

Abstract
In adult cardiomyocytes, T-tubules, junctional sarcoplasmic reticulum (jSR), and mitochondria juxtapose each other and form a unique and highly repetitive functional structure along the cell. The close apposition between jSR and mitochondria creates high Ca2+ microdomains at the contact sites, increasing the efficiency of the excitation-contraction-bioenergetics coupling, where the Ca2+ transfer from SR to mitochondria plays a critical role. The SR-mitochondria contacts are established through protein tethers, with mitofusin 2 the most studied SR-mitochondrial “bridge”, albeit controversial. Mitochondrial Ca2+ uptake is further optimized with the mitochondrial Ca2+ uniporter preferentially localized in the jSR-mitochondria contact sites and the mitochondrial Na+/Ca2+ exchanger localized away from these sites. Despite all these unique features facilitating the privileged transport of Ca2+ from SR to mitochondria in adult cardiomyocytes, the question remains whether mitochondrial Ca2+ concentrations oscillate in synchronicity with cytosolic Ca2+ transients during heartbeats. Proper Ca2+ transfer controls not only the process of mitochondrial bioenergetics, but also of mitochondria-mediated cell death, autophagy/mitophagy, mitochondrial fusion/fission dynamics, reactive oxygen species generation, and redox signaling, among others. Our review focuses specifically on Ca2+ signaling between SR and mitochondria in adult cardiomyocytes. We discuss the physiological and pathological implications of this SR-mitochondrial Ca2+ signaling, research gaps, and future trends.
Keywords: mitofusin 2, mitochondrial Ca2+ uniporter, Calcium, mitochondria, SR-mitochondria contacts, heart
INTRODUCTION
In adult cardiomyocytes, the excitation-contraction-bioenergetics coupling (ECB) process links electrical stimulation of the plasma membrane with the contraction of the muscle and energetics of the mitochondria [1–3]. The excitation of the membrane triggers the opening of L-type Ca2+ channels, initiating the Ca2+-induced Ca2+ release mechanism. The sarcoplasmic reticulum (SR) then liberates a more substantial amount of Ca2+ to the cytosol, resulting in a rapid increase in cytosolic Ca2+ concentration ([Ca2+]c). Free cytosolic Ca2+ binds to troponin C, causing the sliding of the thin and thick filaments to generate sufficient contractile force during systole for the heart to pump the blood. Meanwhile, a certain amount of Ca2+ is taken up by mitochondria to stimulate ATP generation machineries in order to meet the energy demands of the heart. During diastole, the increase in the [Ca2+]c is then quickly returned to basal levels by Ca2+ export through the plasma membrane Na+/Ca2+ exchanger and by re-uptake to SR through the SR Ca2+-ATPase (SERCA). Therefore, the [Ca2+]c fundamentally controls the circulatory function of the heart and thus needs to be regulated tightly.
The transfer of Ca2+ from SR to mitochondria has been described previously[4–6], however, it is worth mentioning that the process not only depends on the Ca2+ transporting proteins’ properties, but also on their spatial distribution along the membranes involved. Furthermore, the apposition of SR-mitochondria (SR-mito) creates localized signals—including reactive oxygen species (ROS), Ca2+, and pH—that can feedback to modulate the nearby ion channels and transporters involved in ECB. Adult cardiac myocytes are highly structured cells, with little room for membrane reorganization or organelle mobility and reconfiguration[7, 8]. Therefore, the cellular components are structured strategically to carry out signaling mechanisms locally. Transverse (T) tubules are deep invaginations of the plasma membrane, located at every junction of two sarcomeres (Z-line). In the dyadic space, the T-tubules are very close to the junctional SR cisternae (jSR) (10-30 nm)[9, 10], optimizing the Ca2+ signal transfer from the L-type Ca2+ to the ryanodine receptors (RyRs). This close association between the elaborated T-tubules and SR is critical to ensure the synchronistic rise of the [Ca2+]c throughout the entire cell for an effective contraction[11, 12]. Defects in this close apposition lead to non-homogeneous Ca2+ transients that make the heart prone to arrhythmias and failure [13].
Mitochondria comprise approximately 30% of the volume in adult cardiomyocytes[14]. This significant amount of mitochondria is essential to satisfy the high demand of the heart for ATP[15, 16]. However, ATP production is not the only function of the mitochondria. Mitochondria also take advantage of their positioning next to the SR Ca2+-releasing sites to uptake cytosolic Ca2+ and, as a final consequence, to sharpen the [Ca2+]c[17–19]. Mitochondrial uptake of Ca2+ during the ECB is widely known to modulate ATP production to ensure the homeostasis of energy demand and supply[20]. This review will focus on what is less well-known: the physical interaction among SR and mitochondria[21–23] in adult cardiomyocytes, how both organelles are connected, and why this connection is critical to control not only a proper Ca2+ signal transfer between SR and mitochondria but also other cellular processes, including bioenergetics, ROS, and mitochondrial dynamics
SR-MITOCHONDRIA CONTACTS IN ADULT CARDIOMYOCYTES.
Cellular organelles are not static and independent structures but rather highly dynamic and interconnected. The connection between endoplasmic reticulum (ER) and mitochondria was reported years ago[24], and recently, fluorescence and EM techniques were used to reveal physical interactions[25, 26]. These techniques revealed that the extent and characteristics of these contacts differ among the different organisms and cell types. Depending on the type of ER involved, the gap distance varies from 10-15 nm for smooth ER to 20-30 nm for rough ER[27]. Regarding the composition, for closer contracts (<30 nm), electron-dense and protein-like structures link the two membranes[28]. The most extensively studied ER-mitochondria (ER-mito) tethering protein complex is the ER-mito encounter structure (ERMES). ERMES complex has been fully characterized in yeast, but no homologous structures have been identified in mammals[29]. Many proteins seem to be involved in the ER-mito contacts in mammals, including the complex IP3R-Grp75-VDAC, VAPB-PTPI51, FUNDC1, and PDZD8[30–33]. However, there is no solid proof that their absence completely abolishes the ER-mito contacts. The complexity of the system is unexpectedly high, and the different components involved may compensate each other upon removal of some individual components.
Cardiac mitochondria can uptake cytosolic Ca2+ most dominantly through a channel called the mitochondrial calcium uniporter complex (MCUC)[34, 35]. However, the reported activation Km for the uniporter is 10 μM, meaning that the channel requires a free Ca2+ concentration of at least 2-5 μM in the sarcoplasm in order to be activated [36]. This concentration is not reached in the bulk sarcoplasm, but it is in Ca2+ microdomains, formed at the mouth of Ca2+-releasing RyR2 channels[37] located in the jSR . In adult cardiomyocytes, the close apposition between jSR and mitochondria favors the Ca2+ transfer from one organelle to the other through these high Ca2+ microdomains[38]. Recent data suggest that the jSR and mitochondria is physically connected through the protein mitofusin 2 (MFN2) as a tether[39]. It is probable that additional tethers are existed for connecting jSR and mitochondria in adult cardiomyocytes. For instance, it has been reported that the mitophagy-related protein FUNDC1 is located in the outer mitochondrial membrane to tether mitochondria for interacting with the IP3R2 on the ER surface of mouse neonatal cardiomyocytes and intact hearts[33]. The role of other classical ER-mito tethers like GRP75 has not been investigated in adult cardiomyocytes.
It is still not clear whether MFN2 acts as a tether or as a spacer in the ER-mito interaction. MFN2 is a dynamin-family GTPase that is mostly present in the outer mitochondrial membrane but can also be found in the ER/SR membranes. It has many functions in the heart, the most well-known being SR-mito tethering and a component of the outer mitochondrial membrane fusion process[40]. The physical structure of MFN2 determines the attachment between SR and mitochondria. Four types of domains configure the MFN2 structure: an N-terminal GTP binding domain; an alpha-helical first heptad repeat (HR1); two following lipophilic transmembrane domains; and a second alpha helical heptad repeated (HR2) C-terminal[41]. It has been broadly accepted that both coiled-coil domains (HR1 and HR2) are exposed to the cytosol, but a recent study showed that in fact the GTPase and HR1 domains are exposed to the cytosol, while the HR2 and the C-terminus are exposed to the intermembrane space[42]. The C-terminal segment of the protein contains the redox-sensitive cysteine residues, which are essential modulators of the MFN2 oligomerization processes. This discovery has challenged the actual view of mitochondrial dynamics and organelle tethering because it means that the whole MFN2 regulation comes from the mitochondrial intermembrane space side instead of from the cytosolic side. In addition to the MFN2 topology controversy, the functional role of MFN2 is also under debate. Two opposite effects have been observed in non-cardiac muscle cells when MFN2 is removed. Scorrano’s lab has shown that the ER-mito distance in MFN2 knockout (KO) cells increases[39, 43] while Pizzo’s lab has shown that is decreases[44, 45].
