

Abstract
Allosteric inhibitors can be more difficult to optimize without an understanding of how their binding influences the conformational motions of the target. Here, we used an integrated computational and experimental approach to probe the molecular mechanism of an allosteric inhibitor of heat shock protein 70 (Hsp70). The anticancer compound, MKT-077, is known to bind a conserved site in members of the Hsp70 family, which favors the ADP-bound state and interferes with a protein-protein interaction (PPI) at long-range. However, the binding site does not overlap with either the nucleotide-binding cleft or the PPI contact surface, so its mechanism is unclear. To this end, we modeled Hsp70’s internal dynamics and studied how MKT-077 alters local sampling of its allosteric states. The results pointed to a set of concerted motions between five loops in Hsp70’s nucleotide-binding domain (NBD), surrounding the MKT-077 binding site. To test this prediction, we mutated key residues and monitored chaperone activities in vitro. Together, the results indicate that MKT-077 interacts with loop 222 to favor a pseudo-ADP bound conformer of Hsp70’s NBD, even when ATP is present. We used this knowledge to synthesize an analog of MKT-077 that would better prevent motions of loop222 and confirmed that it had improved anti-proliferative activity in breast cancer cells. These results provide an example of how to unlock and leverage the complex mechanisms of allosteric inhibitors.
Introduction
Allosteric inhibitors are often powerful probes for chemical biology because they “tune” function without directly competing with the active site. However, it can sometimes be more complicated to optimize them, because their potency depends on both affinity for the target and their ability to shape its conformational preferences.1 MKT-077 is an example of such a chemical probe; it is an allosteric inhibitor of heat shock protein 70 (Hsp70) that advanced to Phase I clinical trials for the treatment of solid tumors.2–4 However, these trials were discontinued, in part because the molecule has relatively modest potency (EC50 ~ 5 µM). Recent efforts to optimize its drug-like properties5, 6 would benefit from a molecular understanding of how it interferes with Hsp70 function. One main hurdle to this goal is that the binding site of MKT-077 does not overlap with known functional regions, making it unclear how the molecule works or how its potency might be improved.
Hsp70 is a molecular chaperone that consists of two domains: a ~44 kDa N-terminal, nucleotide-binding domain (NBD) and a ~25 kDa substrate-binding domain (SBD).7 The NBD is further divided into two lobes (A and B) and four subdomains (IA, IIA, IB and IIB), with an ATP-binding cassette positioned between them (Figure 1A). Likewise, the SBD is divided into a β-sandwich region, which contains the shallow cleft important for interactions with unfolded proteins8, and an α-helical “lid” that regulates access to that cleft.9 Members of the Hsp70 family are widely used as models for the study of dynamic, multi-domain proteins because of the dramatic transitions that are known to accompany ATP cycling in the NBD10 and the large number of both intra- and inter-domain allosteric networks.11–14 For example, recent structures of the prokaryotic ortholog, DnaK, have revealed that the lid is nestled against the NBD in the ATP-bound state,15, 16 allowing access to the b-sandwich (Figure 1B). After ATP hydrolysis, dramatic conformational changes in the NBD release the lid, such that it now “closes” towards the β-sandwich and enhances protein-binding affinity.17–19 In this way, Hsp70 oscillates between two major conformers, ATP- and ADP-bound, causing it to cycle between high- and low-affinity binding states.20 In addition to these intra-molecular motions, protein-protein interactions (PPIs) with co-chaperones further tune Hsp70’s nucleotide cycling.21, 22 Specifically, the J-domain proteins (JDPs) bind to the region between the NBD and SBD to disrupt inter-domain contacts and promote ATP hydrolysis,23, 24 while nucleotide exchange factors (NEFs) “open” the lobes of the NBD to facilitate ADP release.25 Computational studies and evolutionary analyses have identified a number of important allosteric switches that control these major motions within the NBD, including a switch at Pro14726 and a handful of hinge residues (i.e. G229, R155) between the IIB and IIA subdomains.27, 28 Most of these previous efforts were directed at understanding how the conversion from the ATP- to ADP-bound state propagates conformational changes to the rest of the protein. However, with a few notable exceptions,29, 30 less is known about how chemical inhibitors regulate these transitions or how they might interfere with the natural motions.
Figure 1.
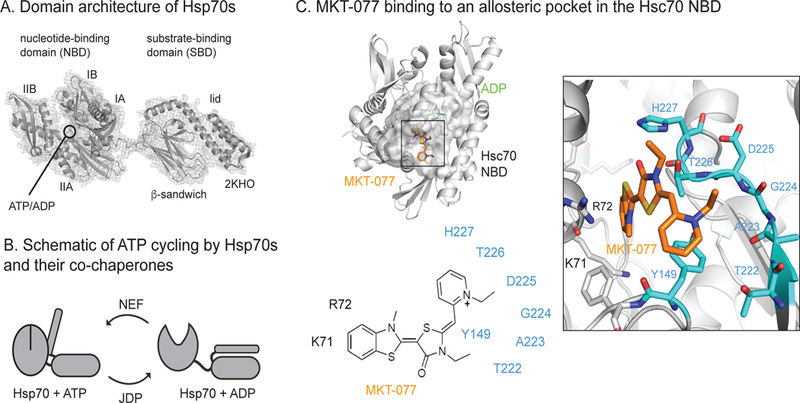
Inhibition of Hsp70 by the allosteric inhibitor, MKT-077. (A) Hsp70s, including Hsc70, are composed of an NBD and SBD, which are connected by a short linker and further divided into subdomains. The structure is of ADP-bound (PDB = 2KHO). (B) Hsp70s undergo dramatic conformational changes in response to ATP cycling, a process that is tuned by J-domain proteins (JDPs) and nucleotide exchange factors (NEFs). (C) MKT-077 is an anti-cancer compound that is known to bind a cryptic, conserved allosteric site in Hsp70s. This site does not overlap with the nucleotide-binding cassette or the NEF PPI. See text for details.
The binding site of MKT-077 was initially identified by nuclear magnetic resonance (NMR) to be located at the interface between lobes IA and IIA in the NBD.31 This binding site is adjacent to, but not overlapping with, the nucleotide-binding cleft (Figure 1C) and the interaction seems to involve two main pockets: one composed of K71, R72, P147 and F150 that surrounds the benzothiazole and another framed by Y149 and a loop from H227 through T222 (loop222) that accepts the pyridinium. In the NMR titrations, MKT-077 only interacted with Hsp70 in its ADP-bound state and not its apo- or ATP-bound states,31 suggesting that it somehow favors the ADP-bound conformer. Later, it was also shown that analogs of MKT-077 block the PPIs between Hsp70 and the BAG family of NEFs.32, 33 That contact surface is ~18 to 20 Å away from the compound’s binding site, so it was not immediately clear why MKT-077 would disrupt the interaction; however, more recent work showed that the ADP-bound state of Hsp70’s NBD has weakened affinity for BAG family NEFs.34 Specifically, human BAG1 binds to Hsp72 (HSPA1A) with an affinity of ~ 8 nM in the apo-state and 14 nM in the ATP-bound state, but only 36 nM in the ADP-bound state. Thus, the key activity of MKT-077 seems to be its ability to trap the ADP conformer, which then has a weaker affinity for NEF PPIs. We reasoned that atomistic knowledge about how MKT-077 interferes with nucleotide-driven allostery might facilitate chemical optimization, as well as clarify how intrinsic control mechanisms regulate this chaperone.
Here, we used molecular dynamic (MD) simulations to model the ATP-to-ADP conformers of Hsp70’s NBD and used these trajectories to probe how MKT-077 alters conformational sampling. More specifically, we focused on interactions of MKT-077 with heat shock cognate 70 (Hsc70; HSPA8), the constitutively expressed form of the chaperone in mammals. This analysis provided a structural rational for why MKT-077 only binds to ADP-bound state and suggested that a concise, coordinated pathway, composed of five loops (loop12, sub69, loop147, loop199 and loop222) links the MKT-077 binding site to the nucleotide-binding cassette. Most strikingly, MKT-077 was predicted to stabilize the ADP-bound conformer by limiting motions of loop222, resisting the influence of ATP binding. To test this prediction, we mutated residues in loop222 and loop199 and found that, like MKT-077, they stabilized the ADP-bound state and blocked NEF-dependent chaperone function. Together, these results suggested that MKT-077 analogs could be improved by resisting nucleotide-driven motions in loop222, a hypothesis that we confirmed by synthesizing an analog of MKT-077, JG-237, with additional bulk pointed in that direction. More broadly, these findings might provide a framework for the optimization of other cryptic, allosteric inhibitors, using a structure-guided understanding of their molecular mechanisms.
