

Abstract
Purpose
To identify the mutation for Volkmann cataract (CTRCT8) at 1p36.33.
Methods
The genes in the candidate region 1p36.33 were Sanger and parallel deep sequenced, and informative single nucleotide polymorphisms (SNPs) were identified for linkage analysis. Expression analysis with reverse transcription polymerase chain reaction (RT-PCR) of the candidate gene was performed using RNA from different human tissues. Quantitative transcription polymerase chain reaction (qRT-PCR) analysis of the GNB1 gene was performed in affected and healthy individuals. Bioinformatic analysis of the linkage regions including the candidate gene was performed.
Results
Linkage analysis of the 1p36.33 CCV locus applying new marker systems obtained with Sanger and deep sequencing reduced the candidate locus from 2.1 Mb to 0.389 Mb flanked by the markers STS-22AC and rs549772338 and resulted in an logarithm of the odds (LOD) score of Z = 21.67. The identified mutation, rs763295804, affects the donor splice site in the long non-coding RNA gene RP1–140A9.1 (ENSG00000231050). The gene including splice-site junctions is conserved in primates but not in other mammalian genomes, and two alternative transcripts were shown with RT–PCR. One of these transcripts represented a lens cell–specific transcript. Meta-analysis of the Cross-Linking-Immuno-Precipitation sequencing (CLIP-Seq) data suggested the RNA binding protein (RBP) eIF4AIII is an active counterpart for RP1–140A9.1, and several miRNA and transcription factors binding sites were predicted in the proximity of the mutation. ENCODE DNase I hypersensitivity and histone methylation and acetylation data suggest the genomic region may have regulatory functions.
Conclusions
The mutation in RP1–140A9.1 suggests the long non-coding RNA as the candidate cataract gene associated with the autosomal dominant inherited congenital cataract from CCV. The mutation has the potential to destroy exon/intron splicing of both transcripts of RP1–140A9.1. Sanger and massive deep resequencing of the linkage region failed to identify alternative candidates suggesting the mutation in RP1–140A9.1 is causative for the CCV phenotype.
Introduction
Congenital cataract (CC) is a problem worldwide, and 34% of all CC cases in Denmark are inherited and caused by genetic modifications [1]. CC is heterogeneous in terms of clinical expression and molecular background, and autosomal dominant inheritance is the most common form [1,2]. More than 44 genetic loci have been mapped, and a causative CC gene has been characterized in 33 of the loci [3]. The mutations characterized in association with CC are found in several different gene families such as the lens crystallins [4,5]; in membrane proteins, such as the gap junction protein genes (connexins) [6]; in transcription factors, such as HSF4 (Gene ID 3299, OMIM 602438) [7] and growth factors [8]; and in intermediate filament proteins, such as MIP (Gene ID 4284, OMIM 154050) [9].
In 1992, we described an autosomal dominantly inherited cataract coined Volkmann cataract (CCV), which segregated in ten generations in a large family of 426 persons [10]. The CCV cataract is characterized by early debut and progressive, central, and zonular cataract with opacities in the embryonic, fetal, and juvenile nucleus and around the anterior and posterior Y-suture. CCV is transmitted as a dominant Mendelian trait with a penetrance of 0.90. Furthermore, few mutation carriers have a hardly recognizable cataract, and 10% have normal vision. Mutation carriers who develop pronounced cataract during the first or second decade of life need surgery [10].
This disease was mapped to a region telomeric to D1S243 (region 1pter-CCV-D1S243) at 1q36.3 with a logarithm of the odds (LOD) score of Zmax = 14.04, θM = 0.025, θF = 0.000, and a penetrance of 0.90 [11]. A total of 107 blood samples were initially collected in the Volkmann family for linkage analysis. One affected person without the disease haplotype in the linkage region lowered the LOD score, and the absence of informative marker systems in the 1pter region hindered further mapping of the CCV locus in the first study. The Volkmann CCV locus is the only cataract locus reported in the 1pter region, and the nearest cataract locus CTRCT6 (OMIM 116600) [12-14] 15 Mb proximal to the CCV locus was excluded by the linkage analysis (Z = –5) and Sanger sequencing of the EPHA2 (Gene ID 1969, OMIM 176946) gene.
We report CCV associated with a mutation in a long non-coding RNA gene (lncRNA), RP1–140A9.1, with an unknown function. The mutation was identified with next-generation sequencing (NGS) of the linkage region 1pter and is predicted to interrupt a donor splice site. The gene and the exon/intron boundaries are conserved in the primate genomes, and bioinformatics analyses suggest the mutation is located in a putative promoter region with several miRNA target sites and may function as an “miRNA sponge” or as competing endogenous RNA (ceRNA) [15]. Furthermore, the mutation interrupts a potential binding site for the RNA binding protein eIF4AIII.
Methods
Clinical data and linkage analysis
The clinical examination including the pedigree of the multigenerational family (107 individuals) and the initial mapping of the CCV locus has been published [10,11]. The follow-up analysis included eight new individuals; therefore, the final linkage analysis comprised 115 family members. The linkage analysis was performed using LIPED software [16] with a penetrance of p = 0.9 and a frequency of 0.001 for the disease. The study protocols adhered to the tenets of the Declaration of Helsinki and the ARVO statement on human subjects. The study received approval from the Danish National Committee on Health Research Ethics in 1990 and 2014 and all participants were informed orally and provided written consent.
