4419-5635-4_4. [DOI] [PubMed] [Google Scholar]
- 43.Smailhodzic D, Klaver CC, Klevering BJ, Boon CJ, Groenewoud JM, Kirchhof B, Daha MR, den Hollander AI, Hoyng CB. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology. 2012;119:339–46. doi: 10.1016/j.ophtha.2011.07.056. [DOI] [PubMed] [Google Scholar]
- 44.Lechner J, Chen M, Hogg RE, Toth L, Silvestri G, Chakravarthy U, Xu H. Higher plasma levels of complement C3a, C4a and C5a increase the risk of subretinal fibrosis in neovascular age-related macular degeneration: Complement activation in AMD. Immun Ageing. 2016;13:4. doi: 10.1186/s12979-016-0060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva AS, Teixeira AG, Bavia L, Lin F, Velletri R, Belfort R, Jr, Isaac L. Plasma levels of complement proteins from the alternative pathway in patients with age-related macular degeneration are independent of Complement Factor H Tyr(4)(0)(2)His polymorphism. Mol Vis. 2012;18:2288–99. [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma NK, Gupta A, Prabhakar S, Singh R, Sharma SK, Chen W, Anand A. Association between CFH Y402H polymorphism and age related macular degeneration in North Indian cohort. PLoS One. 2013;8:e70193. doi: 10.1371/journal.pone.0070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krilis M, Qi M, Madigan MC, Wong JWH, Abdelatti M, Guymer RH, Whitelock J, McCluskey P, Zhang P, Qi J, Hunyor AP, Krilis SA, Giannakopoulos B. Nitration of tyrosines in complement factor H domains alters its immunological activity and mediates a pathogenic role in age related macular degeneration. Oncotarget. 2017;8:49016–32. doi: 10.18632/oncotarget.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, Holz FG, Weber BH, Oppermann M. Systemic complement activation in age-related macular degeneration. PLoS One. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci. 2009;50:5818–27. doi: 10.1167/iovs.09-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lois N, Abdelkader E, Reglitz K, Garden C, Ayres JG. Environmental tobacco smoke exposure and eye disease. Br J Ophthalmol. 2008;92:1304–10. doi: 10.1136/bjo.2008.141168. [DOI] [PubMed] [Google Scholar]
- 51.Chakravarthy U, Augood C, Bentham GC, de Jong PT, Rahu M, Seland J, Soubrane G, Tomazzoli L, Topouzis F, Vingerling JR, Vioque J, Young IS, Fletcher AE. Cigarette smoking and age-related macular degeneration in the EUREYE Study. Ophthalmology. 2007;114:1157–63. doi: 10.1016/j.ophtha.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 52.Mitchell P, Wang JJ, Smith W, Leeder SR. Smoking and the 5-year incidence of age-related maculopathy: the Blue Mountains Eye Study. Arch Ophthalmol. 2002;120:1357–63. doi: 10.1001/archopht.120.10.1357. [DOI] [PubMed] [Google Scholar]
- 53.Tam PO, Ng TK, Liu DT, Chan WM, Chiang SW, Chen LJ, DeWan A, Hoh J, Lam DS, Pang CP. HTRA1 variants in exudative age-related macular degeneration and interactions with smoking and CFH. Invest Ophthalmol Vis Sci. 2008;49:2357–65. doi: 10.1167/iovs.07-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodell A, Coughlin B, Kunchithapautham K, Casey S, Williamson T, Ferrell WD, Atkinson C, Jones BW, Rohrer B. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS One. 2013;8:e67894. doi: 10.1371/journal.pone.0067894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seddon JM, Silver RE, Kwong M, Rosner B. Risk Prediction for Progression of Macular Degeneration: 10 Common and Rare Genetic Variants, Demographic, Environmental, and Macular Covariates. Invest Ophthalmol Vis Sci. 2015;56:2192–202. doi: 10.1167/iovs.14-15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Y, Guan N, Xu J, Yang X, Ma K, Zhou H, Zhang F, Snellingen T, Jiao Y, Liu X, Wang N, Liu N. Association of CFH, LOC387715, and HTRA1 polymorphisms with exudative age-related macular degeneration in a northern Chinese population. Mol Vis. 2008;14:1373–81. [PMC free article] [PubMed] [Google Scholar]
- 58.Nakanishi H, Yamashiro K, Yamada R, Gotoh N, Hayashi H, Nakata I, Saito M, Iida T, Oishi A, Kurimoto Y, Matsuo K, Tajima K, Matsuda F, Yoshimura N. Joint effect of cigarette smoking and CFH and LOC387715/HTRA1 polymorphisms on polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci. 2010;51:6183–7. doi: 10.1167/iovs.09-4948. [DOI] [PubMed] [Google Scholar]
- 59.Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol. 2006;177:6308–16. doi: 10.4049/jimmunol.177.9.6308. [DOI] [PubMed] [Google Scholar]
- 60.Told R, Palkovits S, Haslacher H, Frantal S, Schmidl D, Boltz A, Lasta M, Kaya S, Werkmeister RM, Garhofer G, Schmetterer L. Alterations of choroidal blood flow regulation in young healthy subjects with complement factor H polymorphism. PLoS One. 2013;8:e60424. doi: 10.1371/journal.pone.0060424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mohamed R, Gadhvi K, Mensah E. What Effect Does Ethnicity Have on the Response to Ranibizumab in the Treatment of Wet Age-Related Macular Degeneration? Ophthalmologica. 2018;240:1–6. doi: 10.1159/000486403. [DOI] [PubMed] [Google Scholar]