To elucidate the effects of MFN2 removal in vivo, two independent labs generated cardiac-specific MFN2 KO mouse models. The Dorn lab genereated the mice using a myh6-driven “turbo-cre” which promotes the gene removal only after birth[46] while the Walsh lab using myh6-cre to ablate MFN2 in the embryonic heart[47]. When they analyzed transmission electron microscopic images from their mouse model, the Dorn lab found that the distance between the SR and mitochondria significantly increased (~30%), however, the Walsh lab did not see this change. This discrepancy makes the comparision of phenotypes between these two mouse models challenging, considering the importances of SR-mito associations in dictating cardiac functions. Furthermore, it confirms the complexity of manipulating MFN2 genetically for studying SR-mito associations. It is plausible that this difference is related to the timing of the gene deletion. In Walsh’s studies, MFN2 is removed at the embrionic stage, which may allow other thether mechanims to compensate for the lack of MFN2 and as such render no diference in the SR-mito distance. This difference in the time point for the MFN2 ablation could also be the reason that while the Walsh results showed a significant phenotype in both cardiac and mitochondria in young adults (10-14 weeks), the Dorn lab only showed significant differences between control and MFN2 KO mice in mature animals (16-44 weeks) and not in young mice (6 weeks).
With the detection of a decrease in SR-mito contacts after MFN2 ablation, Dorn’s lab further studied its impacts on Ca2+ signaling between the two organelles. They found that the SR Ca2+ content was increased[46]. The authors interpreted this finding by saying that mitochondria that are physically and functionally linked to SR Ca2+ can act as Ca2+ sponges. In the absence of SR-mito connections, the Ca2+ released by the SR cannot be buffered by mitochondria and will be taken up again by SR, making the SR Ca2+ content higher. Since the authors have previously shown that the Ca2+ uptake by the mitochondria during EC coupling increased the rate of NAD(P)H production and therefore the production of ATP[48], they further confirmed that the decreases in mitochondrial Ca2+ uptake in MFN2 null cardiomyocytes hampered Ca2+-induced stimulation of Krebs cycle dehydrogenases during β-adrenergic stimulation[46]. Surprisingly, the authors also found that MFN2 deficiency cardiomyocytes had similar mitochondrial membrane potential (ΔΨm) and contraction, suggesting that the loss of MFN2 had no direct impact on respiratory chain function or ATP production. It appears that there is a disconnect between the defect in mitochondrial Ca2+ uptake and bioenergetics in MFN2 KO cardiomyocytes. The reasons for diminished mitochondrial Ca2+ uptake without an energetic consequence are not obvious, however, these findings bear some similarities with recent publications showing that the MCU-KO mice don’t experience an energy crisis[49–51].
The major phenotype found in vivo MFN2 null hearts was similar in both models. Cardiac hypertrophy and contractile depression were observed in older mice (30 weeks) but not in younger mice (6 weeks). Only minor hypertrophy was notice for the 6- to 10-week-old mice. This finding points to a progressive increase of the hypertrophy and contractile depression upon MFN2 deletion. The researchers observed also that the MFN2 null hearts are less sensitive to ischemia-reperfusion injury. At the single cardiomycocyte level, the absence of Ca2+ transfer to the mitochondria may prevent mitochondrial matrix overload and therefore avoid mitochondrial permeability transition pore (mPTP) mediated cell death.
Taken together, the structural tethering of SR and mitochondria in adult cardiomyocytes through MFN2 could facilitate the Ca2+ transfer between these two organelles. However, due to the multiple roles of MFN2, the diversity of the ER-mito contact protein family, and the compensatory mechanism between MFN2 and MFN1, the physiological and pathological implications of MFN2 as a SR-mito tether in adult cardiomyocytes are still not yet clear.
MITOCHONDRIAL Ca2+ UPTAKE AND EXTRUSION IN CARDIOMYOCYTES
The central mechanism for the Ca2+ to enter in the mitochondria is the MCUC. The molecular identity of the channel has been elucidated recently, opening a new era for the mitochondrial calcium signaling field[52, 53]. Located in the inner mitochondrial membrane and showing high selectivity for Ca2+, MCUC is a conjunction of several accessories and regulatory subunits. So far, the following proteins have been found to form the MCUC: MCU, MCUb, EMRE, MICUs (1, 2, and 3), EMRE, and MCUR1. MCU is responsible for the pore-forming structure[52, 53], and the most recent studies using Cryo-EM techniques report a tetramer conformation for the channel[54, 55]. MCUb is also a pore-forming component, but its presence in the complex limits the Ca2+ uptake[56]. EMRE is a single-transmembrane subunit essential for the proper function of the whole complex[57]. MICU1 acts both as a gatekeeper at low [Ca2+]c and as an activator for higher [Ca2+]c [58, 59]. MICU2 inhibits Ca2+ at low extramitochondrial Ca2+ concentrations [60]. MICU3, the least studied subunit, appears to act as an activator of the channel[61]. Lastly, MCUR1 binds to MCU and EMRE and functions as a scaffold factor[62]. Early studies reported that the MCUC shows a half-maximal activation (K0.5) in the upper micromolar range[63], and recent patch-clamp experiments have reported an ever higher K0.5, in the millimolar range[64]. These levels are not typically reached in the cardiomyocyte sarcoplasm, so the only plausible explanation for the mitochondrial Ca2+ uptake is that the MCUC is exposed to high Ca2+ microdomains[65]. Taking all of the activation properties of the MCUC into account, the close apposition between SR and mitochondria is necessary to create a high enough Ca2+ microdomain to optimize SR to mitochondria Ca2+ transfer.
The MCUC is not the only mechanism proposed to uptake Ca2+ into cardiac mitochondria. Other described mechanisms include the rapid mode of Ca2+ uptake (RaM) and the ryanodine receptor type 1 (mRyR1). RaM was described in isolated heart mitochondria and found to function in the initial period of repeated extramitochondrial [Ca2+] increases[66]. The Ca2+ uptake is several times faster than the uptake via MCUC, nevertheless, it takes more than 60 s to recover the basal Ca2+ level, making the system unsuitable for a sustained Ca2+ oscillation such as in the heart. Meanwhile the mitochondrial Ca2+ uptake via mRyR1 located in the inner mitochondrial membrane is still controversial. mRyR1 shows similarities with skeletal RyR1 but not with the cardiac RyR2, and several methods prove its presence in the IMM, excluding a potential SR contamination. mRyR1 can be activated at low [Ca2+]c, where the MCUC is still inactive[67, 68]. In addition, a more recent study showed that mRyRl overexpression in H9C2 myoblast increases the ATP amount, consistent with the fact that elevated mitochondrial Ca2+ concentrations ([Ca2+]m) through mRyR1 enhances ATP production[69]. The uncoupler proteins UCP2 and UCP3[70], as well as LETM1[71], have been proposed as possible mitochondrial Ca2+ uptake mechanisms, but their role has not been studied in further detail since the molecular identification of the MCUC.
Two pathways, Na+-dependent and Na+-independent, remove the Ca2+ from the mitochondrial matrix. The Na+-independent extrusion plays little or no role[72] while the Na+-dependent efflux is the predominant pathway in adult cardiomyocytes[73, 74]. The Na+-dependent extrusion occurs via the mitochondrial Na+/Ca2+ exchanger (NCLX), which was recently identified[75]. Early on, researchers debated whether NCLX is electroneutral or electrogenic[74, 76], but then it was found that the exchanger extrudes 1 Ca2+ out of mitochondria per 3 Na+ transported into the mitochondrial matrix[77]. As an electrogenic exchanger, Ca2+ extrusion would lead to the depolarization of mitochondrial membrane potential. It was recently hypothesized that transient opening of the Ca2+-dependent mPTP may prevent Ca2+ overload by providing mitochondria with a fast Ca2+ release channel[78]. Electrophysiological studies have revealed that the mPTP is a large conductance channel with multiple subconductance states, and it may flicker rapidly between a fully closed and subconducted state[79]. Also, oxidative stress has been shown to trigger the repetitive opening and closing of the mPTP in isolated cardiac mitochondria[80]. Finally, during ECB, mitochondria-accumulated Ca2+ can trigger transient mPTP openings and reset matrix Ca2+ and mitochondrial energetics[81].