DNA isolation and sequencing
DNA was extracted from ethylenediamine tetra-acetic acid (EDTA) blood using standard phenol chloroform extraction. Informative sequence-tagged site (STS) markers and single nucleotide polymorphisms (SNPs) used for haplotype construction were found with Sanger sequencing of 41 genes telomeric to D1S243 in IV:1 and III:11 (Appendix 1) and targeted NGS sequencing of the region. Oligonucleotides were designed by Primer3 [17] and purchased from TAG Copenhagen (Copenhagen, Denmark). Chromosome karyotyping and comparative genomic hybridization (CGH) analysis were performed with standard methods, and copy number variation (CNV) analysis was performed using SNP 6.0 arrays (Affymetrix, Santa Clara, CA; Appendix 1). PCR was performed using standard conditions and the primer pair CCV-mut-F/-R for the transcript analysis of RP1–140A9.1 and rs75047691-RNA-F/-R, B2M_11571/11572 and GAPDH_107/108 for the quantative reverse transcriptase PCR (qRT-PCR) analysis of GNB1 (Gene ID 2782, OMIM 139380; Appendix 2). PCR conditions included a 5 min melting step at 98 °C followed by 40 cycles of 96 °C for 30 s, 65 °C for 30 s, and 72 °C for 30 s followed by 72 °C for 5 min. Bidirectional Sanger sequencing (ABI Big Dye version 1.1 and ABI3100 DNA Sequencer, Applied Biosystems, Foster City, CA) was performed in a minimum of two affected individuals. Targeted next-generation sequencing (targeted NGS) was performed for index patient IV:3, and whole genome sequencing (WGS, BGI Europe, ≤800 bp insert normal library with a minimum coverage of 20 reads for the CCV linkage region) was performed for individual IV:7 (Appendix 1). Briefly, targeted NGS was done using 20 µg genomic DNA fragmented by sonication to about 300 bp DNA, blunt ended, followed by adaptor ligation according to the manufacturer’s protocol (Paired-End DNA Sample Prep kit, Illumina, San Diego, CA). The library was hybridized to a custom-made NimbleGen Sequence Capture 385 K microarray (Roche Sequence Capture Service, Madison, WI) designed for the chr1 region 0.5–2.14 Mb according to the manufacturer’s recommendations. The captured fragments were sequenced using a Genome Analyzer IIx (Illumina) and aligned to hg19 using Eland (ABI software package) allowing up to two mismatches in the 32 base seed sequence. Indels analysis was done by applying the Burrows-Wheeler Aligner [18], and inversions where detected by analyzing WGS data for clusters of read-pairs [19] where both reads align in the same orientation. The coverage of the region was continuous with an average sequencing depth of 40 reads, and exons covered by fewer than ten reads were Sanger sequenced.
RNA isolation, cDNA construction, and qPCR
Total RNA was extracted from Epstein Barr transformed B-lymphocytes [19], whole venous blood was taken from five healthy and affected individuals individuals (later frozen) and from human eye [20] using an RNeasy mini kit from Qiagen (Cat No./ID: 74,104; Hilden, Germany). Human universal reference RNA was purchased from Agilent (Cat. No. 740,000; Santa Clara, CA). PCR expression analysis of a human tissue panel of RNA was conducted as described previously [21]. PCR conditions included a 3 min melting step at 95 °C followed by 40 cycles of 95 °C for 30 s, 60 °C of 30 s.
Total RNA treated with DNase I from Agilent was used for the cDNA synthesis employing Superscript II-RT and poly-A priming according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA). cDNA from human lens epithelial cells was purchased from ScienCell (HLEpiC Cat. No. 6554; Carlsbad, CA). qPCR was performed using the Brilliant III Ultra-Fast Sybr qRT-PCR kit following the manufacturer’s protocol from AH-Diagnostics (Denmark) and included GNB1 (OMIM 139380) and the housekeeping genes B2M (OMIM 109700) and GAPDH (OMIM 138400). Data were normalized as described previously [22].
SNP analysis of the NGS data
The targeted NGS and WGS data were analyzed for known and novel SNPs (dbSNP build142), and SNPs with allele frequencies (AFs) greater than 0.01 were excluded. The remaining SNPs were analyzed using Sorting Intolerant From Tolerant (SIFT) for prediction of non-synonymous SNPs [23]. SNPs in coding exons, untranslated regions (UTRs), and regulatory regions were manually curated. Candidate SNPs were genotyped with PCR followed by Sanger sequencing for cosegregation with the cataract trait in the family.
CLIP-seq and miRNA target site analyses of non-annotated cDNA
Cross-Linking-Immuno-Precipitation sequencing (CLIP-Seq) data from multiple cell lines was analyzed with in-depth in silico meta-analysis [24] applying multiple RNA binding protein (RBP) data sets from starBase (starBase, release 2.1) [24]. Searching for RBP partners for the RP1–140A9.1 gene allows a comprehensive exploration of RBP/ncRNA (non-coding RNA) interaction maps from CLIP-Seq and Degradome-Seq data [24]. The predicted RBP and lncRNA interactions in starBase are processed and presented as a summary data set representing data from multiple studies with different cell lines and RBPs. Only RBPs that intersect the CLIP-Seq with RP1–140A9.1 were included. A biologic complexity greater than or equal to 2 was selected, and only those reaching the threshold were regarded as statistically significant [25]. Analysis of the mutation employed a pipeline to predict the effect of the mutations on the RNA secondary structure and the miRNA target sites in the lncRNA gene [26]. The structural effect was predicted using the RNAsnp program [27] employing different folding window sizes. Potential miRNA target sites were predicted for the genome region encoding RP1–140A9.1. TargetScan (v6.1) [28] and miRanda (v3.3a) [29] were applied on wild-type and mutant DNA sequences with default settings using human mature miRNA sequences from miRBase release 19 [30]. Only miRNAs with a predicted target site at the mutation position were retained if the target site was exclusive for the wild-type or the mutant genotype.
Results
Fine mapping of the CCV locus minimized the linkage region
DNA Sanger sequencing of the coding exons and targeted NGS and WGS of the published CCV locus resulted in a series of novel SNPs making refinement of the previously published linkage region possible. Using the new marker systems in a two-point linkage analysis reduced the CCV locus from 2.14 to 0.389 Mb and an LOD score Z = 21.67 (theta = 0, penetrance = 0.9; Figure 1A,B). Recombination events in individuals II:3 and II:5 excluded the region telomeric to STS-22AC, and recombination in individual II:4 excluded the region proximal to rs549772338 (Figure 1A). This new linkage region confirmed the affected individual II:4 and the monozygotic twins IV:9 and IV:10 as carriers of the disease haplotype, which was impossible previously due to the lack of marker systems distal to D1S243 (Figure 1A and Appendix 1) [11].
