

Abstract
Holliday junction (HJ) resolution by its resolving enzymes is essential for chromosome segregation and recombination-mediated DNA repair. HJs undergo two types of structural dynamics that determine the outcome of recombination: conformer exchange between two isoforms and branch migration. However, it is unknown how the preferred branch-point and conformer are achieved between enzyme binding and HJ resolution given the extensive binding interactions seen in static crystal structures. Single molecule fluorescence resonance energy transfer analysis of resolving-enzymes from bacteriophages (T7 endonuclease I), bacteria (RuvC), fungi (GEN1) and humans (hMus81-Eme1) showed that both types of HJ dynamics still occur after enzyme binding. These dimeric enzymes use their multivalent interactions to achieve this, going through a partially-dissociated intermediate in which the HJ undergoes nearly unencumbered dynamics. This evolutionarily conserved property of HJ resolving-enzymes provides previously unappreciated insight on how junction resolution, conformer exchange and branch migration may be coordinated.
Introduction
Holliday junctions (HJs) are structural intermediates in homologous recombination, a ubiquitous DNA metabolic process that is essential both for DNA repair and genetic variation in all forms of life1,2. Once the HJ is formed by the exchange of DNA strands, branch migration extends the heteroduplex, followed by resolution into two duplex DNA molecules by junction resolving enzymes, a group of structure-specific endonucleases3–5. A deficiency in junction resolution leads to impaired DNA replication and repair, chromosome instability and dysfunctional mitoses6.
HJs are highly dynamic DNA structures: the branch point of a junction can migrate spontaneously or through the catalytic function of branch migration enzymes7,8; at a given branch point, HJs also undergo spontaneous conformer exchange between two structural isoforms9–11. Both types of dynamics influence the outcome of HJ resolution. Branch migration extends or shortens the length of DNA heteroduplex and hence determines the length of gene conversion. The two structural isoforms, or conformers, are correlated with the two alternative orientations of HJ cleavage, which dictate whether the HJ resolution results in gene conversion events either with (cross-overs) or without (non-cross-overs) the exchange of flanking parental DNA sequences. However, it is puzzling how the required branch point and isoform are chosen for HJ cleavage because binding of a resolving enzyme to a junction as seen in their static crystal structures4,12–16 would not allow conformer exchange or branch migration, and it is unclear whether there is coordination between HJ resolution and these HJ dynamics.
In this work, we use single molecule Fluorescence Resonance Energy Transfer (smFRET)17 to investigate the HJ dynamics upon binding of a resolving enzyme from diverse organisms, including bacteriophages, bacteria, fungi and humans. We show that initial binding of a resolving enzyme captures the instantaneous structural conformer and branch point at the moment of binding. The resolving enzyme binding does not prevent conformer exchange nor branch migration, and these dimeric enzymes use their multivalency18 to achieve a short-lived partially dissociated intermediate where the HJ can undergo nearly unencumbered dynamics.
Results
Endo I permits conformer exchange of Holliday junction
In the absence of added divalent metal ions, HJs adopt a 4-fold symmetric square structure (open state O; Fig. 1a)19. In the presence of Mg2+, HJs fold into two alternatively stacked conformers 19 (U1 and U2; here ‘U’ stands for ‘unbound’ as opposed to ‘B’ for protein ‘bound’). A single HJ can undergo conformer exchange between U1 and U2 and the exchange rate decreases with increased Mg2+ concentration9–11. The O state, although too short-lived to be detected directly in Mg2+, is considered to be a shared intermediate for both conformer exchange and branch migration8.
Figure 1: Endo I binding to Holliday junctions captures the instantaneous junction conformer and permits exchange between two isoforms (B1 and B2).
