

Abstract
We report label-free detection of single mitochondria with high sensitivity using nanoelectrodes. Measurements of the conductance of carbon nanotube transistors show discrete changes of conductance as individual mitochondria flow over the nanoelectrodes in a microfluidic channel. Altering the bioenergetic state of the mitochondria by adding metabolites to the flow buffer induces changes in the mitochondrial membrane potential detected by the nanoelectrodes. During the time when mitochondria are transiently passing over the nanoelectrodes, this (nano) technology is sensitive to fluctuations of the mitochondrial membrane potential with a resolution of 10 mV with temporal resolution of order milliseconds. Fluorescence based assays (in ideal, photon shot noise limited setups) are shown to be an order of magnitude less sensitive than this nano-electronic measurement technology. This opens a new window into the dynamics of an organelle critical to cellular function and fate.
Keywords: Mitochondria, Resistive sensing, Carbon nanotube, Bioenergetics, Label-free
1. Introduction
Mitochondria regulate calcium homeostasis and generate energy in a cell, control cell fate through the apoptosis cell death pathway, and are believed to be the main producers and targets of reactive oxygen species (Wallace, 2005, 2010). A series of membrane protein complexes (the electron transport chain) converts the chemical energy stored in various metabolic substrates into electrical energy (stored as a membrane potential which varies between 100 and 200 mV in response to physiological conditions) (Nicholls, 2006; Nicholls and Ferguson, 2013), resulting ultimately in the reduction of oxygen to water. The mitochondrial membrane potential energizes the phosphorylation of ADP to ATP through the F0F1 ATP synthase. One of two cell death signaling pathways (the so-called “intrinsic” or mitochondrial signaling pathway) converges at the mitochondrial membrane and causes a dramatic and sudden collapse of the mitochondrial membrane potential, irreversibly committing the cell to death (Galluzzi et al., 2007; Kroemer et al., 2007). This is a highly regulated pathway and clear target for pharmacological manipulation for a variety of diseases including, e.g. cancer (Wallace, 2012; Sarosiek et al., 2013; Rustin and Kroemer, 2008). While the molecular identity of the complex of proteins that leads to this collapse (the so called permeability transition pore (PTP), presumed to be a large channel) is controversial (Bernardi, 2013), the dynamics and statistics of the membrane potential collapse and in particular of the PTP opening and closing are, to date, impossible to measure with time resolution less than about 0.1 s, in spite of evidence of rapid flickering of the membrane potential. New, faster assays of the mitochondrial membrane potential could therefore provide important new information about a biophysical quantity that is critical for cellular energy production and, through apoptosis, cell fate.
The simplest and most convenient method to assay the membrane potential utilizes a charged, lipophilic fluorescence dye (typically Tetramethylrhodamine, Methyl Ester Perchlorate, known as TMRM) that can freely pass through a lipid bilayer membrane. The dye concentration inside the mitochondria (and hence measured fluorescence intensity) changes in response to the membrane potential through the Nernst relationship. Although convenient and prevalent, this technique is limited in spatial resolution since the entire mitochondrion is typically a few pixels in size, and in temporal resolution because of the weak fluorescence signal. Although fluctuations of the membrane potential in time have been demonstrated (“flickering”) (Duchen et al., 1998; Huser and Blatter, 1999; Diaz, 2000; Krippeit-drews et al., 2000; Buckman and Reynolds, 2001; Vergun et al., 2003; Aon et al., 2003; O’Reilly et al., 2004; Falchi et al., 2005; Hattori et al., 2005; Higuchi et al., 2005; Vergun and Reynolds, 2005; Kurz et al., 2010; Nicholls, 2012; Schwarzländer et al., 2012), especially in the context of apoptosis and cell death, limitations on fluorescence assays have prevented investigation of the spatial distribution of the membrane potential along the length of the mitochondria, and flickering/fluctuations with time scales less than about 100 ms (Gerencser and Adam-Vizi, 2005), even though they are believed to be due to the dynamic opening and closing of the PTP.
In this work, we demonstrate proof of concept of an alternative method to measure mitochondrial membrane potential using nanotube electrodes which sense the mitochondria as they flow passed the electrodes one by one. Although we have not applied this technology yet to study the important biophysical problem of flickering, with this prototype system we demonstrate a statistical noise resolution on the membrane potential measurement of better than 10 mV in 3 ms, at least an order of magnitude faster than any published fluorescence membrane potential measurement. In this work this assay is performed during the time when mitochondria are transiently passing over the nanoelectrodes, but in principle this can be applied to permanently attached mitochondria (Pham et al., 2015). In addition, we present a careful, quantitative analysis of the fundamental and practical limits of membrane potential measurements using fluorescence microscopy and show that our prototype electrical assay system provide superior time resolution even when compared against a (hypothetical) perfect, shot noise limited fluorescence configuration, where statistical fluctuations in the number of photons detected per time due to Einstein’s discreteness of the photon energy (photon shot noise) is the dominant noise source.
At this point, this sensing method is entirely phenomenological and at the proof of concept stage only. Future studies will further elucidate the sensing mechanism and sources of fluctuations in the response from one mitochondria to the next. This paper is only the beginning of a journey to develop sensitive, fast, high spatial resolution probes of mitochondrial electrophysiology. We are at a similar stage to when fluorescence probes of membrane potential first appeared on the scene over 40 years ago. Just as it took decades to elucidate quantitatively the application of fluorescence to probes of the membrane potential, we expect substantial additional follow on work (described in detail in the Discussion section below) will be required to explore the limits of this (nano) technology. However, even at this proof of concept stage, assuming our device is indeed probing the membrane potential, we have already demonstrated an enormous improvement in noise as compared to fluorescence probes. Our detailed analysis of the fundamental and practical limits of fluorescence for assays of mitochondrial membrane potential, carried out in this paper for the first time for this particular use case scenario (mitochondrial suspensions) shows that the prototype device concept has inherent value that would benefit from further, future research on this device concept.
Finally, while an obvious advantage of our nanotube potentiometric approach (in addition to dramatically improved time resolution) is the compatibility to scale to high throughput assays on millions of individual mitochondria at a time using modern complementary metal-oxide semiconductor (CMOS) electronics (which fluorescence approaches are not compatible with), the most exciting possibility of this approach is the potential to scale the spatial resolution down to that of a single ion channel. Our recent demonstrations (Wang et al., 2014; Lim et al., 2011a; Lim et al., 2011b; Lim et al., 2011c) of using nanoelectrodes to sense the current through individual ion channels indicates this is an exciting avenue for extending this initial proof of concept to enable qualitatively new methods of investigating mitochondrial electrophysiology using nanotechnology.
