

Abstract
Introduction
Sepsis remains a major health problem with an increasing incidence, high morbidity and high mortality. Apart from treatment with antibiotics and organ support, no approved specific adjunct therapies currently exist. Adrenomedullin (ADM) is a vasoactive peptide. High plasma concentrations of ADM correlate with worse outcome in sepsis patients. Preclinical work with the non-neutralising ADM-binding antibody adrecizumab showed promising effects in animal models of septic shock, including improved vascular barrier function, reduced vasopressor demand and organ dysfunction and increased survival. Therapeutic use of adrecizumab may therefore improve outcome in critically ill patients with septic shock and high ADM plasma concentrations. Phase I studies in healthy volunteers did not reveal any safety concerns. In this biomarker-guided trial, the safety and efficacy of adrecizumab will be investigated in patients with septic shock.
Methods and analysis
We describe a phase II, randomised, double-blind, placebo-controlled, biomarker-guided, proof-of-concept and dose-finding clinical trial in patients with early septic shock and high concentration of circulating ADM. A total of 300 patients will be enrolled at approximately 30 sites within the European Union. Patients are randomised to receive active treatment (2 and 4 mg/kg adrecizumab) or placebo, in a 1:1:2 ratio. Patient selection is guided by clinical parameters, and biomarker-guided by measurement of circulating biologically active ADM concentration at admission. Primary endpoint is safety and tolerability of adrecizumab over a 90-day period. A key secondary endpoint is the Sepsis Severity Index over a 14-day period.
Ethics and dissemination
This study is approved by relevant institutional review boards/independent ethics committees and is conducted in accordance with the ethical principles of the Declaration of Helsinki, the European Medicines Agency guidelines of Good Clinical Practice and all other applicable regulations. Results of this study will be published in a peer-reviewed scientific journal.
Trial registration number
NCT03085758; Pre-results.
Keywords: sepsis, adrecizumab, adrenomedullin, septic shock, vascular integrity, phase Ii clinical trial
Introduction
Worldwide, sepsis is a major health problem, with an increasing incidence and high mortality.1–3 It is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection.4 Septic shock is defined as a subset of sepsis in which profound circulatory, cellular and metabolic abnormalities occur, which are associated with an increased risk of mortality.4 The most prominent abnormalities are vasodilation and loss of vascular integrity, resulting in hypotension, and ultimately, in organ dysfunction and death.5 Besides antibiotics and organ supportive therapies such as vasopressors, mechanical ventilation and renal replacement therapy, there are currently no sepsis-specific adjunctive therapies registered.
Adrenomedullin (ADM) is a vasoactive peptide hormone that plays an important role in sepsis. Circulating ADM exerts endothelial barrier-stabilising effects and maintains vascular integrity.6–10 ADM has vasodilatory properties in the vascular interstitium, and at high concentrations, as observed during sepsis, may contribute to hypotension.11–13 Elevated concentrations of plasma ADM at admission have been reported in septic patients, and these were correlated with vasopressor requirement, organ dysfunction and mortality.14–16 The cut-off value of biologically active ADM (bio-ADM) of 70 pg/mL at admission was found to predict mortality for sepsis patients.14 This cut-off has been validated in independent, large multicentre studies.15 17 18
Based on these data, ADM may be an interesting therapeutic target for sepsis. A potential new adjunctive therapy for the treatment of septic shock is adrecizumab (previously also known as HAM8101). It is a non-neutralising ADM-binding antibody that has shown beneficial effects in preclinical studies. Adrecizumab reduced vascular leakage, organ dysfunction and need for vasopressor treatment during cecal ligation and puncture-induced sepsis in several animal studies and improved urine output and survival.19–21 Importantly, adrecizumab administration was not associated with any safety concerns in the first-in-human phase I study in healthy volunteers (n=24)22–24 and in a follow-up study in healthy volunteers, which were intravenously challenged with lipopolysaccharide (LPS) to induce systemic inflammation (also n=24).23 24 Of note, in the latter study, LPS-induced flu-like symptoms resolved more swiftly in adrecizumab-treated subjects compared with the placebo group. Pharmacokinetic (PK) analysis of adrecizumab showed a half-life of approximately 14 days, indicating that administration of a single dose is sufficient to achieve excess of plasma concentrations of the antibody over ADM for the entire sepsis period.
Based on these preclinical and human phase I data, it is hypothesised that therapeutic use of adrecizumab may improve endothelial dysfunction, restore and maintain vascular integrity and augment haemodynamics in critically ill patients with sepsis and septic shock. In the trial described in the present work, the safety, tolerability and efficacy of adrecizumab is investigated in patients with early septic shock and elevated concentrations of circulating bio-ADM. This will be one of the first precision medicine, biomarker-guided studies in septic patients.
Methods and analysis
Design and setting
AdrenOSS-2 is a phase II, randomised, double-blind, placebo-controlled, biomarker-guided, proof-of-concept and dose-finding clinical trial that is currently being conducted in patients with early septic shock and elevated concentration of circulating bio-ADM (>70 pg/mL). A total of 300 patients will be recruited in medical, surgical and mixed intensive care units (ICU) at approximately 30 sites across Belgium, France, Germany, the Netherlands and Italy (see clinicaltrials.gov of a list of current centres). Patient selection is guided by clinical parameters as well as by biomarker concentrations, by measuring circulating bio-ADM (sphingotest bio-ADM, sphingotec GmbH, Hennigsdorf, Germany).25 Based on preclinical studies, two dosages of adrecizumab will be investigated (2 and 4 mg/kg body weight), in addition to a placebo control arm. After informed consent has been signed by the patient or his/her legal representative, circulating bio-ADM concentrations will be assessed. If bio-ADM concentrations are >70 pg/mL, the clinical coordination centre (CCC) will be contacted for final confirmation of patient eligibility and the patient will be randomised. An interim analysis for futility is planned after 150 patients have completed day 28 of the study. An overview of the study design is depicted in figure 1 and study procedures in figure 2.
Figure 1.