Figure 1.

Figure shows the methods used to narrow down the location of the linkage region. A: Segregation of the CCV phenotype is shown for a small branch of the large multigenerational Volkmann family [10,11]. The fine-mapped CCV linkage region includes eight loci at 1pter, and the disease haplotype is shown boxed and marked in red. The haplotypes for individuals I:1 and I:2 are inferred, and the recombination events in individuals II:3, II:4, II:5, and IV:8 are shown with an arrow. Symbols: square, male; circle, female; filled symbol, affected individual; dot in a symbol, healthy carrier. B: The chromosomal map of 1p36.33–36.32 shows the Volkmann cataract locus found by the initial genome-wide linkage analysis [11] together with the regions covered by direct Sanger sequencing of protein coding genes and with targeted next-generation sequencing (NGS) and whole genome sequencing (WGS). The key sequence-tagged sites (STSs) and single nucleotides (SNPs) are shown for the 2 Mb telomeric chromosome 1 regions together with the Sanger sequenced genes in the linkage region flanked by STS-22AC/ rs761317190 and rs549772338. C: The exon/intron gene structure for the lncRNA gene RP1–140A9.1 (1) includes the position of the CCV splice site mutation, the PCR primer pair (F1 and R1) positions, and the length and structure of the corresponding PCR amplicons (2–4). The CCV mutation is localized distal in exon 1. The cDNA (218 bp) PCR detected (2) from transformed B-leucocytes. The cDNA 324 bp PCR product detected (3) from two different mixed carcinoma cell preparations and from a lens cDNA library. A 500 bp band (4) from gDNA. The alternative splice sites shown in (2,3) are proposed by the NetGene2 predictor. The enhancer-promoter-associated histone mark H3K27ac (5) is active in the lncRNA gene RP1–140A9.1. The positions are shown for the four CAGCTG repeats (6) found in the region. The CAGCTG palindrome is a putative binding site for hsa-miR-1207-3p (seed sequence CAGCUG) and a binding site for the transcription factor AP-4 [35] proposed by TFSEARCH. The palindrome sequence at the exon/intron border is present only in the mutant sequence.
DNA sequencing of the CCV linkage region identified a splice donor variant
Chromosomal rearrangements, duplications, deletions, and inversions in the CCV locus were excluded after chromosomal karyotyping and array CGH analysis of individual II:4 and CNV analysis of individual V:1 (Appendix 1). Sanger sequencing of the coding exons of a total of 41 genes in the 2.14 Mb linkage region in individuals III:11 and IV:1 excluded missense, stop gain/loss, frame shift, and splice site mutations as causative for the CCV cataract. Targeted NGS and WGS of individuals IV:3 and IV:7 confirmed the result obtained with Sanger sequencing, and analysis of the deep sequencing data excluded large indels or inversions in the region.
All SNPs with AFs greater than 0.01 (dbSNP ver142) were excluded from the WGS data (Figure 1B) leaving 41 variations with an AF of less than 0.01 or unknown AFs for further analyses. Of these SNPs, 14, mainly indels, were excluded that they were in repeated regions, ten SNPs were represented in WGS data from six healthy in-house controls, and one SNP with an unknown frequency was later shown to be common. Segregation analysis of the remaining 16 SNPs excluded nine that did not cosegregate with the disease trait, leaving five as putative mutation candidates (Appendix 3). These candidates represented four SNPs in introns and one in an lncRNA gene. All intergenic and intron-located SNPs were excluded, and the SNP (rs763295804) in the lncRNA gene was chosen for further investigation. The SNP represented a G to C transversion (chr1:1,891,852; hg38) in the RP1–140A9.1 gene (ENSG00000231050.1). RP1–140A9.1 comprises two exons and one intron, and the mutation affected the +1 position in the 5′ GT splice consensus sequence of the splice donor site. The functional consequence of the G>C mutation is unproven but will most likely affect processing of the immature RNA transcript by intron skipping and result in a full-length un-spliced product. The RP1–140A9.1 gene is according to Ensembl gene annotation proposed to encode an antisense RNA, and the gene and the exon/intron boundaries are conserved in primates but not in rodents or other mammal genomes (Appendix 4). The mutation tested positive for cosegregation in the 115 members of the CCV family included in the linkage analysis [11] and was found in all affected individuals and absent in all unaffected individuals including spouses and was not present in 200 healthy Danes (AF<0.0025).
RNA analysis revealed two RP1–140A9.1 transcripts
The RP1–140A9.1 gene is annotated with one RNA transcript (ENST00000412228.1). RT–PCR analyses of RNA isolated from a panel human tissues revealed the RP1–140A9.1 gene was not expressed or expressed at low levels. PCR using the primer pair CCV-mut-F/-R and RNA isolated from Epstein Barr virus (EBV)–transformed B-leucocytes from a healthy control individual revealed a 218 bp PCR fragment. Analysis of RNA from cancer cell lines (human universal reference RNA), human fetal eye, and human lens epithelial cells revealed a 324 bp PCR product (Figure 2). These results suggested two different alternative transcripts for the gene. PCR using the same PCR primers and genomic DNA resulted in a 500 bp PCR fragment; this PCR product was seen in RNA samples without DNase I treatment as seen in cDNA from lens epithelial cells. According to the company, the lens epithelial cell cDNA was synthesized without DNase I treated total RNA (personal communication ScienCell). Sanger sequencing of the 500 bp PCR confirmed the genomic sequence, sequencing of the 218 bp PCR fragment confirmed the ENST00000412228.1 transcript using the donor splice site 5′-AGA CCT CCA G^g tga gga agg-3′ and the acceptor splice site ttt cct cca g^G CCT GCA CCA-3′ (genomic sequences in lowercase letters), and Sanger sequencing of the 324 bp PCR product revealed an alternative transcript using the same 5′ donor splice site and a different ctc tcc cca g^C CAG CGC CCT-3′ acceptor site (Figure 1C). The donor and the acceptor splice sites are supported by NetGene2 predictor [31], and the mutation affects both transcripts.