In such extremely structured cells like the adult cardiomyocytes, both Ca2+ uptake and Ca2+ extrusion are expected to be tightly regulated, not only temporally but also spatially. Our group has reported recently that the MCUC and NCLX are reciprocally excluded in the cardiac mitochondrial inner membrane (IMM). MCUC is located specifically in those areas where the mitochondria are in close contact with the jSR, whereas the NCLX is actively excluded from those areas[82, 83]. Having the Ca2+ uptake and Ca2+ extrusion physically distributed in two separated regions, as mentioned, enormously maximizes the mitochondrial Ca2+ signal and minimizes the energy expenses (depolarizations of mitochondrial membrane potential). In this context, it is worth mentioning that MCU absence in the heart has a very mild phenotype[49], while NCLX absence has catastrophic consequences for the heart[84]. These results suggest that Ca2+ overload could be much more harmful to cardiac mitochondria than Ca2+ depletion.
Figure 1 shows all of the mitochondrial Ca2+ uptake and extrusion mechamisms that have been proposed.
Fig. 1. Scheme of the components involved in the SR to mitochondria Ca2+ transfer.
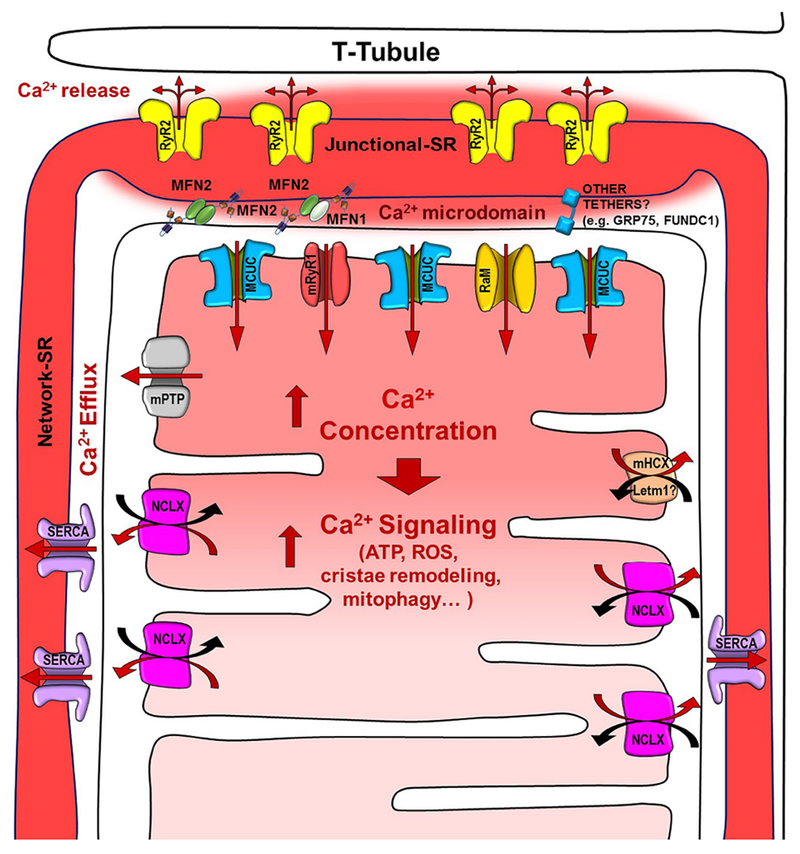
The scheme shows a representation of the T-tubules, sarcoplasmic reticulum (junctional and network), and mitochondria, as well as all of the channels/transporters involved in the Ca2+ signaling: Ryanodine Receptor type 2 (RyR2), Mitochondrial Calcium Uniporter Complex (MCUC) preferentially located in the Junctional SR-mitochondria interface, mitochondrial Ryanodine Receptor type 1 (mRyR1), Rapid Mode of mitochondrial Ca2+ uptake (RaM), mitochondrial Permeability Transition Pore (mPTP), mitochondrial Na+/Ca2+ exchanger (NCLX) excluded from the junctional SR-mitochondria interface, mitochondrial H+/Ca2+ exchanger (mHCX, Letm1), and the SR-mitochondrial tethers MFN1/2. Other alternative SR-mito tethers are also represented. The solid red arrows represent the direction of the Ca+ fluxes.
Ca2+ SIGNALS DURING ECB: CONTROVERSY BETWEEN TWO MODELS
The cardiac MCUC certainly has the capacity to uptake Ca2+ from the high Ca2+ microdomains created at the SR-mito interface. However, how mitochondria decode the rapid cytosolic Ca2+ transients in contracting cardiomyocytes remains controversial[85]. Traditionally two models have been proposed. The first one suggests that, in responding to beat-to-beat cytosolic Ca2+ oscillations, the [Ca2+]m increase is slow and gradual until reaching a new steady state where the Ca2+ influx and the efflux are entirely balanced[86]. Nonetheless, slow increases in [Ca2+]m may not be able to stimulate ATP production fast enough to match the immediate energy demands of the beating heart. The second model suggests that the fast cytosolic Ca2+ oscillations are fully sensed by the mitochondria. Thus, a finite amount of Ca2+ goes rapidly into mitochondria from beat to beat. This second model implies not only that the influx is extremely fast but also that a rapid efflux is required. Furthermore, if the amount of Ca2+ uptake by mitochondria in each heartbeat is large, then in addition to regulating energy production, mitochondria can also play a critical role in shaping the [Ca2+]c pulses. Indeed one group has reported that the magnitude of cytosolic Ca2+ transients increases upon inhibition of the MCUC, but not all studies have found evidence for this[17, 87].
The controversy between the two models relates in part to limitations in the available methods for measuring free mitochondrial [Ca2+], so depending on the method used, the results support one model or the other[88]. Two groups of fluorescent indicators were used to measure free [Ca2+]m. The first group is small molecule fluorescent dyes, such as rhod-2. Although these dyes are designed to be preferentially located in the mitochondria, cytosolic contamination is expected and needs to be removed for accurate [Ca2+]m determinations. The second group is the genetically encoded calcium indicators (GECIs). This group of indicators can be designed genetically to target a specific organelle, however, the application of these indicators to adult cardiomyocytes is technically challenging because expression of GECIs requires either infection of cultured adult cardiomyocytes with a consequent time lapse (a few days) between the infection and the experimental performance, or the delivery of the virus to the heart in vivo through adeno-associated vectors (AAV9). To resolve this controversy, it would be helpful to have a probe that accumulates solely inside the mitochondiral matrix with Ca2+ binding affinity and kinetics that can respond to the dynamic ranges of [Ca2+]m changes.
Model I: mitochondrial free Ca2+ slowly integrates the cytosolic Ca2+ transients.
A significant number of the studies use fluorescence microscopy techniques, specifically fluorescent dyes such us rhod-2 or fluo-3, to measure mitochondrial Ca2+ concentrations ([Ca2+]m). As described above, the main concern about these dyes is their unspecific localization in the cytosol, which can lead to misinterpretation of the mitochondrial Ca2+ kinetics. In order to avoid this cytosolic signal contamination, cells can be treated with manganese or cobalt[86, 89] or the plasma membrane can be permeabilized[90]. In adult rats the data show slow increases in [Ca2+]m occurred upon β-adrenergic stimulation[91] or Ca2+ loading via sarcolemmal NCX [92]. However, in adult cat cardiomyocytes, increases in [Ca2+]m were only barely detectable upon basal electrical stimulation without β-adrenergic stimulation. Another study performed in rats showed a [Ca2+]m increase of only 2-10 nM during a single cytosolic Ca2+ pulse, supporting the idea that mitochondria barely sense the cytosolic Ca2+ oscillations[93]. Lastly, when permeabilized adult cat cardiomyocytes were loaded with fluo-3 in mitochondria and computer-control rapid Ca2+ pulses were added, mitochondria sequestered Ca2+ slowly rather than on the pulse-to-pulse basis. Only after the Ca2+ additions had elapsed for at least 5 min the [Ca2+]m was then able to rise quickly and return slowly to the basal level, showing that the recovery of rapid Ca2+ uptake kinetics is slow[90].
Model II: beat-to-beat mitochondrial Ca2+ oscillations.