Results and Discussion
Molecular dynamic (MD) simulations of nucleotide- and inhibitor-based local motions in the Hsc70 nucleotide-binding domain (NBD)
To unravel the atomistic mechanisms by which MKT-077 regulates Hsc70, we first performed MD simulations of the NBD (PDB 3C7N)35 in the presence or absence of the ligand and in different nucleotide states: (a) ADPonly (no MKT-077, with ADP), (b) ATPonly (no MKT-077, with ATP), (c) MKT-ADP (MKT-077 and ADP) and (d) MKT-ATP (MKT-077 and ATP). For each state, we examined multiple, independent MD simulations (see Methods), totaling more than 650 ns. For the protein backbone atoms of the ADPonly simulation, the time evolution of the average root-mean-square deviation (RMSD) from the initial 3C7N X-ray structure showed that it stably populates the “ADP-like” starting conformation (avg RMSD 2.7 Å) (Figure S1). Conversely, we found that the ATPonly chaperone undergoes the expected conformational changes, with RMSD values up to ~5 Å (Figure S1). Based on the most representative conformations obtained by cluster analysis, this RMSD deviation was largely caused by the ability of ATP to favor a “closing” motion in lobes I and II, which resulted in a 15° rotation between the two axes (Figure S2). Through this motion, the NBD becomes more compact, consistent with recent crystal structures 36 and previous MD simulations 37. Next, we explored the effects of MKT-077 on these conformational transitions. First, we found that MKT-077 stably occupies the known binding site between IA and IIA during the simulations. Moreover, we found that MKT-077 had little effect on the conformations of the ADP-bound state (MKT-ADP, avg. RMSD 2.5 Å), but that it seemed to “trap” the Hsp70-ATP complex in the initial ADP-like state (MKT-ATP, avg. RMSD 2.8 Å) (Figure S1). An analysis of the most representative conformations confirmed that MKT-077 prevented the “closing” motion in response to ATP (Figure S2). This conclusion was also evident in cluster analyses performed on a single meta-trajectory resulting from the concatenation of the ADPonly, ATPonly, MKT-ADP, and MKT-ATP simulations. Specifically, we found that the subsets corresponding to the ADPonly, MKT-ADP and MKT-ATP conformational ensembles were all ADP-like, while ATPonly was the sole system whose conformations were distributed over multiple clusters (Figure S3). Together, these results provide a plausible molecular mechanism for how MKT-077 might favor an ADP-bound conformer.
What are the local conformational changes that allow MKT-077 to resist ATP-dependent transitions? To gain insight into this problem, we repeated the cluster analysis, with a focus on the backbone atoms of residues from the MKT-077 and nucleotide-binding sites. Specifically, these residues included those on loops 12–15, 147–152, 222–231, and the partial helix-loop substructure 69–87. For brevity, these regions will be referred to as loop12, loop147, loop222 and sub69 for the remainder of the manuscript. Examination of these two pockets suggested that they both undergo a series of conformational rearrangements during the ADP to ATP conversion (Figure 2). Most strikingly, loop222 seemed to act as a nucleotide-dependent switch, moving toward the nucleotide in the ATP-bound state in a way that is assisted by the concurrent motion of loop147 and sub69. Because loop222 also forms one side of the MKT-077 binding site, this motion was accompanied by “shrinking” of the compound-binding pocket; namely, the Solvent Accessible Surface Area (SASA) of this region was reduced from 2778 to 2699 Å2. This observation suggests a model in which MKT-077, by occupying this site, might resist the ATP-driven collapse of loop222 and thereby ‘trap’ the ADP-like conformer (see Figure 2).
Figure 2.
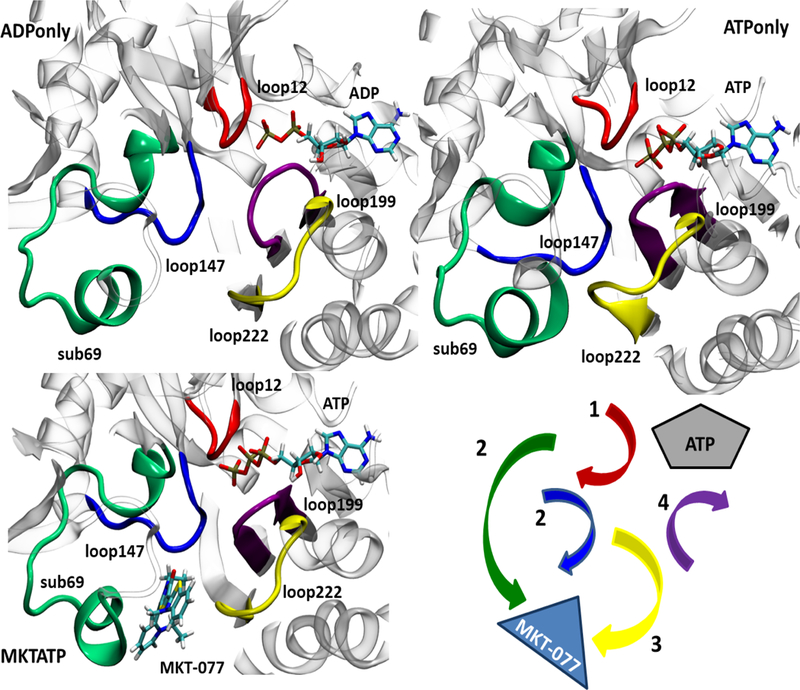
MKT-077 partially prevents ATP-driven conformational transitions in Hsp70’s NBD. MD simulations were performed for Hsp70’s NBD (3C7N; see text). Nucleotide-dependent motions are shown. Binding to ATP favors collapse of the MKT-077 binding pocket, especially mediated by loop222 (yellow), loop199 (purple) and loop 147 (blue) and initiated by changes in loop 12 (red).
Next, we set out to identify specific residues that might underlie the conformational signal. To this end, we calculate the local flexibility (LF) parameter for all of the simulations. This analysis estimates the relative, average deformation that is locally experienced by stretches of residues. Our LF analyses confirmed that the dynamics of loop12, sub69 and loop147 are affected by MKT-077 binding (Figure 3). Moreover, loop199 (residues 199–206) emerged as an important additional modulator because it faces loop12 with respect to the nucleotide and interacts directly with loop222 (see Figure 2). Specifically, ADP seemed to stabilize one network of persistent H-bonds amongst these loops; for example, the persistency of the H-bond between T13 and ADP in ADPonly was calculated to be 49%, with additional bonds between T14-ADP 42% and Y15-ADP 15% (Figure S4A). The presence of ATP pushes the loops apart, with the H-bonds at T13 decreased to only 16% persistency. Rather, binding to ATP favors the interaction of loop12 with sub69 instead (Figure S4A). In turn, this re-arrangement allows sub69 to interact with loop222, stabilizing two salt bridges (ATPonly, Arg72-Glu231 and Asp86-His227), and locking loop222 in the ‘closed conformation’ that is ultimately unable to host MKT-077 (Figure S4B). Finally, these ATP-dependent conformational changes favor coordination between loop199 and the ATP-binding cavity, stabilized by a salt bridge between the ATP γ-PO4 and Asp199 that is mediated by a bound Mg cation (Figure S4C). Together, these observations suggest that ATP binding triggers the rearrangement of a locally compact region. The LF analysis also suggested that most of the loops, with the exception of loop12, move as a rigid, coordinated unit. Most importantly for this study, these motions seem to be silenced by MKT-077 binding (see Figures 2 and S4A-C) largely because loop222 (as well as loop147 and sub69) cannot take part in the necessary structural rearrangements. Therefore, MKT-077 seems to behave as a “rigid stop” to the pivot motions, locking the salt bridges and other contacts in their ADP-like conformations.
Figure 3.