Figure 2.
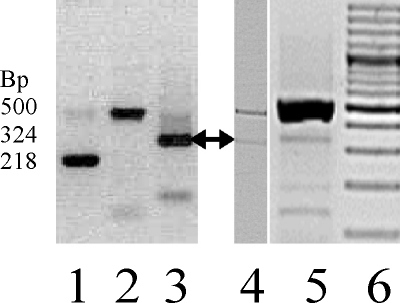
RT–PCR amplification products from different tissues using the primer set CCV-mut-F/-R. Lane 1: Epstein Barr virus–transformed B-lymphocytes cell cDNA preparations showing the 218 bp transcripts. Donor splice site: 5′-AGA CCT CCA G^G TGA GGA AGG and the acceptor splice site: TTT CCT CCA G^G CTG CAC CA-3′. Lane 2: A 500 bp genomic product. Lane 3: Universal Human reference RNA (mixed carcinoma) shows the 324 bp product. Donor splice site: 5′-AGA CCT CCA G^G TGA GGA AGG and acceptor splice site: CTC TCC CCA G^C CAG CGC CCT-3′. Lane 4: cDNA from a human fetal eye from DNase-treated RNA. A 324 bp product (arrow) is seen. Lane 5: cDNA from human lens epithelial cells (HLEpiC cat. No. 6554 lot no. 2531, ScienCell) not DNase treated. A genomic DNA (500 bp) and a 324 bp PCR product (arrow) are seen from the spliced RP1–140A9.1. Lane 6: 100 bp ladder.
The CCV mutation is not involved in regulation of GNB1 in blood
The G>C mutation is located 735 bp upstream in the promoter for the GNB1 gene (NM_002074.4). This promoter region is associated with histone marks shown by data from the ENCODE project (H3K4Me1/H3K4Me3/H3K27Ac marks, UCSC browser hg19) [32], and the mutation may interfere with regulation of GNB1. Employing TFSEARCH [33] for prediction of transcription factor binding sites revealed the wild-type allele of the promoter had the capacity to bind the transcription factor YY1 (DeltaE) [34], and the mutant allele had the capacity to bind the AP-4 [35] and Cap [36] transcription factors (Appendix 5). qRT-PCR expression analysis of GNB1 was performed using RNA from full blood from a mutation carrier and healthy individuals as RNA from affected and healthy lenses was not available (see Methods). qRT-PCR revealed that GNB1 was expressed equally in carriers and non-carriers in the family (Appendix 6).
The mutation affects primate-specific miRNA seed sequences
Bioinformatics analysis of the functional effect of the G>C mutation was made for possible changes in the RNA structure and miRNA binding. An RNAsnp analysis [27,37] showed no statistically significant effect of the mutation (a p value of less than 0.1) on the secondary structure of the un-spliced lncRNA transcript either using the sense or the antisense strand (Appendix 7). The miRNA target site analysis done by miRanda [29] and TargetScan [28] predicted 13 miRNA target sites exclusively in the wild-type sequence, and three miRNA target sites exclusively in the mutant sequence (Appendix 8). Both predictors found miRNA binding sites for hsa-miR-4254 and hsa-miR-619 in the wild-type sequence that were absent in the mutant sequence, and binding sites for hsa-miR-1207-3p and hsa-miR-3135b in the mutant sequence (Appendix 7). In addition to the miRNA target sites covering the mutation, putative binding sites for hsa-miR-1207-3p and hsa-miR-4254 were found in the transcripts.
CLIP-seq analysis: eIF4AIII is a potential RNA binding protein for RP1–140A9.1
Five RBPs (RNA binding proteins) were by CLIP-seq analysis shown to interact in vitro with RP1–140A9.1. Furthermore, 42% of all RP1–140A9.1 CLIP-reads represented eIF4AIII in CLIP-seq experiments conducted in HeLa cells [38]. Using a threshold of 2 in biocomplexity [39], only eIF4AIII is a qualified candidate as a putative binding partner for RP1–140A9.1. We further investigated the three eIF4AIII binding sites found in RP1–140A9.1; one site representing 85% of the reads corresponded to a region at position chr1:1,891,820–1,891,881 (hg38) that included the RP1–140A9.1 mutation. The eIF4AIII RBP is a general core component of the splicing-dependent multiprotein exon junction complex (EJC) [40] deposited at splice junctions on mRNAs [41]. Identification of a putative eIF4AIII binding site supports that the intron in RP1–140A9.1 is spliced out, and the G>C mutation may affect the eIF4AIII binding.
Discussion
The cataract phenotype found in the Volkmann family displays clinical heterogeneity and incomplete penetrance as described by Lund et al. [10]. At least three members of the family are healthy carriers of the mutation as found for the healthy mother III:5 (Appendix 1) and the monozygote twins, who, after clinical reexamination, now have normal vision at the age of 30 years old (Figure 1, individuals IV:9 and IV:10). One of the twins, IV:9, had a mild cataract at the first clinical examination [11].
The multipoint LOD score for the CCV locus was initially calculated as Z = 14 [11] and could with the use of new marker systems be calculated as Z = 21.67 for the updated pedigree now comprising 115 individuals. Chromosome rearrangements were excluded with karyotyping, array CGH, and SNP 6.0 CNV analyses, which suggested a point mutation as the most obvious causative mutation for the CCV phenotype. Sanger sequencing of all genes in the initial 2.14 Mb linkage region excluded missense or protein truncating mutations, and the result was verified with NGS of the region. New DNA variants found with Sanger and next-generation deep sequencing refined the linkage analysis of the CCV locus and reduced it to a 0.388 Mb region. The new linkage region encoded 11 protein coding genes that all were excluded with DNA sequencing and an lncRNA gene with a splice-site mutation (Figure 1B).