Evidence for fast [Ca2+]m transients came from laser scanning confocal microscopy and fluorescent dyes[4, 94]. The experiments were performed in adult rabbit cardiomyocytes upon electrical or β-adrenergic stimulation. The shape of [Ca2+]m transients was almost identical to that of the [Ca2+]c transients, which made the separation of fluorescence signals from mitochondria and from cytosol more challenging. To distinguish these two signals, control experiments were performed to show that MCU blocking with ruthenium red abolished the rhod-2 signal ([Ca2+]m) but did not affect the fluo-3 ([Ca2+]c) signal[95, 96]. As mentioned, to avoid the cytosolic signal contamination, mitochondria-targeted GECIs, such us aequorin or pericam, were used to monitor [Ca2+]m [97] in cardiomyocytes from neonatal rats . Adult rat cardiomyocytes expressing cytosolic and mitochondrial aequorin showed a synchronous elevation in [Ca2+]c and [Ca2+]m upon electrical stimulation. However, the [Ca2+]m decay was much slower, leading to a progressive Ca2+ accumulation within the mitochondria[98]. Other evidence for fast mitochondrial Ca2+ uptake came from a study using adult guinea pig cardiomyocytes, which showed that cytosolic Ca2+ transients were accompanied by fast [Ca2+]m transients, however detection of these trasients depended on elevated Ca2+ cycling (β-adrenergic or increased stimulation frequency). In this study, the mitochondrial Ca2+ increase was detected earlier than the [Ca2+]c increase, proving that mitochondria have privileged uptake of the Ca2+ released by the RyR2, even faster than the global [Ca2+]c increase[48]. This finding supports the previously reported fast local Ca2+ communication between SR and mitochondria through the high Ca2+ microdomains[99]. Maack et al. developed a computation model where 50 ms pulses of 10 to 20 μm Ca2+ (magnitude reported for the Ca2+ microdomains) were applied to adult myocytes. The model predicted characteristics of the [Ca2+]m changes mimicking both model I and II. It predicted a fast mitochondria Ca2+ uptake, however, mitochondrial Ca2+ extrusion is always slower than uptake, so the Ca2+ accumulates in the mitochondria until a new steady-state level is reached[48]. This theoretical model was recently confirmed in experimental results from Wust et al. in which cultured rat adult cardiomyocytes were infected with the FRET-based mitochondrial Ca2+ indicator 4mtD3cpd, MitoCam. As predicted, at 37 °C, mitochondria were able to uptake Ca2+ rapidly (~49 ms) while extrusion was much slower (~1.17 s), reaching a new steady state level where the beat-to-beat Ca2+ oscillations still happen. These results further confirmed that the Ca2+ transients can effectively regulate ATP production upon an increase in the heart workload[100]. The magnitude of mitochondrial matrix free Ca2+ concentrations during these oscillations remains to be determined quantitatively. Furthermore, whether different species have different mitochondrial responses to [Ca2+]c oscillations is not yet clear. With the improvements in mitochondria targeted Ca2+ indicators with optimal Ca2+ sensitivity and kinetics plus the genetic manipulations of mitochondrial Ca2+ tranport proteins, this controversy will be resolved eventually.
PROCESSES REGULATED BY Ca2+ RELEASED TO THE SR-MITO CONTACTS SITES IN ADULT CARDIOMYOCYTES
Bioenergetics and ROS.
As described in the introduction, during ECB, Ca2+ enters the sarcoplasm through L-type Ca2+ channels leading to a greater Ca2+ release by the SR. After the contraction is induced, the reuptake of Ca2+ by SR SERCA pump and the extrusion of Ca2+ across the cell membrane through the NCLX together bring the muscle back to relaxation. A physiological increase in heart workload activates the β-adrenergic pathway, increasing the rate and amplitude of the cytosolic Ca2+ transients that enhance the force generation at the myofilaments and thereby increasing the ATP consumption[15]. Then, the ADP generated as a consequence of the ATP hydrolysis during muscle contraction is transported into the mitochondria, where it can serve as a signal to enhance ATP regeneration. Increased ATP production causes a faster electron flux through the electron transport chain (ETC), oxidizing NADH to NAD+. This process was established as the Pull condition, a term used by O’Rourke[101]. However, the ADP is not the only regulator of the ATP production. Simultaneously, the Ca2+ uptake by the mitochondria activates two Krebs-cycle dehydrogenases plus the pyruvate dehydrogenase to accommodate NADH regeneration (Push condition). Therefore the Ca2+ signal within cardiomyocytes has a dual role: one increasing the energy consumption and another enhancing the energy regeneration. The term “parallel activation” is used to describe this process[1, 102].
The common belief is that Ca2+ entry through MCUC activates the α-ketoglutarate dehydrogenase, the isocitrate dehydrogenase, and the pyruvate dehydrogenase, modulating the ECB. Three models of MCU-deficient mice, all introduced in 2013[49, 50, 103], provided unique model systems to validate this popularly accepted dogma unequivocally. All three models (global constitutive, cardiac-specific, and dominant negative overexpression) showed no rapid mitochondrial Ca2+ uptake, but surprisingly completely normal cardiac function, and only a mild and transient phenotype was observed upon a significant increase in the heart workload (sprint at high speed). Taken together, these results indicate that MCUC is dispensable for physiological ECB coupling and only needed temporally for increased heart activity under “fight and flight” conditions in the mouse. It is worth mentioning that the range of heart workload increases are species-dependent: in humans, the normal resting heart rate is approximately 70 per miniute and can increase 2- to 4-fold during intense exercise[104] and in mice the normal resting heart rate is approximately 500 per minute and can increase only 1.5-fold during intense exercise[105]. It is possible that the energy supply/demand matching meditaed by mitochondrial matrix Ca2+ under physiological sympathetic innervation is not critical on a beat-to-beat basis in mice, but it is in humans.
It is still striking that the hearts lacking MCU are able to keep intact the ECB. The reason why remains in the darkness[106]. Without ATP regeneration mechanisms, the heart should run out of energy supply completely in one minute, so additional ways besides MCU are expected “out there.” Compensation effects are expected, however few of these mechanisms have been elucidated or even proposed yet. In this scenario, it is probable that additional Ca2+ influx mechanisms exist to compensate for the lack of energy production regulation due to Ca2+ uptake through the MCUC. Any alternative mitochondrial Ca2+ entry pathway (mRyR1or one of the other reported Ca2+ influx mechanisms, e.g., rapid mode of mitochondrial Ca2+ uptake, RaM) might be sufficient to maintain adequate ATP production. Seidlmayer et al.[107] reported that the Ca2+ released from the SR through IP3 receptors can enter into the mitochondria through the mRyRl. This fast and high-affinity Ca2+ uptake increased the mitochondrial ATP content without stimulating the mitochondrial ROS production. In the same way that the ADP can modulate the ATP production, several other mediators could also be involved, such as various substrates involved in mitochondrial metabolism and redox environments[108]. A recent report showed that when the mitochondrial fission mediator DRP1 was inhibited, mitochondrial respiration in the heart was reduced in half[109]. The high abundance of DRP1 coupled with the very low frequency of fission events in cardiomyocytes points to a likelihood that DRP1 may have additional functions besides mitochondrial fission.
Reactive oxygen species (ROS) are the by-products of oxidative phosphorylation (Ox-Phos)[110]. The main sites for ROS formation in mitochondria are the complex I and complex III[111]. At both sites, the superoxide radical (O2−) is produced when the reduced forms of flavin mononucleotide or ubiquinone transfer an electron to O2. Under physiological conditions, O2− is converted to H2O2 and then eliminated by glutathione peroxidase and peroxiredoxin[110]. Aon et al. developed a model called redox-optimized ROS balance where a unified scheme integrates the optimization of maximized energy output with minimized ROS overflow by operating in an intermediate redox state of the cell[112]. Minimal amounts of NADPH in this model attenuate H2O2 production while maintaining an intermediate redox state and consequently lowering oxidative stress.
The discovery of inner mitochondrial anion channel (IMAC) activated by ROS[113] raised the possibility of ROS transients, similar to the Ca2+ sparks. In cardiomyocytes, ROS are produced in two phases. Initially, there is a slow increase in ROS followed by a fast ROS release, a phenomenon named ROS-induced ROS release (RIRR)[114]. The propagation of RIRR is mediated by IMAC[115]. As part of the fine tuning of Ca2+ signaling in cardiomyocytes, the ROS produced during the Ox-Phos process also regulates the Ca2+ transport not only at the mitochondrial level but also in the SR[116]. Ca2+-microdomain-localized ROS can serve as a feedback regulator to enhance further Ca2+ release by oxidizing SR-RyR2 cystine residues[117]. However, this positive feedback loop can lead to excess ROS production and mitochondrial dysfunction. Mitochondrial antioxidant defenses are responsible for restoring ROS to basal levels, and the [Ca2+]m is lowered through the mitochondrial Ca2+ efflux mechanisms, avoiding any future mitochondrial damage[118]. In this scenario, the subconductance state of the mPTP is critical. The transient mPTP flicker can expel the excess of Ca2+, avoiding Ca2+ overload, and thus Ca2+-dependent cell death[81].
