

Abstract
The pathophysiology underlying the formation, progression and surgical healing of incisional hernia (IH) that develop as a major complication associated with abdominal laparotomy is poorly understood. The proposed mechanisms include the switch of collagen phenotype and the proliferation of abnormal fibroblasts following surgery. The focus of this article is to critically review the cellular, biochemical and potential molecular events associated with the development of IH. The disturbance in collagen homeostasis with alterations in the expression of collagen subtypes, including type I, type III, type IV and type V, and impairment in the transdifferentiation of fibroblasts to myofibroblasts are discussed. The phenotype switch of wound-repair fibroblasts results in mechanically compromised ECM that triggers the proliferation of abnormal fibroblasts. HMGB-1 could be involved in wound progression, whereas signaling events mediated by TGF-β1, CTGF, LOX, and HIF-1α play significant role in the wound healing response. Thus, the ratio of TGF-β1:HMGB1 could be a critical determinant of the underlying pathology. Potential target sites for therapeutic intervention in the management of IH are recognized.
Keywords: Incisional hernia, Collagen phenotypes, Myofibroblasts, TGF-β1 signaling, Matrix homeostasis
Introduction
Incisional hernias (IH) arise as a major complication associated with laparotomies and has an approximate incidence of 10–20%, although ranges from 5% to as high as 70% 1. This variability is usually the reflective of the comorbidities and the disease process of a particular patient population under study. The multifactorial contributing factors in the pathogenesis of IH, as shown in Fig. 1, are based on the clinical observations but the underlying molecular basis is poorly understood. The IH results from the abnormal wound healing at the abdominal wall where, the laparotomy wounds are most common. Even though this article focuses on laparotomy wounds due to midline vertical incision, the underlying principles could be applicable to any type of abdominal wall injuries. The development of IH following abdominal wall injuries has also been associated with comorbidities such as diabetes, obesity, smoking, and other clinical and habitual factors. Mechanical function of the tissue following an incision is compromised especially at muscle, tendon and fascial layers of abdominal wall due to surgical wound failure. The disturbance in the normal homeostasis of extracellular matrix (ECM), especially switch in collagen phenotypes, is considered a major mechanism underlying hernia formation and this could provide insights in the development of novel strategies to reduce the pathophysiology of IH after laparotomy. Many risk factors that increase the incidence of IH include genetics and co-morbidities, including diabetes, obesity, deep wound infections, perioperative hypotension, aortic aneurism, smoking, steroidal use, and nutritional deficiencies 2, 3. Poor surgical technique in the past has been considered one of the most significant risk factors for IH development but this has been greatly minimized with the widespread acceptance of continuous suture with a 4 to 1 suture length vs wound length ratio 4. Collagen disorganization following surgery has severe impact on the impairment in the function of tissue fibroblast leading to delayed healing response by compromising the mechanical integrity of the abdominal wall 5,6. The existence of abnormal fibroblast population in laparotomy wounds has been reported 2, 7. However, the underlying molecular mechanisms regarding the function, differentiation and phenotype switch of these fibroblasts are largely unknown.
Fig. 1:
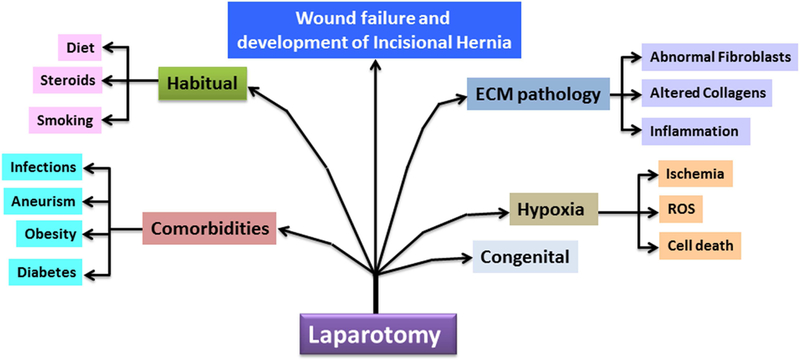
A schematic diagram showing the major contributing factors associated with the development and progression of incisional hernia.
Failure of the load-bearing functions of the abdominal wall as a result of alterations in collagen phenotype following a laparotomy could explain the higher incidence of IH 8. These alterations in mechanical properties activate repair responses in load-bearing tissues, such as tendons and ligaments 9. Interestingly, cells of the load-bearing tendons and ligaments are capable of withstanding hypoxic and ischemic insults following an injury. On the contrary, muscles of the abdominal wall do not tolerate wound ischemia causing fibroblasts to produce abnormal collagen, resulting in a defective ECM 7. In addition, the cause and the underlying mechanisms of the phenotype switch of fibroblasts are unknown and warrant extensive research. Since the main function of fibroblasts is collagen homeostasis, it is likely that the switch of fibroblasts from normal to pathological phenotype results in defective repair biology which forms the basis for IH development. In this article, we critically reviewed the implications of fibroblast biology in the pathogenesis of IH.
Biology of IH Formation
Wound healing after laparotomy is characterized by a well-known cascade of events leading to wound repair. Early in this cascade of events the approximation of the wound depends entirely on the suture used to close the wound. As the cascade proceeds, the wound becomes progressively stronger, depends less upon the suture, and reaches maximum at about 100 days (90% of the original strength before wounding is usually considered the maximum) 10. The cascade is characterized by a complex interaction between platelets, neutrophils, macrophages, lymphocytes and blood supply which ultimately stimulate the fibroblasts to secrete collagen, the basic building block of wound healing 10. Early in wound healing a provisional matrix is produced by platelets and fibrin which acts as a temporary scaffold to recruit other entities necessary for wound repair. Inadequate hemostasis resulting in hematoma can disrupt this matrix and is therefore one of the fundamental causes of IH 10, 11. The provisional matrix attracts inflammatory cells and mediators and induces classical inflammatory pathways. A delayed and /or prolonged inflammatory response results in the activation of pathogenic fibroblasts that finally lead to ECM disorganization 12.
The disturbance in collagen metabolism has been reported to be the primary cause for ECM disorganization in IH tissues in human patients and animal models. The alterations in the quantity, quality and phenotype of matrisome components of IH tissue, especially collagen (the major ECM component), affect the matrisome (an ensemble of genes encoding ECM and ECM-associated proteins) homeostasis. It has been reported that the ratio of collagen type I and type III determines the mechanical integrity of connective tissues 13. The intermolecular cross-linking between collagen type I and type III imparts additional strength to the abdominal wall tissue 14. Interestingly, a drastic decrease in the ratio of collagen type I to type III results in a disorganized ECM in tissues from IH patients 15, 16. However, the molecular mechanisms causing the alteration of collagen type I to type III ratio, and the associated fibroblast biology and their regulatory roles in driving IH development warrant further investigation.