2. Results
2.1. Mitochondrial nanoelectrode sensor: design & fabrication
Our design consists of an array of nanotube transistors fabricated on a 4″ quartz wafer (Fig. 1). A PDMS microfluidic channel (1 µm by 2 µm by 500 µm) enables delivery of electrolyte buffers that act as a liquid “gate” to turn the nanotube channel on and off when a suitable voltage is applied using an Ag/AgCl electrode at the channel entrance (Fig. 1f). The channel dimensions are designed to allow passage of mitochondria (which typically are about 0.5 µm across) one by one across the surface of the nanotube electrodes, which then sense the electrical charge of the mitochondria as they pass by.
Fig. 1.
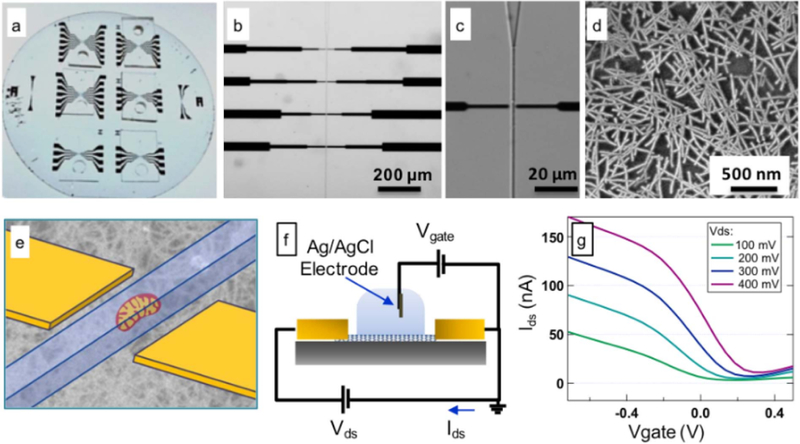
Structure and DC characterization of the mitochondrial nanoelectrode sensor device. (a) Optical image of the quartz wafer with 6 devices; (b) and (c) Bright field microscope images of the device with 20 × and 60 × magnifications; (d) Scanning electron micrograph exhibiting the carbon nanotubes deposited on the wafer surface; (e) Schematic showing the relative location of the carbon nanotube transistor and the flowing mitochondria; (f) Schematic showing the DC measurement set-up; (g) Drain-Source current Ids.vs. (electrolyte) gate voltage for a 3 µm × 3 µm device at different applied drain-source voltages.
The active nanotube channel consists of a random network of purified semiconducting carbon nanotubes with a density of approximately 20 tubes/µm microns2 deposited using methods recently developed by our lab (Rouhi et al., 2011a, 2011b). This provides a simple method to integrate with optical lithography for patterning of the metal contacts to the nanotube network. In our design, only the nanotubes (and not the metal contacts) are exposed to the solution.
The metal contacts to each side of the nanotube network serve as “source” and “drain” electrodes. The conductance from source to drain is measured using either a semiconductor parameter analyzer or a lockin analyzer (see Methods). Consistent with the known properties of nanotube networks with Pd electrical contacts, the nanotube network is conducting at negative electrolyte gate voltages with a mobility of around 0.6 cm2/(V‧s) (Fig. 1g and Supplemental information).
2.2. Resistive flow sensing of vital mitochondria
Vital mitochondria, isolated from HeLa cells using a standard protocol (Trounce et al., 1994), are introduced into the channel using a variety of buffers known to affect the metabolic & bioenergetics status of the mitochondria (Fig. 2). As the mitochondria flow over the nanoelectrode network, distinct changes in the conductance are observed (Fig. 3). Control experiments using buffer only (no mitochondria) show a constant conductance, confirming the changes in the conductance are due to the presence of the mitochondria (Fig. 3). Since a mitochondrion in its vital state sustains a large membrane potential, we hypothesize that the surface has a net charge which “gates” the nanotube devices into a less conducting state (Fig. 3), discussed in more detail below. Prior measurements have shown that the surface charge of the mitochondria (which is responsible for gating our nanotubes) is related to the membrane potential (Fig. S2 in Supporting information) (Kamo et al., 1976). With this system we tested the effect of the metabolic state of the mitochondrion on the measured conductance change pulse heights.
Fig. 2.
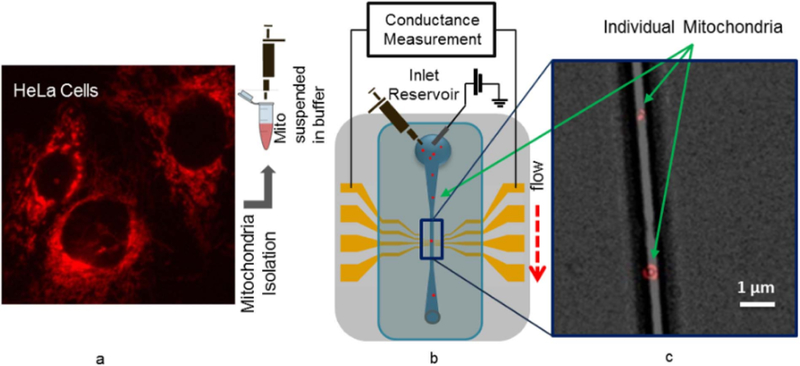
Mitochondria detection set-up. (a) Fluorescence image of Hela cells labeled with TMRM dye. TMRM stains the mitochondria inside the cells. Mitochondria are isolated from Hela cells and suspended in buffer; (b) Schematic depicting the electrical measurement with lock-in amplifier. Buffer containing the isolated mitochondria is pipetted in the inlet reservoir of the channel and flows through the channel; (c) Fluorescence image of TMRM labeled individual mitochondria in the fluidic channel.
Fig. 3.