Mitochondrial dynamics.
In multiple tissues, mitochondria have been described as highly dynamic organelles, constantly varying their position, size, and shape throughout fusion/fission events[119]. However, in adult mammal cardiomyocytes, mitochondria rarely show signs of “dynamism”[7]. This dynamism is important because fusion and fission events ensure proper mitochondria function and act as quality control processes. Mitochondrial dynamics have been extensively described for many tissues and cell types, but few studies have been performed in adult cardiomyocytes. The mitochondrial fusion occurs essentially in three steps: first, two mitochondria are tethered together by GTPase MFN1 or 2 (key proteins initially described as active in the mitochondrial fusion process[120], and later identified as playing a role as SR-mito tethers[39]); second, the mitochondrial OMM are fused thanks to the GTPase activity of the MFNs; and third, GTPase-dependent fusion of the inner mitochondrial membrane occurs through optic atrophy 1 (OPA1)[121].
Mitochondrial fission is driven by the GTPase DRP1, FIS1, mitochondrial fission factor (MFF), and mitochondrial dynamics proteins 49 and 51 (MiD 49, 51)[122]. DRP1 is responsible for constricting the mitochondrial membranes, however it does not possess a mitochondrial targeting sequence. Multiple receptors are capable of recruiting DRP1 to induce the mitochondrial fission however MFF appears be what recruits DRP1 to the OMM[123]. In non-cardiac cells, the diameter of the mitochondrial tubules is around 300 nm, so DRP1 polymeric rings are not sufficiently large to wrap around mitochondria. Pre-constriction of the mitochondrial tubules is needed to reduce the diameter to ~150 nm, allowing DRP1 and the rest of the adaptors to initiate the fission process. ER plays a critical role in this pre-constriction step. The Voeltz lab showed that ER wrap around mitochondria to facilitate the reduction of the initial tubule diameter, marking the potential sites for mitochondrial fission[124]. This ER wrapping occurs upstream and is independent of the fission adaptors. When cells are knocked down or out for the DRP1, MFF, Mid 49, or MiD 51, the ER wrapping effect is still present. Further investigation has shown that the combined action of actin and myosin (IIA and IIB) associated with the ER make the constriction possible. In addition, it was shown that myosins are involved in recruiting DRP1[125]. In adult cardiomyocytes, the SR-mito wrapping effect of marking fission sites has not been studied in detail, however, in adult cardiomyocytes the SR permanently wraps the mitochondria and all the fission machinery is present[126], making the process feasible.
Mitochondrial dynamics may not be as frequent in in adult hearts as in other tissues, but they are present. Their presence is demostrated when ablation of the fusion or fission machinery in adult cardiomyocytes leads to more fragmented or elongated mitochondria, respectively[47, 127, 128]. While the fusion and fission events in mitochondria are not common, high levels of MFN2 and DRP1 have been reported, suggesting non-canonical roles for MFN2 , which plays a crucial role as an SR-mito tether, and DRP1, which plays a role as an energy supply process modulator. The SR-mito tether implicates that the Ca2+ concentrations in microdomains, and the SR plasticity plays a crucial role in the regulation of the mitochondrial fission/fusion process. A recent study showed that the mitochondrial fission and fusion dynamics are more common than initially expected, but because Ca2+ oscillations and contractile activity are required for proper mitochondrial fusion[129], fewer fusion events have been recorded in non-contracting cultured cardiomyocytes.
Whether adult cardiac mitochondria can form extensive networks or only small functional clusters is still under debate. O’Rourke’s lab reported that mitochondria could communicate among themselves by ROS to synchronize clusters of mitochondria within the whole mitochondrial network that are sensitive to events such as substrate availability, antioxidant status, and respiratory chain activity[130]. Balaban’s lab reported that both electrical and physical communication occurs between mitochondria and establishes a network along the cardiomyocyte. Nonetheless, this network is segmented into subnetworks which can be disconnected from the rest to avoid whole cell damage in a case of a single mitochondria malfunction[131]. Demaurex et al. proposed another mechanism in which mitochondrial flashes involve only mitochondrial electrical communications but with no matrix content exchange among them[132]. Then an additional new player involved in the cardiac mitochondrial communication came onto the scene: mitochondrial nanotunnels. Nanotunnels are thin tubular protrusions that connect mitochondria separated by relatively long distances (1 to >5 μm)[133]. Mitochondrial matrix content can be exchanged through nanotunnels, however it is not suitable for mitochondria DNA transport. In addition, communication between mitochondrial nanotunneles is slow compared with the mitochondrial fusion.
State-of-the-art technologies, such as FIB-SEM and multi-organelle 3D reconstructions, will be useful in resolving the enigma of how mitochondria are connected and interact with each other in adult cardiac tissue[134, 135]. The new technologies will also shed some light to the mitophagy process in adult cardiomyocytes. It is known that fusion/fission process is crucial in keeping the mitochondria healthy and in good shape, however just how mitochondria are selected to be degraded upon severe damage is still unclear, especially in adult cardiomyocytes.
CONSEQUENCES OF LOSING THE PROPER ER/SR-MITOCHONDRIAL TETHERING AND Ca2+ SIGNAL.
Since ER/SR and mitochondria are physically and functionally connected, it is not surprising that alteration in the ER/SR-mito tether leads to different pathologies, including neurodegenerative diseases, metabolism syndrome, cancer, and heart failure. In genetically or diet-induced obesity and insulin resistant models, a disrupted ER-mito interaction has been observed in liver and neurons[136]. Indeed, available evidence indicates that the disruption of ER-mito tethers (MFN2 ablation) can induce insulin resistance in pro-opiomelanocortin (POMC) neurons. The Ca2+ represents the mechanistic link between ER-mito, and it has been shown that disruption of Ca2+ homeostasis supresses the hepatic insulin signaling[137]. In cancer cells, the enhanced apoptosis resistance is mediated by ER-mito Ca2+ cross-talk.
The oncogenes Bcl-2 and Bcl-XL decrease the ER Ca2+ content likely increasing the leaking through IP3R, avoiding an eventually mitochondrial Ca2+ overload and thus, inhibiting the mPTP aperture upon stress conditions[138]. The oncogene AKT is responsible for the phosphorylation of IP3Rs and therefore limits ER Ca2+ release, once more avoiding the Ca2+-dependent apoptotic response[139]. In neurons, the endogenous parkin is responsible for the proper ER-mito coupling, increasing the mitochondrial Ca2+ uptake and maintaining the ATP synthesis[140]. In Parkinson’s disease patients displaying parkin mutations, levels of MFN2 are surprisingly higher compare with healthy subjects (MFN2 is a target protein ubiquitinated by parkin) leading to higher ER-mito Ca2+ transfer and therefore elevated chance of mPTP-mediated cell death[141]. Since many proteins are targets of parkin’s, the effect of parkin dysfunction may or may not be MFN2-dependent. In the neurodegenerative disease Charcot Marie-Tooth, over 100 MFN2 mutations have been identified, most of them located in GTPase domain and in the HR1/HR2 motifs[142]. However, the mechanism behind the MFN2 mutations leading to neuropathy is not clear yet. Disrupted ER-mito Ca2+ transfer may alter the proper ATP production, causing function impairment in neurons; defects in the MFN2-mediated mitochondrial fusion process may also be involved in neuronal damage[143].
In adult cardiomyocytes, MFN2 ablation leads to a deficiency in the ATP production upon a high energy demand[144]. By removing the tether, Ca2+ that would normally be transferred into mitochondria is not[145]. Therefore, there is less NADH regeneration and less ATP production. In the whole heart, the consequences of the MFN2 removal range from defects in cardiac development to cardiac hypertrophy in adult mice. However, the role of MFN2 during the ischemia-reperfusion process is still unclear. Papanicolaou et al. reported that MFN2 deficiency may protect against ischemia-reperfusion injury based on the fact that when less Ca2+ enters the mitochondria, the chance of mPTP opening is lower and so is mPTP-mediated cell death[146]. On the contrary, Zhao et al. showed that MFN2 deficiency increases susceptibility to ischemia-reperfusion injury[147]. Ultimately, the timing of MFN2 ablation and the ages of the mice may affect the outcome. The mPTP opening depends lastly on the Ca2+ overload and elevated mitochondrial ROS. In a disrupted SR-mito communication situation, less mitochondrial Ca2+ uptake is expected, however an increase in ROS production is also expected. Since both factors are required for an eventual mPTP opening, ROS production should adequately compensate the decrease in the mitochondrial Ca2+ uptake in order for the mPTP opening process to be initiated. This is the reason why it is still not clear whether the rupture of the SR-mito communication leads to higher chance of mPTP opening. Figure 2 shows a comparison between the normal versus SR-mito disrupted connection in adult cardiomyocytes.