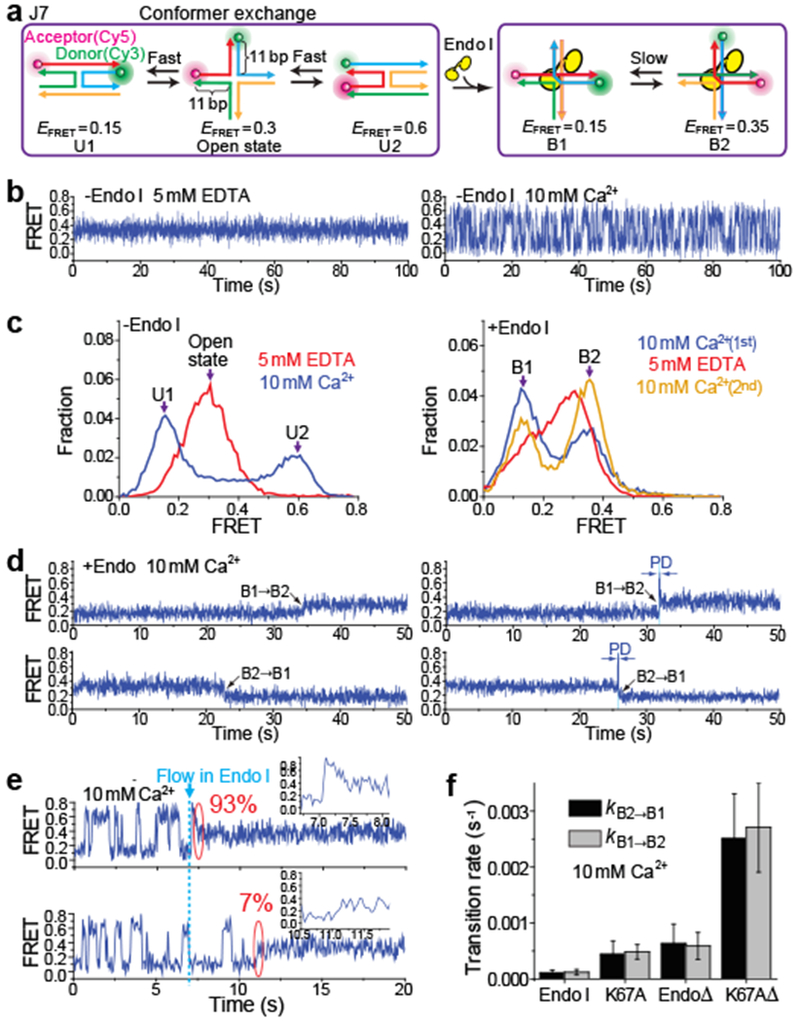
a, Schematic of junction structural dynamics before and after Endo I binding. Junction 7 (J7) comprises four arms of 11 bp. b, smFRET-time traces for unbound J7 obtained at 0 or 10 mM Ca2+. c, EFRET histograms of unbound J7 at 0 or 10 mM Ca2+ (left) and of Endo I-bound J7 obtained in the Ca2+-EDTA-Ca2+ buffer exchange experiments (right). d, smFRET-time traces of Endo I-bound J7 at 10 mM Ca2+, showing the transitions between B1 and B2. Partial dissociation (PD) was observed as an intermediate (blue-shaded region) for ~30% of the transitions between B1 and B2 (right). e, Single molecule time traces of J7 showing an Endo I binding event. The blue dashed line indicates the time when 10 nM Endo I were added. The percentages were analyzed from 1,000 HJ molecules. f, Conformer exchange rates kB1→B2 and kB2→B1 for the four variants of Endo I obtained at 10 mM Ca2+. Data are means ± s.e.m of n = 1,000 HJ molecules. Error bars represent bootstrap estimates of s.e.m..
To study resolving enzyme binding to HJ using smFRET, we first used Junction 7 (J7)9–11, the sequence of which does not allow branch migration (Fig. 1a). We attached Cy3 (donor) and Cy5 (acceptor) to the ends of two adjacent arms such that conformer exchange can be detected as two-state fluctuation in FRET efficiency (EFRET) 2,9,11. To prevent junction cleavage, we replaced Mg2+ with Ca2+. J7 exhibited similar dynamic properties in Ca2+ (Fig. 1b) to those in Mg2+ 9–11. The open state O of HJ in EDTA maintained a steady EFRET of 0.3. With 10 mM Ca2+, we observed exchanges between U1 (EFRET = 0.15) and U2 (EFRET = 0.6) with rates kU1→U2 = 2.1 ± 0.2 s−1 and kU2→U1 = 3.5 ± 0.3 s−1.