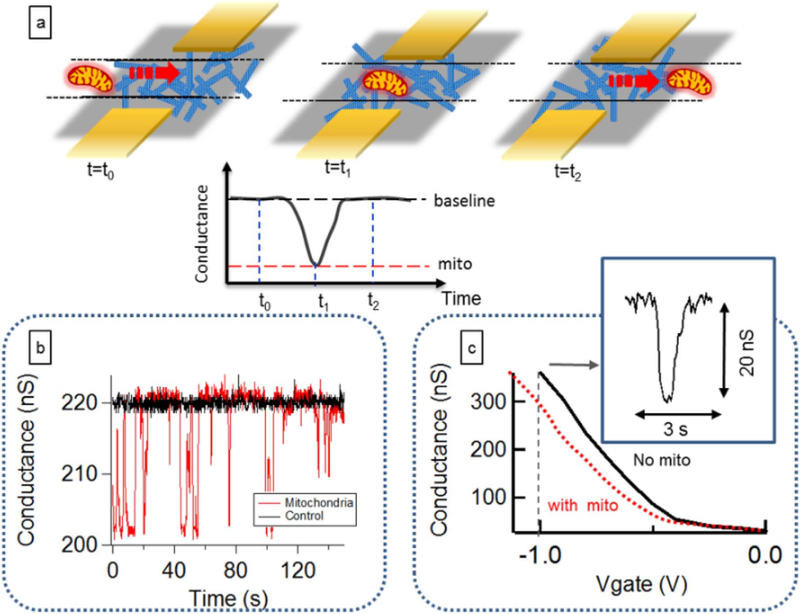
Carbon nanotube detection of vital, individual mitochondria. (a) As a mitochondrion approaches the nanotube electrodes, the source drain current starts to change, and when the mitochondria moves past the transistor channel area, the device current goes back to the baseline; (b) Conductance vs. time data recorded from a device without mitochondria in the flow buffer (black trace) and with mitochondria flowing in the channel (red trace). The spikes in the red curve correspond to mitochondria passing over the sensor area. This device is biased at a (liquid) gate voltage of −1 V; (c) The magnitude and direction of spikes depends on the gate bias voltage. The current change is negative for negative bias voltages, indicating a negative shift of the device threshold voltage as mitochondria approach the device. This has been reproduced nine times on nine separately fabricated devices. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.3. Mitochondrial energetic state can be assayed with nanoelectrodes
In order to further demonstrate the utility of this approach, we next assayed the mitochondrial induced conductance change under three common biochemical conditions (Gnaiger, 2012): 1) Substrate deprived mitochondria. These mitochondria are depleted of their endogenous substrates and the membrane potential is low, due to lack of chemical energy to fuel the electron transport chain proton pumps. This is commonly referred to as Respiratory State 1 (Gnaiger, 2012). 2) Succinate fed mitochondria. Succinate is a substrate that fuels complex II of the electron transport chain, causing a large membrane potential, in the absence of ADP. This is commonly referred to as Respiratory State 2 (Gnaiger, 2012). 3) Depolarized mitochondria. The chemical agent CCCP (carbonyl cyanide m-chloro phenyl hydrazine) is a lipid soluble uncoupling agent that discharges the proton gradient and results in the collapse of the membrane potential of mitochondria.
In state 1 (substrate deprived), the change in the electrode conductance is measurable but small (Fig. 4). Once the mitochondria are provided with substrates (state 2), the concomitant changes in the nanotube conductance are much larger, due to the larger membrane potential. After depolarization of the membrane potential with CCCP, no further pulses are observed. This confirms that “debris” is not detected, since fully depolarized mitochondria do not register as current pulses. This clearly demonstrates that the nanoelec.trode conductance change is a sensitive probe of the mitochondrial membrane potential, and probes the bioenergetics state of individual mitochondria. Thus, we have clearly demonstrated an empirical correlation between the average pulse heights and the energization state of the mitochondria. Since it is known that membrane potential changes for energized and de-energized mitochondria, we have an empirical calibration of the pulse height-membrane potential relationship. Because we also measure the noise of the current, we can thus estimate the noise of our membrane potential measurement, and relate it to the more mature fluorescence technique in detail below.
Fig. 4.
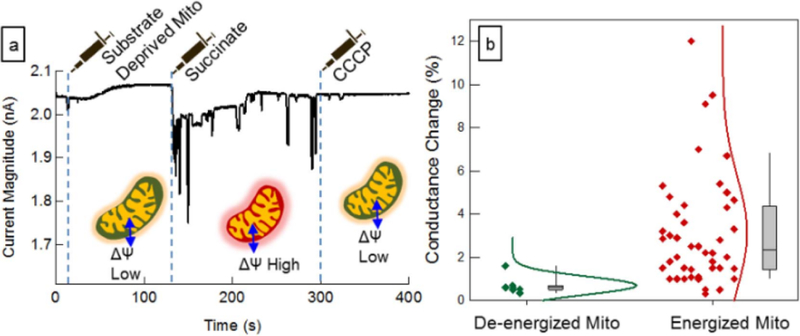
Effect of the metabolic/bioenergetic state of the mitochondria on the conductance change. (a) Addition of different reagents to the reservoir during the measurement alters the membrane potential of mitochondria. Initially mitochondria were suspended in a substrate free buffer and flown into the channel; next mitochondria were energized by adding sodium succinate to a final concentration of 10 mM; finally CCCP was added to the reservoir (final concentration 50 µM) to depolarize the mitochondria; (b) Normalized scatter plot showing the distribution of the events observed for energized and de-energized mitochondria. Detection of energized mitochondria has been reproduced nine times on nine separately fabricated devices.
2.4. Independent membrane potential verification
In a series of control experiments, we verified that the substrate fed mitochondria sustained a large membrane potential compared to substrate deprived mitochondria. This was done using the membrane potential sensitive TMRE dye and several conditions. First, the mitochondria prior to introduction into the channel were imaged inside the inlet reservoir using fluorescence microscopy and then immediately after passing through the channel at the waste reservoir. The TMRE intensity did not change significantly, indicating that the mitochondria sustained their membrane potential while they passed through the channel, and the sufficient oxygen was available in the channel to maintain respiration. Note that the PDMS is permeable to oxygen so no oxygen deficiency is expected (Pham et al., 2016). As a second independent control experiment, we used membrane potential sensitive dye JC1 and TMRE to semi-quantitatively measure the membrane potential of mitochondria trapped at impurities in narrow nanochannels. These indicated that the mitochondria sustain a membrane potential over times longer than our resistive pulse sensing experiments. In a third series of independent experiments, we verified that introduction of substrates just before introduction of mitochondria into the channels increased the membrane potential, as determined by TMRE fluorescence intensity. Thus, three independent experiments using only fluorescence indicators confirm that the mitochondria are energized by substrates, that they sustain a membrane potential, and that the membrane potential is larger in the presence of substrates. These can be taken as independent verification that the mitochondria are indeed “vital” even when inside the nanochannels. Note that a significant variation in the fluorescence intensity of individual mitochondria was observed, indicating significant heterogeneity in the mitochondrial population, independent of the nanoelectrode experiments. These were reported in detail in Zand et al. (2013). In the fluorescence experiments, variations in the fluorescence intensity of JC1 labeled mitochondria were up to a factor of 10 from one mitochondria to the next. Since each mitochondria was one voxel in our microscope, we did not have the capability to ascertain the difference in volume of each mitochondria, and therefore we could not determine of the variation was the membrane potential or the mitochondrial volume.