Fig. 2. Scheme of the ECB in normal SR-mito connections versus SR-mito disrupted connections.
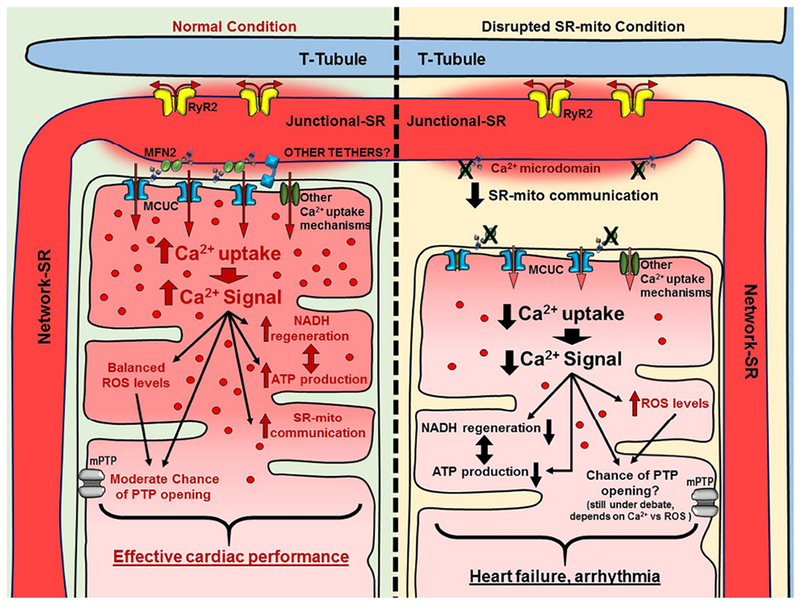
The scheme shows a representation of the Ca2+ transfer from SR to mitochondria and mitochondrial processes that are regulated by variances in the [Ca2+]m. In normal conditions, the Ca2+ is appropriately transferred, enhancing NADH regeneration as well as the ATP production. In this situation ROS production is balanced, keeping ROS levels in a reasonable state and minimizing the chances of mPTP opening. In the absence of MFN2 (or other tethers), SR-mito communication is disrupted. The MCUC (and other Ca2+ influx mechanisms) cannot uptake Ca2+ properly from the Ca2+ microdomains, so the [Ca2+]m reached is low. NADH regeneration and ATP production cannot be adequately adjusted in the absence of Ca2+. ROS production is increased, however, it may not be enough to activate the opening of mPTP because of the low concentration of [Ca2+]m reached.
In the context of heart failure (HF), it has also been reported that the SR-mito tethering is disrupted in guinea pig HF models. The data showed reduced MFN1 and MFN2 protein expression levels compared with controls. The results were confirmed by confocal microscopy colocalization assays, where a population of MFN1 was not associated with RyR2. In this HF model, the decrease in MFN led to more fragmented mitochondria and a less organized mitochondrial network and thus loss the efficiency in conveying signaling molecules/ions and for synchronized energy production in mitochondria in response to changes in workload[148].
SUMMARY
The close connection between SR and mitochondria seems to be critical for controlling several functions in adult cardiomyocytes. Their close connection persists mainly through the mitochondrial fusion mediator MFN2, and several groups have reported its role as an SR-mito tether, enhancing the SR to mitochondria Ca2+ transfer efficiency. When Ca2+ is released at the RyR2 receptors, the high [Ca2+] microdomain that is created is needed to activate the MCUC. Also, MCUC is preferentially located in the SR-mito interface, optimizing maximal Ca2+ signaling. Although it is still not clear how mitochondria decode the beat-to-beat cytosolic Ca2+ transients, the latest reports have shown that mitochondria can rapidly take up Ca2+, making a fast increase in ATP production possible upon a sudden increase in the heart workload. In addition to the MCUC being located where it is most needed, the key Ca2+ extrusion system, the NCLX, is also excluded from the areas where the Ca2+ uptake occurs to minimize the mitochondrial membrane potential consumption during rapid Ca2+ transport in and out of mitochondria. Furthermore, this “Ca2+ zoning” property within one single mitochondrion may differentially regulate local mitochondrial form and function, such as localized cristae remodeling, protein turnover, biogenesis, and selective sites for mitochondrial fission and mitophagy. The lack of severe cardiac phenotype of the MCU KO mice models, albeit quite unexpected, raises opportunities for addressing important questions about whether alternative mitochondrial Ca2+ uptake pathways exist or perhaps some other extra-mitochondrial Ca2+-dependent pathways control ECB coupling in beating hearts[149]. Future advances may come from measuring [Ca2+]m quantitatively and specifically in electrically stimulated intact single cardiomyocytes from species besides rodents (e.g., humans); 3-dimensional imaging of the SR-mito structure, and genetic manipulations of SR-mito tethering distances through artificial linkers. It is hoped that such advances will help to resolve the current controversies in the field and that elucidating the Ca2+ signaling mechanisms between SR and mitochondria will translate the fundamental principles into an improved understanding of the cardiac physiology and pathology.
Acknowledgements
We thank Dr. Celia Femandez-Sanz for her invaluable suggetions and Jennifer Wilson for English editing to this manuscript. This study was suported by: NIH/NHLBI (HL122124, HL093671, HL137426, HL142864).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
REFERENCES
- 1.Balaban RS, Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol, 2002. 34(10): p. 1259–71. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM, Cardiac excitation-contraction coupling. Nature, 2002. 415(6868): p. 198–205. [DOI] [PubMed] [Google Scholar]
- 3.Csordas G, Thomas AP, and Hajnoczky G, Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc Med, 2001. 11(7): p. 269–75. [DOI] [PubMed] [Google Scholar]
- 4.Chacon E, et al. , Mitochondrial free calcium transients during excitation-contraction coupling in rabbit cardiac myocytes. FEBS Lett, 1996. 382(1-2): p. 31–6. [DOI] [PubMed] [Google Scholar]
- 5.Sharma VK, et al. , Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr, 2000. 32(1): p. 97–104. [DOI] [PubMed] [Google Scholar]
- 6.Eisner DA, et al. , Calcium and Excitation-Contraction Coupling in the Heart. Circ Res, 2017. 121(2): p. 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorn G II, Mitochondrial fission/fusion and cardiomyopathy. Curr Opin Genet Dev, 2016. 38: p. 38–44.27061490 [Google Scholar]
- 8.Dorn GW 2nd, Mitochondrial dynamism and heart disease: changing shape and shaping change. EMBO Mol Med, 2015. 7(7): p. 865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forbes MS and van Neil EE, Membrane systems of guinea pig myocardium: ultrastructure and morphometric studies. Anat Rec, 1988. 222(4): p. 362–79. [DOI] [PubMed] [Google Scholar]
- 10.Soeller C and Cannell MB, Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res, 1999. 84(3): p. 266–75. [DOI] [PubMed] [Google Scholar]
- 11.Fawcett DW and McNutt NS, The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J Cell Biol, 1969. 42(1): p. 1–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi T, et al. , Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci, 2009. 122(Pt 7): p. 1005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song LS, et al. , Calcium biology of the transverse tubules in heart. Ann N Y Acad Sci, 2005. 1047: p. 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun N and Finkel T, Cardiac mitochondria: a surprise about size. J Mol Cell Cardiol, 2015. 82: p. 213–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohlhaas M, Nickel AG, and Maack C, Mitochondrial energetics and calcium coupling in the heart. J Physiol, 2017. 595(12): p. 3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams GS, Boyman L, and Lederer WJ, Mitochondrial calcium and the regulation of metabolism in the heart. J Mol Cell Cardiol, 2015. 78: p. 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drago I, et al. , Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A, 2012. 109(32): p. 12986–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yaniv Y, et al. , Crosstalk between mitochondrial and sarcoplasmic reticulum Ca2+ cycling modulates cardiac pacemaker cell automaticity. PLoS One, 2012. 7(5): p. e37582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorn GW 2nd and Maack C, SR and mitochondria: calcium cross-talk between kissing cousins. J Mol Cell Cardiol, 2013. 55: p. 42–9. [DOI] [PubMed] [Google Scholar]
- 20.Yaniv Y, et al. , Matching ATP supply and demand in mammalian heart: in vivo, in vitro, and in silico perspectives. Ann N Y Acad Sci, 2010. 1188: p. 133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rizzuto R, et al. , Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta, 2009. 1787(11): p. 1342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz-Meana M, Fernandez-Sanz C, and Garcia-Dorado D, The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res, 2010. 88(1): p. 30–9. [DOI] [PubMed] [Google Scholar]
- 23.Hwang SJ and Kim W, Mitochondrial dynamics in the heart as a novel therapeutic target for cardioprotection. Chonnam Med J, 2013. 49(3): p. 101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizzuto R, et al. , Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science, 1998. 280(5370): p. 1763–6. [DOI] [PubMed] [Google Scholar]
- 25.Mannella CA, The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta, 2006. 1762(2): p. 140–7. [DOI] [PubMed] [Google Scholar]
- 26.Rowland AA and Voeltz GK, Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol, 2012. 13(10): p. 607–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giacomello M and Pellegrini L, The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ, 2016. 23(9): p. 1417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csordas G, et al. , Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol, 2006. 174(7): p. 915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kornmann B, Osman C, and Walter P, The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A, 2011. 108(34): p. 14151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szabadkai G, et al. , Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol, 2006. 175(6): p. 901–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoica R, et al. , ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun, 2014. 5: p. 3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirabayashi Y, et al. , ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science, 2017. 358(6363): p. 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu S, et al. , Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation, 2017. 136(23): p. 2248–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santo-Domingo J and Demaurex N, Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta, 2010. 1797(6–7): p. 907–12. [DOI] [PubMed] [Google Scholar]