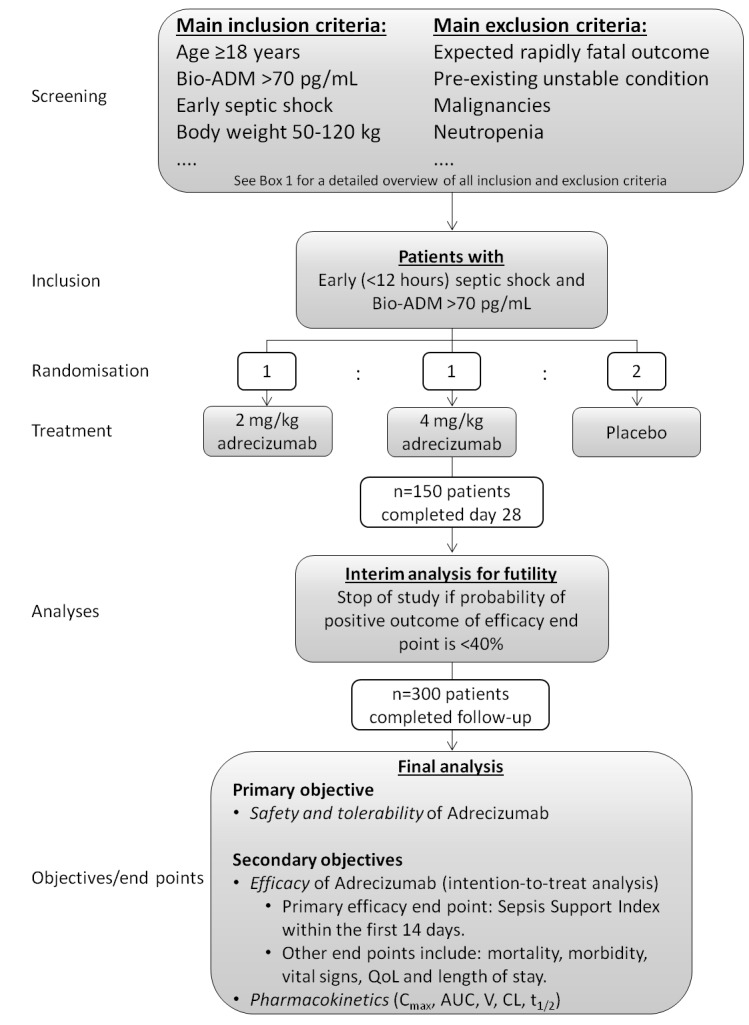
Study design. ADM, adrenomedullin; AUC, area under the curve; CL, systemic clearance; QoL, quality of life.
Figure 2.
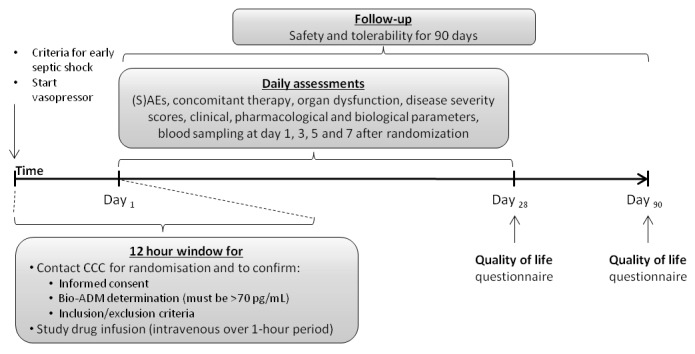
Study timeline. ADM, adrenomedullin; CCC, clinical coordination centre; SAE, severe adverse event.
Primary objective
The primary objective is safety and tolerability, consisting of: mortality possibly related to adrecizumab, interruption of infusion due to suspected intolerability of adrecizumab, new treatment-emergent adverse events (AEs) possibly related to adrecizumab and changes in severity and frequency of treatment-emergent AEs. During the study, an independent Data and Safety Monitoring Board (DSMB) will review safety data on at least a monthly base.
Secondary objectives
The secondary objectives are related to the efficacy and PK of adrecizumab. The primary efficacy end point, the ‘Sepsis Support Index’ (SSI), is a composite end point reflecting organ dysfunction or death within the first 14 days of follow-up. More precisely, within the first 14 days of follow-up, every day on which a vasopressor or mechanical ventilation is used, or renal dysfunction (defined as renal sequential organ failure assessment (SOFA)=4) is apparent, or the patient is not alive anymore, is counted as 1. The sum over the 14-day follow-up period is defined as the SSI score, which can have a maximum of 14 and a minimum of 1 (as vasopressor usage on day 1 is an inclusion criteria). The calculation of the SSI is further illustrated in figure 3.
Figure 3.
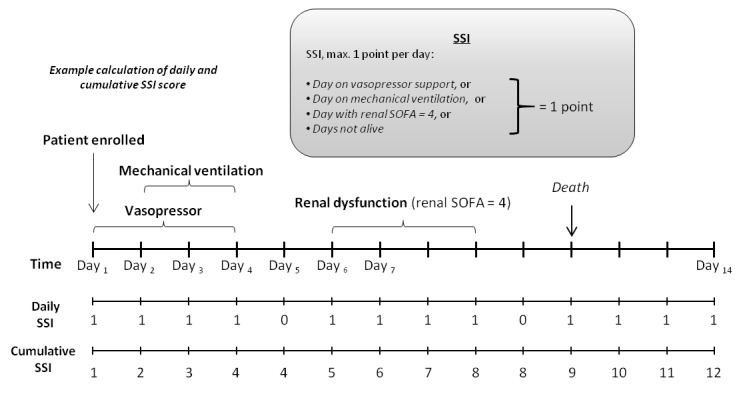
Primary efficacy end point: 14-day Sepsis Support Index (SSI): example calculation.
Additional secondary objectives include: SSI at day 28 of follow-up, penalised SSI (patients who die get penalised with the maximum score), individual SSI components, persistent organ dysfunction or death at days 14 and 28 of follow-up,26 day 28 and day 90 mortality rate and quality of life (Euro-QoL-5), change over time in SOFA and other parameters such as functional parameters (including, but not limited to heart rate, blood pressure, PaO2/FiO2, fluid balance, blood lactate, creatinine, pro-enkephalin, mid-regional-proADM, inflammatory markers, including procalcitonin (PCT) and interleukin-6), total duration of vasopressor/catecholamine use as well as length of stay at ICU/hospital.
For the PK substudy (n=80 patients), end points are key PK parameters, including peak plasma concentrations (Cmax), systemic exposure (area under the curve), volume of distribution (V), systemic clearance and elimination half-life (t1/2) of adrecizumab.