The NGS data discovered a G>C mutation in lncRNA RP1–140A9.1. The gene comprises two exons and one intron, and the G>C mutation is located at position +1 in the GT consensus donor splice site suggesting it causes intron splice defects (Appendix 5). The mutation was found in a non-repeated genomic DNA region, which supports it as a good candidate that all other variants were in the repeated region intronic or intergenic and therefore, were excluded as candidates. In addition, the gene and the exon intron boundary region are conserved in primates but are not present in genomes for rodents or other mammals (Appendix 4). The oldest affected person in the Volkmann family could be traced back ten generations to Germany [10], suggesting the mutation is a central European Caucasian variant.
The mutation was not present in 200 healthy Danes or in the 1000 Genomes Project, but present in the gnomAD browser with a minor allele frequency (MAF) of 0.001335 for the European non-Finnish population. In the TOPMed project, which represents more than 144,000 participants, the five rare SNPs in the candidate region (Appendix 3) all have an MAF of 0.0008. This suggested to us that these five rare alleles that are found in the Volkmann family are in linkage disequilibrium in European and American populations and represent a founder haplotype of about 0.38 Mb that can be found in a population that has European ancestry.
The G>C splice site mutation likely interferes with processing of the transcripts of the RP1–140A9.1 gene. The most likely consequence of the splice-site mutation is intron skipping resulting in a full length unprocessed RNA transcript. We have not been able to verify the consequences of the mutation, and RNA analysis of lens tissue from affected and unaffected individuals has not been available. RT–PCR revealed two alternative transcripts of the lncRNA gene. The Ensembl transcript ENST00000412228.1 was found in leucocytes, and a longer new transcript was found in the fetal eye, lens epithelial cells, and RNA from cancer cell lines (Figure 1 and Figure 2). Both transcripts use the same acceptor splice site and therefore, will be affected by the mutation.
We analyzed possible open reading frames that could be translated into a protein for the two alternative transcripts. None of these coded for an open reading frame with sequence similarity to known proteins in a search of the NCBI protein database. The lack of a protein coding open reading frame is in line with the idea that RP1–140A9.1 is an lncRNA gene that may function as an antisense RNAgene as suggested by Ensembl.
Bioinformatics predictions for the RBP binding sites in the exon/intron boundary suggested eIF4AIII as a possible candidate. The presence of the eIF4AIII binding site supports posttranscriptional processing of the immature RNA leading to the two alternative transcripts, and the introduction of the mutation in the binding site will disturb the processing of the RNA.
The low expression of RP1–140A9.1 in a panel of human tissues is in accordance with the expression profile in the GTEx Portal, where the gene is low or not expressed at all (TPM<1) except in the testis (Gtexportal). The low expression of RP1–140A9.1 is in line with what has been reported for lncRNA genes, and the representation of the gene exclusively in primate genomes corresponds to what has been reported for other lncRNAs [42].
qRT-PCR of the expression of GNB1 showed the mutation has no effects on the expression of the gene in blood. Although the analysis of RNA isolated from full blood could not exclude that the mutation has a regulatory impact on the regulation of GNB1 in lens tissue, GNB1 is not an obvious candidate as a cataract disease gene, and GNB1 has been associated with severe autosomal dominant intellectual disability (MRD42, OMIM 616973) [43].
The function of RP1–140A9.1 is unknown, but it is tempting to suggest it is involved in the early development lens that Volkmann cataract cause opacities seen in the embryonic, fetal, and juvenile nucleus of the lens [10]. Incorrect nuclear processing of pre-mRNA splicing or false miRNA binding capacities can, in general, have a strong impact on downstream functions in the cell [44], and the CCV mutation may disturb miRNA binding. For instance, it is a fourth target site for hsa-miR-1207-3p predicted in the mutant allele of the gene that may then serve as a miRNA sponge for hsa-miR-1207-3p.
We considered whether the miRNAs found to interact with the lncRNA genes are expressed in the lens or cataract. We searched PubMed and GEO, but did not find any direct literature match for these miRNAs or data sets for human lens and cataract. However, Wu et al. [44] examined miR-1207 in the lens and cataract and found that the miR-1207-5p variant in transparent lens samples is more expressed compared to cataractous samples. Lens-specific regulation for lncRNAs and miRNAs is well documented in studies of mouse lenses [45,46] and in humans in healthy as well as cataractous lenses [47,48]. Results have shown that changes in the concentration of different lens miRNAs play a crucial role in the development of age-related cataract [49-55], and it is plausible that the identified mutation not only is involved in the CCV phenotype but also contributes to the development of age-related cataract. This can explain the relative high AF, and the CCV phenotype is a result of more a more complex genetic background. Thus, the present results complement what is described in the literature.
In conclusion, we sequenced all genes in the candidate region for the dominant congenital form of cataracts previously described in 1995 [10,11] without finding any causative pathogenic mutation. Reanalysis of the linkage region using new markers reduced the region to 0.389 Mb, and WGS and targeted NGS identified a splice-site mutation in the lncRNA gene RP1–140A9.1. The mutation segregated with the CCV trait in the family and might represent the first example of a point mutation in an lncRNA causative for an autosomal dominant inherited congenital cataract. Future characterization of the RP1–140A9.1 lncRNA gene will hopefully elucidate the pathogenesis of Volkmann cataract.
Acknowledgments
We thank all family members for their participation in this study. Karen Friis Henriksen is thanked for excellent technical assistance. This work was supported by the Danish Medical Research Council (22-02-0208), The Danish Eye Research Foundation (1994-5:109,119), Fight for Sight, Denmark (2006), CBB, AW, RS, NT, JG by the Innovation Fund Denmark (0603-00320B), LH by the Danish National Research Foundation (DNRF107).
Appendix 1.
