

Abstract
Background
Hair fibre length is an important economic trait of rabbits in fur production. However, molecular mechanisms regulating rabbit hair growth have remained elusive.
Results
Here we aimed to characterise the skin traits and gene expression profiles of short-hair and long-hair rabbits by histological and transcriptome analyses. Haematoxylin-eosin staining was performed to observe the histological structure of the skin of short-hair and long-hair rabbits. Compared to that in short-hair rabbits, a significantly longer anagen phase was observed in long-hair rabbits. In addition, by RNA sequencing, we identified 951 genes that were expressed at significantly different levels in the skin of short-hair and long-hair rabbits. Nine significantly differentially expressed genes were validated by quantitative real-time polymerase chain reaction. A gene ontology analysis revealed that epidermis development, hair follicle development, and lipid metabolic process were significantly enriched. Further, we identified potential functional genes regulating follicle development, lipid metabolic, and apoptosis as well as important pathways including extracellular matrix-receptor interaction and basal cell carcinoma pathway.
Conclusions
The present study provides transcriptome evidence for the differences in hair growth between short-hair and long-hair rabbits and reveals that lipid metabolism and apoptosis might constitute major factors contributing to hair length.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5503-x) contains supplementary material, which is available to authorized users.
Keywords: Skin, Hair fibre length, Histological analysis, Gene expression, RNA sequencing, Rabbit
Background
Rabbits are small mammals providing not only meat, but also hair. Hair is produced from follicles as skin appendages unique to mammals that are characterised by periodic regrowth [1, 2]. Rabbit hair is one of the most favourite natural fibres used in textile industries. The key traits contributing to the economic value of rabbit hairs include fibre diameter, density, and length, which are determined by both genetics and the environment [3–6]. The mechanisms regulating hair traits are complicated. Understanding the genetic principles of hair traits could help to elucidate the mechanism of hair development, thus, promoting rabbit breeding.
Hair fibre length is a critical economic trait in wool production from Angora rabbits, as it is closely associated with wool quality and yield. In addition, hair length influences textile quality, and also determines the system of manufacture. Different types of hairs in rabbits possess different growth rates, which vary with breed, nutrition, and season [4]. For Angora rabbits in the same environment conditions, gender, body site, and the month age after plucking are closely related to growth rates of rabbit hairs [4]. Hair follicles are the bases of hair growth. An intact hair coat is maintained by lifelong cycling of the hair follicles through periodic stages of growth (anagen), regression (catagen), and relative quiescence (telogen) [2, 7–9]. During the anagen phase, hair follicles grow rapidly by cell proliferation and reach their longest length. After regression by apoptosis in the catagen phase, they enter the telogen phase [10, 11]. Apoptosis is a critical event in the regulation of the hair cycle as anagen hairs normally grow for 4–7 years in humans before the cycle enters into the resting phases of catagen and telogen [12, 13]. During the process of hair follicles development, Wnt signalling plays a crucial role in hair follicle morphogenesis and regeneration [14, 15]. In addition, some signalling molecules, such as bone morphogenetic protein (BMP), sonic hedgehog (SHH), fibroblast growth factor (FGF), and transforming growth factor (TGF)-β, are involved in regulating the time of entry into the anagen phase of the hair follicle cycle [16–18]. Hair growth is also regulated by insulin-like growth factor 1 (IGF-1) affecting human follicular proliferation, hair growth cycle, and follicular differentiation [19]. FGF5, a member of the FGF family, is the leading candidate for hair length variation, and it is considered a regulator determining the duration of active hair growth (anagen) and the time of hair follicle regression (catagen) [20–22]. Recent studies have showed that VEGFR-2 and transcription factor NF-κB are involved in hair cycle regulation and NF-κB is indispensable for hair follicle stem cell activation, maintenance, and/or growth in mice [23, 24].
With the rapid development of molecular biological techniques, especially next-generation sequencing, large-scale gene expression detection has become possible. Rabbit genome sequencing has been used to study the polygenic basis of phenotypic variation during rabbit domestication [25] and differential gene expression in parthenogenetic and normal embryos cultured under the same conditions [26]. The landscape of long non-coding RNAs has also been analysed in Trichophyton mentagrophytes-induced dermatophytosis lesional skin and normal skin of rabbit by RNA sequencing (RNA-Seq) [27]. De novo transcriptome sequencing of Rex rabbit has been performed, and some novel genes have been found to play important roles during skin growth and development [28]. In the present study, to gain a better understanding of the growth rhythm of rabbit hair and molecular mechanisms regulating hair length in rabbits, a histological comparison of hair follicle phenotypes and transcriptome analysis were performed between short-hair and long-hair rabbits. Our results elucidate the molecular mechanism of hair growth and provide valuable information for future studies.
Results
Histological analysis of skin and follicle morphology of short-hair and long-hair rabbits
Complete hair follicle structure was observed in short-hair and long-hair rabbits at the fourth week of hair growth (Fig. 1a, b, g, h), which indicated that the hair follicles were in the anagen phase in short-hair and long-hair rabbits. Compared with that at the fourth week, no obvious changes were observed in long-hair rabbits at the sixth week (Fig. 1c, i). However, the hair follicles of short-hair rabbits appeared shrunk at the sixth week (Fig. 1d), and the area of primary and secondary hair follicles and thickness of inner root sheaths obviously decreased when compared with those at the fourth week (Fig. 1d, j). The hair follicle structure remained intact at the eighth week in long-hair rabbits (Fig. 1e, k), whereas, the hair follicles of short-hair rabbits significantly shrunk and finger-like papillae atrophied up to the arrectores pilorum, which indicated that the hair follicles of short-hair rabbits at the eighth week had entered the telogen phase, resulting in the cessation of hair growth (Fig. 1f, l). However, the hair follicles of long-hair rabbits were still in the anagen phase until at the eighth week, leading to continuous growth of hair.
Fig. 1.

The histological observation of skin tissue after plucking out hairs from long-hair and short-hair rabbits. a Transverse section of long-hair at the fourth week. b Transverse section of short-hair rabbit at the fourth week. c Transverse section of long-hair at the sixth week. d Transverse section of short-hair rabbit at the sixth week. e Transverse section of long-hair at the eighth week. f Transverse section of short-hair rabbit at the eighth week. g Longitudinal section of long-hair at the fourth week. h Longitudinal section of short-hair rabbit at the fourth week. i Longitudinal section of long-hair at the sixth week. j Longitudinal section of short-hair rabbit at the sixth week. k Longitudinal section of long-hair at the eighth week. l Longitudinal section of short-hair rabbit at the eighth week. Phf-Primary hair follicles, Shf-Secondary hair follicles, Bars = 200 μm
Transcriptome of rabbit skin by RNA sequencing
Six cDNA libraries of the long-hair rabbit groups (L1, L2, and L3) and short-hair rabbit groups (S1, S2, and S3) were constructed. After removing low-quality reads and adaptor sequences, more than 36 M clean reads were obtained from each sample, and Q30 was ≥91.74% (Table 1; Additional file 1: Figure S1). The map rates of unique reads from the sequenced skin RNA of long-hair and short-hair rabbits were > 60.45% (Table 2). After the standard analysis, 16,931 genes were detected from the six samples. Interestingly, the number of genes found among all the samples was similar with respect to the FPKM value (Additional file 2: Figure S2). The largest proportion of genes exhibited low (0 < FPKM < 10) and moderate (10 < FPKM < 100) expression, and a small fraction of the genes were expressed at high levels (FPKM > 100), further confirming the advantage of detecting low-abundance genes by high-throughput sequencing. In addition, 951 DEGs (FDR < 0.01, |log2Ratio| ≥ 1) were filtered between the groups comprising 539 genes expressed highly in the skin of long-hair rabbits and 412 genes expressed highly in the skin of short-hair rabbits (Fig. 2a; Additional file 3: Figure S3). A number of genes that were identified as being more highly expressed in long-hair rabbits were from the keratin (KRT) families, including the 10 genes (KRT23, KRT25, KRT26, KRT28, KRT34, KRT38, KRT39, KRT40, KRT7, and KRT84) (Table 3), which suggested that the 10 genes are important candidates regulating hair length in rabbits. In addition, a principal component analysis (PCA) was performed on DEGs, which showed that the short-hair and long-hair rabbits were clearly differentiated in the direction of the component 1 (Fig. 2b). Moreover, PCA identified several genes functioning in hair growth regulation, such as KRT25, S100A3, COL3α1, COL1α2, LOC100340949, LOC100338342, LOC100359226 (Fig. 2c).