Patient selection
A total of 300 adult patients with early septic shock and elevated bio-ADM concentration will be randomised. Early septic shock is defined as sepsis with hypotension (mean arterial pressure<65 mm Hg) refractory to fluid resuscitation and requiring vasopressor therapy.4 Patients with a measurement of circulating bio-ADM >70 pg/mL will be eligible to be randomised. The cut-off point for bio-ADM of 70 pg/mL was selected based on the specific needs and purpose of this study. Per patient data available for this evaluation included data from the Albumin Replacement in Patients with Severe Sepsis or Septic Shock (ALBIOS), Frog-ICU and AdrenOSS-1 studies, to name the largest and most relevant, as well as data from healthy normal individuals. Specific needs to be met for the study were that patients with normal bio-ADM, as well as low severity and low expected mortality were to be excluded, to maximise the observable treatment effect, while keeping the eligible population as large as possible. The window for inclusion and infusion of study medication is 12 hours following initiation of vasopressor therapy. A lactate concentration >2 mmol/L is not an inclusion criteria, as concentrations may change quickly in response to initial therapy. Patients will be screened for clinical inclusion and exclusion criteria (Box 1). Screening and enrolment logs will be maintained for all patients. For patients not enrolled in the study, the reason for non-enrolment is documented. Patients will undergo various screening assessments, including recording of information on hospital and ICU admission (date, time, location before admission, diagnosis, origin of sepsis), documenting of relevant ongoing conditions, relevant medical history and comorbidities present or treated within the last year (cardiovascular and non-cardiovascular), concomitant medication use, age, gender, ethnic origin, physical examination including weight and height, blood sampling for laboratory examinations and bio-ADM measurement, pregnancy test (urine or serum), recording of 12-lead ECG, and calculation of Acute Physiology And Chronic Health Evaluation (APACHE) II and SOFA score. Eligibility will be confirmed by the CCC in Brussels, Belgium. Patients that fulfil all inclusion criteria and none of the exclusion criteria will be eligible to be randomised.
Box 1. Inclusion and exclusion criteria.
Inclusion criteria
Written informed consent by patient or legal representative (according to country-specific regulations).
Male and female patient, age ≥18 years.
Body weight 50–120 kg.
Biologically active adrenomedullin concentration >70 pg/mL.
Patient with early septic shock (start of vasopressor therapy <12 hours).
Women of childbearing potential must have a negative serum or urine pregnancy test before randomisation and have to use a highly effective method of contraception.
Exclusion criteria
Moribund.
Pre-existing unstable condition (eg, a recent cerebral haemorrhage or infarct, a recent acute unstable myocardial infarction (all <3 months), congestive heart failure—New York Heart Association class IV).
Patients who required cardiopulmonary resuscitation in the last 4 weeks prior to evaluation for enrolment.
Severe chronic obstructive pulmonary disease with chronic oxygen need at home (Global Initiative for Chronic Obstructive Long Disease (GOLD) IV).
Any organ or bone marrow transplant within the past 24 weeks.
Uncontrolled serious haemorrhage (≥2 units of blood/platelets in the previous 24 hours). Patients may be considered for enrolment if bleeding has stopped and patient is otherwise qualified.
Uncontrolled haematological/oncological malignancies.
Absolute neutropenia <500/µL.
Severe chronic liver disease (Child-Pugh C).
Systemic fungal infection or active tuberculosis.
Neuromuscular disorders that impact breathing/spontaneous ventilation.
Burns >30% of body surface.
Plasmapheresis.
Women who are pregnant or nursing.
Participation in a clinical trial involving another investigational drug within 4 weeks prior to inclusion.
Unwilling or unable to be fully evaluated for all follow-up visits.
Strengths and limitations of this study.
Patient selection is guided by clinical parameters, and biomarkerguided by measurement of circulating biologically active plasma adrenomedullin, allowing to select patients with an impaired outcome who may benefit most from adrecizumab therapy.
Patients will be recruited in medical, surgical and mixed intensive care units at approximately 30 sites across four countries in Europe, promoting the studies generalisability.
The study has appropriate randomisation using random block sequence generation, good allocation concealment as well as blinding of treating and research personnel.
The key secondary end point and primary efficacy end point is the composite Sepsis Support Index, which combines all-cause mortality and organ dysfunction, aimed to be more sensitive to assess the efficacy of the treatment.
Strict inclusion and exclusion criteria, as well as the brief time-window for inclusion (within 12 hours following the initiation of vasopressor therapy) may limit generalisation of the results for the entire population of critically ill patients with sepsis, although this may facilitate detection of an efficacy signal.
Measuring bio-ADM
For measurement of bio-ADM, 5 mL EDTA blood will be collected after written informed consent is obtained. After centrifugation (2500G, 15 min, 20°C), bio-ADM levels are determined using a fully validated, CE-marked, commercially available immunoluminometric assay (sphingotest bio-ADM assay). This assay is performed locally by trained personnel. The assay is highly specific for C-terminally amidated ADM (bio-ADM). Each patient sample will be measured in duplicate, and in parallel two calibrators (one with a concentration around the decision-making point (70 pg/mL)) will be run in triplicate along with each patient sample. The functionality of the measuring system will be checked on a monthly basis at each site. Finally, bio-ADM will be remeasured from banked aliquots in batch at a central lab to verify locally gained results. Further details about the assay are described elsewhere.25
Randomisation
Patients are randomly assigned to receive active treatment (2 and 4 mg/kg adrecizumab) or placebo, using a block randomisation scheme (1:1:2 treatment allocation ratio). A randomisation code list will be generated by an independent statistician not involved in the study. For each centre, study medication is provided in boxes containing 4 pairs of vials according to the four-block randomisation list, allowing stratification by centre.
Informed consent
Prior to any study-related procedures, patients must provide informed consent in accordance with the EU Clinical Trial Directive, the Declaration of Helsinki and International Countcil for Harmonisation (ICH)-Good Clinical Practise (GCP) requirements. Informed consent is obtained according to local requirements in Belgium, France, Germany and the Netherlands. Written informed consent is obtained by trained investigators after providing adequate verbal and written information about the study (in order to fully understand the study and any risks it entails), and giving the patient opportunity to ask questions and appropriate time to decide on participation in the study. For patients unable to provide consent themselves due to their medical condition, written informed consent is to be obtained by the patient’s legal representative or by other accepted procedures according to applicable national law and local regulations, for example, consent by relatives or family members. In addition, retrospective patient consent to voluntarily continue the study will be obtained once the patient has sufficiently recovered. Patient and/or the patient’s legal representatives can withdraw their consent on study participation at any time without providing an explanation.