Selected sub-pedigree for the Volkmann family modified from Eiberg et al. [1]. The key persons II:4 and III:8 show recombination events (shown by an arrow) that delimit the linkage region. Note that the disease haplotype in II:4 is telomeric to D1S243 and individuals III:5 and IV:10 are unaffected carriers. One of the twins (IV:9) had a mild cataract at age 16, but after reexamination at age 30 had normal vision. Individual III:3 had vision problems, but no cataract at the time of examination. Inferred haplotypes are shown in italics, and the disease haplotype is boxed. The extended pedigree for the 426 members large family can be found in [1]. The filled symbols represent cataractous affected individuals, squares are males and circles are females. III:11 and IV:1 (marked S) were Sanger sequenced for 44 genes in the region 1pter-D1S243, IV:3 (marked T) was target-NGS and IV:7 (marked WGS) was whole genome sequenced. 1. Eiberg H, Lund AM, Warburg M, Rosenberg T. Assignment of congenital cataract Volkmann type (CCV) to chromosome 1p36. Hum Genet 1995; 96: 33-8. To access the data, click or select the words “Appendix 1.”
Appendix 2. PCR-primers.
To access the data, click or select the words “Appendix 2.”
Appendix 3. MAF values for identified SNPs.
a MAF data is for the non-Finnish Europeans population, gnomAD browser. b MAF data is for the American population, TOPMed. The American population is very heterogenous, and TOPMed. represent a cohort of >144k participants representing 40% European ancestry, 32% African, 16% Hispanic/Latino, 10% Asian etc. (https://www.nhlbiwgs.org/). To access the data, click or select the words “Appendix 3.”
Appendix 4. The genomic region around the splice site mutation is conserved in 8 primate genomes and less or not conserved in other mammals.
No alignments for: Mouse, Chicken, Marmoset, Tarsier, Elephant, Hedgehog, Dolphin. To access the data, click or select the words “Appendix 4.”
Appendix 5.
Transcription factor binding site analysis (TFSEARCH, I. Ovcharenko unpublished data) discovered the recognition site CCTCACCTGG for YY1 (previously named DeltaE) at the minus strand. The mutant allele sequence does not match the YY1 recognition site but recognition sites for the transcription factors AP-4 and Cap. To access the data, click or select the words “Appendix 5.”
Appendix 6.
qPCR of GNB1 using cDNA made from RNA isolated from fetal eye, and whole blood from five healthy individuals and five heterozygote mutant carriers. To access the data, click or select the words “Appendix 6.”
Appendix 7. RNA binding protein CLIP-Seq data
RNA binding protein (RBP) CLIP-Seq data subtracted and filtered using starBase V2.0. RP1-140A9.1 and the protein partners are mapped. Column 1 shows the name of the RBP identified; column 2 shows the number of identified target sites; column 3 shows the number of individual experiments where an RNA–RBP interaction has been observed. Last column shows the read count from CLIP-Seq experiments where a given RBP is interacting with RP1-140A9.1. To access the data, click or select the words “Appendix 7.”
Appendix 8.
Results of miRNA target prediction. The genomic region around the RP1-140A9.1 SNP (chr1: 1,891,852 G/C, hg38) showing the following tracks from top to bottom: “miRNA target predictions by TargetScan” shows miRNA target (miRT) sites as predicted by TargetScan [1]. Note that TargetScan requires an exact complement match to the seed sequence (only the seed sequence is marked). The set of miRNAs is limited to those were the mutation is predicted to create or destroy a binding site by either TargetScan or miRanda. For the predictions that overlap the mutation position, it is indicated whether the binding site is specific to wild-type “(WT)” or mutant “(MUT)”. Additional target sites for those miRNAs predicted along the transcript are also shown. Different miRNAs are highlighted with individual colors and labeled with miRT-XXX which refers to predicted target site for hsa-miR-XXX. “miRNA target predictions by miRanda” same details as above but for miRanda [2] predictions. “Repeat region” displays the location of the self-complementary “CAGCTG” repeats in the lncRNA transcript. The repeats that are common in wild-type and mutant sequence are shown in dark blue; the new repeat formed by the mutation is highlighted in light blue color. “Mutation” indicates the position of the G to C mutation in the lncRNA gene.“Ensembl Gene Predictions” shows Gencode v24, transcript of RP1-140A9.1. “cDNA_1: Found in B-lymphocyte cDNA in this study (Figure 2 lane 1), cDNA_2: Found in universal RNA, fetal eye and lens cDNA in this study /Figure 2 lane 3,4 and 5). 1. TargetScan software for prediction of microRNA targets: http://www.targetscan.org/ 2. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol 2003; 5:R1.1-R14. To access the data, click or select the words “Appendix 8.”
References
- 1.Jensen S, Goldschmidt E. Genetics counselling in sporadic cases of congenital cataract. Acta Ophthalmol. 1971;49:572–6. doi: 10.1111/j.1755-3768.1971.tb02963.x. [DOI] [PubMed] [Google Scholar]
- 2.Haugaard B, Wohlfahrt J, Fledelius HC, Rosenberg T, Melbye M. A nationwide Danish study of 1027 cases of congenital/infantile cataracts: etiological and clinical classifications. Ophthalmol. 2004;111:2292–8. doi: 10.1016/j.ophtha.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 3.Shiels A, Hejtmancik JF. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2017;156:95–102. doi: 10.1016/j.exer.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lubsen NH, Renwick JH, Tsui L-C, Breitman ML, Schoenmakers JGG. A locus for a human hereditary cataract is closely linked to the gamma-crystallin gene family. Proc Natl Acad Sci USA. 1987;84:489–92. doi: 10.1073/pnas.84.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen L, Mikkelsen A, Nürnberg P, Nürnberg G, Anjum P, Eiberg H, Rosenberg T. Comprehensive Mutational Screening in a Cohort of Danish Families with Hereditary Congenital Cataract. Inv Ophth Vis Sci. 2009;50:3291–303. doi: 10.1167/iovs.08-3149. [DOI] [PubMed] [Google Scholar]