2.5. Variability
The high variability in the conductance change from one mitochondrion to the next in the succinate stimulated case could be due to three sources: 1) Flickering of membrane potential and the PTP or depolarization through damage during isolation, or 2) variation in the distance between the mitochondria and the Nanoelectrodes, or 3) differing size of mitochondria, causing a varying conversion factor from conductance change to membrane potential. For this reason, the absolute membrane potential of a single mitochondrion will be difficult to calibrate in the existing configuration. Therefore, the significance of this experiment is that it demonstrates the ability to assay ensemble averages in response to different chemical environments. What is even more significant is that fast fluctuations (even if not calibrated absolutely) can be assayed with unprecedented temporal resolution. We turn to the calibration and sensitivity next.
2.6. Mitochondrial membrane potential fluctuations can be quantitatively assayed
Our measurements above clearly show the conductance change is sensitive to the metabolic state of the individual mitochondria (i.e. to its membrane potential). While an absolute calibration of the membrane potential is challenging, it is clear that we can sensitively assay changes in the membrane potential from one mitochondrion to the next, and as a function of the metabolic state of the mitochondria in response to its biochemical environment. We now discuss how to calibrate the sensitivity of this technique to differences in membrane potential from the measured conductance change.
The change in the conductance of the nanoelectrode is larger at larger mitochondrial membrane potentials. Due to the capacitive nature of the detection mechanism, the assumption of a linear signal relationship between the magnitude of change in conductance and the membrane potential of mitochondria is reasonable. For purposes of calibration we assume a resting membrane potential of 150 mV (Nicholls and Ferguson, 2013). In practice, this may vary between 120 and 200 mV (Nicholls and Ferguson, 2013). Although not the main focus of this work, the absolute calibration, which rests on this assumption, therefore may be off by up to 50%.
With this caveat, we can calibrate the sensitivity of the device by measuring the current change from baseline. In our proof of concept devices, at a gate bias voltage of −1V, an average of 0.1 nA (4.5%) current change from the base line occurs as the energized mitochondria pass over the detector. This gives a current to membrane potential conversion factor of typically 0.1 nA per 150 mV of mitochondrial membrane potential. Using this, an estimate for the statistical noise (not the systematic noise) in the membrane potential measurement can be ascertained.
The conductance is measured as a function of time. In order to reduce the noise on the conductance measurement, a low pass filter is used at the output of the lock in analyzer (see Methods).
The magnitude of the measured signal noise depends on the integration time of this filter. With an integration time of 3 ms, the measured standard deviation of the current in the absence of mitochondria ranged from as low as 1.2 pA to as high as 8 pA. For error calculations we assumed a standard deviation of 6 pA. This corresponds to a maximum of 9 mV of error in the measurement of a membrane potential of −150 mV for a measurement time of 3 ms. This is an unprecedented measurement as the prior best result has a measurement time of almost 2 orders of magnitude longer (100 ms) based on fluorescence, and most fluorescence measurements are even longer than that (second). It should be pointed out that the noise sources in our electronic measurement have not been analyzed in detail and this measurement represents an initial proof of concept spot measurement of the noise that could be improved with further effort. We next compare the prototype device realized in our lab with the fundamental limit that fluorescence microscopy can be expected to provide under optimum conditions, as well as practical conditions with various commercially available camera systems.
2.7. Comparing the fundamental and practical limits of fluorescen ce membrane potential assays
Fluorescence imaging is the established method for the study of membrane potential of single mitochondria using membrane potential sensitive dyes (“voltage dyes”) (Loew et al., 1993; Scaduto and Grotyohann, 1999). In this method the membrane potential indicator is generally a lipophilic cationic fluorescence dye that is taken up by mitochondria in proportion to the membrane potential according to the Nernst equation. Mitochondria with higher membrane potential accumulate more dye in the matrix. The fluorescence intensity of the dye is dependent (usually linearly) on the concentration of dye. Fluorescence intensity of the dye is detected inside and outside of the mitochondrion and the ratio is used to calculate the membrane potential.
Although this is a powerful tool, it has some disadvantages associated with it. First, high intensity illumination generates reactive oxygen species in mitochondria and leads to phototoxicity which in many cases actually causes ROS induced depolarization of the membrane potential one is trying to study (Zorov et al., 2000). This effect (in addition to photobleaching of the fluorescence dye) constrains the maximum illumination intensity that can be used, and hence the maximum brightness that can be expected from the fluorophores.
In the supporting information, we provide a detailed analysis of the noise sources of fluorescence microscopy, by first determining the noise on the measurement of the fluorescence intensity, and then converting that to a noise on the measured membrane potential. Noise sources on the fluorescence intensity measurement can come from many sources, but in a properly optimized lab setup, are dominated by three main contributions:
Shot noise of the emitted light due to the discrete nature of the photon
Dark current noise on the photodetector
Read noise on the camera readout electronics.
These noise sources are indicated schematically in Fig. 5a. The effect of the photon shot noise on the membrane potential error was analyzed in Gerencser and Nicholls (2008) for their particular experimental conditions. In this work, we generalize this analysis to provide a comprehensive understanding of noise vs. integration time for typical and best case experimental conditions (Peterka et al., 2011). With mitochondria, since the organelles are so small (in fact in many cases diffraction limited to one pixel/voxel), and since in mitochondria the dynamics are not well understood (in contrast to neurons, where Hodgkin-Huxley model explains the important aspects of action potential dynamics), this analysis is even more critical. Therefore, a detailed analysis of these noise sources is presented in the supplemental info (Table 1).
Fig. 5.