The inflammatory response following the incisional wound and the repair signaling by endogenous mediators result in fibroproliferative burst 17. However, the balance between matrisome homeostasis and fibroblast proliferation is believed to be disturbed in the laparotomy wound matrix resulting in IH and its reoccurrence. Xing et al. 18 demonstrated the existence of an abnormal fibroblast population which is responsible for the secretion of altered collagen phenotype in early laparotomy wound failure. The phenotype selection of fibroblasts is determined by the loss of mechanical integrity of abdominal wall 18. The tendons and fascia of abdominal wall respond to intrinsic and/or extrinsic mechanical stimuli to maintain fibroblast homeostasis. The signaling associated with mechano-transduction transmits mechanical stimuli to cells via integrin receptors and cytoskeleton anchoring proteins. The subsequent activation of focal adhesion complex triggers cytoskeletal rearrangements and activates c-Src and MAP kinases for cell proliferation 19, 20.
Following the incision, the inflammatory cells migrate and marginalize to the site of injury and activate the inflow of other leukocytes and plasma proteins. The phagocytosis by neutrophils is the first line of defense which is followed by monocytes. The migration of monocytes occurs within 2–3 days and the cells differentiate to tissue macrophages. Apart from the phagocytic activity, the neutrophils and macrophages act as reservoir for growth factors and regenerative mediators 21. A large body of evidence support the association of inflammation with laparotomy healing 22, 23; however, information on alterations in the inflammatory responses resulting in IH is lacking. In general, the inflammatory cells release several cytokines in the vicinity of the injury to increase vascular permeability and fibroblast activity 24. Therefore, the major hallmarks associated with IH pathology are alterations in collagen type I to type III ratio, loss of mechanical integrity and abnormal fibroblast proliferation. The key biological events associated with the development of IH are shown in Fig. 2.
Fig. 2:
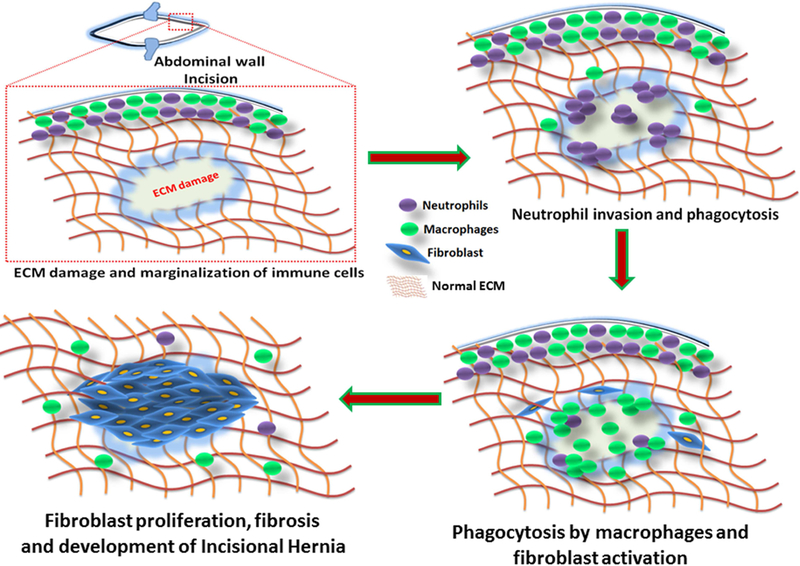
The pathological events associated with the development of IH. Following the incision at the abdominal wall, the leukocytes and plasma protein get marginalized surrounding the wound. The neutrophils act as first line of defense and elicit acute inflammatory responses followed by the recruitment of macrophages. The phagocytosis and macrophage signaling sustain the inflammation and activate MMPs and fibroblasts. The proliferation of fibroblasts results in fibrosis and leads to altered ECM composition which in turn activates MMPs and inflammation. This process continues to build up truncated ECM due to the decreased collagen type I to type III ratio and excessive proliferation of fibroblasts that eventually leads to IH formation.
Altered Fibroblast Biology – A Door to IH Formation
Fibroblasts are considered to be the repair cells that maintain ECM collagen homeostasis facilitating recovery of tissue function following surgical incision. The biomarkers for fibroblasts from various tissues are shown in Table 1. The migration and proliferation of fibroblasts at the wound site begin within 2 days of incision and constitute the major portion of granulation tissue within 4 days. The fibroblast migration and proliferation are regulated by soluble growth factors, inflammatory cytokines, and elements of the provisional matrix 2. However, very little is known why fibroblast function is impaired in IH wound failure. In other disease processes, a reduction in the level of growth factors can drive the fibroblast cells to a quiescence state, such as in ulcerative wounds 25. The fascial or tendon ischemia has also been shown to induce cell cycle arrest in fibroblasts 2. This could play a role in the development IH but has not been thoroughly investigated.
Table 1:
Biomarkers for fibroblast characterization
| Biomarkers | Abbreviation | References |
|---|---|---|
| Podoplanin | PDPN | 79 |
| Cadherin-11 | CDH11 | 79 |
| Thymocyte differentiation antigen 1/ Cluster of differentiation-90 |
THY1/CD90 | 79 |
| Cluster of differentiation-34 | CD34 | 79 |
| Fibroblast specific protein-1 | FSP1 | 83 |
| Platelet-derived growth factor receptor-α | PDGFR-α | 83 |
Degenerative changes taking place in the abdominal wall following a laparotomy explain the development of IH. Diaz et al. 26 reported histological alterations in the fascia of IH patients. The tissue sections displayed a significant thinning of ECM and decreased fibroblast density with negligible immune cells. The fibroblasts were round or oblong with minimal cell-matrix interaction. The fibroblasts from IH tissue displayed a spindle bipolar morphology with reduced surface area and expressed more network of vimentin than actin. The data from electron microscopy revealed vacuolated cytoplasm and mitochondrial swelling. These fibroblasts displayed an increased proliferation, decreased adhesion and faster migration in the culture in response to fibronectin and collagen type I. Also, the IH fibroblast cells revealed increased sensitivity to apoptosis and autophagy 26.
The resident fibroblasts regulate hernia tissue remodeling independent of inflammatory cells such as macrophages. Also, the excessive secretion of collagen type III by skin fibroblasts has been associated with the development of IH. Moreover, the disorganized hernia tissue matrix has been associated with increased activity of MMPs, especially MMP-1, MMP-2, MMP-9 and MMP-13 27, 28. The fibroblasts from different areas in an IH complex (for example Sac vs. fascia vs. fat) exhibit difference in chemotactic responses. However, the exact histological origin of these fibroblasts is largely unknown 18. In addition, changes in load bearing properties of the abdominal wall contribute to phenotype selection of fibroblasts 8. The mechanical stimuli are transmitted to the fibroblasts through integrin receptors and affect cellular metabolism 19.