- 6.Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S. Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet. 1999;64:1357–64. doi: 10.1086/302383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bu L, Jin YP, Shi YF, Chu RY, Ban AR, Eiberg H, Andres L, Jiang H, Zheng G, Qian M, Cui B, Xia Y, Liu J, Hu L, Zhao G, Hayden MRO, Kong X. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet. 2002;31:276–8. doi: 10.1038/ng921. [DOI] [PubMed] [Google Scholar]
- 8.Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: Putting cataract on the map. Mol Vis. 2010;16:2007–15. [PMC free article] [PubMed] [Google Scholar]
- 9.Berry V, Francis P, Kaushal S, Moore A, Bhattacharya S. Missense mutations in MIP underlie autosomal dominant ‘polymorphic’ and lamellar cataracts linked to 12q. Nat Genet. 2000;25:15–7. doi: 10.1038/75538. [DOI] [PubMed] [Google Scholar]
- 10.Lund AM, Eiberg H, Rosenberg T, Warburg M. Autosomal dominant congenital cataract; linkage relations; clinical and genetic heterogeneity. Clin Genet. 1992;41:65–9. doi: 10.1111/j.1399-0004.1992.tb03634.x. [DOI] [PubMed] [Google Scholar]
- 11.Eiberg H, Lund AM, Warburg M, Rosenberg T. Assignment of congenital cataract Volkmann type (CCV) to chromosome 1p36. Hum Genet. 1995;96:33–8. doi: 10.1007/BF00214183. [DOI] [PubMed] [Google Scholar]
- 12.Ionides ACW, Berry V, Mackay DS, Moore AT, Bhattacharya SS, Shiels A. A locus for autosomal dominant posterior polar cataract on chromosome 1p. Hum Mol Genet. 1997;6:47–51. doi: 10.1093/hmg/6.1.47. [DOI] [PubMed] [Google Scholar]
- 13.McKay JD, Patterson B, Craig JE, Russell-Eggitt IM, Wirth MG, Burdon PK, Hewitt AW, Cohn AC, Kerdraon Y, Mackey DA. The telomere of human chromosome 1p contains at least two independent autosomal dominant congenital cataract genes. Br J Ophthalmol. 2005;89:831–4. doi: 10.1136/bjo.2004.058495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang T, Hua R, Xiao W, Burdon KP, Bhattacharya SS, Craig JE, Shang D, Zhao X, Mackey DA, Moore AT, Luo Y, Zhang J, Zhang X. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract. Hum Mutat. 2009;30:E603–11. doi: 10.1002/humu.20995. [DOI] [PubMed] [Google Scholar]
- 15.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ott J. A computer program for linkage analysis of general human pedigrees. Am J Hum Genet. 1976;28:528–9. [PMC free article] [PubMed] [Google Scholar]
- 17.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics Methods and Protocols: Methods in Molecular Biology. Humana Press, Totowa, NJ; 2000. P. 365–86. [DOI] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minocherhomji S, Seemann S, Mang Y, El-schich Z, Bak M, Hansen C, Papadopoulos N, Josefsen K, Nielsen H, Gorodkin J, Tommerup N, Silahtaroglu A. Sequence and expression analysis of gaps in human chromosome 20. Nucleic Acids Res. 2012;40:6660–72. doi: 10.1093/nar/gks302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Møllgård K, Jacobsen M. Immunohistochemical identification of some plasma proteins in human embryonic and fetal forebrain with particular reference to the development of the neocortex. Brain Res. 1984;15:49–63. doi: 10.1016/0165-3806(84)90076-2. [DOI] [PubMed] [Google Scholar]
- 21.Seemann SE, Mirza AH, Hansen C, Bang-Berthelsen CH, Garde C, Christensen-Dalsgaard M, Torarinsson E, Yao Z, Workman CT, Pociot F, Nielsen H, Tommerup N, Ruzzo WL, Gorodkin J. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res. 2017;27:1371–83. doi: 10.1101/gr.208652.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bang-Berthelsen CH, Holm TL, Pyke C, Simonsen L, Søkilde R, Pociot F, Heller RS, Folkersen L, Kvist PH, Jackerott M, Fleckner J, Vilién M, Knudsen LB, Heding A, Frederiksen KS. GLP-1 Induces Barrier Protective Expression in Brunner’s Glands and Regulates Colonic Inflammation. Inflamm Bowel Dis. 2016;22:2078–97. doi: 10.1097/MIB.0000000000000847. [DOI] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res. 2011;39:D202–9. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winther TN, Jacobsen KS, Mirza AH, Heiberg IL, Bang-Berthelsen CH, Pociot F, Hogh B. Circulating MicroRNAs in Plasma of Hepatitis B e Antigen Positive Children Reveal Liver-Specific Target Genes. Int J Hepatol. 2014;2014:791045. doi: 10.1155/2014/791045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sabarinathan R, Wenzel A, Novotny P, Tang X, Kalari KR, Gorodkin J. Transcriptome-wide analysis of UTRs in non-small cell lung cancer reveals cancer-related genes with SNV-induced changes on RNA secondary structure and miRNA target sites. PLoS One. 2014;9:e82699. doi: 10.1371/journal.pone.0082699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabarinathan R, Tafer H, Seemann SE, Hofacker IL, Stadler PF, Gorodkin J. RNAsnp: Efficient Detection of Local RNA Secondary Structure Changes Induced by SNPs. Hum Mutat. 2013;34:546–56. doi: 10.1002/humu.22273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–46. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunak S, Engelbrecht J, Knudsen S. Prediction of Human mRNA Donor and Acceptor Sites from the DNA Sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- 32.Celniker SE, Dillon LAL, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, Micklem G, Piano F, Snyder M, Stein L, White KP, Waterston RH. Unlocking the secrets of the genome. Nature. 2009;459:927–30. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel A, Kel O, Ignatieva E, Ananko E, Podkolodnaya O, Kolpakov F, Podkolodny N, Kolchanov N. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–7. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung S, Perry RP. The importance of downstream delta-factor binding elements for the activity of the rpL32 promoter. Nucleic Acids Res. 1993;21:3301–8. doi: 10.1093/nar/21.14.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu YF, Lüscher B, Admon A, Mermod N, Tjian R. Transcription factor AP-4 contains multiple dimerization domains that regulate dimer specificity. Genes Dev. 1990;4:1741–52. doi: 10.1101/gad.4.10.1741. [DOI] [PubMed] [Google Scholar]