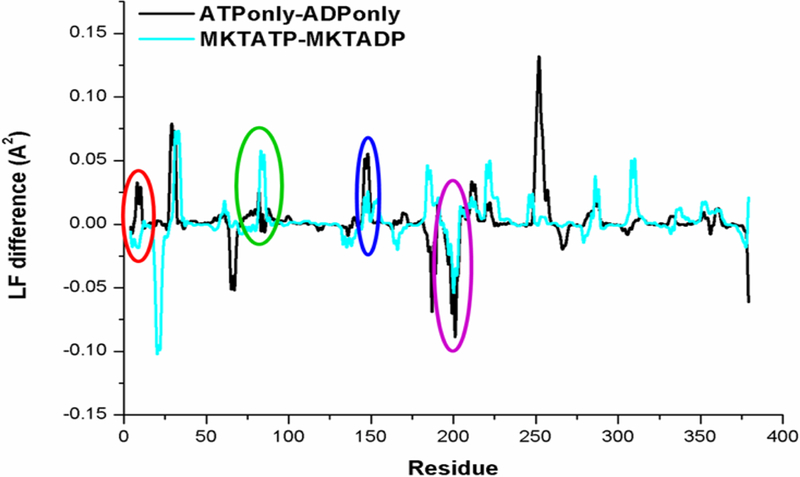
Local flexibility analysis highlights the ability of MKT-077 to suppress ATP-dependent conformational transitions. Many of the ATP-driven deformations (black) were limited by MKT-077 (light blue), especially in the local binding environment. The circles use the same color scheme as figure 2.
MD simulations suggest a model for how MKT-077 partially disrupts a distant protein- protein interaction
The PPI interface between Hsp70 and the NEF’s BAG-domain involves a series of important electrostatic interactions.38, 39 For example, residues E212, Q245, D222 and R237 in the BAG domain of BAG1 make contact with R261, R262, D285 and E283 of Hsc70’s IIB subdomain and mutations of R261A or R262A ablate the interaction.40, 41 Also, it is known that the affinity of Hsp70 for BAG1 is strongest in the apo-state state (Kd ~8 nM), while it is weaker in the ADP- or ATP-bound states (12 and 36 nM, respectively).34 Because analogs of MKT-077 have been found to partially weaken the Hsp70-BAG3 interaction in vitro and in cells,6, 32 we wondered whether the MD simulations might provide insight into how MKT-077 impacts a PPI nearly 20 Å away.
We hypothesized that there might be allosteric coupling between the MKT-077 binding site, the nucleotide-binding cleft and the key residues of the BAG-binding surface. We focused this search on loop 12 (Figure 4A) because our work had identified it to be important in MKT-077 and nucleotide cycling, while Sondermann et al. had shown that packing of this region was important for BAG1 binding.38 Consistent with this hypothesis, we examined the MD simulations and found that nucleotide-dependent motions in loop 12 (residues 12, 13, 14 and 15) seemed to be coupled to movement of both the MKT-077 binding site (residues 72, 76, 82, 86, 149–150, 205–207, 222 and 224–226) and the helix involved in the BAG-binding surface (residues 257–258, 263–278, 284 and 286–289) (Figure 4B). Next, we compared the position of key polar residues in the apo- (PDB 1HX1) state to their position in the ADPonly, ATPonly and MKT-ATP states. First, we confirmed that electrostatic interactions of E212 with R261, Q245 with R262 and R237 with E283 are observed in the apo-state (Figure 4C). Consistent with the weakened affinity of the complex in the ADP-bound state,34 we found that the many of the predicted contacts, except possibly E212-R262 and R237-E283, were disrupted upon ADP binding. Similarly, only a few contacts, such as E212-R262, remained in the intermediate, ATP-bound state. To understand how MKT-077 might shift this complex, we compared these structures to the MKT-ATP state. We found that the MKT-ATP state resembled aspects of both the weak, ADP- and ATP-bound states, with only the R237-E263 contact predicted to remain. These results provide a possible mechanistic model for understanding how MKT-077 can partially disrupt a PPI at long distance, through regulating the motions of loop12. Specifically, the compound seems to favor the ADP-bound state of Hsp70, which places loop 12 in a conformer that propagates a relatively weak-binding state to the IIB subdomain.
Figure 4.
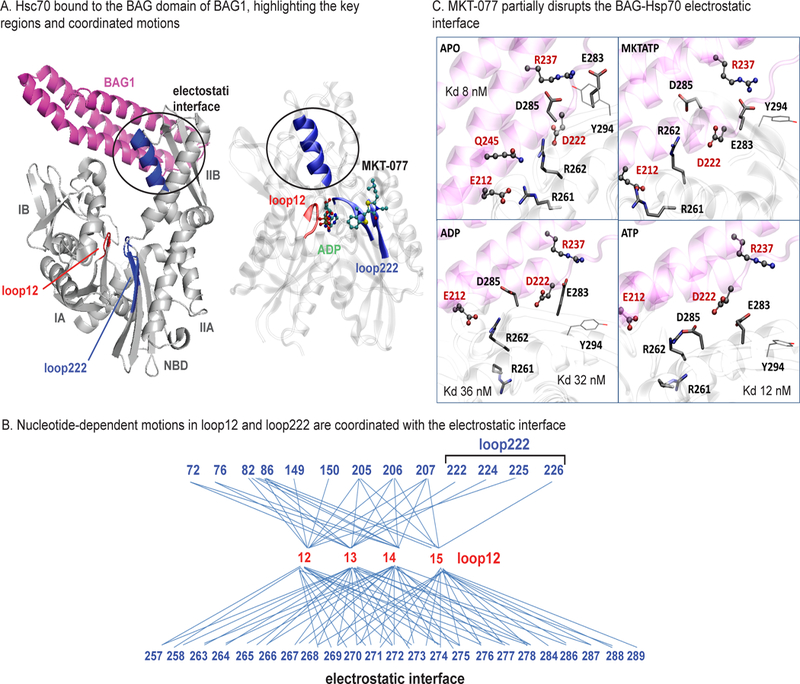
The binding site of MKT-077 is linked to the electrostatic interface with the BAG domain. (A) The structure of Hsc70 bound to the BAG domain of BAG1 (pdb 1HX1) is shown, highlighting the location of the subdomains, loops12 (red) and 222 (blue). A helix involved in electrostatic binding the BAG domain is nestled at the interface of loop12, nucleotide (ADP), MKT-077 and loop 222 (right). Similar coordination was found in the MKT-ATP state (no shown). The BAG domains of other NEFs bind similarly (not shown). (B) Coordinated motions link the position of loop12 to both the MKT-077 binding site (top) and the BAG-binding region (bottom). (C) Close-up of the BAG-binding interface, showing key residues from BAG1 (E212, D222, Q245, R237) and Hsc70 (R261, R262, D285, E283) in the apo-, ADPonly, ATPonly and MKT-ATP states. MKT-077 seems to partially disrupt polar contacts by re-positioning R261, R262, D285 and E283. VMD Licorice and CPK representations pinpoint the main residues involved in the interaction for Hsp70 and BAG1 respectively.
Mutations in key, predicted residues suggest that MKT-077 locks local motions in an allosteric site to favor the ADP-bound state.
The MD simulations suggested that MKT-077 acts as a “rigid stop” to nucleotide-dependent motions in Hsp70’s NBD. To test these predictions, we selected residues predicted to be involved in the nucleotide-dependent motions for mutagenesis. We focused on the prominent motions in residues T226 and D206, which collapse towards the MKT-077 binding pocket upon ATP binding (Figure 5A). We reasoned that alanine mutations (T226A or D206A) might partially disrupt side-chain packing in this locally compact region and limit the ability of ATP to activate its allosteric program. These residues were also selected, in part, because it was previously shown that they partly weaken affinity for MKT-077 analogs6 (Figure S5A). As a control, we also mutated a residue (T222A) at the far opposite end of loop222, because it did not dramatically change position during transition to the ATP-bound state (see Figure 5A). To test whether the purified point mutants of Hsp70, T226A, D206A and T222A, were properly folded, we performed malachite green (MG) ATP turnover assays, as previously described,42 and compared their activities to that of wild type, full length, human Hsc70. This experiment confirmed that each mutant had similar, albeit weak, enzymatic activity to WT (~2 pmol/µM Hsc70/min) (Figure 5B).
Figure 5.