Collagen Homeostasis – Outcome of Fibroblast Function
Collagen constitutes more than 80% of abdominal wall fascia and is the product of fibroblasts. Collagen turnover is maintained by the balance between its synthesis and degradation. Defective collagen homeostasis results in too little collagen type I at the wound site and leads to mechanical weakness and wound failure 2. Among the 28 phenotypes of collagen, the abdominal wall fascia consisted of collagen type I and little amounts of type III and IV. Collagen type III imparts weaker mechanical properties to the tissue while collagen type IV initiates ECM formation. The collagen molecules undergo extensive modifications before maturation and subsequent deposition to ECM. The non-standard amino acids, hydroxyproline and hydroxylysine, are the unique features of collagen triple helix. The enzymes proline hydroxylase and lysine hydroxylase catalyze the cross linking of collagens to impart mechanical integrity and stability 29. Patients with IH displayed a reduction in hydroxyproline content in the collagen associated with their structural tissues. Also, the fibroblasts from these patients exhibited a defective proliferation rate and failed to translocate hydroxyproline which resulted in decreased cross linking and increased solubility of collagen and thus decreasing its mechanical integrity 30, 31.
The collagen type I to type III ratio is known to be an important determinant for the development of an incisional hernia. Collagen type III has also been found to be associated with collagen type I in several tissue types 32, 33, 34. Type I collagen is fibrous, mechanically strong and thicker in diameter when compared to collagen type III. If an overall content of type III compared to type I is increased, the collagen fibers will have a decreased diameter and will be more disorganized 35. Also, the basement membrane composed of collagen type IV is important 36. In addition, collagen type V associates with IH formation; however, the level of type V collagen remains unaltered in scar tissues of IH patients. Collagen type V will also interact with type I and is involved in fibrillogenesis. Mutation studies revealed that decreased level of collagen type V leads to poor quality ECM 37. On the other hand, a drastic decrease in type IV collagen was observed in the fascia of IH tissues 38. This suggests that the alteration of basement membrane leads to IH development and the disturbances in collagen homeostasis and turnover produce truncated ECM/scar tissue following laparotomy. However, the exact ratios of collagen type I to type III and type IV to type V have not been established. The alterations in the composition and phenotype of collagen paves the way to the abdominal wall weakening and IH development. Therefore, it is reasonable to speculate that altered collagen phenotype results from the switch of healthy fibroblasts to abnormal phenotype. However, this requires further investigation in relation to the pathogenesis of IH.
The degradation products of collagen in the ECM released into blood stream following laparotomy and tissue remodeling are considered to be the serum biomarkers for collagen turnover 39. Such degradation fragments are referred as neo-epitopes and their circulating levels reflect collagen homeostasis 40, 41. Based on estimating the collagen degradation fragments in circulation, Henriksen et al. 41 reported the predominance of collagen type IV turnover preoperatively in IH patients when compared to collagen type V. The serum level of N-terminal pro-peptide of type IV collagen 7S domain (P4NP-7S), a fragment of collagen type IV degradation, was reported to be elevated in IH patients and is considered to be associated with IH development 42. These studies suggest the diagnostic value of measuring the degradation products of collagen. Also, the alterations in collagen phenotypes and poor quality of ECM associated with IH could be due to abnormal metabolism of the resident fibroblast cells. Thus, the implications of collagen degradation products in fibroblast signaling leading to IH development open up new avenues for further investigation.
Transdifferentiation of Fibroblasts to Myofibroblasts – The key to Abdominal Wound Healing
The resident fibroblasts of fascial tissues are potentially active in collagen synthesis when compared to dermal fibroblasts, especially after laparotomy. The fascia is more hypovascular than the dermis and is similar to ligaments and tendons. The wound environment stimulates proliferation and migration of the fibroblasts and facilitates the maturation of the ECM. It is likely that the functional activities of fascial fibroblasts and dermal fibroblasts are different 7. Apart from the deposition of collagen subtypes, these fibroblasts secrete other components of matrisome including proteoglycans, fibronectin and hyaluronic acid 43. In addition, the fibroblasts are well known sources for MMP-1, MMP-2, MMP-9 and MMP-19 and tissue inhibitors of MMPs, especially TIMP-1, TIMP-2 and TIMP-3 44. The fibroblasts present in the wound differentiate to a contractile phenotype – the alpha smooth muscle actin ( α-SMA)-positive myofibroblasts. The mechanical forces generated by the myofibroblasts result in the contraction of the neo-matrisome and subsequent healing of the wound 45. The endogenous tension exerted by the fibroblasts within the wound matrix triggers the upregulation of a battery of mechanoresponsive genes which includes the genes for matrisome components and cytoskeletal proteins. Also, this mechanotransduction has been considered to activate the proliferation and differentiation of fibroblasts to myofibroblasts 45.
The myofibroblasts have been described as a marker for scarring and fibrosis 46. The ECM components deposited by the myofibroblasts replace the provisional wound matrix and results in wound contraction due to the formation of stress fibers. Stress fiber formation is due to the expression of α-SMA in the microfilaments of fibroblast cells. The differentiation is characterized by the appearance of protomyofibroblasts which express β- and γ-cytoplasmic actins 47. However, following the resolution phase of wound healing the number of myofibroblasts drastically decreases due to apoptosis 48. The local connective tissues are considered to be the major sources for myofibroblasts 49, and the skin and/or fascial fibroblasts may acquire myofibroblastic phenotype following the incisional wound. Heterogeneous subpopulations of fibroblasts reside in different locations of the abdominal wall tissue. For instance, skin consists of papillary fibroblasts that are superficial usually present 300–400µm deep in the dermis, reticular fibroblasts deep in the dermis, and the fibroblasts of hair follicles 50.
Fibrocytes differ from fibroblast by virtue of the fact that they originate in blood and can be distributed in circulation. These circulating fibrocytes express fibroblast components like collagen subtypes, vimentin, fibronectin and have a role in wound repair 51. The fibrocytes are recruited to the wound site through general circulation and in association with inflammatory cells, differentiate to myofibroblasts 52. Also, the fibrocytes trigger local inflammation at the wound site and upregulate the expression of ECM proteins through TGF-β1 signaling 53, 54. Bone marrow-derived circulating progenitor cells that differentiate to myofibroblasts were also identified in human and animal models 55, 56. In addition, epithelial/endothelial-to-mesenchymal transition generates ECM-secreting fibroblasts and myofibroblasts 57. The myofibroblasts derived from circulation signify an alternative source of myofibroblasts when local fibroblasts are not sufficient for ECM remodeling 58. The major sources of myofibroblasts are shown in Table 2.