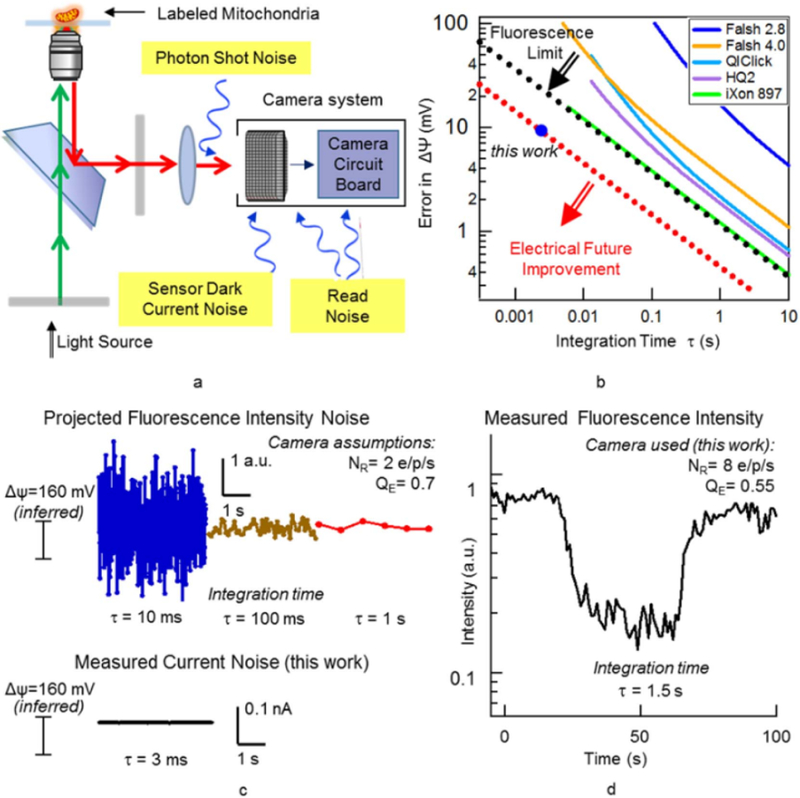
Fluorescence measurement of mitochondrial membrane potential. (a) System schematics and sources of measurement noise; (b) Membrane Potential Measurement error versus integration time for different cameras compared to the shot noise limit (black dotted line) and electrical measurement noise of the fabricated devices (red dotted line); (c) Simulated (projected) time traces for measured fluorescence intensity with different integration times and the electrically measured current of the nanoelectrodes; (d) Measured Fluorescence time trace for a single flickering mitochondria. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Specifications and coefficients used to drive the accuracy of membrane potential measurements by fluorescence microscopy.
| Microscope system | TMRM characteristics | ||
|---|---|---|---|
| Illumination | 120 W-mercury lamp | Absorption/emission | 549/574 nm |
| Filter cube Objective lensa |
TRITC Oil immersion 60 × Plan-Apochromat NA = 1.4 |
Quantum yield Extinction coefficient Absorption cross section |
0.34 1.1 × 105 M−1 cm−1 (kenmoku et al., 2007) 3.824 × 10−19 cm2 |
| Field number: 22 mm FOI = 0.001 cm2 | Concentration (nonquench) | 1–30 nM | |
Mitochondrial matrix volume: 0.04–0.08 µm −3 (Kaasik et al., 2007).
Example: http://objectives.nikoninstruments.com/compare.php?c[] = 93.
In general, longer measurement (i.e. integration) times result in lower noise. In order to compare apples to apples, in Fig. 5b we present the statistical error in the membrane potential for our measured devices (electronic), as well as the fundamental limits of the accuracy of fluorescence measurement of membrane potential changes and typical practical use cases, all as a function of integration time. The plot clearly indicates that the nanoelectrode device in the set-up reported in this work achieves a dramatically lower measurement error compared to fluorescence microscopy. In order to interpret this in the time domain, in Fig. 5c we compare normalized simulated time traces for measured fluorescence intensity for different integration times and the electrically measured current versus time of the nanoelectrodes from this work. The difference in noise levels is striking, and confirms that this nanoelectronic assay provides a new window into mitochondrial dynamics at the ms timescale.
3. Discussion
3.1. Sensing mechanism: surface charge
Prior measurements have shown that the surface charge of the mitochondria (which is responsible for gating our nanotubes) is related to the membrane potential (Fig. S2 in Supporting information) (Kamo et al., 1976). However, studies with similar membrane potential probes have indicated that these measurements generally have to be corrected for probe binding in a manner that is sensitive to surface charge effects (Rottenberg, 1984) and other investigators have found that mitochondrial membrane surface charge is generally unrelated to the transmembrane potential difference that constitutes the major component of the proton motive force (Hashimoto and Rottenberg, 1983; Wojtczak et al., 1987). In future work, with additional measurements (e.g. zeta potential measurements, or more systematic experiments to quantify the effect of buffer ionic strength on the pulse heights), it will be important to further determine the relative effects of surface charge from transmembrane potential on the response of the device. Note that regardless of the detailed zeta potential, there is always some charge present on any membrane that sustains a potential, and if this is within the debye length of the nanotube, the nanotube will detect it electrically.
3.2. Significance & future work
Although our prototype device is first in class proof of concept, it is still superior to the fundamental limit of fluorescence microscopy. By optimizing the device design and measurement conditions, we anticipate that further improvement in the electrical measurement. Ultimately, the electronic sampling frequency can be increased as high as the cut-off frequency of the nanotube transistor, which can be in the GHz range.
In this work, the mitochondria are not trapped permanently on top of the nanoelectrodes, but rather are transiently analyzed as they flow past, similar to how flow cytometry analyzes fluorescence properties in a flow system, hence the term resistive flow cytometry. Although our noise floor is superior to fluorescence, the next obvious step would be to develop a technology to attach the mitochondria to the nanoelectrodes and analyze them permanently, rather than transiently. Such a technology, while beyond the scope of this manuscript, it currently under development in our labs.
At this point, we have no indication that we have been capturing fluctuations of mitochondrial membrane potential, as opposed to device noise. If the mitochondria are indeed “silent”, this would not show up in our measurements. The only conclusion we can draw is that the detectable noise limit (mitochondrial flicker PLUS measurement apparatus noise) is quantitatively improved as a technology using nanoelectronics as compared to fluorescence.
Finally, we note that this technology is reproducible and robust, with multiple repeats of measurements. In the supplemental information we provide statistics about the yield of device fabrication. In some cases, the fabrication process failed prior to testing the device on vital mitochondria. However, in all cases in which the device fabrication process succeeded, we observed current pulses as vital mitochondria traversed the device. This has been reproduced nine times on nine separately fabricated devices.
In this work, the measurement is on vital mitochondria, with both the inner and out membrane intact. This has the potent advantage of preserving biological function that relies on both membranes. A potential increase in sensitivity could come by using mitoplasts which unfold the inner membrane. In fact, prior work initially by Zoratti/Sorgato (Sorgato et al, 1987; Petronilli et al., 1989) and followed up on by other groups (Kirichok et al., 2004; Fedorenko et al., 2012; Fieni et al., 2012) has been able to use mitoplast patch-clamp, and have been successfully applied in the past to resolve biological questions. In comparison to this work, the patch-clamp allows direct control of the membrane potential, with the added complication of impaling the membrane.