Table 1.
Summary of RNA-seq data for each sample
| Samples | Clean reads | Clean bases | ≥ Q30 (%) |
|---|---|---|---|
| L1 | 47,547,114 | 13,997,256,514 | 91.76% |
| L2 | 36,468,426 | 10,495,999,462 | 92.39% |
| L3 | 57,393,736 | 16,932,383,874 | 92.02% |
| S1 | 46,632,332 | 13,639,996,064 | 93.44% |
| S2 | 46,105,321 | 13,567,869,214 | 92.01% |
| S3 | 49,671,512 | 14,615,589,684 | 91.74% |
Table 2.
Mapping statistics for each sample
| Samples | Clean reads | Mapped reads | Unique mapped reads | Multiple mapped reads |
|---|---|---|---|---|
| L1 | 95,094,228 | 62,621,046 (65.85%) | 59,482,256 (62.55%) | 3,138,790 (3.30%) |
| L2 | 72,936,852 | 46,536,580 (63.80%) | 44,088,800 (60.45%) | 2,447,780 (3.36%) |
| L3 | 114,787,472 | 77,804,317 (67.78%) | 73,059,536 (63.65%) | 4,744,781 (4.13%) |
| S1 | 93,264,664 | 69,770,638 (74.81%) | 67,443,108 (72.31%) | 2,327,530 (2.50%) |
| S2 | 92,210,642 | 60,012,231 (65.08%) | 57,464,532 (62.32%) | 2,547,699 (2.76%) |
| S3 | 99,343,024 | 65,785,338 (66.22%) | 62,960,030 (63.38%) | 2,825,308 (2.84%) |
Clean reads are single-end Reads
Fig. 2.
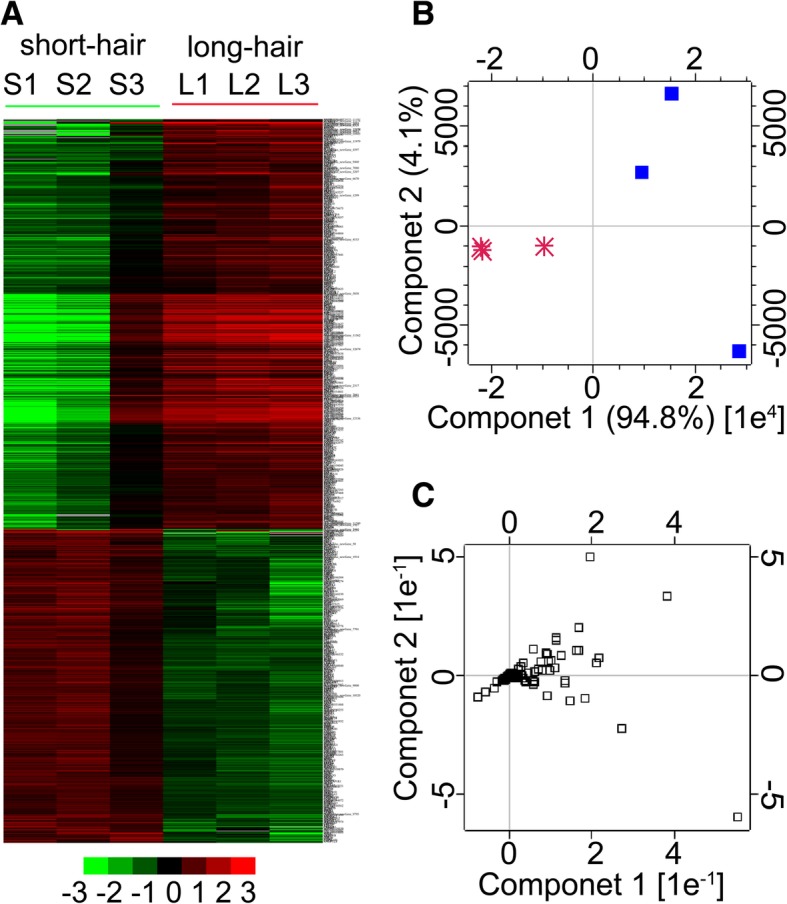
A heatmap and Principal Component Analysis of the DEGs between short-hair and long-hair rabbit. a Cluster analysis of DEGs. The red colour indicates increased expression and the green colour indicates decreased expression. S represents short-hair rabbits; L represents long-hair rabbits. b PCA of DEGs between short-hair and long-hair rabbit. The two rabbit types were clearly separated along the direction of component 1. The star represents long-hair rabbit; the solid square represents short-hair rabbit. c PCA of DEGs. The hollow box represents DEGs. The genes crucial for hair length regulation were identified by PCA
Table 3.
KRT genes from KRT family differentially expressed in the skin of short-hair and long-hair rabbits. The keratin filament (GO:0045095) and intermediate filament (GO:0005882) in the cellular component category were enriched by KRT7 and KRT34, respectively. The epithelial cell differentiation (GO:0030855) in the biological process category was enriched by KRT40
| Gene symbol | Type | log2FoldChange | FDR | GO term |
|---|---|---|---|---|
| KRT7 | II | 2.390336 | 4.68E-05 | keratin filament (GO:0045095) |
| KRT23 | I | 1.250867 | 0.005003 | NG |
| KRT25 | I | 2.695538 | 4.30E-05 | NG |
| KRT26 | I | 3.158961 | 2.08E-05 | NG |
| KRT28 | I | 2.377374 | 0.002598 | NG |
| KRT34 | I | 3.512258 | 8.31E-10 | intermediate filament (GO:0005882) |
| KRT38 | I | 3.924441 | 3.98E-07 | NG |
| KRT39 | I | 2.660676 | 1.92E-06 | NG |
| KRT40 | I | 2.310793 | 4.14E-05 | epithelial cell differentiation (GO:0030855) |
| KRT84 | II | 3.162653 | 1.44E-13 | NG |
Verification of DEGs with quantitative real-time PCR
To evaluate our DEG library, the relative expression levels of five DEGs from the KRT family (KRT25, KRT28, KRT39, KRT40, and KRT84) as well as FGF5 and ECM-related genes (COL3α1, TNXB, and VWF) were analysed by q-PCR. As shown in Fig. 3 and Additional file 4: Figure S4, the results of RNA-Seq and q-PCR were similar. In general, the results of q-PCR validated the RNA-Seq results and demonstrated the reliability of our data. Furthermore, we also found that the expression profiles of the selected genes at the eighth week after plucking (Additional file 5: Figure S5) were similar to those at the sixth week.