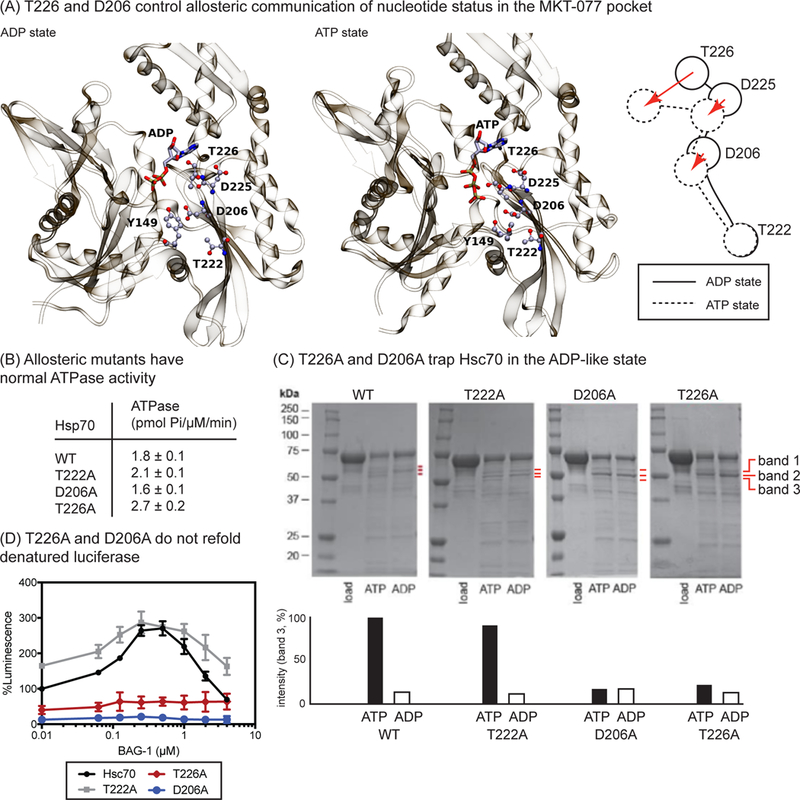
Point mutations in loop222 and loop199 compromise ATP-dependent transitions. (A) Comparison of ATP- and ADP-bound states of Hsc70, highlighting specific residues that undergo major conformational transitions in the MKT-077 binding site. (B) Hsc70 mutants are functional in ATPase assays. (C) The allosteric mutants T226A and D206A are ‘trapped’ in the ADP-like state, as judged by partial proteolysis. Results are representative of experiments performed in duplicate. Bands 1, 2 and 3 are indicated by the red lines. (D) Mutants T226A and D206A are defective in BAG1-stimulated luciferase refolding. Results are the average of experiments performed in triplicate and error bars are standard error measurement (SEM).
Based on the MD simulations, we predicted that mutations at T226A and D206A might interrupt the ability of ATP to trigger conformational transitions. To test this idea, we took advantage of partial proteolysis as a convenient way to discern between the ATP- and ADP-like structural states in this system.43 Briefly, in an Hsp70 partial proteolysis experiment, the appearance of specific bands, including one corresponding to NBD at ~ 44 kDa and bands 1, 2 and 3 located between 50 and 65 kDa, is diagnostic of the nucleotide status.10 Band 3 is particularly informative, as it has been shown to appear largely in the ATP-bound state.10 Likewise, free NBD (~44 kDa) and band 2 are more prominent in the ADP-bound state, likely because the two domains become dissociated from one another in that conformation,18 revealing a trypsin site.44 Thus, the relative changes in band intensity are often used to estimate whether Hsp70 is predominantly in the ATP-or ADP-like states.31, 44 In our experiments, we first we confirmed that limited treatment of human Hsc70 with trypsin in the ATP-bound state (1 mM) generates the expected set of three fragments (bands 1, 2 and 3) between 50 and 65 kDa (Figure 5C), while band 3 largely disappears in the presence of ADP (Figure 5C). As expected, the nucleotide-dependent change in band 3 intensity was retained in the control mutant, T222A. However, both D206A and T226A remained trapped in the ADP-like conformation, even if ATP was present (Figure 4C). This result is strikingly similar to previous observations, in which treatment with an analog of MKT-077 stabilizes an ADP-like state by partial proteolysis (also see below).31 Together, these results support the model in which MKT-077 communicates with the nucleotide-binding cleft through coordinated motions.
To understand the functional implications of trapping the ADP-like conformer, we measured the luciferase refolding activity of WT Hsc70 and its mutants. Briefly, Hsc70 is known to require ATP, as well as JDPs, such as DnaJA2, and NEFs, such as BAG1, to restore denatured firefly luciferase in vitro.45 Moreover, analogs of MKT-077 are known to interrupt this activity, by blocking the required PPIs with BAG1.32 To see if the mutants might share this inhibitory activity, we first confirmed that both WT and T222A are able to work with BAG1 to refold luciferase (Figure 5D). In this assay, the Hsc70 and DnaJA2 concentrations are held constant, while the levels of BAG1 are increased, resulting in a characteristic curve shape with a maximum at ~0.4 µM of BAG1. Next, we tested whether D206A and T226A would be able to support chaperone activity, but found that they were both unable to catalyze the refolding of luciferase (Figure 5D). These results suggest that disruption of the local allosteric network in Hsc70, mirroring treatment with MKT-077 analogs, interrupts functional collaboration with NEFs.
Finally, we envisioned that this atomistic, mechanistic insight might enable rational design of MKT-077 analogs that better resisted the conformational changes associated with ATP binding. In that effort, we started with JG-98 (Figure 6A), an MKT-077 analog with improved metabolic stability.5 Guided by docking and observations from previous analogs (Figure S5B), we predicted that appending a bulky group to the ortho-position of the pendant phenyl ring, to create JG-237 (Figure 6A), might interfere with motions of loop222 by encumbering residues such as T226 and D225 (Figure 6B). To test this idea, we also moved the substitution around the ring to the less optimal, meta- and para-positions (JG-115 and JG-118, respectively). Finally, the negative control, JG-258, which cannot bind Hsc70, and the positive controls, JG-231 and JG-294, were prepared (Figure S5B)6 Consistent with the model, we found that JG-237 (16 µM; 1 equivalent) ‘trapped’ the ADP-like state, as measured by partial proteolysis (Figure 6C). Further, JG-231 was able to partially suppress binding of BAG3 to Hsp70 in MCF7 cell lysates, as measured by co-immunoprecipitation (Figure 6D). Moreover, we found that it was 2-fold better than JG-98 in suppressing proliferation of MCF7 (0.37 ± 0.22 µM) and MDA-MB-231 (0.14 ± 0.02 µM) breast cancer cells, as measured by MTT assays, while being ~10-fold less toxic to normal, healthy mouse embryonic fibroblasts (MEFs; Figure 6E). This potency was similar to that of optimized molecules, JG-231 and JG-294, which resulted from a recent property-guided, medicinal chemistry campaign.6 Moreover, consistent with the model, both JG-115 and JG-118 had activity that was similar to the parent molecule, JG-98 (EC50 ~ 0.6 µM in MCF7 cells; EC50 0.4 to 1.2 µM in MDA-MB-231 cells). Finally, we wondered if the improved cell-based activity of JG-237 may be due to better binding to Hsp70. Because the molecules work through an allosteric mechanism, we considered two possibilities; specifically, JG-237 might bind tighter to Hsp70 than JG-98 or, alternatively, it may have a similar affinity but improved ability to resist motions in loop222. Using a direct binding assay,6 we found that JG-237 had a slightly improved affinity (1.5 ± 0.3 µM) for human Hsc70 when compared to JG-98, JG-115 or JG-118 (Kd ~ 4.5, 3.1 and 2.6 µM, respectively). The control molecule, JG-258, had unmeasurable affinity (Kd > 10 µM). Thus, JG-237 did seem to have slightly improved affinity. Together, these results suggest that accentuating the “rigid stop” ability of MKT-077 analogs, by better binding the pocket and resisting motions of loop222, may be an effective way to drive their potency. More broadly, this knowledge seems especially important for allosteric molecules, where potency is a product of both affinity and conformational sampling.
Figure 6.