- 35.De Stefani D, Rizzuto R, and Pozzan T, Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu Rev Biochem, 2016. 85: p. 161–92. [DOI] [PubMed] [Google Scholar]
- 36.Fonteriz R, et al. , Modulation of Calcium Entry by Mitochondria. Adv Exp Med Biol, 2016. 898: p. 405–21. [DOI] [PubMed] [Google Scholar]
- 37.Peskoff A and Langer GA, Calcium concentration and movement in the ventricular cardiac cell during an excitation-contraction cycle. Biophys J, 1998. 74(1): p. 153–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohlhaas M and Maack C, Calcium release microdomains and mitochondria. Cardiovasc Res, 2013. 98(2): p. 259–68. [DOI] [PubMed] [Google Scholar]
- 39.de Brito OM and Scorrano L, Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature, 2008. 456(7222): p. 605–10. [DOI] [PubMed] [Google Scholar]
- 40.Schrepfer E and Scorrano L, Mitofusins, from Mitochondria to Metabolism. Mol Cell, 2016. 61(5): p. 683–694. [DOI] [PubMed] [Google Scholar]
- 41.Koshiba T, et al. , Structural basis of mitochondrial tethering by mitofusin complexes. Science, 2004. 305(5685): p. 858–62. [DOI] [PubMed] [Google Scholar]
- 42.Mattie S, et al. , A new mitofusin topology places the redox-regulated C terminus in the mitochondrial intermembrane space. J Cell Biol, 2018. 217(2): p. 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naon D, et al. , Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulummitochondria tether. Proc Natl Acad Sci U S A, 2016. 113(40): p. 11249–11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Filadi R, et al. , Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc Natl Acad Sci U S A, 2015. 112(17): p. E2174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leal NS, et al. , Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid beta-peptide production. J Cell Mol Med, 2016. 20(9): p. 1686–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y, et al. , Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res, 2012. 111(7): p. 863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papanicolaou KN, et al. , Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res, 2012. 111(8): p. 1012–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maack C, et al. , Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res, 2006. 99(2): p. 172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan X, et al. , The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol, 2013. 15(12): p. 1464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luongo TS, et al. , The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep, 2015. 12(1): p. 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rasmussen TP, et al. , Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A, 2015. 112(29): p. 9129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Stefani D, et al. , A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature, 2011. 476(7360): p. 336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baughman JM, et al. , Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature, 2011. 476(7360): p. 341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fan C, et al. , X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature, 2018. 559(7715): p. 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen NX, et al. , Cryo-EM structure of a fungal mitochondrial calcium uniporter. Nature, 2018. 559(7715): p. 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raffaello A, et al. , The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. Embo j, 2013. 32(17): p. 2362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sancak Y, et al. , EMRE is an essential component of the mitochondrial calcium uniporter complex. Science, 2013. 342(6164): p. 1379–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Csordas G, et al. , MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab, 2013. 17(6): p. 976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de la Fuente S, et al. , Dynamics of mitochondrial Ca2+ uptake in MICU1-knockdown cells. Biochem J, 2014. 458(1): p. 33–40. [DOI] [PubMed] [Google Scholar]
- 60.Patron M, et al. , MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell, 2014. 53(5): p. 726–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patron M, et al. , MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mallilankaraman K, et al. , MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol, 2012. 14(12): p. 1336–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gunter TE and Pfeiffer DR, Mechanisms by which mitochondria transport calcium. Am J Physiol, 1990. 258(5 Pt 1): p. C755–86. [DOI] [PubMed] [Google Scholar]
- 64.Michels G, et al. , Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation, 2009. 119(18): p. 2435–43. [DOI] [PubMed] [Google Scholar]
- 65.Rizzuto R and Pozzan T, Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev, 2006. 86(1): p. 369–408. [DOI] [PubMed] [Google Scholar]
- 66.Buntinas L, et al. , The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta, 2001. 1504(2-3): p. 248–61. [DOI] [PubMed] [Google Scholar]
- 67.Beutner G, et al. , Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem, 2001. 276(24): p. 21482–8. [DOI] [PubMed] [Google Scholar]
- 68.Beutner G, et al. , Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta, 2005. 1717(1): p. 1–10. [DOI] [PubMed] [Google Scholar]
- 69.J, O.U., et al. , Overexpression of ryanodine receptor type 1 enhances mitochondrial fragmentation and Ca2+-induced ATP production in cardiac H9c2 myoblasts. Am J Physiol Heart Circ Physiol, 2013. 305(12): p. H1736–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trenker M, et al. , Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol, 2007. 9(4): p. 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang D, Zhao L, and Clapham DE, Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science, 2009. 326(5949): p. 144–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosier RN, et al. , Ca2+ transport against its electrochemical gradient in cytochrome oxidase vesicles reconstituted with mitochondrial hydrophobic proteins. Arch Biochem Biophys, 1981. 210(2): p. 549–64. [DOI] [PubMed] [Google Scholar]
- 73.Gunter KK and Gunter TE, Transport of calcium by mitochondria. J Bioenerg Biomembr, 1994. 26(5): p. 471–85. [DOI] [PubMed] [Google Scholar]
- 74.Gunter TE, et al. , Calcium and mitochondria. FEBS Lett, 2004. 567(1): p. 96–102. [DOI] [PubMed] [Google Scholar]
- 75.Palty R, et al. , NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A, 2010. 107(1): p. 436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jung DW, Baysal K, and Brierley GP, The sodium-calcium antiport of heart mitochondria is not electroneutral. J Biol Chem, 1995. 270(2): p. 672–8. [DOI] [PubMed] [Google Scholar]
- 77.Murphy E and Eisner DA, Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res, 2009. 104(3): p. 292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bernardi P and von Stockum S, The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell Calcium, 2012. 52(1): p. 22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zoratti M and Szabo I, Electrophysiology of the inner mitochondrial membrane. J Bioenerg Biomembr, 1994. 26(5): p. 543–53. [DOI] [PubMed] [Google Scholar]
- 80.Huser J and Blatter LA, Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem J, 1999. 343 Pt 2: p. 311–7. [PMC free article] [PubMed] [Google Scholar]
- 81.Gong G, et al. , Mitochondrial flash as a novel biomarker of mitochondrial respiration in the heart. Am J Physiol Heart Circ Physiol, 2015. 309(7): p. H1166–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De La Fuente S, et al. , Strategic Positioning and Biased Activity of the Mitochondrial Calcium Uniporter in Cardiac Muscle. J Biol Chem, 2016. 291(44): p. 23343–23362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De La Fuente S, et al. , Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart. Cell Rep, 2018. 24(12): p. 3099–3107.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Luongo TS, et al. , The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature, 2017. 545(7652): p. 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dedkova EN and Blatter LA, Calcium signaling in cardiac mitochondria. J Mol Cell Cardiol, 2013. 58: p. 125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miyata H, et al. , Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol, 1991. 261(4 Pt 2): p. H1123–34. [DOI] [PubMed] [Google Scholar]
- 87.Andrienko TN, Picht E, and Bers DM, Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J Mol Cell Cardiol, 2009. 46(6): p. 1027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dedkova EN and Blatter LA, Measuring mitochondrial function in intact cardiac myocytes. J Mol Cell Cardiol, 2012. 52(1): p. 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Davidson SM, Yellon D, and Duchen MR, Assessing mitochondrial potential, calcium, and redox state in isolated mammalian cells using confocal microscopy. Methods Mol Biol, 2007. 372: p. 421–30. [DOI] [PubMed] [Google Scholar]
- 90.Sedova M, Dedkova EN, and Blatter LA, Integration of rapid cytosolic Ca2+ signals by mitochondria in cat ventricular myocytes. Am J Physiol Cell Physiol, 2006. 291(5): p. C840–50. [DOI] [PubMed] [Google Scholar]
- 91.Griffiths EJ, Stern MD, and Silverman HS, Measurement of mitochondrial calcium in single living cardiomyocytes by selective removal of cytosolic indo 1. Am J Physiol, 1997. 273(1 Pt 1): p. C37–44. [DOI] [PubMed] [Google Scholar]