Fig. 3.

A q-PCR analysis of nine DEGs in the skin of short-hair and long-hair rabbits. a KRT25 b KRT28 c KRT39 d KRT40 e KRT84 f FGF5 g COL3α1 h TNXB i VWF. The skin tissues were from short-hair and long-hair rabbits at the sixth week after plucking. GAPDH was used as a reference gene to normalize q-PCR data. Bars represent the standard error. ** P < 0.01, * P < 0.05
GO and KEGG analyses for DEGs between short-hair and long-hair rabbits
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were used for the functional analysis and signalling pathway annotations of these DEGs. The GO terms with the Q value ≤0.05 were considered to be significantly enriched. Several GO terms related to hair follicle development and lipid metabolism in the biological process category were found (Fig. 4, Additional file 6: Table S1). A comparison between short-hair and long-hair rabbits showed some DEGs involved in the biological processes including epidermis development (9 genes, Q = 0.00427), molting cycle (4 genes, Q = 0.01352), hair cycle (4 genes, Q = 0.01352), lipid biosynthetic process (8 genes, Q = 0.01829), hair follicle development (4 genes, Q = 0.02563), and so on (Additional file 6: Table S1). The GO analysis demonstrated large differences in hair follicle development and lipid metabolism between short-hair and long-hair rabbits. In addition, six important pathways related to the regulation of hair follicle development were identified (Table 4). Among them, the extracellular matrix (ECM)-receptor interaction and basal cell carcinoma pathway were significantly enriched between short-hair and long-hair rabbits (P < 0.05). Therefore, we focused on these two pathways for further characterisation. The DEGs enriched in the both pathways are listed in Table 4. In the ECM-receptor interaction, the expression of 15 DEGs, including the collagen genes (such as COL1A2, COL3A1, COL5A2, and COL5A3), laminin genes (such as LAMA4 and LAMC3), integrin genes (such as ITGB3 and LOC103346304), and tenascin genes (such as TNN and TNXB) was down-regulated in long-hair rabbits (Table 4). In the basal cell carcinoma pathway, 11 up-regulated genes, including GLI1, PTCH1, and SHH, were identified (Table 4).
Fig. 4.

The significantly enriched GO terms of DEGs between short-hair and long-hair rabbits (P < 0.05). The hierarchical category of the GO terms is biological process. The x-axis represents the number of DEGs enriched in biological processes; y-axis represents the GO terms enriched by DEGs
Table 4.
The DEGs enriched in the pathways related to skin development between short-hair and long-hair rabbits
| Pathway | DEGs | |
|---|---|---|
| Up | Down | |
| ECM-receptor interaction | CHAD, COL1α2, COL3α1, COL5α2, COL5α3, FN1, ITGB3, LAMA4, LAMC3, LOC103346304, SPP1, THBS2, TNN, TNXB, VWF | |
| Basal cell carcinoma | FZD5, GLI1, HHIP, LEF1, LOC100355594, PTCH1, PTCH2, SHH, TCF7, WNT11, WNT5A | |
| Hedgehog signalling pathway | GLI1, HHIP, LOC100341681, PTCH1, PTCH2, SHH, WNT11, WNT5A | GAS1, |
| TGF-beta signalling pathway | BAMBI, BMP7, LOC100008826, PPP2R1B, LOC100341360, LOC103347033, SMAD6, SMAD7, | SMAD9, TGFBR2, TGFB3, INHBA, |
| Wnt signalling pathway | BAMBI, FZD5, LEF1, LOC100341681, LOC100355594, TCF7, WNT11, WNT5A | DAAM2, DKK2, |
| Notch signalling pathway | DTX3L, DTX4 | |
Alternative splicing and single nucleotide polymorphisms analyses
In the present study, we detected various splicing events, including alternative 5′ first exon (TSS), alternative 3′ last exon (TTS), alternative exon ends (AE), intron retention (IR), and skipped exon (SKIP) (Additional file 7: Figure S6). The number of alternative splicing events and types was similar between the long-hair and short-hair rabbits. The TSS and TTS, which occur when two or more splice sites at 5′ first or 3′ last exon are present, are the most frequently observed events, accounting for approximately 41 and 31% of alternative splicing events in rabbits, respectively. SKIP is the next most common event in AS accounting for approximately 17% of alternative splicing events. IR, whereby an intron is retained in the resulting mature mRNA, is barely detected, accounting for approximately 1.6% of the AS events. These results showed that the number of each AS event was similar between the short-hair and long-hair rabbits.
In addition, we compared the sequence data with known reference sequences using the TopHat2 software, and then identified potential SNPs. A total of 726,852 and 852,149 SNPs were detected in long-hair and short-hair rabbits, respectively (Additional file 8: Table S2). Compared with that in the long-hair rabbits, more SNPs were identified in short-hair rabbits, demonstrating the differences in SNPs during skin development in short-hair and long-hair rabbits. Approximately 75% SNP sites belonged to the transition type and 25% SNP sites were transversion type. The proportion of heterozygosity SNPs was different in each sample, accounting for 30.90–34.98%. Meanwhile, more SNP sites were found in genic regions in each sample. As presented in Additional file 9: Figure S7, the proportion of SNPs in the intron region was the highest.
Discussion
Large phenotypic differences exist with respect to hair length between short-hair and long-hair rabbits. The cycle of hair follicles comprises three distinct phases—growth, regression, and resting phases [2, 8, 9]. These stages can be distinguished according to hair follicle morphology. In our previous studies, we reconstructed the follicle cycle of rabbit hair growth by plucking hairs and analysed the hair growth rhythm of short-hair and long-hair rabbits [29, 30]. The results showed a linear upward trend from the fourth to eleventh week in different types of hair from long-hair rabbits, and the hair of short-hair rabbits exhibited rapid growth before the sixth week and remained stable after 6 weeks and suggested different hair growth rates and hair cycles between the two populations. In the present study, we determined the follicle morphology of short-hair and long-hair rabbits at week four, six, and eight after plucking. The results demonstrated that the hair follicles were still in the anagen phase until the eighth week. The regression phase of hair follicle development in long-hair rabbits needs further studies; the hair follicles in short-hair rabbits began to degenerate from the fourth to sixth week and entered the resting phase from the sixth to eighth week. Hair growth is fuelled by bulge stem cells, which are activated at the start of the anagen phase by the dermal papilla [31]. In the present study, long growth phase of hair follicles in long-hair rabbits continuously promoted hair growth. During the catagen phase, hair follicle stem cells in the outer root sheath and the germinal matrix undergo apoptosis, and the lower two-third of hair follicles regress [9]. In the telogen phase, hair follicle stem cells are relatively quiescent, inhibiting hair growth [32]. These findings were consistent with our results, which showed that hair follicles at the sixth week were in the catagen phase and entered the telogen phase when they were at the eighth week after plucking in short-hair rabbits, resulting in the cessation of hair growth.