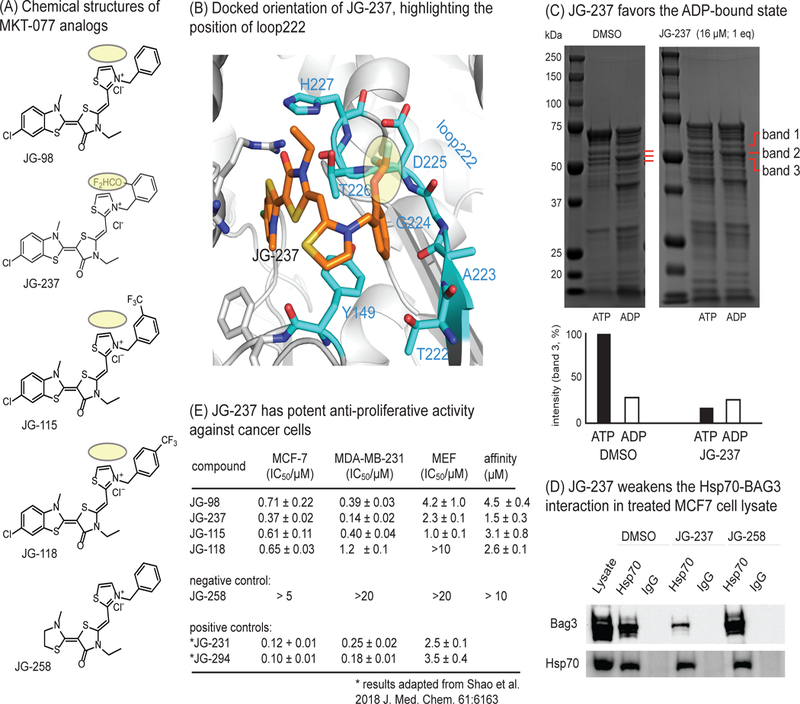
Development of an improved allosteric inhibitor of Hsc70. (A) Chemical structures of MKT-077 analogs. The bulky group addition in JG-237 is highlighted in yellow. (B) Docked structure of JG-237 bound to Hsc70’s NBD, with the position of loop222 shown. Appending a bulky group to the ortho-position of the phenyl group (yellow) is predicted to improve steric blockade of the loop222 movement. (C) Partial proteolysis confirms that JG-237 traps the ADP-like state, even when ATP is present. (D) MCF7 cell lysates were treated with JG-237 (10 µM), followed by co-immunoprecipitation with anti-Hsp70 and Western blotting for BAG3. Results are representative of duplicates. (E) JG-237 has improved anti-proliferative activity, as measured by MTT assays. Results are the average of experiment performed in triplicate and the error bars represent SEM. For reference, the activity values for positive controls (labeled with an asterisk) from a medicinal chemistry campaign are shown. The affinity of analogs for purified Hsc70 were measured by monitoring a quench in compound fluorescence upon binding. The results are average of two independent experiments performed in triplicate and error is SEM. Affinity values are typically weaker than apparent potency values for this series due to accumulation in the cancer cells. Also see Figure S5
Conclusions
Together, our findings define a local allosteric network in Hsc70 that involves cross-talk between the nucleotide- and MKT-077 binding pockets. It is known that ATP normally stabilizes a relatively closed and rigid conformation of the NBD.37, 46 However, we found that MKT-077 limits conversion to this ATP-bound state by occupying a pocket adjacent the nucleotide-binding cleft and preventing residues, such as T226 and D206, from collapsing into this space. Thus, MKT-077 seems to be a ‘rigid-stop’ to the scissor motions of the NBD’s A and B lobes. It is also important to mention that MKT-077 might disrupt other, established allosteric pathways in Hsc70 at the same time. For example, the switch residue, Pro147, is known to communicate nucleotide-dependent conformations to the SBD.26 This residue is located on nearby loop147, so it is likely impacted by binding to MKT-077. Similarly, Tyr149, also located in loop147, was recently reported47–49 as a molecular switch that connects loop12 and loop199. Our work extends these previous observations by providing specific insight into how a chemical probe, MKT-077, might limit the coordinated motions in this system by placing itself at an allosteric “crossroads”. For example, a major hypothesis of this work is that better MKT-077 analogs might be assembled by counter-acting the ATP-driven motions, a model that we tested with JG-237 (see Figure 6). In further support of this idea, a number of the most potent, previously reported MKT-077 analogs also have bulky substitutions near loop222 (see Figure S5A).6 Together, these findings suggest that atomistic knowledge of the allosteric mechanisms of MKT-077 can be leveraged to rationally guide the design of improved analogs. This concept may provide a broader framework for improving allosteric inhibitors of other targets, such as kinases.50
Another interesting aspect of MKT-077 function is that it perturbs the PPIs between Hsc70 and the BAG family of NEFs.32, 41 This observation was initially puzzling because the contact surface of the Hsc70-NEF interaction is ~ 18 to 20 Å away from the compound’s binding site.25 Thus, MKT-077 seemed to be an example of a molecule that acts allosterically to break a distal PPI. A recent analysis of all reported PPI inhibitors suggested that molecules with an allosteric, rather than orthosteric, mechanism-of-action might be more suited to “difficult” PPIs51; such as those with large buried surface area (BSA) and/or weak affinity (Kd > 200 nM). In turn, this makes it important to establish ways of studying the mechanisms of such allosteric PPI inhibitors. Here, we found that MKT-077 acts on a local allosteric network, but one that links nucleotide status to broader conformational motions through the position of loop12 (see Figure 4). The reorientation of this loop is needed to stabilize the BAG-binding conformation,38 so we envision that, by locking loop12 in its ADP-like state, MKT-077 may be able to shift the equilibrium of the accessible ensembles towards the weaker BAG-binding states. Thus, using a combination of computational and experimental approaches, these results suggest how a small molecule works on a topologically challenging PPI by enacting local and long-distance allosteric programs.
Materials and Methods
Molecular dynamic simulations
MD simulations of Hsc70 NBD (PDB code: 3C7N)35 were performed using the AMBER 12.0 package52 with the AMBER ff99SB.53 The PDB code 3C7N was used as the starting point for these studies because it is the structure initially used to map the binding site of MKT-077.31 Further, we found that the residues in this pocket are nearly identical in the other available NBD structures (RMSD of heavy atoms < 1 Å), so, at least in terms of the MKT-077 pocket, 3C7N was considered representative. The simulations were run in the presence or in the absence of the MKT-077 (bound at its NMR-determined binding site, see Figure 1C). The ATP-state was generated by building an ATP molecule from the ADP scaffold in the nucleotide-binding site of Hsp70. The parametrization of the ligands was obtained by means of a GAFF force field. In order to improve the description of the Mg2+ ion bound at the nucleotide cavity, the magnesium-cationic dummy atom model (MD62+) was employed.54 All the structures were solvated with TIP3P water molecules55, counter-ions were randomly added to ensure overall charge neutrality. The system was first minimized using the steepest descendent scheme. Secondly a 0.4 ns simulation was run in a NVT (constant Number, Volume and Temperature) ensemble in which the positions of alpha carbons were weakly restrained (force constant 10 kcal mol−1 Å−2) and slowly increasing the temperature up to 300 K. Later a 0.2 ns simulation in a NPT (constant Number, Pressure and Temperature) ensemble maintaining the restraints on alpha carbons was performed. Finally, the whole system was simulated under NPT conditions for 100 ns at (300 K, 1 atm) with a 1 fs time-step. All the simulations were performed in periodic boundary condition (PBC). Short-range electrostatic and van der Waals interactions were calculated within a 10 Å cutoff, whereas long-range electrostatic interactions were assessed using the particle Ewald method.56 Shake algorithm was used to treat all bonds involving hydrogen atoms.57 All simulations were run in two replicates. A total sampling time of more than 650ns for each complex was analyzed. The docked co-structures in Figures 1C and5B were generated as previously described.6 Briefly, MKT-077 or JG-237 were docked to bovine HSPA8 NBD crystal structure (PDB accession code: 3HSC)58 using both InducedFit and Glide docking software (Software Suite 2017–3, Schrodinger Inc).
Cluster analysis.
Cluster analysis was carried out using g_cluster module of Gromacs to evaluate the structural effects on Hsc70 NBD driven by the nucleotides and/or MKT-077 binding. Clustering of the trajectories was obtained with gromos method fitting backbone atoms with a RMSD cutoff of 0.2 nm. Two different types of inspections were performed. The cluster analysis was performed fitting the 4 Å cavity of the MKT-077 to characterize the conformational rearrangement induced by the ligand on its binding pocket. To study the global motions instead the analysis was carried on fitting the whole protein.