Blinding
The study will be performed in a double-blinded fashion. All study personnel, including the investigator and site staff, patients, monitors, sponsor and Contract Research Organisation (CRO) staff will be blinded to treatment assignment until study closure. The randomisation list is kept strictly confidential by the data management vendor and accessible only to authorised persons who are not involved in the conduct of the study. In case of emergency, blinding will only be broken if specific emergency treatment would be indicated by knowing the treatment status of the patient. Specific emergency envelopes will be available at each site. The investigator is required to notify the sponsor within 24 hours following the code break reporting the reason for unblinding. The investigational drug and its matching placebo are indistinguishable and all study drug kits will be packed in the same way. Unblinding will be authorised by the sponsor after completion of the study, locking of the database and performance of a blinded data review.
Study intervention
A single dose of the study drug (2 or 4 mg/kg adrecizumab, or placebo) is administered over a 1-hour period by continuous intravenous infusion, as soon as possible, but at the latest, within 12 hours following start of vasopressor therapy. Study drug is administered separately from any concomitant drugs using a dedicated lumen of a central venous catheter or a separate peripheral line. Study medication is provided in boxes according to the four-block randomisation list. Each box contains four pairs of vials for a 1:1:2 treatment allocation ratio. The following pairs of vials are supplied in the box, in a blinded fashion: a set of two vials of adrecizumab (for reconstitution of the 4 mg/kg dose), a set of one vial of adrecizumab and one vial of placebo (for reconstitution of the 2 mg/kg dose) and two sets of two placebo vials. All vials are indistinguishable from each other, containing the same volume of solution, the same aqueous buffer and identical packaging. The study drug, adjusted to the patient’s body weight, has to be reconstituted from a pair of vials. All study drug are stored in a secure and adequately temperature-monitored pharmacy storage facility at 2°C–8°C.
Concomitant medication
There are no specific restrictions regarding use of concomitant medication or other therapies. All patients will be treated according to ‘International Guidelines for Management of Severe Sepsis and Septic Shock’.27 All concomitant medical treatments and medication will be recorded from inclusion until day 28 or ICU discharge (whichever comes first).
Patient and public involvement
Patients and the public were not involved in elaboration of the study protocol. There is no plan to disseminate the results directly to the study participants. Results will be published in a peer-reviewed journal and presented on conferences.
Statistical and analytical plan
Sample size calculations
The sample size was calculated for the primary efficacy end point (SSI up to day 14). A sample size of n=150 patients is planned for the combined treatment groups receiving 2 and 4 mg/kg adrecizumab. As both dosages result in an excess of antibody over the target peptide ADM, no difference in treatment effect is expected between the dosage groups. Therefore, the two dosage groups are pooled together for the final analysis, unless either dose is insufficient or safety and tolerability analysis indicate that one dose is not safe or tolerable. Power calculation was based on simulation analyses. The distribution of the SSI was based on real patient data from the ALBIOS study (n=539)15 and underlying assumptions were re-evaluated using results from the AdrenOSS-1 observational study.18 Based on the previously conducted observational AdrenOSS-1 study18 performed in septic patients, we anticipate a median SSI in the control group of 4 (IQR 2–11), while in the ALBIOS study15 the median was 7 (IQR 4–14) (these medians reflect a selection of patients with septic shock and bio-ADM larger 70 pg/mL). However, due to the non-normal distribution of the SSI, the median is still highly volatile (the majority of patients have either a low SSI (1–3 days, if improving and discharged early), or a high SSI (14 days, as patients that die within the first 14 days are usually on organ support while alive and in ICU)). For the simulations, a sample size of n=150 per group (treatment or placebo), and an effect size resulting in an approximately 10% decrease in SSI in the adrecizumab-treatment group (compared with the simulated control group) resulted in a power of the study of >80% to demonstrate an improvement of SSI of >0 with at least 80% probability. The 80% probability corresponds to the lower limit of the 60% CI of the effect estimate, delta SSI, which is based on the estimated difference of location from the Wilcoxon test. If the simulated lower limit of delta SSI was >0, the simulation run reached the end point
Statistical analyses
Continuous variables will be summarised by the number of patients, mean, SD or median, quartile and range, as appropriate. Categorical variables will be summarised using number and percentage by category. Demographic and medical background data, secondary end points and safety variables will be analysed by means of descriptive and exploratory methods. Regarding the primary end point (safety), all AEs will be listed. The number and percentage of patients experiencing one or more AEs will be summarised by treatment arm/control group, relationship to study drug and severity/grade. Severe adverse event (SAE)-specific listings for each patient population will be generated on reported SAEs, but not as Suspected Unexpected Serious Adverse Reaction (SUSARs). The same will be made for related SAEs. Mortality analysis is described below.
The primary analysis for efficacy will be performed as an intention-to-treat analysis based on the combined dosage groups of adrecizumab (n=150 patients total) versus placebo. A secondary analysis will compare the two doses for differences in efficacy. In case patients did not receive the treatment they were randomised to, an analysis based on the actual treatment will also be performed (as-treated analysis). The primary efficacy end point, 14-day SSI, will be analysed using the non-parametric Wilcoxon test, to estimate the treatment effects (based on the Wilcoxon estimate for difference in location) as well as its CI. First, it will be determined whether the improvement in SSI due to treatment is >0 with at least 80% probability (based on the lower limit of the one-sided CI of the effect estimate of the Wilcoxon test). If this is achieved, the classical p value from the Wilcoxon test will also be calculated. All-cause mortality will be evaluated using Kaplan-Meier plots comparing treatment (separate for each dose, as well as a comparison combining both doses into one group) versus placebo (log-rank test) and Cox regression modelling including covariates to adjust for potential confounders. Potential confounders include age, gender, MAP, HR, source of infection, blood culture, comorbidities and initial SOFA score as well as variables showing significant between-group differences (despite randomisation). In order to identify subgroups which may possibly benefit more from adrecizumab treatment, interactions with other drugs, as well as exploratory subgroup analyses are planned in patients defined by disease severity, biomarkers, concomitant medication or other clinical data. The subgroup analyses is nevertheless purely exploratory. Subgroups will be defined by tertiles for continuous variables. For categorical variables, categories will be summarised such that they best represent tertiles if more than three categories are available. Statistical analysis of secondary end points is exploratory, and will be specified in a separate statistical analysis plan, which is to be finished before conclusion of the study.