Table 2:
Precursors of myofibroblasts
| Cells | Sources | References |
|---|---|---|
| Fibroblasts | Wound tissue | 44 |
| Fibrocytes | blood | 51 |
| Circulating progenitor cells |
Bone marrow and blood | 55 |
| Epithelial | Epithelial/endothelial-to-mesenchymal transition |
56 |
| Endothelial | Epithelial/endothelial-to-mesenchymal transition |
56 |
Myofibroblast Signaling
Myofibroblasts provide mechanical integrity to the tissues following injury. After the completion of the repair, the myofibroblasts are removed from the wound site by apoptosis to reduce the scar formation. On the other hand, the reduction in apoptotic signals in the tissue leads to the sustenance of myofibroblasts and subsequent scarring. The differentiation and apoptosis of myofibroblasts are regulated primarily by mechanotransduction 59. TGF-β1 signaling induces the differentiation of fibroblasts to myofibroblasts and the contractile nature of the latter increases the mechanical stress and stiffness of the wound matrix 60. The myofibroblasts release TGF-β1 complexed with latency-associated peptide (LAP) and a binding peptide and this complex acts as a reservoir for TGF-β1 which is activated as healing progresses 61. Moreover, the integrins secreted by myofibroblasts activate TGF-β1 without releasing the LAP and facilitates the binding to its receptors 62. This acts as a positive feedback regulation for the differentiation and survival of myofibroblasts 63. Wound stress activates the upregulation of α-SMA and collagen which facilitates ECM maturation by the upregulation of TIMPs with a concomitant downregulation of MMPs 64, 65. The major molecular events associated with the transdifferentiation of fibroblasts to myofibroblasts are depicted in Fig. 3.
Fig. 3:
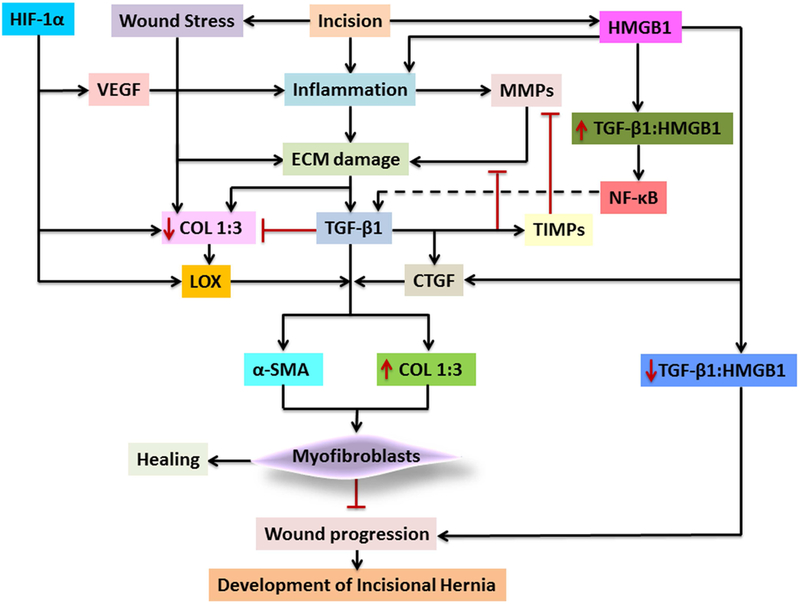
Transdifferentiation of fibroblasts to myofibroblasts. The TGF-β signaling upregulates α-SMA expression resulting in the differentiation to myofibroblasts. The integrins secreted by myofibroblasts activate TGF-β resulting in a positive feedback regulation. The increase in myofibroblast causes wound contraction, which leads to their apoptosis and paves ways for wound healing with minimum scaring.
The abdominal wound experiences severe hypoxic stress immediately after surgical incision, which has been considered as a major risk factor for wound failure 66. The center of the wound is hypoxic which interferes with angiogenesis and healing process 67. The surviving tissue responds to hypoxia by altering the metabolic flux by upregulating the expression of HIF-1α. HIF-1α facilitates angiogenesis by upregulating VEGF to recover the tissues from the deleterious effects of hypoxia 68. The revascularization at the wound site is critical to improve the healing responses as the oxygen tension determines the deposition of collagen to the wound matrix 69. In addition, the hypoxia inhibits the activation of myofibroblasts and reduces the expression of collagen and α-SMA. Furthermore, HIF-1α directly influences the collagen deposition and migration of fibroblasts and their differentiation to myofibroblasts. However, the cross talk between HIF-1α and TGF-β signaling worsens the matrix renovation and results in fibrosis 58. The understanding of the effects of hypoxia on wound healing and the development of IH may help unravel the underlying molecular pathology and allow the development of therapeutic strategies to reduce the effect.
The expression of connective tissue growth factor (CTGF) has been reported to be increased during ECM remodeling which acts in connection with TGF-β signaling. CTGF is overexpressed in the fibroblasts present in the wound and is a potential biomarker for profibrotic fibroblasts 70. TGF-β directly induces the transcription of CTGF suggesting that CTGT may act as a downstream mediator for TGF-β signaling to sustain the fibrotic response. Interestingly, TGF-β and CTGF alone result in transient fibrosis; however the synergistic effects of TGF-β and CTGF are required for sustained fibrosis 71. Moreover, CTGF acts as an extracellular adapter protein for TGF-β via the cysteine-rich domain and this binding aids to present the TGF-β to its receptors to stimulate fibrosis 72. CTGF also has been shown to contribute to the transdifferentiation of fibroblasts to myofibroblasts 70, 73.
The LOX family of enzymes are another important component of the ECM responsible for its mechanical integrity and is associated with fibroblast proliferation 74. The myofibroblasts from several tissues were characterized with the increased secretion of the crosslinking lysyl oxidase (LOX) family of enzymes, including LOX and LOX-like 1, −2, −3 and −4 (LOXL1, −2, −3 and −4). LOX and LOXL2 also associate with the transdifferentiation of fibroblast to myofibroblasts 75, 76. Apart from the crosslinking function, LOX prevents inflammation by reducing the levels of CCL2 77. In contrast, LOX acts as a chemoattractant for monocytes suggesting their interplay between inflammation and ECM remodeling 78. Tissue hypoxia is a well-known inducer of LOX expression which is prevalent in tumor cells. LOXL2 mediates the fibrosis-associated collagen crosslinking and catalyzes the oxidative deamination of ε-NH2 group of Lys residues in the collagens and elastin 79. However, the potential role of LOX family of enzymes in the transdifferentiation of myofibroblasts and fibrosis associated with IH are largely unknown.
Recently, the existence of heterogeneous population of fibroblasts was reported in synovial tissue using single-cell proteomics and is classified into seven subsets based on their surface proteins. These fibroblasts are expected to differ in their function, phenotype, proliferation and exhibit different response to injury and inflammation 80. In addition, the damage-associated molecular pattern molecules (DAMPs), including high-mobility group box 1 (HMGB1), heat shock proteins (HSPs), purine metabolites, hyaluronic acid, heparin sulfate, and others are well known inducers of inflammation. Specifically, HMGB1 is involved in the fibrosis associated with multiple pathologies 32. The exogenous administration of HMGB1 resulted in proliferation of dermal fibroblasts with a concomitant upregulation of the transcripts of collagen, TIMPs and α-SMA and subsequent downregulation of MMPs, especially MMP-1 81. Interestingly, HMGB1 triggers the differentiation of fibroblasts to myofibroblasts by activating NF-κB-mediated release of TGF-β1 82. This suggests the possible role of HMGB1 in transdifferentiation of fibroblasts to myofibroblasts and HMGB1 signaling is similar to that of TGF-β which warrant further investigation in regard to the development of IH. Moreover, HMGB1 activates the expression of CTGF in several tissues suggesting its role in myofibroblast activation 83, 84.