Principal components analysis (PCA)
PCA of MD-trajectories was used to assess the effects of ligand-induced perturbations on the collective coordinates that best account for the proteins’ global structural fluctuations. Gromacs module g_covar and g_anaeig were employed to calculate the covariance matrix on Cα atoms and performed the PCA analysis.
Local flexibility (LF)
Local flexibility was used to assess the intrinsic plasticity properties of the structural fluctuations. LF is obtained by calculating the time-dependent mean square fluctuation of the distance rij between Cα atoms of neighboring residues j comprised in the interval (i − 2, i + 2) along the sequence.14, 59
Hydrogen bond network
Hydrogen bonds analysis was performed by means of ptraj module of AMBER12. Each residue of the MKT-077 cavity that established a hydrogen bond for at least 15% of the whole simulation time was considered.
Protein expression and purification
Full-length human Hsc70 (HSPA8), BAG1, DnaJA2 and mutants were expressed in E. coli BL21 (DE3) cells and purified as described.34 Briefly, cultures of Terrific Broth were grown at 37 °C until an OD600 of 0.6, cooled to 25 °C and induced with isopropyl β-D-1-thiogalactopyranoside (IPTG; final concentration of 500 μM) and then grown overnight at 25 °C. After sonication and centrifugation, pellets were re-suspended in His-binding buffer (50 mM TRIS, 10 mM Imidazole, 500 mM NaCl, pH 8) supplemented with protease inhibitor cocktail. The supernatant was then applied to Ni-NTA His-Bind Resin (Novagen, Darmstadt, Germany) and washed twice. Finally, the proteins were eluted using a His-elution buffer (50 mM TRIS, 300 mM Imidazole, 300 mM NaCl, pH 8). Eluted proteins were concentrated and stored in 50 mM TRIS, 300 mM NaCl, pH 7.4 buffer without removing the N-terminal His-tag.
Biochemical assays
The ATPase rate of Hsc70 and its mutants was measured using malachite green (MG) assays, as previously described.42 Background signal from non-specific ATP hydrolysis was subtracted. A phosphate standard curve of potassium dibasic phosphate was used to calculate pmol Pi/µM Hsc70/min. Stimulation curves were fit to a modified Michaelis-Menten equation. All experiments were performed at least twice in triplicate. All experimental data were analyzed using GraphPad Prism 6 software. Luciferase refolding was performed according the method previously reported.34 Briefly, guanadinium hydrochloride-denatured firefly luciferase stocks were prepared and stored at −80 °C. In 96-well plates, denatured luciferase (100 nM), Hsp70 (1 µM), DnaJA2 (0.1 µM) and BAG1 were added to give a final volume of 25 µL in refolding buffer (23 mM HEPES, 120 mM KAc, 1.2 mM MgAc, 15 mM DTT, 60 mM creatine phosphate, 35 U/mL creatine kinase, 5 ng/µL BSA, pH 7.4). The reaction was initiated by adding 10 μL of 2.5 mM ATP. Plates were incubated at 37 °C for 1 h and Steady-Glo reagent (Promega) was added (25 µL). Luminescence values were measured using a Molecular Devices Spectramax M5 plate reader (Sunnyvale, CA). Partial proteolysis experiments were performed using the procedure described.31 Briefly, samples of Hsc70 were prepared in a partial proteolysis buffer (40 mM HEPES, 20 mM NaCl, 8 mM MgCl2, 20 mM KCl, 0.3 mM EDTA, pH 8.0) with either 1 mM ATP or ADP. Samples were incubated at rt for 30 minutes prior to addition of trypsin (EC 3.4.21.4, Sigma) at a 1:4 molar ratio (trypsin:Hsc70). Proteolysis was carried out for 40 minutes and quenched with 25 µL of SDS loading buffer (240 mM TRIS, 6% (w/v) SDS, 30% (v/v) glycerol, 16% (v/v) β-mercaptoethanol, 0.6 mg/mL bromophenol blue, pH 6.8) and then heated at 95 °C for 3 minutes. Samples were separated by SDS-PAGE using 10% Mini-PROTEAN TGX Precast Gels (cat # =4561036, BioRad) and stained with Coomassie blue. Cells (MCF7, MDA-MB-231 and MEFs) were cultured and MTT assays were performed as described.6
Cell viability
MCF-7, MDA-MB-231 and MEF viabilities were calculated using an MTT cell viability kit from ATCC (ATCC number: 30–1010 K). IC50 values were derived from dose-response curves plotted and fitted using Prism v.6.0c (GraphPad).
Co-immunoprecipitations
MCF7 cell extracts were prepared in M-PER lysis buffer (Thermo Scientific) and adjusted to 5 mg / mL of total protein. Then, equal 500 μL samples were incubated with either a rabbit polyclonal anti-Hsp70 or Goat IgG and treated with either DMSO or JG-237 (10 µM). After rotating samples overnight at 4 °C and a 4 hr incubation with protein A/G-Sepharose Beads (Santa Cruz), the samples were centrifuged (1000 xg), washed with PBS pH 7.4, and eluted with SDS-loading dye. After separation, the gels were transferred and membranes were blocked in nonfat milk (5% milk in TBS, 0.1% Tween) for 1 hr, incubated with primary antibodies for Hsp70 and BAG3 overnight at 4 °C. Imaging was performed using a horseradish peroxidase-conjugated secondary antibody (Anaspec). Finally, membranes were developed using chemiluminescence (Thermo Scientific, Supersignal® West Pico).6
Hsp70 binding assays
Hsc70 (25 µL) in buffer (25 mM HEPES, 5 mM MgCl2, 10 mM KCl, pH 7.5) were added to 384-well black, low volume plates. Then, 1 µL of test compound in DMSO was added and the solution incubated for ~ 2 hours (rt). Fluorescence readings were measured (Excitation 470 nM; Emission 580 nM) and quenching data fit using Prism v.6.0c (GraphPad).6
Synthesis and characterization
JG-98, JG-258, JG-237, JG-115 and JG-118 were synthesized using a previously reported procedure.6 All compounds were characterized for identity by 1H NMR and LC/MS-MS and for purity by HPLC (>95%).
2-((Z)-((E)-5-(6-chloro-3-methylbenzo[d]thiazol-2(3H)-ylidene)-3-ethyl-4-oxothiazolidin-2-ylidene)methyl)-3-(2-(difluoromethoxy)benzyl)thiazol-3-ium chloride (JG-237). Anal. RP-HPLC: tR 2.48 min, purity 97.0 %. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (d, J = 4.0 Hz, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.88 (d, J = 4.0 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.55 (dd, J = 8.8 , 2.4 Hz, 1H), 7.49 (td, J = 8.0, 1.2 Hz, 1H), 7.42 (dd, J = 8.4, 1.6 Hz, 1H), 7.34 – 7.29 (m, 2H), 7.26 (s, J = 73.2 Hz, 1 H), 6.49 (s, 1H), 5.77 (s, 2H), 4.10 (s, 3H), 4.05 (q, J = 7.2 Hz, 2H), 1.08 (t, J = 7.2 Hz, 3H). ESI-MS: calculated for C25H21ClF2N3OS3+ 564.04; found 564.0.
2-((Z)-((E)-5-(6-chloro-3-methylbenzo[d]thiazol-2(3H)-ylidene)-3-ethyl-4-oxothiazolidin-2-ylidene)methyl)-3-(3-(trifluoromethyl)benzyl)thiazol-3-ium chloride (JG-115). Anal. RP-HPLC: tR 2.55 min, purity 95.0 %. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (d, J = 4.0 Hz, 1H), 8.10 (s, 1H), 7.92 (s, 1H), 7.89 (s,1H), 7.78–7.63 (m, 3H), 7.56 (t, J = 8.8 Hz, 2H), 6.53 (s, 1H), 5.86 (s, 2H), 4.17–4.03 (m, 5H), 0.94 (t, J = 7.2 Hz, 3H). ESI-MS: calculated for C25H20ClF3N3OS3+ 566.04; found 566.0.