Interim analysis with futility stop
An unblinded interim analysis is planned after 50% of patients completed the study on day 28. The study will be terminated if the probability of a positive outcome after of all patients is below 40%, based on the primary efficacy end point 14-day SSI. In case the futility stop is reached, but if some of the other efficacy end points show a promising outcome for the full study, the futility stop may be suspended. Statistical consequence of applying the futility analysis was included in the power simulation. An independent statistician is responsible for analysing the data at interim analysis, and the steering committee, as well as the sponsor, will remain blinded until the end of the study. Note that the interim analysis focuses on futility only, potential termination of the trial based on harm is based on the reviewing and evaluation of unblinded data on safety and mortality by the DSMB (described further below).
Data quality assurance
All data management activities are done according to ICH-GCP as required by regulatory agencies. A commercial CRO, M.A.R.C.O. GmbH & Co (M.A.R.C.O), will be responsible for data management. All sites will maintain source documentation and enter patient data into an electronic case report form (eCRF). The clinical centre is responsible for the secure and restrictive archiving of source data for at least 15 years or until the written notification from the sponsor that the documents are no longer required. During the required period, the clinical centre will ensure that archived data and documents will be undamaged, legible and accessible to the sponsor and/or for regulatory purposes, if required. The study master file, the eCRFs, code envelopes and other material supplied for the performance of the study will be retained by the sponsor according to applicable regulations and laws, including the new GDPR (see also the section on ‘Confidentiality’). Regarding the eCRF, automated and manual checks will be performed to ensure completeness and consistency of the data, and investigator site personnel seeking access must go through training processes before access to the system is granted. The eCRF was designed by M.A.R.C.O. in the Amedon system. Validation checks are implemented in the system or programmed with SAS, V.9.1 or higher, according to the data validation plan set up by M.A.R.C.O.
Missing data
In general, missing data in clinical variables will not be replaced or imputed. If missing data should occur in variables required for secondary efficacy end points (eg, SOFA score or other secondary efficacy end points), a sensitivity analysis will be conducted assigning missing end point data with the worst possible value (as defined for withdrawals), in addition to the analysis based on valid data only. In addition, an analysis will be conducted where missing data points will be imputed using interpolation or extrapolation, with the exception that missing bilirubin will be set to normal (liver SOFA component=0). Missing follow-up time information will not be replaced for mortality analysis, but rather treated as respective methods for survival analysis intend.
Safety assessments
Medication error
Adequately trained hospital staff will prepare, double-check and administer study medication. The dose levels that are administered in the study have not caused any safety concerns in previous studies in healthy volunteers22–24 or in preclinical safety and toxicological studies in animals and non-human primates. The risk for adverse health effects due to medication errors are thought to be minimal.
Overdose risks
No drug-specific antidote for adrecizumab is available. An overdose is defined as any dose higher than the assigned treatment dose. However, if by accident, the maximum volume would be withdrawn from a pair of adrecizumab vials during preparation of study medication, this would not exceed the tested maximum dose of 8 mg/kg adrecizumab in healthy volunteers, which did not result in any safety concerns.22–24
AE reporting
All patients are monitored for AEs. AEs are defined as any untoward medical occurrence in a patient administered a product and which does not necessarily have a causal relationship with this treatment. Investigators must document all AEs (whether serious or non-serious and judged related or unrelated to the study drug) that occur during the study period extending from day 1 (inclusion) until 90 days after study drug administration in the eCRF. If the AE is serious, a ‘serious AE report form’ must also be sent to the safety contact of the sponsor (spm,2 Safety Projects & more GmbH, Hirschberg an der Bergstraße, Germany) within 24 hours of becoming aware of the SAE. The severity of the AE will be rated as ‘mild’, ‘moderate’, ‘severe’, ‘life-threatening’, ‘disabling’ or ‘death related to event’. Investigators will use medical judgement to determine whether there is evidence for a causal relationship and will describe this causality using terms such as ‘certain’, ‘probably/likely’, ‘possible’, ‘unlikely’ or ‘unrelated’. All AEs will be followed-up until they have abated, or until a stable situation has been reached, and will be reported as such.
External data monitoring committee
An independent DSMB has been established to monthly review safety data including SAEs and, overall safety data, and will judge the relevance of events for patient safety. DSMB members will have no direct relationship to the study or to the study sponsor. The DSMB, composed by two clinical experts in the field of sepsis, a biostatistician and a pharmacovigilance representative, will operate independently. The DSMB is empowered to recommend changes in the design of the study to ensure the safety of the patients and scientific integrity of the study.
Withdrawal
Participation is strictly voluntary and a patient or their legal representative may withdraw the patient from the study at any time without providing an explanation. This will not affect his/her right for future medical care. If a patient would withdraw from the study, the date, circumstances and any reason provided will be documented on the withdrawal page of the eCRF. No study-specific data or patient material will be collected after withdrawal of consent. No data obtained after withdrawal of consent will be recorded on eCRFs, unless the patient consents to the use thereof. For safety analysis, the patient’s outcome status (dead or alive) at day 90 will be collected. For the main efficacy analysis, these patients will be excluded. In order to rule out that patient withdrawal is linked to treatment, a sensitivity analysis will be conducted assigning missing end point data with the worst possible value (ie, worst possible value for patients in the treatment group, the best possible value for patients in the control group). In addition, an analysis will be conducted where missing data points will be imputed using interpolation or extrapolation, if applicable.
Study period
The study started enrolling patients in December 2017. The estimated study enrolment completion date is anticipated in the first half of 2019. Please note that this manuscript was finalised prior to the interim analysis.
Ethics and dissemination
Ethics
The study is performed in accordance with the Declaration of Helsinki, ICH-GCP, Code of Federal regulations and all other applicable regulations. Collection of personal data is performed according to country-specific regulations.