In contrast, HMGB1 is well known for inducing inflammation and delayed healing responses via TLRs, RAGE and TREM-1-mediated activation of NLRP3 inflammasome in different tissues 32. Thus, HMGB1 has a dual function of facilitating wound healing via TGF-β1 signaling and inducing wound progression through NLRP3 inflammasome. These contrasting effects suggest that the ratio of TGF-β1 and HMGB1 determines wound healing or wound progression. Thus, it is reasonable to speculate that the increased TGF-β1:HMGB1 ratio drives the healing response and vice versa. Obviously, detailed investigations are required.
Summary and Future Perspectives
The alteration of matrisome components and the proliferation of abnormal fibroblasts are the most significant molecular events leading to IH formation. The scar tissue formed as a result of an abdominal wall incision results in poor mechanical properties which could be the trigger for proliferation of abnormal fibroblasts. The hypoxia and ischemia associated with the incision also trigger the deposition of a truncated ECM resulting in delayed wound healing. The alteration in the levels and ratios of collagen type 1, type 3, type 4 and type 5 are the root cause of ECM disorganization which is due to the impairments in MMPs and TIMPs. Since fibroblasts function in collagen homeostasis, it is reasonable to hypothesize that the phenotypic switch of fibroblasts is a critical factor in ECM disorganization following laparotomy. The phenotype selection of fibroblasts is determined by the loss of mechanical integrity of abdominal wall and the persistence of ECM damage. However, the underlying molecular mechanisms that drive the phenotype selection of fibroblasts are yet to be discovered. The cytokines released by inflammatory cells manipulate the activity of fibroblasts that determine the status of wound healing. In addition, the prolonged inflammatory responses lead to the fibroproliferative burst and ECM damage. There is a drastic increase in the density of fibroblasts in the hernia tissue, but it is not clear whether these are quiescent fibroblasts or precursor cells which also warrants further investigation. The disturbances in collagen homeostasis due to the phenotype switch of fibroblasts results in mechanically compromised ECM which allows further proliferation of abnormal fibroblasts. The measurement of degradation products of collagen phenotype in serum shows promise as a diagnostic tool. However, the signaling effects of such fragments in the development of IH also need further research. The transdifferentiation of fibroblasts to myofibroblasts is crucial for abdominal wound healing and the impairment in the formation of myofibroblasts leads to abnormal proliferation and herniation of the wound. The in-depth analysis of the precursor cells for myofibroblasts other than fibroblasts warrants further attention. Signaling by TGF-β1, CTGF, LOX, and HIF-1α plays a significant role in the wound healing responses. The DAMP, HMGB1, plays contrasting roles in wound healing and wound progression. It is proposed that the TGF-β1:HMGB1 ratio determines the role of HMGB1 in the IH tissue. Based on the critical review of the literature, we conclude that the TGF-β1:HMGB1 ratio and myofibroblast proliferation are major targets for intervention for the prevention of IH. To prove this, the underlying molecular mechanisms for myofibroblast proliferation and their regulation in regard to IH development warrant further investigation. A possible signaling pathway has been proposed in this article; however, this remains to be proven (Fig. 4). Currently, there is a significant gap in our knowledge of basic biology in the pathogenesis of IH. A thorough screening of the cellular and molecular mediators associated with the changes in the fibroblast phenotype in the tissues of patients with incisional hernia using immunohistological approaches could reveal the molecular pathology as well as therapeutic targets. The unavailability of a clinically-relevant animal model for IH limits the understanding of the underlying molecular mechanisms in the pathogenesis of incisional hernia. Indeed, the identification of novel molecular targets would open opportunities for the development of knock-out animal models for IH. Overall, an understanding of the molecular and cellular events associated with the development of incisional hernia would pave the way for the development of novel therapeutic strategies for the prevention of IH.
Fig. 4:
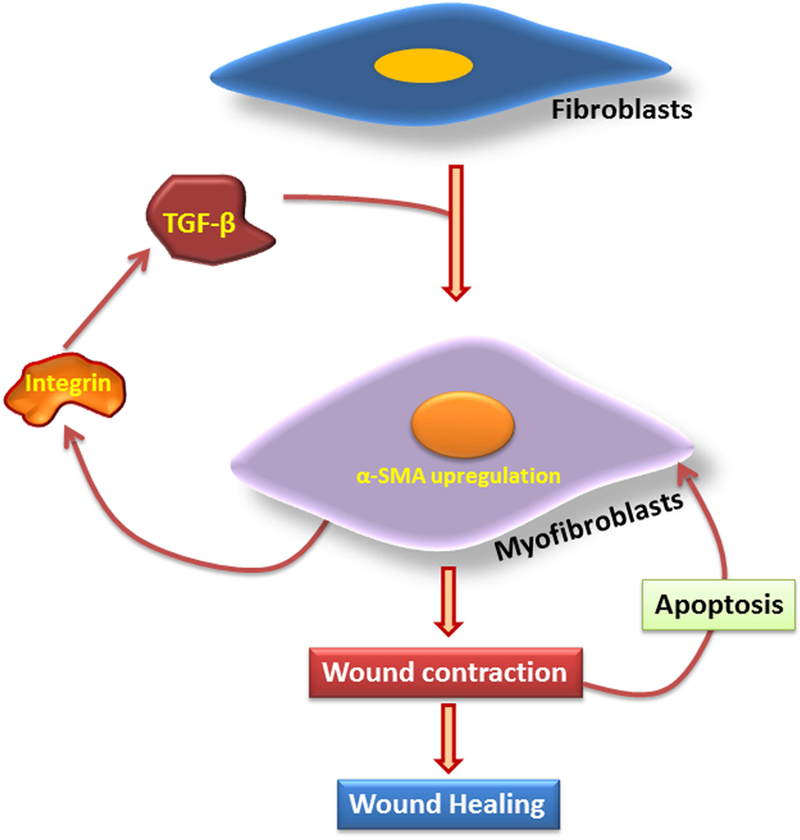
The proposed mechanism for IH formation in the abdominal wall: The incision triggers classical inflammatory responses where the persistence of inflammation induces the ECM damage by decreasing the ratio of collagen type 1 to type 3. This is due to the hyperactivity of MMPs and inhibition of TIMPs. The inflammation and ECM damage triggers the expression and activation of TGF-β1 which reduces inflammation by increasing the ratio of collagen type 1 to type 3, and inhibition of MMPs by TIMP activation. TGF-β1 modulates the transdifferentiation of fibroblasts to myofibroblasts by upregulating α-SMA and collagen type 1. TGF-β1 also activates CTGF and LOX, which induces the transdifferentiation of fibroblasts to myofibroblasts. The wound stress and HIF-1α trigger LOX activity which functions in collagen maturation. The HMGB1 triggers inflammation via the activation of inflammasome. The high ratio of TGF-β1-to-HMGB1 drives TGF-β1 signaling to trigger healing responses, whereas the low ratio results in delayed healing by the persistence of inflammation that leads to the formation of incisional hernia.