The present study also provides an opportunity to compare the transcriptome of skin tissue between short-hair and long-hair rabbits by high-throughput sequencing to analyse the molecular mechanism associated with differences in hair growth. The transcriptome data obtained are useful to elucidate the different mechanisms of hair growth in the skin of long-hair and short-hair rabbits. It has been reported that > 18 M reads from RNA-Seq are required for each sample to attain a saturated state for expression analysis [32, 33]. More than 36 M clean reads were obtained from each sample in the present study, and the quality control assay of our data revealed that the RNA-Seq data were well qualified (Table 1, Additional file 1: Figure S1). The analyses of heatmaps and PCA of DEGs revealed a larger number of significantly DEGs in the skin tissue of short-hair and long-hair rabbits at the sixth week after plucking hairs. These genes might play crucial roles in regulating hair follicle development and hair growth, and their differential expression might be the reason for difference in hair length between short-hair and long-hair rabbits. Ten genes encoding the structural proteins of wool were detected (Table 3). Keratin intermediate filament (KRT-IF) and keratin-associated protein genes encode the majority of wool and hair proteins [34, 35]. The wool follicle-related genes KRT34, KRT38, and KRT39 were expressed only in the cortex, and KRT40 and KRT84 were expressed in the fibre cuticle [34]. Ovine KRT25-KRT28, which encode type I KRTs in the inner root sheath, have also been described [34]. The primary function of KRTs is to protect epithelial cells from apoptosis and mechanical or nonmechanical stresses that can lead to cell death [36, 37]. However, in the present study, a few genes encoding keratin-associated protein were differentially expressed in the two populations. Interestingly, the expression of 10 genes of the KRT family was up-regulated in the skin tissue of long-hair rabbit, suggesting that the KRT genes might contribute to the continuous activity of hair follicle in the long-hair rabbit. We postulated that the 10 KRT genes might also be useful in promoting hair growth. In earlier reports, FGF5 has been identified as an inhibitor of hair growth in various mammals. FGF5 induced regression of the human hair follicle [38] and knock-out and disruption of FGF5 using the CRISPR/Cas9 system in goat resulted in longer fibres [39, 40]. However, there is a recent report on rabbits indicating that FGF5 was also up-regulated in the long wool group (Wan line Angora rabbit) and down-regulated in the short wool group (Chinchilla Rex rabbit and White Rex rabbit) [41], which was consistent with our findings of RNA-seq and q-PCR, showing that the FGF5 gene was up-regulated in the skin of long-hair rabbit. Previous studies also showed that the polypeptide of FGF5s translated from an alternatively spliced variant of FGF5 mRNA suppressed the activity of FGF5, increasing hair growth [42, 43]. We hypothesized that FGF5s of FGF5 mRNA suppressed the activity of FGF5 and promoted hair growth in the rabbit. Further studies are required to investigate the roles of alternatively spliced variants of the FGF5 genes in hair length. In addition, the nine genes were also selected for validation by q-PCR to evaluate our DEG library. All the selected genes showed similar expression patterns between RNA-Seq and q-PCR, demonstrating the reliability of our data. An additional expression analyses of the selected genes showed that their expression profiles at the eighth week after plucking were similar to those at the sixth week. The hair follicles of short-hair and long-hair rabbits at the eighth week were in the telogen and the anagen phase, respectively. The finding further demonstrated that the genes played important roles in regulating hair follicle development and hair growth.
The GO analysis showed that a large proportion of DEGs were significantly enriched in the biological processes such as epidermis development, epithelial cell differentiation, hair follicle development, and lipid metabolism process. The results demonstrated that the DEGs are potential regulators of hair follicle development and lipid metabolism in rabbits.
In addition, a number of DEGs were also found to be significantly enriched in extracellular matrix (ECM)-receptor interaction and basal cell carcinoma pathway. The extracellular matrix in the ECM-receptor interaction is a complex mixture of structural and functional macromolecules, including fibrous proteins (collagen, fibronectin, and laminin) [44, 45]. Different collagens form different ECM components, for example, collagens 1, 3, and 5 form the fibrils, collagen 6 forms the microfibril, and collagen 4 forms the basal membrane [46]. The amount of ECM per cell contributes to the volume of the dermal papilla [47]. The previous study showed that many of the ECM-related genes, including collagen, laminin, and fibronectin were differentially expressed between primary and secondary hair follicle dermal papilla cells, resulting in an enlarged dermal papilla [48]. Dermal papillas control the number of matrix cells and, thus, the character and size of the hair follicle and its shaft [49]. In the present study, the differential expression of COL1α2, COL3α1, COL5α2, and COL5α3 suggested a relation to the size difference of the hair follicle and its shaft between the two populations and the PCA further showed that COL1α2 and COL3α1 were important regulators of hair follicle development. The differential expression of other ECM-related genes, such as ITGB3, LAMA4, LAMC3, VWF, and TNXB, also shows the difference in the ECM structure between short-hair and long-hair rabbits. The results of the present study also showed that the expression of GLI1, SHH, and PTCH1 involved in the basal cell carcinoma pathway was up-regulated in short-hair rabbits. It has been reported that the inhibition of GLI1 induces cell-cycle arrest and enhances apoptosis in brain glioma cell lines [50]. SHH and PTCH1 are also key components of the Hedgehog pathway, which primarily regulates the genes involved in cell growth, proliferation, survival, and apoptosis [51]. Hair follicle genes were also found to be involved in cell differentiation, proliferation, apoptosis, and growth in Hu sheep lambskin [52]. Therefore, the differential expression of GLI1, SHH, and PTCH1 suggests the difference in hair follicle cycle and apoptosis in the two populations, and their roles in the regulation of hair follicle cycle and apoptosis in rabbits. WNT signals are required for the initiation of hair follicle development [14]. It has also been reported that LEF-1 is involved in controlling the development of whisker follicles [53], and WNT5a inhibits the growth of hair follicles in mice [54]. It has been reported that Wnt, Shh, TGF-b and Notch signalling pathways may be involved in hair follicle development and cycling in cashmere goats [55]. The results of the present study showed that LEF-1, WNT5a, and TCF7 involved in the WNT signalling pathway were significantly up-regulated in long-hair rabbits. Further, the Notch signalling pathway regulates hair cell differentiation in the cochlea of mammals [56] and mediates hair cell regeneration partially depending on Wnt signalling [57]. In the present study, we found the upregulation of BAMBI and SMAD7 in the TGF-β signalling pathway in long-hair rabbits; they are the key regulators of hair cycle [7]. Overall, the candidate genes identified in the present study might therefore serve as potential regulators of hair length in rabbits.
Alternative splicing and SNPs can increase protein complexity and cause diversity in biological traits. Alternative splicing is one of the major mechanisms contributing to protein diversity [58]. Although AS is widespread in eukaryotes, we tend to underestimate its proportion. The changes in AS might therefore represent a major source of species-specific differences [59–61]. In this study, we detected five primary alternatively spliced events and analysed the number of each AS event (Additional file 7: Figure S6). The results showed that the number of TSS and TTS events was the highest and the IR event was the least in rabbits. Most genomic variations are attributable to the SNPs, and therefore, offer the highest resolution to track disease-related genes and population history [62, 63]. A total of 231,653–317,941 SNPs were found to specifically match the genome of Oryctolagus cuniculus in six rabbit samples. Compared with the intergenic SNPs, more SNPs were found in genic regions where most SNPs are located in the intron regions. The SNPs in or near genes explain more variations than the SNPs between genes for complex traits [64, 65]. These results imply that the diversity in regulation of complex traits, which can be by either genic regions or nearby gene regions, and even by intergenic region. The specific roles of AS and SNPs in regulating hair length of short-hair and long-hair rabbits need further study.