Confidentiality
After written informed consent has been obtained, patients will be assigned a unique six-digit patient identification number. This allows identification of patients, while maintaining patient confidentiality. The investigators, designated CRO and sponsor and all other involved parties will preserve the confidentiality of all patients taking part in the study, in accordance with ICH-GCP and local regulations. Confidentiality of all patient identities will be maintained, except during source data verification when monitors, auditors and other authorised agents of the sponsor or its designee, the ethics committee or any other applicable regulatory authorities are granted direct access to the study patient’s original medical records. No material bearing a patient’s name will be kept on file by the CRO or sponsor. The code list with treatment allocations (randomisation list) is stored separately from the sponsor at the data management vendor (CRO) during the course of the study. These data management vendors will provide all relevant data (pseudonymised) to the sponsor after the end of the study. In addition, sets of sealed envelopes with randomisation codes are kept at the site for emergency unblinding, with the DSMB and with the party responsible for reporting SUSARs as required by regulatory agencies. Data retained from this study will be protected in accordance with all applicable legal requirements. Information about study patients will be kept confidential and managed according to the requirements of EU-directives 2001/20/EC, 2005/28/EC and 2003/63/EC, and relevant national and local legislation. All ongoing subjects signed the ICF (including the data protection part) and additionally the ‘Information letter for ongoing patients’ regarding the new GDPR/(DSGVO, Germany). All patients have been informed by investigators before they signed these documents.
Data access
The following parties have access to the data: sponsor, sites and selected vendors (data management, pharmacovigilance). Individual patient data may be used by site investigators for publication in agreement with the sponsor. Please note that the confidentiality section also specifies some external parties that may access data (regulatory authorities, etc).
Sample storage
A biobank for biomarkers is implemented and samples are stored for potential future use.
Study monitoring
The study is monitored by a clinical monitor, who will visit the investigator and study sites at periodic intervals in addition to phone, letter and email contact. The monitor will follow the study closely through reviewing of study records and source documents, and will determine if the reported data are accurate and complete.
Dissemination policy
The data of the study will be reported at scientific meetings and published in a peer-reviewed scientific journal, regardless of the results on outcome, in accordance with the good publication practice guideline of the international society for medical publication professionals. The sponsor and the investigator and other individuals who have expertise in the area and who are willing to interpret the data and write or review articles and presentations will form a publication steering committee to oversee the preparation of articles and presentations from this study.
Discussion
The development of new therapies for the treatment of sepsis and septic shock has proven to be a challenging task over the last decades. Many trials have investigated potential adjunctive therapies, predominantly focussing on anti-inflammatory agents. Unfortunately, this enormous effort put into dozens of clinical trials has not yielded compounds with clinically relevant beneficial effects. This can be explained by many factors, such as heterogeneous study populations and difficulties in selecting patients who may best benefit from an intervention. Also, the timing of the intervention, inappropriate outcome measures and the complexity of the disease with multiple pathways of injury hamper clinical research in sepsis patients.5 28
Importantly, when antibodies were used, most interventions were based on complete neutralisation of the target. However, physiology probably is more balanced as some targets can exert both beneficial and detrimental effects, often even simultaneously. This may also represent a major contributing factor to the failure of many therapies to improve outcome witnessed in the last decades. Along these lines, it might be argued that a partially neutralising therapy is more effective than total neutralisation. The AdrenOSS-2 trial is an innovative, biomarker driven trial with a novel, supposedly clinically relevant efficacy end point.
Patient heterogeneity is a substantial contributor to the difficulties in identifying effective therapies for sepsis. Patient selection is innovative in this study for two reasons. First, a more homogeneous subgroup of sepsis patients is selected, based on the combination of presence of early signs of shock, that is, requiring vasopressor support, as well as elevated concentration of the biomarker bio-ADM. Selecting patients in the early phase of septic shock should select patients with preventable organ dysfunction compared with patients for whom septic shock and need of vasopressors lasted >12 hours. Furthermore, as previously described, measuring bio-ADM at baseline correlates strongly with the need for organ supporting therapy and mortality.14 15 17 18 Therefore, including bio-ADM as an inclusion criteria likely allows for better selection of patients who need vasopressors and have a poor outcome. Combining need of vasopressor and high bio-ADM may contribute to obtaining a more homogeneous population of patients who may benefit most from this adjunctive sepsis therapy. To our knowledge, this is one of the first precision medicine study in sepsis patients.29
ADM is a key vasoactive peptide involved in several important pathways in sepsis, which makes it an attractive therapeutic target in sepsis.10 It has previously been described as a double-edged sword in sepsis.30 On vascular smooth muscle cells, ADM exerts vasodilatory effects and thereby induces vasodilation and hypotension.11–13 This effect of interstitial ADM may exacerbate the severity of shock and may lead to organ hypoperfusion and organ dysfunction. In contrast, ADM present in the circulation exerts potent endothelial barrier stabilising effects, reducing vascular leakage that may improve survival, as was demonstrated in vitro6 7 31 32 and in vivo in animal models of sepsis and systemic inflammation.8 9 33 34 However, direct administration of ADM during sepsis poses several limitations. Because of a short half-life,11 continuous infusion of ADM would be required. In addition, due to ADM’s potent vasodilative effects, ADM-induced hypotension might be an issue, which might further aggravate shock in septic patients. A non-neutralising antibody might attenuate ADM’s vasodilatory effects on vascular smooth muscle cells (VSM) and potentiate ADM’s effects on endothelial cells.
Adrecizumab, a non-neutralising ADM-binding antibody, is one of the first therapies specifically aimed at improving vascular endothelial barrier function, and represents a new candidate drug for the treatment of septic shock. A detailed description of adrecizumab’s supposed mode of action is described elsewhere.35 Briefly, during sepsis, increased concentrations of ADM in the interstitial compartment are thought to contribute to hypotension. Adrecizumab, which is confined to the blood compartment, shifts the distribution of ADM away from the interstitium towards the blood, by preventing diffusion of bound ADM.35 This results in a strong increase of (bound) ADM concentrations in the blood,22–24 where it, being bound to a non-neutralising antibody, interacts with receptors on endothelial cells and reduces vascular leakage and tissue oedema. At the same time, concentrations in the interstitium are reduced through this mechanism, leading to less vasodilation and subsequent hypotension. This increase in plasma ADM concentration was observed in a rapid and dose-dependent manner on intravenous administration of adrecizumab, both in animals and in humans.21–24 Through reducing vascular leakage, tissue oedema and hypotension, adrecizumab could increase tissue perfusion and improve the prognosis of sepsis patients, whereas it might also reduce the use of vasopressors, thereby limiting potential adverse effects of vasopressors.36 37
Adrecizumab, administered as a single intravenous dose (due to its long half-life of 14 days), showed promising results in preclinical studies of systemic inflammation and septic shock, including attenuation of vascular leakage, lower vasopressor infusion rates and less organ dysfunction, related to improved survival.19–21
Substantial effort has been directed at reducing mortality in sepsis patients. Nevertheless, all major sepsis trials have failed to improve survival. Although survival is a clear and relevant end point, it may be too insensitive to demonstrate a beneficial effect of a novel intervention. Therefore, novel end points beyond all-cause mortality should be considered.38 The use of composite end points allows for a more nuanced assessment of morbidity and mortality. A new composite end point, the SSI, is used in the present study as the primary efficacy end point. The SSI is a composite index reflecting days on organ supportive therapy (haemodynamics, pulmonary), days with organ dysfunction (renal) as well as all-cause mortality. These organ systems were improved by adrecizumab administration in preclinical models, and support of these organ systems defines ICU care, indicating that a therapeutic effect is of clinical relevance. The SSI is thought to allow for earlier and more sensitive observations of possible clinically relevant beneficial effects of adrecizumab compared with more traditional primary efficacy end points.