Acknowledgments
Funding: This work was supported by research grants of the Department of Surgery, Creighton University School of Medicine to RJF. The research work of DKA is supported by R01 HL120659 and R01HL144125 grants from the National Heart, Lung and Blood Institute, National Institutes of Health, USA. The content of this review article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. All authors have read the journal’s authorship agreement.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
All authors have read the journal’s policy on disclosure of potential conflicts of interest. The authors have no other relevant affiliations or financial or non-financial involvement with any organization or entity with financial or non-financial interest or conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
References
- 1.Sevinç B, Oku ş A, Ay S, Aksoy N, Karahan Ö. Randomized prospectiv e comparison of long-term results of onlay and sublay mesh repair techniques for incisional hernia. Turk J Surg 2018;34(1):17–20. doi: 10.5152/turkjsurg.2017.3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franz MG. The Biology of Hernia Formation. Surg Clin North Am 2008;88(1):1–15. doi: 10.1016/j.suc.2007.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anthony T, Bergen PC, Kim LT, et al. Factors affecting recurrence following incisional herniorrhaphy. World J Surg 2000;24(1):95–100; discussion 101. [DOI] [PubMed] [Google Scholar]
- 4.Millbourn D, Cengiz Y, Israelsson LA. Effect of stitch length on wound complications after closure of midline incisions: a randomized controlled trial. Arch Surg 2009;144(11):1056–1059. doi: 10.1001/archsurg.2009.189.) [DOI] [PubMed] [Google Scholar]
- 5.Flum DR, Horvath K, Koepsell T. Have outcomes of incisional hernia repair improved with time? A population-based analysis. Ann Surg 2003;237(1):129–135. doi: 10.1097/01.SLA.0000041042.86225.9C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katsumi A, Naoe T, Matsushita T, Kaibuchi K, Schwartz MA. Integrin Activation and Matrix Binding Mediate Cellular Responses to Mechanical Stretch. J Biol Chem 2005;280(17):16546–16549. doi: 10.1074/jbc.C400455200 [DOI] [PubMed] [Google Scholar]
- 7.DuBay DA, Wang X, Adamson B, Kuzon WM, Dennis RG, Franz MG. Progressive fascial wound failure impairs subsequent abdominal wall repairs: A new animal model of incisional hernia formation. Surgery 2005;137(4):463–471. doi: 10.1016/j.surg.2004.12.016 [DOI] [PubMed] [Google Scholar]
- 8.Benjamin M, Hillen B. Mechanical Influences on Cells, Tissues and Organs ? ?Mechanical Morphogenesis? Eur J Morphol 2003;41(1):3–7. doi: 10.1076/ejom.41.1.3.28102 [DOI] [PubMed] [Google Scholar]
- 9.Fouda MB, Thankam FG, Dilisio MF, Agrawal DK. Alterations in tendon microenvironment in response to mechanical load: potential molecular targets for treatment strategies. Am J Transl Res 2017;9(10):4341–4360. [PMC free article] [PubMed] [Google Scholar]
- 10.Dubay DA, Wang X, Kuhn MA, Robson MC, Franz MG. The prevention of incisional hernia formation using a delayed-release polymer of basic fibroblast growth factor. Ann Surg 2004;240(1):179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robson MC, Steed DL, Franz MG. Wound healing: Biologic features and approaches to maximize healing trajectories. Curr Probl Surg 2001;38(2):A1–140. doi: 10.1067/msg.2001.111167 [DOI] [PubMed] [Google Scholar]
- 12.Robson MC, Shaw RC, Heggers JP. The reclosure of postoperative incisional abscesses based on bacterial quantification of the wound. Ann Surg 1970;171(2):279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Junge K, Klinge U, Klosterhalfen B, Rosch R, Stumpf M, Schumpelick V. Review of Wound Healing with Reference to an Unrepairable Abdominal Hernia. Eur J Surg 2002;168(2):67–73. doi: 10.1080/11024150252884269 [DOI] [PubMed] [Google Scholar]
- 14.Friedman DW, Boyd CD, Mackenzie JW, Norton P, Olson RM, Deak SB. Regulation of Collagen Gene Expression in Keloids and Hypertrophic Scars. J Surg Res 1993;55(2):214–222. doi: 10.1006/jsre.1993.1132 [DOI] [PubMed] [Google Scholar]
- 15.Klosterhalfen B, Lynen P, Schumpelick V, et al. Decreased collagen type I/III ratio in patients with recurring hernia after implantation of alloplastic prostheses. Langenbecks Arch Surg 2004;389(1):17–22. doi: 10.1007/s00423-003-0429-8 [DOI] [PubMed] [Google Scholar]
- 16.Koruth S, Narayanaswamy Chetty YV. Hernias- Is it a primary defect or a systemic disorder? Role of collagen III in all hernias- A case control study. Ann Med Surg 2017;19:37–40. doi: 10.1016/j.amsu.2017.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franz M, Kuhn M, Nguyen K, et al. A biological approach to prevention and treatment of incisional hernias. J Am Coll Surg 2000;191(4):S48. doi: 10.1016/S1072-7515(00)00519-6 [DOI] [Google Scholar]
- 18.Xing L, Culbertson EJ, Wen Y, Franz MG. Early laparotomy wound failure as the mechanism for incisional hernia formation. J Surg Res 2013;182(1):e35–e42. doi: 10.1016/j.jss.2012.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt C, Pommerenke H, Dürr F, Nebe B, Rychly J. Mechanical stressing of integrin receptors induces enhanced tyrosine phosphorylation of cytoskeletally anchored proteins. J Biol Chem 1998;273(9):5081–5085. [DOI] [PubMed] [Google Scholar]
- 20.Skutek M, van Griensven M, Zeichen J, Brauer N, Bosch U. Cyclic mechanical stretching modulates secretion pattern of growth factors in human tendon fibroblasts. Eur J Appl Physiol 2001;86(1):48–52. doi: 10.1007/s004210100502 [DOI] [PubMed] [Google Scholar]
- 21.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016;44(3):450–462. doi: 10.1016/j.immuni.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koduru P, Abraham BP. P185 INCISIONAL HERNIA REPAIR COMPLICATING INFLAMMATORY BOWEL DISEASE. Inflamm Bowel Dis 2018;24(suppl_1):S73–S73. doi: 10.1093/ibd/izy038.023 [DOI] [Google Scholar]
- 23.Clarke J, Thomson H, Windsor A, McCullough J. PWE-230 The exaggerated inflammatory response to large incisional hernia and enterocutaneous fistula repair with biological mesh. Gut 2015;64(Suppl 1):A313–1-A313.. doi: 10.1136/gutjnl-2015-309861.676 [DOI] [Google Scholar]
- 24.Di Vita G, Patti R, Vetri G, et al. Production of cytokines at the operation site. Il G Chir 2005;26(6–7):241–245. [PubMed] [Google Scholar]