We first studied endonuclease I from bacteriophage T712,14,20–22 (termed ‘Endo I’ here) (Fig. 1a and Supplementary Fig. 1a). Endo I cleaved surface-immobilized HJs in Mg2+ but not in Ca2+ (Supplementary Fig. 1b, c), confirming that the enzyme is active under our experimental conditions. In general, the junction resolving enzymes bind in dimeric form with high affinity (Kd ~ 1 nM)4. Endo I binding induces either of the two alternative complexes (termed B1 and B2) that differ in coaxial pairing of arms12,14. After incubating 10 nM Endo I with surface-immobilized J7 in Ca2+, we flushed out the unbound proteins so that all subsequently observed dynamics can be attributed to the pre-formed complex rather than protein dissociation and binding (Supplementary Fig. 1a; referred to as “flush condition”). The resulting EFRET histogram determined from ~10,000 molecules contained two peaks at EFRET = 0.15 and 0.35, assigned to B1 and B2, respectively, based on structural considerations 12,14 (Fig. 1c). In the flush condition, most molecules were bound with proteins, as is evident by the nearly complete disappearance of U2 population. The smFRET-time traces showed slow exchanges between B1 and B2 (Fig. 1d) at rates kB1→B2 = (1.1 ± 0.4)×10−4 s−1 and kB2→B1 = (1.0 ± 0.4)×10−4 s−1. Interestingly, ~30 % of such transitions showed a short-lived intermediate (Fig. 1d and Supplementary Fig. 2) which we will discuss in the next section.
After observing the complexes in Ca2+, we flushed the sample chamber with buffer containing EDTA. A broad peak was observed in the EFRET histogram (Fig. 1c), and correlation analysis23 revealed that the time traces contained anti-correlated fluctuations of donor and acceptor intensities (ID and IA) with the time scale of 0.11 ± 0.02 s (Supplementary Fig. 3), likely due to fast exchange between B1 and B2 previously hypothesized to occur in EDTA14. When Ca2+ buffer was reintroduced, the relative populations of B1 and B2 were different from those prior to the EDTA pulse. This redistribution probably occurs because the U1/U2 equilibrium is different from that of B1/B2. It is likely that the initial Endo I binding captures the U1/U2 equilibrium which only later relaxes to the B1/B2 equilibrium. Indeed, single molecule time traces capturing the moment of Endo I binding showed that for J7 molecules that were locked into B2, 93% had been in U2 (E0.6), and only 7% had started from U1 (E0.15) (Fig. 1e and Supplementary Fig. 4a).
Population redistribution after EDTA pulse was also observed (Supplementary Fig. 5) for three mutants of Endo I12,20,24: 1) EndoΔ, lacking the 16 amino-acids N-terminal tail and possessing slower HJ cleavage, 2) K67A, a catalytically-impaired mutant, and 3) K67AΔ, combining both mutations. All three showed increased exchange rates between B1 and B2 (Fig. 1f) with K67AΔ having the highest rate, suggesting that their bound states are less stable than the wild type.
Endo I permits branch migration through an intermediate
To investigate branch migration we used two previously described HJ constructs 7: J5m contains a central 5-bp homologous sequence so it can migrate over six branch points, and J0m is an otherwise identical junction that lacks homology so its branch point cannot migrate (Fig. 2a). We attached the fluorophores to the diametrically-opposed arms so that EFRET is sensitive to the branch point movement but not to conformer exchange (Fig. 2a and Supplementary Fig.6). Any EFRET dynamics seen in J5m but not in J0m can be attributed to branch migration. smFRET data for J0m by itself confirmed that labeling configuration is insensitive to conformer exchange because O, U1 and U2 merged into one degenerate state (EFRET=0.3 or E0.3) (Fig. 2b, c).
Figure 2: Endo I binding captures the instantaneous branch position and permits branch migration through a partially dissociated intermediate.