Just as the development of fluorescent means to detect membrane potential gave rise do decades of experimental data on the electrophysiology of mitochondria, we also expect this new technology has the potential to provide a plethora of new knowledge about mitochondrial dynamics. For example, addition of rotenone will make the succinate-induced energization more consistent. Also, if the variability is in part due to ‘flickering’ or transient opening of a permeability transition pore, the range of measured conductance changes may be restricted by chelating calcium with 0.1 mM EGTA or by addition of cyclosporine A. In addition, future possible experiments to more quantitatively calibrate the system could include a partial depletion of the membrane potential (e.g. by addition of ADP (State 3), or by using a lower concentration of uncoupler,or by partial inhibition of succinate oxidation with malonate or an ETC inhibitor.
4. Conclusions
This work shows for the first time that single isolated mitochondria can be directly detected by changes in conductance of carbon nanotube devices, as mitochondria flow one by one over the carbon nanotubes in a microfluidic channel. The surface charge of mitochondria induces a change in the threshold voltage of the carbon nanotubes that leads to changes in the transistor conductance. The magnitude of this conductance change depends on the energization state of the mitochondria and therefore can be used as a method for rapid, label free assessment of mitochondria membrane potential. Our analysis reveals that electrical measurements of membrane potential provide orders of magnitude higher time resolution and accuracy compared to the conventional method of fluorescence microscopy for study of the membrane potential of isolated mitochondria. Thus, this work is a step towards high resolution, high throughput measurements of single mitochondria bioenergetics.
5. Methods
5.1. Device fabrication
Four inch quartz wafer is cleaned with hot Piranha solution for 1h, followed by carbon nanotube deposition according to the procedure described in Rouhi et al. (2011a). In short, the cleaned quartz wafer is immersed in 1% 3-aminopropyltriethoxy silane (APTES) in isopropanol solution for 1 h. This treatment creates a monolayer of amine terminated silane on the surface. This monolayer helps with selective absorption of semiconductive carbon nanotubes. The quartz wafer is then rinsed with isopropanol and purified 99% semiconducting carbon nanotube solution (IsoNanotubes-S 99%, Nanointegris Inc.) is drop-casted on the surface. The carbon nanotube (CNT) solution is left on the surface for 1h. Subsequently the wafer is rinsed with DI water and baked in a 60 °C oven overnight. Alternatively, a vacuum filtration method was used for nanotube deposition. In this method, single-walled nanotubes (SWNT) network was obtained by vacuum filtration of 99% purity semiconducting carbon nanotube ink onto mixed cellulose membrane with 25 nm pore size (MFMillipore VSWP04700). 600 µI of CNT ink (IsoNanotubes -S 99%,diluted in DI water to a concentration of 1µg/ml) was filtered through the membrane resulting in a uniform coated CNT network film. Followed by 200 µI DI water rinse to remove residual surfactant, a CNT network film on mixed cellulose membrane was made and stored for transferring. Soda lime glass was used as the substr.ate and treated with 1:3 (v/v) H2O2:H2SO4 solution for 40 min at 140 °C to achieve a clean surface. Premade SWNT network film was then moistened with ethanol and placed in contact with the cleaned.substrate. After 1 h immersion of the device in acetone vapor, most of the mixed cellulose membrane was dissolved and the SWNT network was bonded to the substrate. The residual cellulose was removed in acetone and methanol step by step under carefully tuned condition (30 min in 50 °C warmed up acetone with stir speed 60 rpm and 20 min in methanol at 50 °C with stir speed 60 rpm). After IPA rinse and N2 gas blow dry, a large area uniform SWNT network on glass substrates was made.
The Ti(5 nm)/Pd (15 nm)/Au (30 nm) electrodes are deposited by E-beam evaporation and patterned by liftoff procedure using polymethylglutarimide (PMGI) SF6 and Microposit SC 1827 (MicroChem Corp) photoresists. Fluidic channels are fabricated by soft lithography of Polydimethylsiloxane (PDMS). Silicone elastomer and curing agent (Sylgard® 184, Dow Corning Co.) are mixed at a 10:1 ratio, degassed and poured over a silicon mold. The mold is placed in a 60 °C curing oven overnight. After curing, PDMS is cut and peeled off from the mold. Inlet reservoir and outlet hole are punched, followed by exposure of the PDMS channels to 70 W oxygen plasma. This treatment takes 30 s and turns the PDMS from hydrophobic to hydrophilic; this improves the bond with the quartz wafer and also enhances the fluid flow by capillary force. The PDMS chip is immersed in methanol immediately after the oxygen plasma treatment. Then the PDMS channel is placed on the quartz wafer and manually aligned under a microscope. Methanol lowers the friction between the CNT device and the PDMS, enabling the alignment without PDMS sticking to the substrate and damaging the transistor device. After the alignment the methanol evaporates and the PDMS bonds to the quartz wafer. The device is baked at 60 °C for 30 min to improve the bonding.
The PDMS channel consists of a 2 µm wide, 1 µm deep, 500 µm long flow channel, connected to two wider (100 µm) access channels with a 70 µl inlet reservoir and an outlet hole to introduce the liquid. The access channel lengths are 2 cm. Different source-drain electrodes widths and gaps ranging from 3 to 20 µm have been fabricated. Alternatively, device arrays with various channel lengths were then patterned on the SWNT network by a standard photolithography. Ti (2 nm)/Pd (20 nm)/Au (50 nm) were deposited by ebeam evaporation, followed by a liftoff process to form contact elect.rodes. Final step of ebeam lithography was used to open windows (2 µm by 10 µm) in the channel region, leaving the electrodes protected under photoresist and SWNT network exposed. PDMS reservoir was aligned on the device array for delivering aqueous solution.
5.2. Electrical measurement
For conductance versus time measurements, 10 mv AC voltage at 10 kHz is applied between the drain-source electrodes and the current is measured using an SR-830 lock-in amplifier. By increasing the integration time and the order of the lock-in amplifier low pass filter the output becomes less noisy. The time constant and the filter order also determine how long it takes for the filter output signal to respond to changes in the device conductance. The integration time was set at 3 ms. Therefore, for a first order filter, 3 ms is the 1/e time to detect abrupt changes in the nanoelectrode conductance, and is the rate limiting step in the conductance measurement. In our set-up we used a second order filter, the integration time for each stage of the filter was 3 ms. This resulted in a delay time (time it takes for the measured current to reach 50% of its final value) of 5 ms. A custom built semiconductor parameter analyzer was used to measure the DC transport curve of the devices.