Potential limitations of the study include strict inclusion and exclusion criteria and a short window for patient inclusion (within 12 hours following vasopressor therapy). These limitations result in a more homogenous study population, but they may make recruitment more difficult and limit the generalisability of the results.
In conclusion, despite the exponential increase of knowledge gathered in the last decades pertaining the pathophysiology of septic shock, this has not translated to effective therapeutic interventions and as a consequence, this condition remains to have an unacceptable high morbidity and mortality. The AdrenOSS-2 trial is one of the first personalised medicine trial in patients with septic shock, aimed at characterising the safety and efficacy of the ADM-binding antibody adrecizumab in patients with septic shock with elevated concentrations of bio-ADM. The trial incorporates a number of innovative features such as biomarker-guided patient selection and a novel efficacy end point in its design to avoid pitfalls of previous sepsis trials. Adrecizumab represents a promising approach to treat this lethal syndrome. The results of this proof-of-concept and dose-finding phase II trial are eagerly awaited, and will importantly aid the design of future trials with this drug.
Supplementary Material
Acknowledgments
The authors would like to thank the staff and patients participating in the study. The authors would also like to thank Joachim Struck for critically reviewing the manuscript.
Footnotes
Patient consent for publication: Not required.
CG and AB contributed equally.
Contributors: CG and AB drafted the manuscript. The manuscript was critically reviewed by MK, OH, PS, JZ, GM, P-FL, AM and PP. All authors read and approved the final manuscript for publication.
Funding: This work was supported by Adrenomed AG (the study sponsor; contact JZ, jzimmermann@adrenomed.com).
Competing interests: CG and AB received travel reimbursements from Adrenomed AG. MK declares to have no competing interests. OH, PS and JZ are employed by Adrenomed AG. GM received travel reimbursements and consultancy fees from Adrenomed AG. P-FL received travel reimbursements and consultancy fees from Adrenomed AG AM received travel reimbursements from Adrenomed AG. UMR-S 942 Inserm received a research grant from Adrenomed AG. PP received travel reimbursements and consultancy fees from Adrenomed AG. PP institution received a research grant from Adrenomed AG. Adrenomed AG reviewed this manuscript. Adrenomed AG holds patent rights on anti-ADM antibodies.
Ethics approval: This study is approved by relevant institutional review boards/independent ethics committees and is conducted in accordance with the ethical principles of the Declaration of Helsinki, the European Medicines Agency guidelines of Good Clinical Practice and all other applicable regulations.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Gaieski DF, Edwards JM, Kallan MJ, et al. . Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 2013;41:1167–74. 10.1097/CCM.0b013e31827c09f8 [DOI] [PubMed] [Google Scholar]
- 2. Fleischmann C, Scherag A, Adhikari NK, et al. . Assessment of global incidence and mortality of hospital-treated sepsis. current estimates and limitations. Am J Respir Crit Care Med 2016;193:259–72. 10.1164/rccm.201504-0781OC [DOI] [PubMed] [Google Scholar]
- 3. Vincent JL, Marshall JC, Namendys-Silva SA, et al. . Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med 2014;2:380–6. 10.1016/S2213-2600(14)70061-X [DOI] [PubMed] [Google Scholar]
- 4. Singer M, Deutschman CS, Seymour CW, et al. . The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016;315:801–10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ 2016;353:i1585 10.1136/bmj.i1585 [DOI] [PubMed] [Google Scholar]
- 6. Hippenstiel S, Witzenrath M, Schmeck B, et al. . Adrenomedullin reduces endothelial hyperpermeability. Circ Res 2002;91:618–25. 10.1161/01.RES.0000036603.61868.F9 [DOI] [PubMed] [Google Scholar]
- 7. Hocke AC, Temmesfeld-Wollbrueck B, Schmeck B, et al. . Perturbation of endothelial junction proteins by Staphylococcus aureus alpha-toxin: inhibition of endothelial gap formation by adrenomedullin. Histochem Cell Biol 2006;126:305–16. 10.1007/s00418-006-0174-5 [DOI] [PubMed] [Google Scholar]
- 8. Temmesfeld-Wollbrück B, Brell B, Dávid I, et al. . Adrenomedullin reduces vascular hyperpermeability and improves survival in rat septic shock. Intensive Care Med 2007;33:703–10. 10.1007/s00134-007-0561-y [DOI] [PubMed] [Google Scholar]
- 9. Müller-Redetzky HC, Will D, Hellwig K, et al. . Mechanical ventilation drives pneumococcal pneumonia into lung injury and sepsis in mice: protection by adrenomedullin. Crit Care 2014;18:R73 10.1186/cc13830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geven C, Kox M, Pickkers P. Adrenomedullin and adrenomedullin-targeted therapy as treatment strategies relevant for sepsis. Front Immunol 2018;9:292 10.3389/fimmu.2018.00292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meeran K, O’Shea D, Upton PD, et al. . Circulating adrenomedullin does not regulate systemic blood pressure but increases plasma prolactin after intravenous infusion in humans: a pharmacokinetic study. J Clin Endocrinol Metab 1997;82:95–100. 10.1210/jc.82.1.95 [DOI] [PubMed] [Google Scholar]
- 12. Lainchbury JG, Troughton RW, Lewis LK, et al. . Hemodynamic, hormonal, and renal effects of short-term adrenomedullin infusion in healthy volunteers. J Clin Endocrinol Metab 2000;85:1016–20. 10.1210/jc.85.3.1016 [DOI] [PubMed] [Google Scholar]
- 13. Troughton RW, Lewis LK, Yandle TG, et al. . Hemodynamic, hormone, and urinary effects of adrenomedullin infusion in essential hypertension. Hypertension 2000;36:588–93. 10.1161/01.HYP.36.4.588 [DOI] [PubMed] [Google Scholar]