Conclusions
In conclusion, we identified differences in the histology and transcriptome profile of skin between short-hair and long-hair rabbit. The histological analysis showed different hair cycles and growth rate between the two populations. The differences in the transcriptome profile of the two populations were consistent with the differences observed in morphological traits. RNA-Seq enabled us to obtain candidate genes involved in hair follicle development, lipid metabolism, and apoptosis. The expression of differentially expressed lipid metabolic and apoptotic genes in the skin of rabbits can be used as a novel trait with the potential to alter hair length. Further studies are required to investigate the roles of candidate genes in hair length to improve rabbit breeding programmes.
Methods
Animals
All rabbits were procured from the rabbit farm in the Animal Husbandry Institute of Anhui Academy of Agriculture Sciences, Hefei, Anhui, China. The rabbits were raised and managed under the same condition, feeding to appetite; drinking water was supplied via an automatic drinking bowl. A cross between the Wan Strain Angora rabbit and Rex rabbit was carried out to produce the F1 generation (Fig. 5). The F1 generation was then backcrossed (BC) with the Wan Strain Angora rabbit, which produced F2 rabbits. Two phenotypic traits segregated in the families; two hair length characters (586 short-hair and 613 long-hair rabbits). The segregation ratio of hair length in F2 generation was consistent with Mendel heredity principles (1:1, χ2 = 0.564, P = 0.436). Normal rabbit fur is composed of three different types of hairs including coarse hairs, awn hairs, and fine hairs [66, 67]; the length of the different hair types was measured at the tenth week after plucking (Additional file 10: Figure S8). Finally, three female short-hair and long-hair rabbits were selected for the present study, respectively.
Fig. 5.

Hair length trait segregation in the family. Two phenotypic traits segregate in the F2 population: long-hair, with recessive-type allele and short-hair, with wild-type and mutated alleles. All the images of rabbits are our own and photographed by our researchers
Sample collection, preparation, and histological examination
Rabbits were given anesthesia through an ear vein injection of 0.7% pentobarbital sodium (6 ml/kg) before sampling. Skin tissue samples (1 cm2) from the back of each rabbit were collected at week four, six and eight after plucking hairs for histological analysis. The skin samples were fixed with 4% formaldehyde (40% formaldehyde solution and distilled water mixed at a 1:9 ratio) solution. Cross sections of the fixed samples were washed with running water, dehydrated using an ethyl alcohol series, cleaned in xylene, and embedded in paraffin wax. The specimens were sectioned to a thickness of 4 μm using a Leica RM2235 microtome (Leica, Wetzlar, Germany). Transverse and vertical cross-sections of the fixed and paraffin-embedded samples were stained with HE, examined and photographed using an Olympus BX51 biomicroscope (Olympus Optical Company, Tokyo, Japan).
cDNA library construction and sequencing
Under anesthesia, skin samples from the back were collected at the sixth week after plucking the hair, when the wool cycle of long-hair rabbit is expected to be in the anagen phase and that of short-hair rabbit is expected to be in catagen phase, frozen in liquid nitrogen immediately, and stored at − 80 °C prior to RNA extraction. The total RNA was extracted from the skin samples of long-hair rabbits (designated as L1, L2, and L3) and short-hair rabbits (designated as S1, S2, and S3) using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The samples were sent to the Beijing Biomarker Technologies Company. The quality of RNA was evaluated using the RNA integrity number value using the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). The mRNA was purified, fragmented, and converted to cDNA, adapters were ligated to the end of double-stranded cDNA, and libraries were created by polymerase chain reaction (PCR) using the Illumina TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA) according to manufacturer’s protocols. The libraries constructed were quantified with Qubit2.0 and the insert size library was determined using Agilent 2100. Six independent paired-end libraries were sequenced on an Illumina HiSeq 2500 (Illumina, San Diego, CA, USA) system according to the manufacturer’s instructions. Above 10.50 Gb clean reads per sample were generated for the genome-wide transcriptomic analyses.
Mapping and assembling
The trimmed reads were assembled and mapped to the reference genome (https://www.ncbi.nlm.nih.gov/genome/?term=Rabbit) by performing alignments using the TopHat2 [68] software. The AS and SNPs were analysed using the Cufflinks [69] and GATK [70] software, respectively.
Differential gene expression analysis
The values of fragments per kilobase of transcript per million fragments mapped (FPKM) [71] were generated and used to identify the expression level of genes in each rabbit sample. Differential expression analysis was performed with DESeq, an R package [72]. The P-value corresponds to a differential gene expression test in which the false discovery rate (FDR) was used to determine the threshold of P-value in multiple tests. We filtered the differentially expressed genes (DEGs) with the standards (Fold Change ≥2 and FDR < 0.01). The gene ontology (GO) analysis was employed to analyse the functions of the DEGs. All the DEGs were mapped to the GO terms in the database (http://www.geneontology.org/). The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to perform pathway enrichment analysis of the DEGs. Cluster 3.0 [73] was used for the clustering analysis.
Quantitative real-time PCR
To verify the RNA-Seq results, a set of six candidate genes were selected from the list of DEGs and the relative expression level of each gene was evaluated by quantitative real-time PCR (q-PCR). The PCRs were performed for each individual using 1.0 μg of residual RNA from the original extraction described above. The first-strand cDNA was synthesised using EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen, Beijing, China). The q-PCR was carried out in a LightCycler 96 (Roche, Basel, Switzerland) using TransStart Green qPCR SuperMix (Transgen, Beijing, China). The following conditions were used for amplification: 94 °C for 30 s, followed by 40 cycles of 94 °C for 5 s, 62 °C for 35 s, and a melt curve stage of 97 °C for 10 s, 65 °C for 1 min, and 97 °C for 1 s. The primers for the q-PCR are listed in Additional file 11: Table S3. The GAPDH gene was used as a constitutive expression control in these experiments. The relative expression level of each gene was estimated by the 2-ΔΔCT method [74]. The q-PCR analysis was performed in triplicates for each sample.
Statistical analyses
Excel 2010 (Microsoft) was used to analyse the data. The data are presented as mean ± standard deviation (SD). Student’s t-test was used for statistical comparisons. The results with a P value < 0.05 were considered to be statistically significant.