5.3. Cell culture and mitochondria isolation
Mitochondria were isolated from the human cervical cancer cell line HeLa (ATCC, CCL-2).The adherent cells were cultured and maintained in log growth phase in media consisting of EMEM (ATCC, 30–2003) supplemented with 10% FBS (Invitrogen, 10438–018) and 1% Penicillin-Streptomycin (ATCC, 30–2300). All other chemicals were obtained from Sigma Aldrich, unless otherwise noted. The mitochondrial isolation protocol was adapted from Lim et al (2010). Briefly, cells were harvested at 100% confluence in a T-75 flask. Approximately 8 × 106 cells were pelleted and washed in Phosphate Buffered Saline. Ice-cold H-buffer (210 mM mannitol, 70 mM sucrose, 1 mM EGTA, 5 mM HEPES, 0.5% BSA, pH adjusted to 7.2 with 1 M KOH) was used in all of the following isolation steps. The cells were physically sheared with 20 passes in an ice-cold dounce homogenizer and centrifuged at low speed (800 × g for 5 min) at 4 °c in an Eppendorf 5417R centrifuge. The cell lysate was further purified for the removal of cell debris through 2 additional rounds of low speed spins. The resulting supernatant was subjected to 2 rounds of high speed centrifugation (10,000 × g for 20 min). BSA-free H-buffer was used to re-suspend the resulting pellet, which was spun again at high speed. The isolated mitochondrial sample was pelleted and diluted in ice-cold respiration buffer (225 mM Mannitol, 75 mM Sucrose, 10 mM KCl, 10 mM TrisHCl, 5 mM KH2P04, pH adjusted to 7.2 with 1 M KOH) and used. 1:1, 1:2, 1:10 dilutions of the suspension were used in protein determination with the BCA Protein Assay Kit (Thermo Scientific, 23227).
5.4. Fluorescence imaging
For Fluorescence assays of membrane potential mitochondria were labeled with TMRM (Life Technologies) in non-quench mode. TMRM was dissolved in DMSO and added to the mitochondrial respiration buffer to a final concentration of 30 nM. Mitochondria were incubated with TMRM for 20 min prior to imaging. Mitochondria were imaged with Olympus IX71 inverted fluorescence microscope, equipped with a 12 bit monochromatic CCD camera (QIClick-F-M-12), a LUCPLFLN Semi-Apochromat 60 ×, 0.7 NA objective, 120 W Mercury vapor excitation light source and standard FITC (490 nm–525 nm) and TRITC (557 nm–576 nm) filters. Image analysis was performed with ImageJ software.
5.5. Mitochondrial injection into channel
50 µl respiration buffer is pipetted inside the reservoir, the capillary force carries the buffer inside the channel resulting in a flow rate of approximately 0.01 µl/h Mitochondria suspension and other reagents are pipetted into the same reservoir. To reduce the electrical measurement noise a syringe pump is not used. The channel experiments were performed at room temperature. The mitochondrial protein concentration was titrated as in our recent work (Fig. S10 in that reference) to enable only on average one mitochondria at a time to flow through the channel. Typical concentrations were 1–10 µg/ml.
The exposure time required to image the flowing mitochondria is long, therefore it is not possible to image the mitochondria as they are flowing past the CNT device; but we had previously developed a method to decrease the height of the flow channel to 500 nm and trap the mitochondria inside the flow channel (Zand et al., 2013). This method allowed us to verify that mitochondria enter the channel one by one and they remain vital and functional.
Supplementary Material
Acknowledgements
This work was supported in part by the following grants awarded to D.C. Wallace: NS21328 and CA143351. P. Burke acknowledges support of this work from NIH National Cancer Institute Grant 1R21CA143351–0l, and R33 CA182384 as well as support from the ARO (MURI W911NF-11–1-0024). T. Pham is supported by the National Science Foundation Lifechips Integrative Graduate Education and Research Trainee.ship 0 549 479.
References
- Aon MA, Cortassa S, Marbán E, O’Rourke B, 2003. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem 278, 44735–44744. [DOI] [PubMed] [Google Scholar]
- Bernardi P, 2013The mitochondrial permeability transition pore: a mystery solved? Front. Physiol 4, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman JF, Reynolds IJ, 2001. Spontaneous changes in mitochondrial membrane potential in cultured neurons. J. Neurosci 21, 5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz G, 2000. Homogeneous longitudinal profiles and synchronous fluctuations of mitochondrial transmembrane potential. FEBS Lett 475, 218–224. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Leyssens A, crompton M, 1998. Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium Release in single rat cardiomyocytes. J. Cell Biol 142, 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchi AM, Isola R, Diana A, Putzolu M, Diaz G, 2005. Characterization of depolarization and repolarization phases of mitochondrial membrane potential fluctuations induced by tetramethylrhodamine methyl ester photoactivation. FEBS J 272, 1649–1659. [DOI] [PubMed] [Google Scholar]
- Fedorenko A, Lishko PV, Kirichok Y,2012. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieni F, Lee SB, Jan YN, Kirichok Y, 2012. Activity of the mitochondrial calcium uniponer varies greatly between tissues. Nat. Commun 3, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G, 2007. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 14, 1237–1243. [DOI] [PubMed] [Google Scholar]
- Gerencser A, Adam-Vizi V, 2005. Mitochondrial Ca2+ dynamics reveals limited intramitochondrial Ca2+ diffusion. Biophys. J 88, 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Nicholls DG, 2008. Measurement of instantaneous velocity vectors of organelle transport: mitochondrial transport and bioenergetics in hippocampal neurons. Biophys. J 95, 3079–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnaiger E, 2012. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. In: Mitochondr Physiol Network 17 18, 3rd ed. OROBOROS MiPNet Publications. [Google Scholar]
- Hashimoto K, Rottenberg H, 1983. Surface potential in rat liver mitochondria: terbium ion as a phosphorescent probe for surface potential. Biochemistry 22, 5738–5745. [DOI] [PubMed] [Google Scholar]
- Hattori T, Watanabe K, Uechi Y, Yoshioka H, Ohta Y, 2005. Repetitive transient depolarizations of the inner mitochondrial membrane induced by proton pumping. Biophys. J 88, 2340–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi Y, Miura T, Kajimoto T, Ohta Y, 2005Effects of disialoganglioside GD3 on the mitochondrial membrane potential. FEBS Lett 579, 3009–3013. [DOI] [PubMed] [Google Scholar]
- Huser J, Blatter LA, 1999. F1uctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J 343,311–317. [PMC free article] [PubMed] [Google Scholar]