2-((Z)-((E)-5-(6-chloro-3-methylbenzo[d]thiazol-2(3H)-ylidene)-3-ethyl-4-oxothiazolidin-2-ylidene)methyl)-3-(4-(trifluoromethyl)benzyl)thiazol-3-ium chloride (JG-118). Anal. RP-HPLC: tR 2.56 min, purity 98.0 %. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (d, J = 4.0 Hz, 1H), 8.07 (d, J = 2.4 Hz, 1H), 7.94 (d, J = 4.0 Hz, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.8 Hz, 1H), 7.58–7.52 (m, 3H), 6.49 (s, 1H), 5.93 (s, 2H), 4.09 (s, 3H), 4.08 (q, J = 7.2 Hz, 2H), 0.91 (t, J = 7.2 Hz, 3H). ESI-MS: calculated for C25H20ClF3N3OS3+ 566.04; found 566.0.
Supplementary Material
Acknowledgements
Our work is supported by grants from the National Institutes of Health (NS059690) to J.E.G. and from AIRC (Associazione Italiana Ricerca sul Cancro) through Grant IG 20019 to G.C. and through a personal fellowship to S.R. The authors thank Isabelle Taylor (UCSF) for helpful comments and Chakrapani Kalyanaraman (UCSF) for assistance with the molecular docking.
Footnotes
Supporting Information
Supporting information (Figures S1–4), including additional analyses of the MD simulations, is available on-line.
Citations
- 1.Ferraro M; D’Annessa I; Moroni E; Morra G; Paladino A; Rinaldi S; Compostella F; Colombo G Allosteric modulators of hsp90 and hsp70: Dynamics meets function through structure-based drug design. J Med Chem 2018, 10.1021/acs.jmedchem.8b00825. [DOI] [PubMed] [Google Scholar]
- 2.Tatsuta N; Suzuki N; Mochizuki T; Koya K; Kawakami M; Shishido T; Motoji N; Kuroiwa H; Shigematsu A; Chen LB Pharmacokinetic analysis and antitumor efficacy of mkt-077, a novel antitumor agent. Cancer Chemother Pharmacol 1999, 43, 295–301 [DOI] [PubMed] [Google Scholar]
- 3.Evans CG; Chang L; Gestwicki JE Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem 2010, 53, 4585–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wadhwa R; Sugihara T; Yoshida A; Nomura H; Reddel RR; Simpson R; Maruta H; Kaul SC Selective toxicity of mkt-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res 2000, 60, 6818–6821 [PubMed] [Google Scholar]
- 5.Li X; Srinivasan SR; Connarn J; Ahmad A; Young ZT; Kabza AM; Zuiderweg ER; Sun D; Gestwicki JE Analogs of the allosteric heat shock protein 70 (hsp70) inhibitor, mkt-077, as anti-cancer agents. ACS Med Chem Lett 2013, 4, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shao H; Li X; Moses MA; Gilbert LA; Kalyanaraman C; Young ZT; Chernova M; Journey SN; Weissman JS; Hann B; Jacobson MP; Neckers LM; Gestwicki JE Exploration of benzothiazole-rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (hsp70). J Med Chem 2018, 10.1021/acs.jmedchem.8b00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer MP Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci 2013, 38, 507–514 [DOI] [PubMed] [Google Scholar]
- 8.Pellecchia M; Montgomery DL; Stevens SY; Vander Kooi CW; Feng HP; Gierasch LM; Zuiderweg ER Structural insights into substrate binding by the molecular chaperone dnak. Nat Struct Biol 2000, 7, 298–303 [DOI] [PubMed] [Google Scholar]
- 9.Buczynski G; Slepenkov SV; Sehorn MG; Witt SN Characterization of a lidless form of the molecular chaperone dnak: Deletion of the lid increases peptide on- and off-rate constants. J Biol Chem 2001, 276, 27231–27236 [DOI] [PubMed] [Google Scholar]
- 10.Buchberger A; Theyssen H; Schroder H; McCarty JS; Virgallita G; Milkereit P; Reinstein J; Bukau B Nucleotide-induced conformational changes in the atpase and substrate binding domains of the dnak chaperone provide evidence for interdomain communication. J Biol Chem 1995, 270, 16903–16910 [DOI] [PubMed] [Google Scholar]
- 11.Kityk R; Vogel M; Schlecht R; Bukau B; Mayer MP Pathways of allosteric regulation in hsp70 chaperones. Nature communications 2015, 6, 8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smock RG; Rivoire O; Russ WP; Swain JF; Leibler S; Ranganathan R; Gierasch LM An interdomain sector mediating allostery in hsp70 molecular chaperones. Mol Syst Biol 2010, 6, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhuravleva A; Clerico EM; Gierasch LM An interdomain energetic tug-of-war creates the allosterically active state in hsp70 molecular chaperones. Cell 2012, 151, 1296–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiappori F; Merelli I; Milanesi L; Colombo G; Morra G An atomistic view of hsp70 allosteric crosstalk: From the nucleotide to the substrate binding domain and back. Sci Rep 2016, 6, 23474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kityk R; Kopp J; Sinning I; Mayer MP Structure and dynamics of the atp-bound open conformation of hsp70 chaperones. Mol Cell 2012, 48, 863–874 [DOI] [PubMed] [Google Scholar]
- 16.Yang J; Nune M; Zong Y; Zhou L; Liu Q Close and allosteric opening of the polypeptide-binding site in a human hsp70 chaperone bip. Structure 2015, 23, 2191–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison CJ; Hayer-Hartl M; Di Liberto M; Hartl F; Kuriyan J Crystal structure of the nucleotide exchange factor grpe bound to the atpase domain of the molecular chaperone dnak. Science 1997, 276, 431–435 [DOI] [PubMed] [Google Scholar]
- 18.Bertelsen EB; Chang L; Gestwicki JE; Zuiderweg ER Solution conformation of wild-type e. Coli hsp70 (dnak) chaperone complexed with adp and substrate. Proc Natl Acad Sci U S A 2009, 106, 8471–8476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlecht R; Erbse AH; Bukau B; Mayer MP Mechanics of hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol 2011, 18, 345–351 [DOI] [PubMed] [Google Scholar]
- 20.Swain JF; Gierasch LM The changing landscape of protein allostery. Curr Opin Struct Biol 2006, 16, 102–108 [DOI] [PubMed] [Google Scholar]
- 21.Liberek K; Marszalek J; Ang D; Georgopoulos C; Zylicz M Escherichia coli dnaj and grpe heat shock proteins jointly stimulate atpase activity of dnak. Proc Natl Acad Sci U S A 1991, 88, 2874–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zuiderweg ER; Hightower LE; Gestwicki JE The remarkable multivalency of the hsp70 chaperones. Cell Stress Chaperones 2017, 22, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmad A; Bhattacharya A; McDonald RA; Cordes M; Ellington B; Bertelsen EB; Zuiderweg ER Heat shock protein 70 kda chaperone/dnaj cochaperone complex employs an unusual dynamic interface. Proc Natl Acad Sci 2011, 108, 18966–18971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kityk R; Kopp J; Mayer MP Molecular mechanism of j-domain-triggered atp hydrolysis by hsp70 chaperones. Mol Cell 2018, 69, 227–237 [DOI] [PubMed] [Google Scholar]
- 25.Bracher A; Verghese J The nucleotide exchange factors of hsp70 molecular chaperones. Frontiers Mol Biosci 2015, 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vogel M; Bukau B; Mayer MP Allosteric regulation of hsp70 chaperones by a proline switch. Mol Cell 2006, 21, 359–367 [DOI] [PubMed] [Google Scholar]