Additional files
Figure S1. Quality control of RNA-seq data. A L1 B L2 C L3 D S1 E S2 F S3. (PDF 236 kb)
Figure S2. The numbers of annotated genes with different expression levels against the range of FPKM values. (PDF 36 kb)
Figure S3. List of up-regulated and down-regulated genes in the comparison of short-hair and long-hair rabbits. (PDF 118 kb)
Figure S4. The results of RNA-seq between long-hair and short-hair rabbits. A KRT25 B KRT28 C KRT39 D KRT40 E KRT84 F FGF5 G COL3α1 H TNXB I VWF. (PDF 154 kb)
Figure S5. A q-PCR analysis of the relative expression levels of nine DEGs in the skin of short-hair and long-hair rabbits at the eighth week after plucking by q-PCR. A KRT25 B KRT28 C KRT39 D KRT40 E KRT84 F FGF5 G COL3α1 H TNXB I VWF. (PDF 175 kb)
Table S1. Significantly enriched GO terms in the biological process category of DEGs between short-hair and long-hair rabbits (P < 0.05). (PDF 13 kb)
Figure S6. The number of alternative splicing events of six samples. A L1 B L2 C L3 D S1 E S2 F S3. (PDF 144 kb)
Table S2. Analyses of SNP sites of six samples. (PDF 12 kb)
Figure S7. Analyses of SNPs annotation. A L1 B L2 C L3 D S1 E S2 F S3. (PDF 355 kb)
Figure S8. Hair length of the short-hair and long-hair rabbits at the tenth week after plucking. Fibres are divided into three categories, including coarse, fine, and awn fibres. (PDF 107 kb)
Table S3. Primers for qPCR. F1, forward primer. R2, reverse primer. (PDF 95 kb)
Acknowledgments
Not Applicable
Funding
This work was supported by the earmarked fund for Modern Agro-industry Technology Research System from the Ministry of Agriculture of P.R. China (Grant No.: CARS-43-A-4) and Anhui Provincial Natural Science Foundation (Grant No.: 1708085MC62).
Availability of data and materials
The data was presented in the manuscript and the supporting materials. The raw reads data was submitted to the Short Read Archive (SRA) under the accession number SRP173735 and BioProject accession number PRJNA509998 (https://www.ncbi.nlm.nih.gov/sra/PRJNA509998).
Abbreviations
- AS
Alternative splicing
- DEGs
Differentially expressed genes
- ECM
Extracellular matrix
- FDR
False discovery rate
- FPKM
Fragments per kilobase of transcript per million fragments mapped
- GO
Gene ontology
- HE
Haematoxylin-eosin
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KRT
Keratin
- PCA
Principal component analysis
- q-PCR
Quantitative real-time PCR
- RNA-Seq
RNA sequencing
- SD
Standard deviation
- SNP
Single nucleotide polymorphisms
Authors’ contributions
HLZ and DWH conceived the study. HLZ, DWH, XFW and GLC performed sample collection and total RNA preparation. XWZ and YXQ performed the q-PCR validation. DWH and HSD conducted the data analysis and prepared figures and tables. HSD, DWH and YXY wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was carried out in strict accordance with relevant guidelines and regulations by the Ministry of Agriculture of the People’s Republic of China. All experimental protocols were approved by the Ethics Committee of Anhui Academy of Agricultural Sciences.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Haisheng Ding, Email: dinghs123@163.com.
Huiling Zhao, Email: zhl11988@163.com.
Guanglong Cheng, Email: cgl0126@126.com.
Yongxin Yang, Email: yyongxin@yahoo.com.
Xiaofei Wang, Email: 274923128@qq.com.
Xiaowei Zhao, Email: xiaowei1986mm@163.com.
Yunxia Qi, Email: qiyx09@163.com.
Dongwei Huang, Email: hdwscience@163.com.
References
- 1.Nagorcka B, Dollin A, Hollis D, Beaton C. A technique to quantify and characterize the density of fibres and follicles in the skin of sheep. Aust J Agric Res. 1995;46(8):1525–1534. [Google Scholar]
- 2.Stenn KS, Paus R. Controls of hair follicle cycling. Physiol Rev. 2001;81(1):449. doi: 10.1152/physrev.2001.81.1.449. [DOI] [PubMed] [Google Scholar]
- 3.Rougeot J, Thebault RG. Variations saisonnières de la composition et de la structure du pelage: exemple de la toison du lapin angora. Ann Zootech. 1983;32(3):287–314. [Google Scholar]
- 4.Lanszki J, Thébault RG, Allain D, Szendro Z, Eiben C, et al. The effects of melatonin treatment on wool production and hair follicle cycle in angora rabbits. Anim Res. 2001;50(1):79–89. [Google Scholar]
- 5.Rafat SA, Allain D, De Rochambeau H. Genetic description of a divergent selection experiment in angora rabbits with overlapping generations. J Anim Breed Genet. 2015;126(3):189–197. doi: 10.1111/j.1439-0388.2008.00769.x. [DOI] [PubMed] [Google Scholar]
- 6.Rahman SU, Wang X, Yu L. Observations on biotic parameters of angora rabbit breed under controlled conditions in different housing systems. Vet World. 2018;11(1):88–92. doi: 10.14202/vetworld.2018.88-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunner M, Jagannathan V, Waluk DP, Roosje P, Linek M, Panakova L, et al. Novel insights into the pathways regulating the canine hair cycle and their deregulation in alopecia X. PLoS One. 2017;12(10):e0186469. doi: 10.1371/journal.pone.0186469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider MR, SchmidtUllrich, Paus R. The hair follicle as a dynamic Miniorgan. Curr Biol. 2009;19(3):132–142. doi: 10.1016/j.cub.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Tamura Y, Takata K, Eguchi A, Kataoka Y. In vivo monitoring of hair cycle stages via bioluminescence imaging of hair follicle NG2 cells. Sci Rep. 2018;8(1):393. doi: 10.1038/s41598-017-18763-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller-Röver S, Handjiski B, van der Veen C, Eichmüller S, Foitzik K, McKay IA, et al. A comprehensive guide for the accurate classification of murine hair follicles in distinct hair cycle stages. J Invest Dermatol. 2001;117(1):3–15. doi: 10.1046/j.0022-202x.2001.01377.x. [DOI] [PubMed] [Google Scholar]
- 11.Ishihara T, Yamashita H, Sakurai T, Morita J, Sakamoto K, Ishii A, et al. Morphological analysis of patchy thickening and reddish discoloration of active hair growth areas in the skin of New Zealand white rabbits. J Toxicol Pathol. 2017;30(4):315–322. doi: 10.1293/tox.2017-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawaya ME, Blume-Peytavi U, Mullins DL, Nusbaum BP, Whiting D, Nicholson DW, et al. Effects of finasteride on apoptosis and regulation of the human hair cycle. J Cutan Med Surg. 2002;6(1):1–9. doi: 10.1007/s10227-001-0024-y. [DOI] [PubMed] [Google Scholar]
- 13.Sawaya ME. Regulation of the human hair cycle. Curr Probl Dermatol. 2001;13(3):206–210. [Google Scholar]
- 14.Andl T, Reddy ST, Gaddapara T, Millar SE. WNT signals are required for the initiation of hair follicle development. Dev Cell. 2002;2(5):643–653. doi: 10.1016/s1534-5807(02)00167-3. [DOI] [PubMed] [Google Scholar]
- 15.Li YH, Zhang K, Yang K, Ye JX, Xing YZ, Guo HY, et al. Adenovirus-mediated Wnt10b overexpression induces hair follicle regeneration. J Invest Dermatol. 2013;133(1):42–48. doi: 10.1038/jid.2012.235. [DOI] [PubMed] [Google Scholar]