- Kaasik A, Safiulina D, Zharkcovsky A, Veksler V, 2007. Regulation of Mitochondrial Matrix Volume pp. 157–163. [DOI] [PubMed]
- Kamo N, Muratsugu M, Kurihara K, Kobatake Y, 1976. Change in surface charge density and membrane potential of intact mitochondria during energization. FEBS Lett 72, 247–250. [DOI] [PubMed] [Google Scholar]
- Kenmoku S, Urano Y, Kojima H, Nagano T, 2007. Development of a highly specific rhodamine-based fluorescence probe for hypochlorous acid and its application to real-time imaging of phagocytosis. J. Am. Chem. Soc 129, 7313–7318. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G,Clapham DE, 2004. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 23722–23728. [DOI] [PubMed] [Google Scholar]
- Krippeit-drews P, Du M, Drews G, Dfer M, 2000. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem. Biophys. Res. Commun 267, 179–183. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C, 2007. Mitochondrial membrane permeabilization in cell death. Physiol. Rev 87, 99–163. [DOI] [PubMed] [Google Scholar]
- Kurz FT, Aon MA, O’Rourke B, Armoundas AA, 2010. Spatio-temporal oscillations of individual mitochondria in cardiac myocytes reveal modulation of synchronized mitochondrial clusters. Proc. Natl. Acad. Sci. U.S. A 107, 14315–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim T-S, Davila A, Wallace DC, Burke PJ, 2010. Assessment of mitochondrial membrane potential using an on-chip microelectrode in a microfluidic device. Lab Chip 10, 1683–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim T-S, Jain D, Burke PJ, 2011a. Biomembrane-gated carbon nanotube transistor as a sensing platform. In: Proc. 15th Int. Conf. Miniaturized Syst. Chem. Life Sci, pp. 1770–1772. [Google Scholar]
- Lim TS, Jain D, Burke PJ, 2011b. Fabrication of supported lipid bilayer (SLB) and nanotube transistor hybrid biosensing platform using microfluidic channels. In:Proc. 11th IEEE Conf. Nanotechnol, pp. 371–373. [Google Scholar]
- Lim T-S, Jain D, Burke PJ, 2011c. Protein nanopore-gated bio-transistor for membrane ionic current recording. In: Proc. 69th Device Res. Conf. 26 pp. 131–132. [Google Scholar]
- Loew LM, Tuft RA, Carrington W, Fay FS, 1993. Imaging in five dimensions: time-dependent membrane potentials in individual mitochondria. Biophys. J 65, 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, 2006. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J. Biol. Chem 281, 14864–14874. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, 2012. Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. Methods 810, 119–133. [DOI] [PubMed] [Google Scholar]
- Nicholls D, Ferguson S, 2013. Bioenergetics, 4th ed.. [Google Scholar]
- O’Reilly CM, Fogarty KE, Drummond RM, Tuft RA, Walsh JV, 2004. Spontaneous mitochondrial depolarizations are independent of SR Ca2+ release. Am. J. Cell Physiol 286, C1139–C1151. [DOI] [PubMed] [Google Scholar]
- Peterka DS, Takahashi H, Yuste R, 2011. Imaging voltage in neurons. Neuron 69, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Szabò I, Zoratti M, Szabo I, 1989. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett 259, 137–143. [DOI] [PubMed] [Google Scholar]
- Pham T, Burke P, Wallace DC, 2015An Electronic Assay of Cell Death. Prep
- Pham TD, Wallace DC, Burke PJ, 2016. Microchambers with solid-state phosphorescent sensor for measuring single mitochondrial respiration. Sensors 1065, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg H, 1984. Membrane potential and surface potential in mitochondria: uptake and binding of lipophilic cations. J. Membr. Biol 81, 127–138. [DOI] [PubMed] [Google Scholar]
- Rouhi N, Jain D, Zand K, Burke PJ, 2011a. Fundamental limits on the mobility of nanotube-based semiconducting inks. Adv. Mater 23, 94–99. [DOI] [PubMed] [Google Scholar]
- Rouhi N,Jain D, Burke PJ, 2011b. High-performance semiconducting nanotube inks: progress and prospects. ACS Nano 5, 8471–8487. [DOI] [PubMed] [Google Scholar]
- Rustin P, Kroemer G, 2008. Mitochondria and cancer. Ernst Shering Found. Symp. Proc 4, 1–21. [DOI] [PubMed] [Google Scholar]
- Sarosiek KA, Chonghaile TN, Letai A, 2013Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol 23, 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto RC, Grotyohann LW, 1999. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J 76, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzländer M, Logan DC, Johnston IG, Jones NS, Meyer AJ, Fricker MD, Sweetlove LJ, 2012. Pulsing of membrane potential in individual mitochondria: a stress-induced mechanism to regulate respiratory bioenergetics in Arabidopsis. Plant Cell 24, 1188–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorgato MC, Keller BU, Stuhmer W, 1987. Patch-clamping of the inner mitochondrial-membrane reveals a voltage-dependent ion channel. Nature 330, 498–500. [DOI] [PubMed] [Google Scholar]
- Trounce I, Neill S, Wallace DC, 1994. Cytoplasmic transfer of the mtDNA Nt 8993 T→G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc. Natl. Acad. Sci 91, 8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Reynolds IJ, 2005. Distinct characteristics of Ca(2 +)-induced depolarization of isolated brain and liver mitochondria. In:Biochimica et Biophysica Acta vol. 1709 pp. 127–137. [DOI] [PubMed] [Google Scholar]
- Vergun O, Votyakova TV, Reynolds IJ, 2003Spontaneous changes in mitochondrial membrane potential in single isolated brain mitochondria. Biophys. J 85, 3358–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, 2005A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet 39, 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, 2010. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man. Proc. Natl. Acad. Sci. U.S. A 107, 8947–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, 2012. Mitochondria and cancer. Nat. Rev. cancer 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Pham TD, Zand K, Li J, Burke PJ, 2014. Charging the quantum capacitance of graphene with a single biological ion channel. ACS Nano 8, 4228–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtczak L, Nał\kecz MJ, Famulski KS, Dygas A, Szetwczyk A, 1987. Does the energy state of mitochondria influence the surface potential of the inner mitochondrial membrane? A critical appraisal. Acta Biochim. Pol 34, 299–318. [PubMed] [Google Scholar]
- Zand K, Pham T, Davila A, Wallace DC, Burke PJ, 2013Nanofluidic platform for single mitochondria analysis using fluorescence microscopy. Anal. Chem 85, 6018–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ, 2000. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med 192, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.