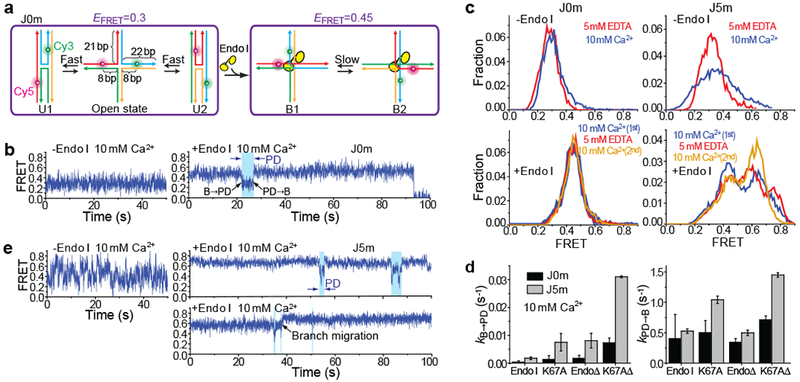
a, Schematic of J0m exhibiting different EFRET states before and after Endo I binding. b, smFRET-time traces of unbound and Endo I-bound J0m in 10 mM Ca2+. Partial dissociation (PD) of Endo I was observed as transient reduction in EFRET (blue-shaded region). c, smFRET histograms of unbound J0m or J5m obtained at 0 or 10 mM Ca2+ (top), and of Endo I-bound J0m or J5m obtained in the Ca2+-EDTA-Ca2+ buffer exchange experiments (bottom). d, kB→PD and kPD→B for four variants of Endo I obtained at 10 mM Ca2+. Data are means ± s.e.m. of n = 1,000 HJ molecules. Error bars represent bootstrap estimates of s.e.m.. e, smFRET-time traces of unbound and Endo I-bound J5m in 10 mM Ca2+. Visits to PD were observed as transient reduction in EFRET (blue-shaded regions). A representative branch migration event is marked (black arrow).
Upon Endo I binding, J0m exhibited an E0.45 state which represents a degenerate mixture of B1 and B2 (Fig. 2c) but also underwent brief excursions to an E0.3 state (Fig. 2b; blue-shaded regions). The brief excursions to E0.3 are unlikely due to complete dissociation and binding of another Endo I molecule because unbound proteins were washed out. E0.3 excursions occurred more frequently with the Endo I mutants (Fig. 2d and Supplementary Fig. 5c, e). Such enhanced transition rates for Endo I mutants were also observed for the B1↔B2 conformer exchange of Endo I-bound J7 (Fig. 1f) where a short-lived state with a EFRET=0.6 value was often observed as an intermediate (Fig. 1d and Supplementary Fig. 2), indicating this intermediate represents a loosely bound mode. Therefore, we assigned E0.45 to the fully bound (B) state of J0m and E0.3 to a partially-dissociated (PD) state through which conformer exchange occurs.
Unlike J0m which showed a constant EFRET value with Ca2+, J5m showed fluctuations in a broad range of 0.1-0.7, likely due to branch migration7 (Fig. 2e). Upon Endo I binding, the EFRET peak shifted toward higher values while maintaining a broad range of 0.2-0.8, probably reflecting instantaneous branchpoint positions trapped by Endo I binding (Fig. 2c and Supplementary Fig. 4b and 5d). Most time traces (Fig. 2e and Supplementary Fig. 7a, b) comprised constant EFRET values, but with occasional episodes, which we attribute to PD, of EFRET fluctuations similar to what were observed for bare J5m. Consistent with this assignment, kB→PD rates were higher for the mutant proteins (Fig. 2d), sharing the trend of kB→PD observed for enzyme-bound J0m. Similar smFRET time traces containing PD could also be observed in Mg2+ with the catalytically-impaired Endo I (K67A) (Supplementary Fig. 8). kPD→B did not change even in the presence of saturating (100 nM) Endo I, further ruling out the possibility that B↔PD transitions were due to full dissociation and binding of the enzyme (Supplementary Fig. 7c).
We could directly observe branch migration as an abrupt change between two different steady EFRET values via PD (black arrows, Fig. 2e and Supplementary Fig. 7e-g), showing that PD is indeed an intermediate for branch migration. An EDTA pulse redistributed the EFRET populations (Fig.2c and Supplementary Fig. 5), suggesting that an Endo I-bound junction can undergo extensive branch migration in EDTA. Endo I-bound J5m could also undergo slow branch migration in Ca2+ (Supplementary Fig. 7d).
Taken together, our data suggest the following model. PD serves as an intermediate that allows a resolving-enzyme-bound junction to undergo both branch migration and conformer exchange. Immediately after binding to a HJ, the enzyme fixes the instantaneous branch position and conformer. The equilibrium population distribution for branch position and conformer is different with and without the enzyme, and the enzyme-bound junction approaches the new equilibrium over time. Indeed, we found direct evidence in smFRET time traces that PD acts as an intermediate for branch migration (black arrows, Fig. 2e and Supplementary Fig. 7e-g) and conformer exchange (Fig. 1d and Supplementary Fig. 2a).