- 25.Vande Berg JS, Rudolph R, Hollan C, Haywood-Reid PL. Fibroblast senescence in pressure ulcers. Wound Repair Regen Off Publ Wound Heal Soc Eur Tissue Repair Soc 1998;6(1):38–49. [DOI] [PubMed] [Google Scholar]
- 26.Diaz R, Quiles MT, Guillem-Marti J, et al. Apoptosis-Like Cell Death Induction and Aberrant Fibroblast Properties in Human Incisional Hernia Fascia. Am J Pathol 2011;178(6):2641–2653. doi: 10.1016/j.ajpath.2011.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jansen PL, Mertens PR, Klinge U, Schumpelick V. The biology of hernia formation. Surgery 2004;136(1):1–4. doi: 10.1016/j.surg.2004.01.004 [DOI] [PubMed] [Google Scholar]
- 28.Bellón JM, Bajo A, Ga-Honduvilla N, et al. Fib roblasts from the transversalis fascia of young patients with direct inguinal hernias show constitutive MMP-2 overexpression. Ann Surg 2001;233(2):287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henriksen NA, Jensen KK, Jorgensen LN. The Biology of Hernia Formation. In: Textbook of Hernia Springer, Cham; 2017:1–5. doi: 10.1007/978-3-319-43045-4_1 [DOI] [Google Scholar]
- 30.Jorgensen LN, Kallehave F, Karlsmark T, Gottrup F. Reduced collagen accumulation after major surgery. Br J Surg 1996;83(11):1591–1594. [DOI] [PubMed] [Google Scholar]
- 31.Zheng H, Si Z, Kasperk R, et al. Recurrent inguinal hernia: disease of the collagen matrix? World J Surg 2002;26(4):401–408. doi: 10.1007/s00268-001-0239-5 [DOI] [PubMed] [Google Scholar]
- 32.Thankam FG, Roesch ZK, Dilisio MF, et al. Association of Inflammatory Responses and ECM Disorganization with HMGB1 Upregulation and NLRP3 Inflammasome Activation in the Injured Rotator Cuff Tendon. Sci Rep 2018;8(1). doi: 10.1038/s41598-018-27250-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thankam FG, Boosani CS, Dilisio MF, Dietz NE, Agrawal DK. MicroRNAs Associated with Shoulder Tendon Matrisome Disorganization in Glenohumeral Arthritis. Kukreja R, ed. PLOS ONE 2016;11(12):e0168077. doi: 10.1371/journal.pone.0168077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chenicheri S,RU, Ramachandran R, Thomas V, Wood A. Insight into Oral Biofilm: Primary, Secondary and Residual Caries and Phyto-Challenged Solutions. Open Dent J 2017;11(1):312–333. doi: 10.2174/1874210601711010312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radu P, Brătucu M, Garofil D, et al. The Role of Collagen Metabolism in the Formation and Relapse of Incisional Hernia. Chir Buchar Rom 1990 2015;110(3):224–230. [PubMed] [Google Scholar]
- 36.Kruegel J, Miosge N. Basement membrane components are key players in specialized extracellular matrices. Cell Mol Life Sci 2010;67(17):2879–2895. doi: 10.1007/s00018-010-0367-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Symoens S, Syx D, Malfait F, et al. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat 2012;33(10):1485–1493. doi: 10.1002/humu.22137 [DOI] [PubMed] [Google Scholar]
- 38.Rosch R, Junge K, Knops M, Lynen P, Klinge U, Schumpelick V. Analysis of collagen-interacting proteins in patients with incisional hernias. Langenbecks Arch Surg 2003;387(11–12):427–432. doi: 10.1007/s00423-002-0345-3 [DOI] [PubMed] [Google Scholar]
- 39.Henriksen NA, Mortensen JH, Lorentzen L, et al. Abdominal wall hernias—A local manifestation of systemically impaired quality of the extracellular matrix. Surgery 2016;160(1):220–227. doi: 10.1016/j.surg.2016.02.011 [DOI] [PubMed] [Google Scholar]
- 40.DuBay DA, Choi W, Urbanchek MG, et al. Incisional herniation induces decreased abdominal wall compliance via oblique muscle atrophy and fibrosis. Ann Surg 2007;245(1):140–146. doi: 10.1097/01.sla.0000251267.11012.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henriksen NA, Mortensen JH, Sorensen LT, et al. The collagen turnover profile is altered in patients with inguinal and incisional hernia. Surgery 2015;157(2):312–321. doi: 10.1016/j.surg.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 42.Kayashima H, Maeda T, Harada N, et al. Risk factors for incisional hernia after hepatic resection for hepatocellular carcinoma in patients with liver cirrhosis. Surgery 2015;158(6):1669–1675. doi: 10.1016/j.surg.2015.06.001 [DOI] [PubMed] [Google Scholar]
- 43.Clark RAF, Ghosh K, Tonnesen MG. Tissue Engineering for Cutaneous Wounds. J Invest Dermatol 2007;127(5):1018–1029. doi: 10.1038/sj.jid.5700715 [DOI] [PubMed] [Google Scholar]
- 44.Toriseva M, Kähäri V- M. Proteinases in cutaneo us wound healing. Cell Mol Life Sci 2009;66(2):203–224. doi: 10.1007/s00018-008-8388-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rolin G, Binda D, Tissot M, et al. In vitro study of the impact of mechanical tension on the dermal fibroblast phenotype in the context of skin wound healing. J Biomech 2014;47(14):3555–3561. doi: 10.1016/j.jbiomech.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 46.Desmoulière A, Darby IA, Gabbiani G. Normal and pathologic soft tissue remodeling: role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab Investig J Tech Methods Pathol 2003;83(12):1689–1707. [DOI] [PubMed] [Google Scholar]
- 47.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3(5):349–363. doi: 10.1038/nrm809 [DOI] [PubMed] [Google Scholar]
- 48.Desmoulière A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol 1995;146(1):56–66. [PMC free article] [PubMed] [Google Scholar]
- 49.Higashiyama R, Nakao S, Shibusawa Y, et al. Differential Contribution of Dermal Resident and Bone Marrow–Derived Cells to Collagen Production during Wound Healing and Fibrogenesis in Mice. J Invest Dermatol 2011;131(2):529–536. doi: 10.1038/jid.2010.314 [DOI] [PubMed] [Google Scholar]
- 50.Mine S, Fortunel NO, Pageon H, Asselineau D. Aging Alters Functionally Human Dermal Papillary Fibroblasts but Not Reticular Fibroblasts: A New View of Skin Morphogenesis and Aging. Sullivan B, ed. PLoS ONE 2008;3(12):e4066. doi: 10.1371/journal.pone.0004066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blakaj A, Bucala R. Fibrocytes in health and disease. Fibrogenesis Tissue Repair 2012;5(Suppl 1):S6. doi: 10.1186/1755-1536-5-S1-S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol Baltim Md 1950 2001;166(12):7556–7562. [DOI] [PubMed] [Google Scholar]
- 53.Kamel RA, Ong JF, Eriksson E, Junker JPE, Caterson EJ. Tissue Engineering of Skin. J Am Coll Surg 2013;217(3):533–555. doi: 10.1016/j.jamcollsurg.2013.03.027 [DOI] [PubMed] [Google Scholar]