- 16.Sato N, Leopold PL, Crystal RG. Induction of the hair growth phase in postnatal mice by localized transient expression of sonic hedgehog. J Clin Invest. 1999;104(7):855–864. doi: 10.1172/JCI7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100(4):423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- 18.Paladini RD, Saleh J, Qian C, Xu GX, Rubin LL. Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J Invest Dermatol. 2005;125(4):638–646. doi: 10.1111/j.0022-202X.2005.23867.x. [DOI] [PubMed] [Google Scholar]
- 19.Trüeb RM. Further clinical evidence for the effect of IGF-1 on hair growth and alopecia. Skin Appendage Disord. 2018;4(2):90–95. doi: 10.1159/000479333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenquist TA, Martin GR. Fibroblast growth factor signalling in the hair growth cycle: expression of the fibroblast growth factor receptor and ligand genes in the murine hair follicle. Dev Dyn. 2010;205(4):379–386. doi: 10.1002/(SICI)1097-0177(199604)205:4<379::AID-AJA2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 21.Cho YM, Woodard GL, Dunbar M, Gocken T, Jimenez JA, Foley J. Hair-cycle-dependent expression of parathyroid hormone-related protein and its type I receptor: evidence for regulation at the anagen to catagen transition. J Invest Dermatol. 2003;120(5):715–727. doi: 10.1046/j.1523-1747.2003.12147.x. [DOI] [PubMed] [Google Scholar]
- 22.Pethö-Schramm A, Müller HJ, Paus R. FGF5 and the murine hair cycle. Arch Dermatol Res. 1996;288(5–6):264. doi: 10.1007/BF02530098. [DOI] [PubMed] [Google Scholar]
- 23.Wu X, Jing J, Lu Z, Zheng M. Expression and localization of VEGFR-2 in hair follicles during induced hair growth in mice. Arch Dermatol Res. 2018;310(7):591–598. doi: 10.1007/s00403-018-1843-7. [DOI] [PubMed] [Google Scholar]
- 24.Krieger K, Millar SE, Mikuda N, Krahn I, Kloepper JE, Bertolini M, Scheidereit C, Paus R, Schmidt-Ullrich R. NF-κB participates in mouse hair cycle control and plays distinct roles in the various pelage hair follicle types. J Invest Dermatol. 2017;138(2):256–264. doi: 10.1016/j.jid.2017.08.042. [DOI] [PubMed] [Google Scholar]
- 25.Carneiro M, Rubin C-J, Di Palma F, Albert FW, Alföldi J, Barrio AM, et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science. 2014;345(6200):1074–1079. doi: 10.1126/science.1253714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naturil-Alfonso C, Saenz-De-Juano MD, Peñaranda DS, Vicente JS, Marco-Jiménez F. Transcriptome profiling of rabbit parthenogenetic blastocysts developed under in vivo conditions. PLoS One. 2012;7(12):e51271. doi: 10.1371/journal.pone.0051271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao W, Hu Y, Tong Y, Cai M, He H, Liu B. Landscape of long non-coding RNAs in Trichophyton mentagrophytes-induced rabbit dermatophytosis lesional skin and normal skin. Funct Integr Genomic. 2018;18(4):1–10. doi: 10.1007/s10142-018-0601-4. [DOI] [PubMed] [Google Scholar]
- 28.Pan L, Liu Y, Wei Q, Xiao C, Ji Q, Bao G. Solexa-sequencing based transcriptome study of plaice skin phenotype in rex rabbits (Oryctolagus cuniculus) PLoS One. 2015;10(5):e0124583. doi: 10.1371/journal.pone.0124583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu Q, Huang D, Cheng G, Zhao X, Yang Y, Zhao H, et al. Study on hair growth regularity of short-hair rabbit. China Herbivore Sci. 2017;37(3):12–15. [Google Scholar]
- 30.Huang D, Zhao H, Cheng G, Zhao X, Yang Y, Wang X, et al. Study on hair growth regularity of angora rabbit. China Herbivore Sci. 2016;36(5):12–14. [Google Scholar]
- 31.Hsu YC, Pasolli HA, Fuchs E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell. 2011;144(1):92–105. doi: 10.1016/j.cell.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8(6):469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 33.Sang YM, Brichalli W, Rowland RRR, Blecha F. Genome-wide analysis of antiviral signature genes in porcine macrophages at different activation statuses. PLoS One. 2014;9(2):e87613. doi: 10.1371/journal.pone.0087613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu Z, Wildermoth JE, Wallace OA, Gordon SW, Maqbool NJ, Maclean PH, et al. Annotation of sheep keratin intermediate filament genes and their patterns of expression. Exp Dermatol. 2011;20(7):582–588. doi: 10.1111/j.1600-0625.2011.01274.x. [DOI] [PubMed] [Google Scholar]
- 35.Yu Z, Gordon SW, Nixon AJ, Bawden CS, Rogers MA, Wildermoth JE, et al. Expression patterns of keratin intermediate filament and keratin associated protein genes in wool follicles. Differentiation. 2009;77(3):307–316. doi: 10.1016/j.diff.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Sulayman A, Tursun M, Sulaiman Y, Huang X, Tian K, Tian Y, et al. Association analysis of polymorphisms in six KRT genes with wool traits in sheep. Asian Austral J Anim. 2018;31(6):775–783. doi: 10.5713/ajas.17.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coulombe PA, Omary MB. ‘Hard’ and ‘soft’ principles defining the structure, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol. 2002;14(1):110–122. doi: 10.1016/s0955-0674(01)00301-5. [DOI] [PubMed] [Google Scholar]
- 38.Higgins CA, Petukhova L, Harel S, Ho YY, Drill E, Shapiro L, et al. FGF5 is a crucial regulator of hair length in humans. Proc Natl Acad Sci U S A. 2014;111(29):10648–10653. doi: 10.1073/pnas.1402862111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C, Zhou S, Li Y, Li G, Ding Y, Li L, et al. Trio-based deep sequencing reveals a low incidence of off-target mutations in the offspring of genetically edited goats. Front Genet. 2018;9:449. doi: 10.3389/fgene.2018.00449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Cai B, Zhou J, Zhu H, Niu Y, Ma B, et al. Correction: disruption of FGF5 in cashmere goats using CRISPR/Cas9 results in more secondary hair follicles and longer fibers. PLoS One. 2016;11(11):e0167322. doi: 10.1371/journal.pone.0167322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao B, Chen Y, Hao Y, Yang N, Wang M, Mei M, et al. Transcriptomic analysis reveals differentially expressed genes associated with wool length in rabbit. Anim Genet. 2018;49(5):428–437. doi: 10.1111/age.12701. [DOI] [PubMed] [Google Scholar]
- 42.He X, Chao Y, Zhou G, Chen Y. Fibroblast growth factor 5-short (FGF5s) inhibits the activity of FGF5 in primary and secondary hair follicle dermal papilla cells of cashmere goats. Gene. 2016;575(2 Pt 2):393–398. doi: 10.1016/j.gene.2015.09.034. [DOI] [PubMed] [Google Scholar]
- 43.Ito C, Saitoh Y, Fujita Y, Yamazaki Y, Imamura T, Oka S, et al. Decapeptide with fibroblast growth factor (FGF)-5 partial sequence inhibits hair growth suppressing activity of FGF-5. J Cell Physiol. 2003;197(2):272–283. doi: 10.1002/jcp.10369. [DOI] [PubMed] [Google Scholar]