- 27.Chang L; Thompson AD; Ung P; Carlson HA; Gestwicki JE Mutagenesis reveals the complex relationships between atpase rate and the chaperone activities of escherichia coli heat shock protein 70 (hsp70/dnak). J Biol Chem 2010, 285, 21282–21291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y; Gierasch LM; Bahar I Role of hsp70 atpase domain intrinsic dynamics and sequence evolution in enabling its functional interactions with nefs. PLoS Comp Biol 2010, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodina A; Patel PD; Kang Y; Patel Y; Baaklini I; Wong MJ; Taldone T; Yan P; Yang C; Maharaj R; Gozman A; Patel MR; Patel HJ; Chirico W; Erdjument-Bromage H; Talele TT; Young JC; Chiosis G Identification of an allosteric pocket on human hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem Biol 2013, 20, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liebscher M; Roujeinikova A Allosteric coupling between the lid and interdomain linker in dnak revealed by inhibitor binding studies. J Bacteriol 2009, 191, 1456–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rousaki A; Miyata Y; Jinwal UK; Dickey CA; Gestwicki JE; Zuiderweg ER Allosteric drugs: The interaction of antitumor compound mkt-077 with human hsp70 chaperones. J Mol Biol 2011, 411, 614–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young ZT; Rauch JN; Assimon VA; Jinwal UK; Ahn M; Li X; Dunyak BM; Ahmad A; Carlson GA; Srinivasan SR; Zuiderweg ER; Dickey CA; Gestwicki JE Stabilizing the hsp70-tau complex promotes turnover in models of tauopathy. Cell Chem Biol 2016, 23, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X; Colvin T; Rauch JN; Acosta-Alvear D; Kampmann M; Dunyak B; Hann B; Aftab BT; Murnane M; Cho M; Walter P; Weissman JS; Sherman MY; Gestwicki JE Validation of the hsp70-bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol Cancer Ther 2015, 14, 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rauch JN; Gestwicki JE Binding of human nucleotide exchange factors to heat shock protein 70 (hsp70) generates functionally distinct complexes in vitro. J Biol Chem 2014, 289, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuermann JP; Jiang J; Cuellar J; Llorca O; Wang L; Gimenez LE; Jin S; Taylor AB; Demeler B; Morano KA; Hart PJ; Valpuesta JM; Lafer EM; Sousa R Structure of the hsp110:Hsc70 nucleotide exchange machine. Mol Cell 2008, 31, 232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi R; Sarbeng EB; Liu Q; Le KQ; Xu X; Xu H; Yang J; Wong JL; Vorvis C; Hendrickson WA; Zhou L; Liu Q Allosteric opening of the polypeptide-binding site when an hsp70 binds atp. Nat Struct Mol Biol 2013, 20, 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo HJ; Jiang J; Lafer EM; Sousa R Atp-induced conformational changes in hsp70: Molecular dynamics and experimental validation of an in silico predicted conformation. Biochemistry 2009, 48, 11470–11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sondermann H; Scheufler C; Schneider C; Hohfeld J; Hartl FU; Moarefi I Structure of a bag/hsc70 complex: Convergent functional evolution of hsp70 nucleotide exchange factors. Science 2001, 291, 1553–1557 [DOI] [PubMed] [Google Scholar]
- 39.Briknarova K; Takayama S; Brive L; Havert ML; Knee DA; Velasco J; Homma S; Cabezas E; Stuart J; Hoyt DW; Satterthwait AC; Llinas M; Reed JC; Ely KR Structural analysis of bag1 cochaperone and its interactions with hsc70 heat shock protein. Nat Struct Biol 2001, 8, 349–352 [DOI] [PubMed] [Google Scholar]
- 40.Bimston D; Song J; Winchester D; Takayama S; Reed JC; Morimoto RI Bag-1, a negative regulator of hsp70 chaperone activity, uncouples nucleotide hydrolysis from substrate release. EMBO J 1998, 17, 6871–6878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colvin TA; Gabai VL; Gong J; Calderwood SK; Li H; Gummuluru S; Matchuk ON; Smirnova SG; Orlova NV; Zamulaeva IA; Garcia-Marcos M; Li X; Young ZT; Rauch JN; Gestwicki JE; Takayama S; Sherman MY Hsp70-bag3 interactions regulate cancer-related signaling networks. Cancer Res 2014, 74, 4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang L; Bertelsen EB; Wisén S; Larsen EM; Zuiderweg ER; Gestwicki JE High-throughput screen for small molecules that modulate the atpase activity of the molecular chaperone dnak. Anal Biochem 2008, 372, 167–176 [DOI] [PubMed] [Google Scholar]
- 43.Kamath-Loeb AS; Lu CZ; Suh WC; Lonetto MA; Gross CA Analysis of three dnak mutant proteins suggests that progression through the atpase cycle requires conformational changes. J Biol Chem 1995, 270, 30051–30059 [DOI] [PubMed] [Google Scholar]
- 44.Needham PG; Patel HJ; Chiosis G; Thibodeau PH; Brodsky JL Mutations in the yeast hsp70, ssa1, at p417 alter atp cycling, interdomain coupling, and specific chaperone functions. J Mol Biol 2015, 427, 2948–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levy EJ; McCarty J; Bukau B; Chirico WJ Conserved atpase and luciferase refolding activities between bacteria and yeast hsp70 chaperones and modulators. FEBS Lett 1995, 368, 435–440 [DOI] [PubMed] [Google Scholar]
- 46.Bhattacharya A; Kurochkin AV; Yip GN; Zhang Y; Bertelsen EB; Zuiderweg ER Allostery in hsp70 chaperones is transduced by subdomain rotations. J Mol Biol 2009, 388, 475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shida M; Arakawa A; Ishii R; Kishishita S; Takagi T; Kukimoto-Niino M; Sugano S; Tanaka A; Shirouzu M; Yokoyama S Direct inter-subdomain interactions switch between the closed and open forms of the hsp70 nucleotide-binding domain in the nucleotide-free state. Acta Crystallogr D Biol Crystallogr 2010, 66, 223–232 [DOI] [PubMed] [Google Scholar]
- 48.Sousa MC; McKay DB The hydroxyl of threonine 13 of the bovine 70-kda heat shock cognate protein is essential for transducing the atp-induced conformational change. Biochemistry 1998, 37, 15392–15399 [DOI] [PubMed] [Google Scholar]
- 49.Johnson ER; McKay DB Mapping the role of active site residues for transducing an atp-induced conformational change in the bovine 70-kda heat shock cognate protein. Biochemistry 1999, 38, 10823–10830 [DOI] [PubMed] [Google Scholar]
- 50.Wylie AA; Schoepfer J; Jahnke W; Cowan-Jacob SW; Loo A; Furet P; Marzinzik AL; Pelle X; Donovan J; Zhu W; Buonamici S; Hassan AQ; Lombardo F; Iyer V; Palmer M; Berellini G; Dodd S; Thohan S; Bitter H; Branford S; Ross DM; Hughes TP; Petruzzelli L; Vanasse KG; Warmuth M; Hofmann F; Keen NJ; Sellers WR The allosteric inhibitor abl001 enables dual targeting of bcr-abl1. Nature 2017, 543, 733–737 [DOI] [PubMed] [Google Scholar]
- 51.Ran X; Gestwicki JE Inhibitors of protein-protein interactions (ppis): An analysis of scaffold choices and buried surface area. Curr Opin Chem Biol 2018, 44, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Case DA, T. A. D., Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Götz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, and Kollman PA (2012), AMBER 12, University of California, San Francisco. [Google Scholar]
- 53.Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the amber ff99sb protein force field. Proteins 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oelschlaeger P; Klahn M; Beard WA; Wilson SH; Warshel A Magnesium-cationic dummy atom molecules enhance representation of DNA polymerase beta in molecular dynamics simulations: Improved accuracy in studies of structural features and mutational effects. J Mol Biol 2007, 366, 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jorgensen WL, Chandrasekhar J, Madura JD, Impey W, Klein ML Tip3p water model: Comparison of simple potential functions for simulating water. J . Chem. Phys 1983, 79, 926 [Google Scholar]
- 56.Salomon-Ferrer R; Gotz AW; Poole D; Le Grand S; Walker RC Routine microsecond molecular dynamics simulations with amber on gpus. 2. Explicit solvent particle mesh ewald. J Chem Theory Comput 2013, 9, 3878–3888 [DOI] [PubMed] [Google Scholar]
- 57.Ryckaert JP, Ciccotti G, Berendsen HJC Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Physics 1977, 23, 327–341 [Google Scholar]
- 58.Flaherty KM; DeLuca-Flaherty C; McKay DB Three-dimensional structure of the atpase fragment of a 70k heat-shock cognate protein. Nature 1990, 346, 623–628 [DOI] [PubMed] [Google Scholar]
- 59.Morra G; Potestio R; Micheletti C; Colombo G Corresponding functional dynamics across the hsp90 chaperone family: Insights from a multiscale analysis of md simulations. PLoS Comp Biol 2012, 8, e1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.