- 36.Gebhardt A, Habjan M, Benda C, Meiler A, Haas DA, Hein MY, Mann A, Mann M, Habermann B, Pichlmair A. mRNA export through an additional cap-binding complex consisting of NCBP1 and NCBP3. Nature Commun 2015; 6: 8192. Nat Commun. 2015;6:8192. doi: 10.1038/ncomms9192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabarinathan R, Tafer H, Seemann SE, Hofacker IL, Stadler PF, Gorodkin J. The RNAsnp web server: predicting SNP effects on local RNA secondary structure. Nucleic Acids Res. 2013;•••:W475–79. doi: 10.1093/nar/gkt291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saulière J, Murigneux V, Wang Z, Marquenet E, Barbosa I, Le Tonquèze O, Audic Y, Paillard L, Roest Crollius H, Le Hir H. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat Struct Mol Biol. 2012;11:1124–31. doi: 10.1038/nsmb.2420. [DOI] [PubMed] [Google Scholar]
- 39.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 40.Tange TØ, Nott A, Moore M. The ever-increasing complexities of the exon junction complex. Curr Opin Cell Biol. 2004;3:279–84. doi: 10.1016/j.ceb.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Ferraiuolo MA, Lee CS, Ler LW, Hsu JL, Costa-Mattioli M, Luo MJ, Reed R, Sonenberg N. A nuclear translation-like factor eIF4AIII is recruited to the mRNA during splicing and functions in nonsense-mediated decay. Proc Natl Acad Sci USA. 2004;101:4118–23. doi: 10.1073/pnas.0400933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigó R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrovski S, Kury S, Myers CT, Anyane-Yeboa K, Cogne B, Bialer M, Xia F, Hemati P, Riviello J, Mehaffey M, Besnard T, Becraft E, Wadley A, Politi AR, Colombo S, Zhu X, Ren Z, Andrews I, Dudding-Byth T, Schneider AL, Wallace G. University of Washington Center for Mendelian Genomics, Rosen ABI, Schelley S, Enns GM, Corre P, Dalton J, Mercier S, Latypova X, Schmitt S, Guzman E, Moore C, Bier L, Heinzen EL, Karachunski P, Shur N, Grebe T, Basinger A, Nguyen JM, Bézieau S, Wierenga K, Bernstein JA, Scheffer IE, Rosenfeld JA, Mefford HC, Isidor B, Goldstein DB. Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am J Hum Genet. 2016;98:1001–10. doi: 10.1016/j.ajhg.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu C, Lin H, Wang Q, Chen W, Luo H, Chen W, Zhang H. Discrepant expression of microRNAs in transparent and cataractous human lenses. Invest Ophthalmol Vis Sci. 2012;53:3906–12. doi: 10.1167/iovs.11-9178. [DOI] [PubMed] [Google Scholar]
- 45.Chen W, Yang S, Zhou Z, Zhao X, Zhong J, Reinach PS, Yan D. The Long Noncoding RNA Landscape of the Mouse Eye. Invest Ophthalmol Vis Sci. 2017;58:6308–17. doi: 10.1167/iovs.17-22178. [DOI] [PubMed] [Google Scholar]
- 46.Khan SY, Hackett SF, Riazuddin SA. Non-coding RNA profiling of the developing murine lens. Exp Eye Res. 2016;145:347–51. doi: 10.1016/j.exer.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen Y, Dong LF, Zhou RM, Yao J, Song YC, Yang H, Jiang Q, Yan B. Role of long non-coding RNA MIAT in proliferation, apoptosis and migration of lens epithelial cells: A clinical and in vitro study. J Cell Mol Med. 2016;20:537–48. doi: 10.1111/jcmm.12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wan P, Su W, Zhuo Y. Precise long non-coding RNA modulation in visual maintenance and impairment. J Med Genet. 2017;54:450–9. doi: 10.1136/jmedgenet-2016-104266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng CH, Liu JH, Woung LC, Lin TJ, Chiou SH, Tseng PC, Du WY, Cheng CK, Hu CC, Chien KH, Chen SJ. MicroRNAs and cataracts: correlation among let-7 expression, age and the severity of lens opacity. Br J Ophthalmol. 2012;96:747–51. doi: 10.1136/bjophthalmol-2011-300585. [DOI] [PubMed] [Google Scholar]
- 50.Wu C, Liu Z, Ma L, Pei C, Qin L, Gao N, Li J, Yin Y. MiRNAs regulate oxidative stress related genes via binding to the 3′ UTR and TATA-box regions: a new hypothesis for cataract pathogenesis. BMC Ophthalmol. 2017;17:142. doi: 10.1186/s12886-017-0537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Liu S, Zhang F, Jiang P, Wu X, Liang Y. Expression of the microRNAs hsa-miR-15a and hsa-miR-16–1 in lens epithelial cells of patients with age-related cataract. Int J Clin Exp Med. 2015;8:2405–10. [PMC free article] [PubMed] [Google Scholar]
- 52.Li G, Song H, Chen L, Yang W, Nan K, Lu P. TUG1 promotes lens epithelial cell apoptosis by regulating miR-421/caspase-3 axis in age-related cataract. Exp Cell Res. 2017;356:20–7. doi: 10.1016/j.yexcr.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 53.Gu S, Rong H, Zhang G, Kang L, Yang M, Guan H. Functional SNP in 3′-UTR MicroRNA- Binding Site of ZNF350 Confers Risk for Age-Related Cataract. Hum Mutat. 2016;37:1223–30. doi: 10.1002/humu.23073. [DOI] [PubMed] [Google Scholar]
- 54.Qin Y, Zhao J, Min X, Wang M, Luo W, Wu D, Yan Q, Li J, Wu X, Zhang J. MicroRNA-125b inhibits lens epithelial cell apoptosis by targeting p53 in age-related cataract. Biochim Biophys Acta. 2014;1842:2439–47. doi: 10.1016/j.bbadis.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 55.Yu X, Zheng H, Chan MT, Wu WKK. MicroRNAs: new players in cataract. Am J Transl Res. 2017;9:3896–903. [PMC free article] [PubMed] [Google Scholar]