- 44.Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci. 2010;67(8):1277–1292. doi: 10.1007/s00018-010-0263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29(6):1575–1591. doi: 10.1128/MCB.01300-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B, Qiao L, An L, Wang W, Liu J, Ren Y, et al. Transcriptome analysis of adipose tissues from two fat-tailed sheep breeds reveals key genes involved in fat deposition. BMC Genomics. 2018;19(1):338. doi: 10.1186/s12864-018-4747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elliott K, Stephenson TJ, Messenger AG. Differences in hair follicle dermal papilla volume are due to extracellular matrix volume and cell number: implications for the control of hair follicle size and androgen responses. J Invest Dermatol. 1999;113(6):873–877. doi: 10.1046/j.1523-1747.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 48.Zhu B, Xu T, Zhang Z, Guo X, Liu D. Transcriptome sequencing reveals differences between anagen and telogen secondary hair follicle-derived dermal papilla cells of the cashmere goat (Capra hircus) PLoS One. 2013;8(9):e76282. doi: 10.1371/journal.pone.0076282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med. 1999;341(7):491–497. doi: 10.1056/NEJM199908123410706. [DOI] [PubMed] [Google Scholar]
- 50.Wang K, Pan L, Che X, Cui D, Li C. Gli1 inhibition induces cell-cycle arrest and enhanced apoptosis in brain glioma cell lines. J Neuro-Oncol. 2010;98(3):319. doi: 10.1007/s11060-009-0082-3. [DOI] [PubMed] [Google Scholar]
- 51.Tam M, Lin P, Hu P, Lennon PA. Examining hedgehog pathway genes GLI3, SHH, and PTCH1 and the p53 target GLIPR1/GLIPR1L1/GLIPR1L2 gene cluster using fluorescence in situ hybridization uncovers GLIPR1/GLIPR1L1/GLIPR1L2 deletion in 9% of patients with multiple myeloma. J Assoc Genet Technol. 2010;36(3):111–114. [PubMed] [Google Scholar]
- 52.Sun W, Ni R, Yin J, Musa H, Ding T, Chen L. Genome Array of hair follicle genes in lambskin with different patterns. PLoS One. 2013;8(7):e68840. doi: 10.1371/journal.pone.0068840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kratochwil K, Dull M, Farinas I, Galceran J, Grosschedl R. Lef1 expression is activated by BMP-4 and regulates inductive tissue interactions in tooth and hair development. Genes Dev. 1996;10(11):1382–1394. doi: 10.1101/gad.10.11.1382. [DOI] [PubMed] [Google Scholar]
- 54.Fang DR, Lv ZF, Qiao G. Dynamic Wnt5a expression in murine hair follicle cycle and its inhibitory effects on follicular in vivo. Asian Pac J Trop Med. 2014;7(4):285–288. doi: 10.1016/S1995-7645(14)60039-0. [DOI] [PubMed] [Google Scholar]
- 55.Geng R, Yuan C, Chen Y. Exploring differentially expressed genes by RNA-Seq in cashmere goat (Capra hircus) skin during hair follicle development and cycling. PLoS One. 2013;8(4):e62704. doi: 10.1371/journal.pone.0062704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Powell BC, Passmore EA, Nesci A, Dunn SM. The notch signalling pathway in hair growth. Mech Develop. 1998;78(1–2):189–192. doi: 10.1016/s0925-4773(98)00177-4. [DOI] [PubMed] [Google Scholar]
- 57.Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, Kelley MW. Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat Genet. 1999;21(3):289–292. doi: 10.1038/6804. [DOI] [PubMed] [Google Scholar]
- 58.Barbosa-Morais NL, Blencowe BJ. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2013;338(6114):1587–93. [DOI] [PubMed]
- 59.Calarco JA, Xing Y, Cáceres M, Calarco JP, Xiao X, Pan Q, et al. Global analysis of alternative splicing differences between humans and chimpanzees. Genes Dev. 2007;21(22):2963. doi: 10.1101/gad.1606907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gracheva EO, Cordero-Morales JF, González-Carcacía JA, Ingolia NT, Manno C, Aranguren CI, et al. Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature. 2011;476(7358):88–91. doi: 10.1038/nature10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science. 2012;338(6114):1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 62.Lander ES. The new genomics: global views of biology. Science. 1996;274(5287):536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- 63.Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, et al. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407(6803):513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- 64.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43(6):519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Do DN, Janss LL, Jensen J, Kadarmideen HN. SNP annotation-based whole genomic prediction and selection: an application to feed efficiency and its component traits in pigs. J Anim Sci. 2015;93(5):2056–2063. doi: 10.2527/jas.2014-8640. [DOI] [PubMed] [Google Scholar]
- 66.Castle WE, Nachtsheim H. Linkage interrelations of three genes for rex (short) coat in the rabbit. Proc Natl Acad Sci U S A. 1993;19(12):1006–1011. doi: 10.1073/pnas.19.12.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Diribarne M, Mata X, Rivière J, Bouet S, Vaiman A, Chapuis J, et al. LIPH expression in skin and hair follicles of normal coat and rex rabbits. PLoS One. 2012;7(1):e30073. doi: 10.1371/journal.pone.0030073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ. Transcript assembly and quantification by RNA Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Florea L, Song L, Salzberg SL. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2013;2:188. doi: 10.12688/f1000research.2-188.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Quality control of RNA-seq data. A L1 B L2 C L3 D S1 E S2 F S3. (PDF 236 kb)
Figure S2. The numbers of annotated genes with different expression levels against the range of FPKM values. (PDF 36 kb)
Figure S3. List of up-regulated and down-regulated genes in the comparison of short-hair and long-hair rabbits. (PDF 118 kb)
Figure S4. The results of RNA-seq between long-hair and short-hair rabbits. A KRT25 B KRT28 C KRT39 D KRT40 E KRT84 F FGF5 G COL3α1 H TNXB I VWF. (PDF 154 kb)
Figure S5. A q-PCR analysis of the relative expression levels of nine DEGs in the skin of short-hair and long-hair rabbits at the eighth week after plucking by q-PCR. A KRT25 B KRT28 C KRT39 D KRT40 E KRT84 F FGF5 G COL3α1 H TNXB I VWF. (PDF 175 kb)
Table S1. Significantly enriched GO terms in the biological process category of DEGs between short-hair and long-hair rabbits (P < 0.05). (PDF 13 kb)
Figure S6. The number of alternative splicing events of six samples. A L1 B L2 C L3 D S1 E S2 F S3. (PDF 144 kb)
Table S2. Analyses of SNP sites of six samples. (PDF 12 kb)
Figure S7. Analyses of SNPs annotation. A L1 B L2 C L3 D S1 E S2 F S3. (PDF 355 kb)
Figure S8. Hair length of the short-hair and long-hair rabbits at the tenth week after plucking. Fibres are divided into three categories, including coarse, fine, and awn fibres. (PDF 107 kb)
Table S3. Primers for qPCR. F1, forward primer. R2, reverse primer. (PDF 95 kb)
Data Availability Statement
The data was presented in the manuscript and the supporting materials. The raw reads data was submitted to the Short Read Archive (SRA) under the accession number SRP173735 and BioProject accession number PRJNA509998 (https://www.ncbi.nlm.nih.gov/sra/PRJNA509998).