- 14. Marino R, Struck J, Maisel AS, et al. . Plasma adrenomedullin is associated with short-term mortality and vasopressor requirement in patients admitted with sepsis. Crit Care 2014;18:R34 10.1186/cc13731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caironi P, Latini R, Struck J, et al. . Circulating biologically active adrenomedullin (bio-ADM) predicts hemodynamic support requirement and mortality during sepsis. Chest 2017;152:312–20. 10.1016/j.chest.2017.03.035 [DOI] [PubMed] [Google Scholar]
- 16. Nishio K, Akai Y, Murao Y, et al. . Increased plasma concentrations of adrenomedullin correlate with relaxation of vascular tone in patients with septic shock. Crit Care Med 1997;25:953–7. 10.1097/00003246-199706000-00010 [DOI] [PubMed] [Google Scholar]
- 17. Blet A, et al. . Bioactive adrenomedullin, organ support therapies and survival in the critically ill: Results from the FROG-ICU study, 2018. Submitted. [DOI] [PubMed] [Google Scholar]
- 18. Mebazaa A, Geven C, Hollinger A, et al. . Circulating adrenomedullin estimates survival and reversibility of organ failure in sepsis: the prospective observational multinational Adrenomedullin and Outcome in Sepsis and Septic Shock-1 (AdrenOSS-1) study. Crit Care 2018;22:354 10.1186/s13054-018-2243-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Struck J, Hein F, Karasch S, et al. . Epitope specificity of anti-adrenomedullin antibodies determines efficacy of mortality reduction in a cecal ligation and puncture mouse model. Intensive Care Med Exp 2013;1:3 10.1186/2197-425X-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wagner K, Wachter U, Vogt JA, et al. . Adrenomedullin binding improves catecholamine responsiveness and kidney function in resuscitated murine septic shock. Intensive Care Med Exp 2013;1:2 10.1186/2197-425X-1-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geven C, Peters E, Schroedter M, et al. . Effects of the humanized anti-adrenomedullin antibody adrecizumab (HAM8101) on vascular barrier function and survival in rodent models of systemic inflammation and sepsis. Shock 2018;50:648–54. 10.1097/SHK.0000000000001102 [DOI] [PubMed] [Google Scholar]
- 22. Geven C, Kox M, Scigalla P, et al. . Safety, tolerability and pharmacokinetics/-dynamics of the anti-adrenomedullin antibody Adrecizumab: a first in man study. Intensive Care Med Exp 2017;5. [Google Scholar]
- 23. Geven C, van Lier D, ten Elzen B, et al. . The safety, tolerability and pharmacokinetics/- dynamics of the selective anti-adrenomedullin antibody Adrecizumab during experimental human endotoxemia. Br J Clin Pharmacol 2018;26:30–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geven C, van Lier D, Blet A, et al. . Safety, tolerability and pharmacokinetics/pharmacodynamics of the adrenomedullin antibody adrecizumab in a first-in-human study and during experimental human endotoxaemia in healthy subjects. Br J Clin Pharmacol 2018;84:2129–41. 10.1111/bcp.13655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber J, Sachse J, Bergmann S, et al. . Sandwich immunoassay for bioactive plasma adrenomedullin. J of Applied Laboratory Medicine: An AACC Publication 2017;2:222–33. 10.1373/jalm.2017.023655 [DOI] [PubMed] [Google Scholar]
- 26. Heyland DK, Muscedere J, Drover J, et al. . Persistent organ dysfunction plus death: a novel, composite outcome measure for critical care trials. Critical Care 2011;15:R98 10.1186/cc10110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rhodes A, Evans LE, Alhazzani W, et al. . Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med 2017;43:304–77. 10.1007/s00134-017-4683-6 [DOI] [PubMed] [Google Scholar]
- 28. Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov 2014;13:741–58. 10.1038/nrd4368 [DOI] [PubMed] [Google Scholar]
- 29. Pickkers P, Kox M. Towards precision medicine for sepsis patients. Crit Care 2017;21:11 10.1186/s13054-016-1583-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kox M, Pickkers P. Adrenomedullin: its double-edged sword during sepsis slices yet again. Intensive Care Med Exp 2014;2:1–2. 10.1186/2197-425X-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brell B, Temmesfeld-Wollbrück B, Altzschner I, et al. . Adrenomedullin reduces staphylococcus aureus alpha-toxin-induced rat ileum microcirculatory damage. Crit Care Med 2005;33:819–26. 10.1097/01.CCM.0000159194.53695.7A [DOI] [PubMed] [Google Scholar]
- 32. Temmesfeld-Wollbrück B, Brell B, zu Dohna C, et al. . Adrenomedullin reduces intestinal epithelial permeability in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol 2009;297:G43–51. 10.1152/ajpgi.90532.2008 [DOI] [PubMed] [Google Scholar]
- 33. Itoh T, Obata H, Murakami S, et al. . Adrenomedullin ameliorates lipopolysaccharide-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol 2007;293:L446–52. 10.1152/ajplung.00412.2005 [DOI] [PubMed] [Google Scholar]
- 34. Müller HC, Witzenrath M, Tschernig T, et al. . Adrenomedullin attenuates ventilator-induced lung injury in mice. Thorax 2010;65:1077–84. 10.1136/thx.2010.135996 [DOI] [PubMed] [Google Scholar]
- 35. Geven C, Bergmann A, Kox M, et al. . Vascular effects of adrenomedullin and the anti-adrenomedullin antibody adrecizumab in sepsis. Shock 2018;50:132–40. 10.1097/SHK.0000000000001103 [DOI] [PubMed] [Google Scholar]
- 36. Stolk RF, van der Poll T, Angus DC, et al. . Potentially Inadvertent Immunomodulation: norepinephrine use in sepsis. Am J Respir Crit Care Med 2016;194:550–8. 10.1164/rccm.201604-0862CP [DOI] [PubMed] [Google Scholar]
- 37. Dünser MW, Hasibeder WR. Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med 2009;24:293–316. 10.1177/0885066609340519 [DOI] [PubMed] [Google Scholar]
- 38. Mebazaa A, Laterre PF, Russell JA, et al. . Designing phase 3 sepsis trials: application of learned experiences from critical care trials in acute heart failure. J Intensive Care 2016;4:24 10.1186/s40560-016-0151-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.