RuvC permits both types of HJ dynamics through PD
To test whether cellular (i.e. non-phage) HJ resolving enzymes exhibit similar behaviour, we investigated E. coli RuvC25–27. Unlike Endo I which is in the restriction endonuclease superfamily, RuvC belongs to the integrase superfamily28. Junction resolving enzymes of the integrase superfamily exhibit marked sequence specificity for cleavage (Supplementary Fig. 1c) though they can bind equally well to HJs of any sequence3,29. RuvC binding induces a 2-fold symmetrical X-shaped HJ structure with two alternative conformers15,30 (Fig. 3a). Indeed, we observed two populations for RuvC-bound J7 (Fig. 3b; EFRET = 0.15 for B1 and 0.35 for B2) with a strong preference for B1. Time traces for RuvC-bound J7 exhibited B1↔B2 exchanges through PD. PD is relatively long-lived and had imbedded within it rapid exchange between U1 (E0.15) and U2 (E0.6) (Fig. 3c and Supplementary Fig. 9a), with rates similar to those of protein-free J7 (Fig. 3d). Therefore, upon partial dissociation of RuvC, J7 undergoes conformer exchange nearly unencumbered by the still bound RuvC. PD was also observed in a flipped experimental scheme where RuvC was immobilized (Supplementary Fig. 9b), and the addition of saturating concentration (500 nM) of RuvC did not significantly change its average lifetime (τPD) (Fig. 3e, Supplementary Fig. 9c, d), confirming that PD does not represent full dissociation.
Figure 3: RuvC binding permits conformer exchange and branch migration through PD.
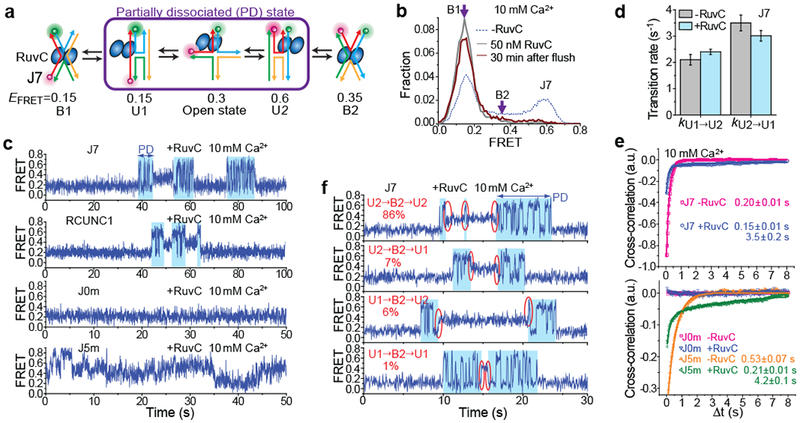
a, Schematic of HJ structural dynamics for RuvC-bound J7. b, EFRET histograms of J7, with and without RuvC bound, at 10 mM Ca2+. c, smFRET-time traces of RuvC-bound J7, RCUNC1, J0m and J5m at 10 mM Ca2+. Partial dissociation of RuvC was observed as an intermediate (blue-shaded region) between B1 and B2. d, The conformer exchange rates between U1 and U2 for bare J7 and for RuvC-bound J7 within PD. Data are means ± s.e.m. of n = 1,000 HJ molecules. Error bars represent bootstrap estimates of s.e.m.. e, Cross-correlations of ID and IA for J7E, J0m, J5m and n-J5m, with and without RuvC bound, are fit to single (for free HJs) or double (for RuvC-bound HJs) exponential functions (see also Supplementary Table 1). Means ± s.e.m are indicated (n = 1,000 HJ molecules). f, smFRET-time traces of RuvC-bound J7 with different types of PD→ B2→ PD transitions. 86% of PD→ B2→ PD events occur as U2→ B2→ U2 (red circles). The percentages were analyzed from 1,000 HJ molecules.