- 54.Yang L, Scott PG, Dodd C, et al. Identification of fibrocytes in postburn hypertrophic scar. Wound Repair Regen Off Publ Wound Heal Soc Eur Tissue Repair Soc 2005;13(4):398–404. doi: 10.1111/j.1067-1927.2005.130407.x [DOI] [PubMed] [Google Scholar]
- 55.Opalenik SR, Davidson JM. Fibroblast differentiation of bone marrow-derived cells during wound repair. FASEB J Off Publ Fed Am Soc Exp Biol 2005;19(11):1561–1563. doi: 10.1096/fj.04-2978fje [DOI] [PubMed] [Google Scholar]
- 56.Direkze NC, Forbes SJ, Brittan M, et al. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells Dayt Ohio 2003;21(5):514–520. doi: 10.1634/stemcells.21-5-514 [DOI] [PubMed] [Google Scholar]
- 57.Nakamura M, Tokura Y. Epithelial-mesenchymal transition in the skin. J Dermatol Sci 2011;61(1):7–13. doi: 10.1016/j.jdermsci.2010.11.015 [DOI] [PubMed] [Google Scholar]
- 58.Darby IA, Laverdet B, Bonté F, Desmoulière A.Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol 2014;7:301–311. doi: 10.2147/CCID.S50046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hinz B The myofibroblast: paradigm for a mechanically active cell. J Biomech 2010;43(1):146–155. doi: 10.1016/j.jbiomech.2009.09.020 [DOI] [PubMed] [Google Scholar]
- 60.Goffin JM, Pittet P, Csucs G, Lussi JW, Meister J-J, Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol 2006;172(2):259–268. doi: 10.1083/jcb.200506179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buscemi L, Ramonet D, Klingberg F, et al. The single-molecule mechanics of the latent TGF-β1 complex. Curr Biol CB 2011;21(24):2046–2054. doi: 10.1016/j.cub.2011.11.037 [DOI] [PubMed] [Google Scholar]
- 62.Wipff P-J, Rifkin DB, Meister J-J, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol 2007;179(6):1311–1323. doi: 10.1083/jcb.200704042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aarabi S, Bhatt KA, Shi Y, et al. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J Off Publ Fed Am Soc Exp Biol 2007;21(12):3250–3261. doi: 10.1096/fj.07-8218com [DOI] [PubMed] [Google Scholar]
- 64.Wang J, Chen H, Seth A, McCulloch CA. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol 2003;285(5):H1871–1881. doi: 10.1152/ajpheart.00387.2003 [DOI] [PubMed] [Google Scholar]
- 65.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J Off Publ Fed Am Soc Exp Biol 2004;18(7):816–827. doi: 10.1096/fj.03-1273rev [DOI] [PubMed] [Google Scholar]
- 66.Chang N, Goodson WH, Gottrup F, Hunt TK. Direct measurement of wound and tissue oxygen tension in postoperative patients. Ann Surg 1983;197(4):470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knighton DR, Silver IA, Hunt TK. Regulation of wound-healing angiogenesis-effect of oxygen gradients and inspired oxygen concentration. Surgery 1981;90(2):262–270. [PubMed] [Google Scholar]
- 68.Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol Cell Physiol 2001;280(6):C1367–1374. doi: 10.1152/ajpcell.2001.280.6.C1367 [DOI] [PubMed] [Google Scholar]
- 69.Lisiecki J, Rinkinen J, Eboda O, et al. Adipose-Derived Mesenchymal Stem Cells from Ventral Hernia Repair Patients Demonstrate Decreased Vasculogenesis. BioMed Res Int 2014;2014:1–7. doi: 10.1155/2014/983715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsai C-C, Wu S-B, Kau H-C, Wei Y-H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci Rep 2018;8(1). doi: 10.1038/s41598-018-25370-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol 2003;81(6):355–363. doi: 10.1139/o03-069 [DOI] [PubMed] [Google Scholar]
- 72.Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol 2002;4(8):599–604. doi: 10.1038/ncb826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bruno G, Cencetti F, Pertici I, et al. CTGF/CCN2 exerts profibrotic action in myoblasts via the up-regulation of sphingosine kinase-1/S1P3 signaling axis: Implications in the action mechanism of TGFβ. Biochim Biophys Acta 2015;1851(2):194–202. doi: 10.1016/j.bbalip.2014.11.011 [DOI] [PubMed] [Google Scholar]
- 74.Marinković A, Liu F, Tschumperlin DJ. Matrices of Physiologic Stiffness Potently Inactivate Idiopathic Pulmonary Fibrosis Fibroblasts. Am J Respir Cell Mol Biol 2013;48(4):422–430. doi: 10.1165/rcmb.2012-0335OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pappritz K, Savvatis K, Koschel A, Miteva K, Tschöpe C, Van Linthout S. Cardiac (myo)fibroblasts modulate the migration of monocyte subsets. Sci Rep 2018;8(1). doi: 10.1038/s41598-018-23881-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tjin G, White ES, Faiz A, et al. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis Model Mech 2017;10(11):1301–1312. doi: 10.1242/dmm.030114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Onoda M, Yoshimura K, Aoki H, et al. Lysyl oxidase resolves inflammation by reducing monocyte chemoattractant protein-1 in abdominal aortic aneurysm. Atherosclerosis 2010;208(2):366–369. doi: 10.1016/j.atherosclerosis.2009.07.036 [DOI] [PubMed] [Google Scholar]
- 78.Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res 2014;102(2):258–269. doi: 10.1093/cvr/cvu062 [DOI] [PubMed] [Google Scholar]
- 79.Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis: The myofibroblast matrix. J Pathol 2013;229(2):298–309. doi: 10.1002/path.4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mizoguchi F, Slowikowski K, Wei K, et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat Commun 2018;9(1). doi: 10.1038/s41467-018-02892-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee WJ, Song SY, Roh H, et al. Profibrogenic effect of high-mobility group box protein-1 in human dermal fibroblasts and its excess in keloid tissues. Sci Rep 2018;8(1). doi: 10.1038/s41598-018-26501-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Q, Wang J, Wang J, et al. HMGB1 induces lung fibroblast to myofibroblast differentiation through NF-κB-mediated TGF-β1 release. Mol Med Rep 2017;15(5):3062–3068. doi: 10.3892/mmr.2017.6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheng M, Liu H, Zhang D, et al. HMGB1 Enhances the AGE-Induced Expression of CTGF and TGF-β via RAGE-Dependent Signaling in Renal Tubular Epithelial Cells. Am J Nephrol 2015;41(3):257–266. doi: 10.1159/000381464 [DOI] [PubMed] [Google Scholar]
- 84.Kanisicak O, Khalil H, Ivey MJ, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 2016;7:12260. doi: 10.1038/ncomms12260 [DOI] [PMC free article] [PubMed] [Google Scholar]