- 92.Zhou Z, Matlib MA, and Bers DM, Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. J Physiol, 1998. 507 (Pt 2): p. 379–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Di Lisa F, et al. , Intramitochondrial free calcium in cardiac myocytes in relation to dehydrogenase activation. Cardiovasc Res, 1993. 27(10): p. 1840–4. [DOI] [PubMed] [Google Scholar]
- 94.Ohata H, et al. , Mitochondrial Ca2+ transients in cardiac myocytes during the excitation-contraction cycle: effects of pacing and hormonal stimulation. J Bioenerg Biomembr, 1998. 30(3): p. 207–22. [DOI] [PubMed] [Google Scholar]
- 95.Trollinger DR, Cascio WE, and Lemasters JJ, Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca(2+)-indicating fluorophores. Biophys J, 2000. 79(1): p. 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trollinger DR, Cascio WE, and Lemasters JJ, Selective loading of Rhod 2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochem Biophys Res Commun, 1997. 236(3): p. 738–42. [DOI] [PubMed] [Google Scholar]
- 97.Robert V, et al. , Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. Embo j, 2001. 20(17): p. 4998–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bell CJ, et al. , ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J Biol Chem, 2006. 281(38): p. 28058–67. [DOI] [PubMed] [Google Scholar]
- 99.Garcia-Perez C, Hajnoczky G, and Csordas G, Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem, 2008. 283(47): p. 32771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wust RC, et al. , Rapid frequency-dependent changes in free mitochondrial calcium concentration in rat cardiac myocytes. J Physiol, 2017. 595(6): p. 2001–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cortassa S, et al. , An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J, 2003. 84(4): p. 2734–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cortassa S, et al. , A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophys J, 2006. 91(4): p. 1564–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kwong JQ, et al. , The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep, 2015. 12(1): p. 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chapman CB, Fisher JN, and Sproule BJ, Behavior of stroke volume at rest and during exercise in human beings. J Clin Invest, 1960. 39: p. 1208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Desai KH, et al. , Cardiovascular indexes in the mouse at rest and with exercise: new tools to study models of cardiac disease. Am J Physiol, 1997. 272(2 Pt 2): p. H1053–61. [DOI] [PubMed] [Google Scholar]
- 106.Wang P, et al. , Why don’t mice lacking the mitochondrial Ca(2+) uniporter experience an energy crisis? J Physiol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seidlmayer LK, et al. , Inositol 1,4,5-trisphosphate-mediated sarcoplasmic reticulummitochondrial crosstalk influences adenosine triphosphate production via mitochondrial Ca2+ uptake through the mitochondrial ryanodine receptor in cardiac myocytes. Cardiovasc Res, 2016. 112(1): p. 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang W, Fernandez-Sanz C, and Sheu SS, Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim Biophys Acta, 2018. 1864(5 Pt B): p. 1991–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang H, et al. , A novel fission-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc Res, 2017. 113(2): p. 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nickel A, Kohlhaas M, and Maack C, Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol, 2014. 73: p. 26–33. [DOI] [PubMed] [Google Scholar]
- 111.Murphy MP, How mitochondria produce reactive oxygen species. Biochem J, 2009. 417(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Aon MA, Cortassa S, and O’Rourke B, Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta, 2010. 1797(6-7): p. 865–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aon MA, et al. , Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem, 2007. 282(30): p. 21889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zorov DB, et al. , Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med, 2000. 192(7): p. 1001–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhou L, et al. , A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol, 2010. 6(1): p. e1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arieli Y, et al. , Gender modulation of Ca(2+) uptake in cardiac mitochondria. J Mol Cell Cardiol, 2004. 37(2): p. 507–13. [DOI] [PubMed] [Google Scholar]
- 117.Xi Q, Cheranov SY, and Jaggar JH, Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res, 2005. 97(4): p. 354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feissner RF, et al. , Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci (Landmark Ed), 2009. 14: p. 1197–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tilokani L, et al. , Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem, 2018. 62(3): p. 341–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chen H and Chan DC, Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet, 2005. 14 Spec No. 2: p. R283–9. [DOI] [PubMed] [Google Scholar]
- 121.Cipolat S, et al. , OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A, 2004. 101(45): p. 15927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hall AR and Hausenloy DJ, The shape of things to come: mitochondrial fusion and fission in the adult heart. Cardiovasc Res, 2012. 94(3): p. 391–2. [DOI] [PubMed] [Google Scholar]
- 123.Bossy-Wetzel E, et al. , Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol, 2003. 15(6): p. 706–16. [DOI] [PubMed] [Google Scholar]
- 124.Friedman JR, et al. , ER tubules mark sites of mitochondrial division. Science, 2011. 334(6054): p. 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hatch AL, Gurel PS, and Higgs HN, Novel roles for actin in mitochondrial fission. J Cell Sci, 2014. 127(Pt 21): p. 4549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hom J and Sheu SS, Morphological dynamics of mitochondria--a special emphasis on cardiac muscle cells. J Mol Cell Cardiol, 2009. 46(6): p. 811–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kageyama Y, et al. , Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo j, 2014. 33(23): p. 2798–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Song M, et al. , Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab, 2015. 21(2): p. 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Eisner V, et al. , Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci U S A, 2017. 114(5): p. E859–e868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kurz FT, et al. , Functional Implications of Cardiac Mitochondria Clustering. Adv Exp Med Biol, 2017. 982: p. 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Glancy B, et al. , Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep, 2017. 19(3): p. 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Santo-Domingo J, et al. , OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. Embo j, 2013. 32(13): p. 1927–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lavorato M, et al. , Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc Natl Acad Sci U S A, 2017. 114(5): p. E849–e858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cohen S, Valm AM, and Lippincott-Schwartz J, Interacting organelles. Curr Opin Cell Biol, 2018. 53: p. 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu Y, et al. , Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A, 2017. 114(24): p. E4859–e4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schneeberger M, et al. , Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell, 2013. 155(1): p. 172–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ozcan L, et al. , Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab, 2013. 18(6): p. 803–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bittremieux M, et al. , ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca(2+) signaling. Biochim Biophys Acta, 2016. 1863(6 Pt B): p. 1364–78. [DOI] [PubMed] [Google Scholar]
- 139.Szado T, et al. , Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A, 2008. 105(7): p. 2427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cali T, et al. , Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim Biophys Acta, 2013. 1832(4): p. 495–508. [DOI] [PubMed] [Google Scholar]
- 141.Gautier CA, et al. , The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet, 2016. 25(14): p. 2972–2984. [DOI] [PubMed] [Google Scholar]
- 142.Filadi R, Pendin D, and Pizzo P, Mitofusin 2: from functions to disease. Cell Death Dis, 2018. 9(3): p. 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Franco A, et al. , Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature, 2016. 540(7631): p. 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dorn GW 2nd, Song M, and Walsh K, Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol, 2015. 78: p. 123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Beikoghli Kalkhoran S, et al. , Assessing the effects of mitofusin 2 deficiency in the adult heart using 3D electron tomography. Physiol Rep, 2017. 5(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Papanicolaou KN, et al. , Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol, 2011. 31(6): p. 1309–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhao T, et al. , Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem, 2012. 287(28): p. 23615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goh KY, et al. , Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc Res, 2016. 109(1): p. 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jhun BS, et al. , The mitochondrial Ca2+ uniporter: regulation by auxiliary subunits and signal transduction pathways. Am J Physiol Cell Physiol, 2016. 311(1): p. C67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]