The cross-correlation analysis suggests anti-correlated fluctuations between IA and ID with two time components 0.15 ± 0.01 and 3.5 ± 0.2 s (Fig. 3e). The faster component is likely related to the conformer exchange of J7 within PD, and the slower component to the transitions between PD and B. From the real time traces, we could also deduce that when RuvC-bound J7 enters and exits a B state, it does so while maintaining coaxial partners. For example, 86% of PD→B2→PD events occurred as U2→B2→U2 (red circles, Fig. 3f).
We further examined the possibility for branch migration proceeding within a RuvC-bound junction. RuvC binding only slightly reduced EFRET for both J5m and J0m (Supplementary Fig. 10), but unlike the control J0m, RuvC-bound J5m exhibited anti-correlated fluctuations between IA and ID, with two time components 0.21 ± 0.01 and 4.2 ± 0.1 s (Fig. 3c, e, and Supplementary Fig. 11), consistent with branch migration. The fast component is likely related to the branch migration rate within PD and is slightly slower than that obtained from protein-free J5m, indicating that branch migration persists in PD but with a slower rate. The slow component is likely due to the B↔PD transitions, and is indeed similar to the slow component observed in RuvC-bound J7. In addition, because our junctions do not contain the consensus RuvC cleavage sequence, making HJ cleavage in Mg2+ inefficient29,31 (Supplementary Fig. 1c), we could further show that RuvC-bound junction in Mg2+ also undergoes conformer exchange and branch migration via PD (Supplementary Fig. 9d, e and Supplementary Fig. 11b, c). The observed rates for these processes were 100-fold higher for RuvC compared to wild type Endo I as a result of more frequent visits to PD. The greater tendency to visit PD may help RuvC in the search for its consensus cleavage sequence.
Next, we examined the B↔PD transitions for a HJ construct containing one RuvC cleavage site 5’-(A/T)TT↓(G/C)-3’32 and labelled to report on conformer exchange as in J7 (referred to as RCUNC1). RCUNC1 can be cleaved by RuvC, but more slowly than with both cleavage sites as previously reported32 (Supplementary Figs. 12a, b). At 10 mM Ca2+, RuvC-bound RCUNC1 showed single molecule behaviours similar to those obtained for RuvC-bound J7 (Fig. 3c and Supplementary Fig. 12c, d). Therefore, introducing a cleavage site does not change the overall behaviour except for small differences in the rates of visiting PD (Supplementary Fig. 12e).
Increasing ionic strength makes PD more frequent
Because many DNA-protein interactions can be weakened by increases in ionic strength, we investigated the ionic strength dependence of B↔PD transitions. For Endo I (K67A)-bound J5m, kB→PD was at the minimum at 10 mM Ca2+, and increased as [Ca2+] was further increased (Supplementary Fig. 13a). This [Ca2+] dependence is well aligned with the [Ca2+] dependence measured by gel shift assays: in the low concentration range (<1 mM), Ca2+ stabilizes the Endo I binding, whereas in the higher concentration range (≥ 10 mM), increasing [Ca2+] decreases the binding stability (Supplementary Fig. 14). Similarly, for RuvC-bound J7, kB1→PD was at the minimum at 1 mM Ca2+, and increased as [Ca2+] increased above 1 mM (Supplementary Fig. 13b and Supplementary Fig. 9f, g). When [Ca2+] was kept at 10 mM, increasing [NaCl] also increased kB1→PD (Supplementary Fig. 13c). Overall, PD is more frequently visited at increased ionic strengths and therefore should correspond to a binding mode with lower binding stability compared to the B states.
GEN1 binding permits conformer exchange
To test if our observations can be generalized to a eukaryotically conserved junction resolving enzyme GEN15, we examined GEN1 from a thermophilic fungus. The current structural model of GEN1-bound HJ is that one pair of opposite arms of the HJ are coaxially aligned, while the other pair are rotated toward each other around the axis of the coaxial arms to include an angle of close to 90°16. This structural model predicts that B1 and B2 would be merged into one degenerate EFRET state when Cy3 and Cy5 are attached to the ends of two adjacent HJ arms, making J7 a suitable HJ construct to monitor the transitions between B and PD. Because GEN1 binding requires junctions with longer arms16, we extended the arms of J7 from 11 to 20 bp to create J7E (Fig. 4a and Supplementary Fig. 9h).
Figure 4: GEN1 and hMus81-Eme1 binding both permit conformer exchange and hMus81-Eme1 binding also permits branch migration.
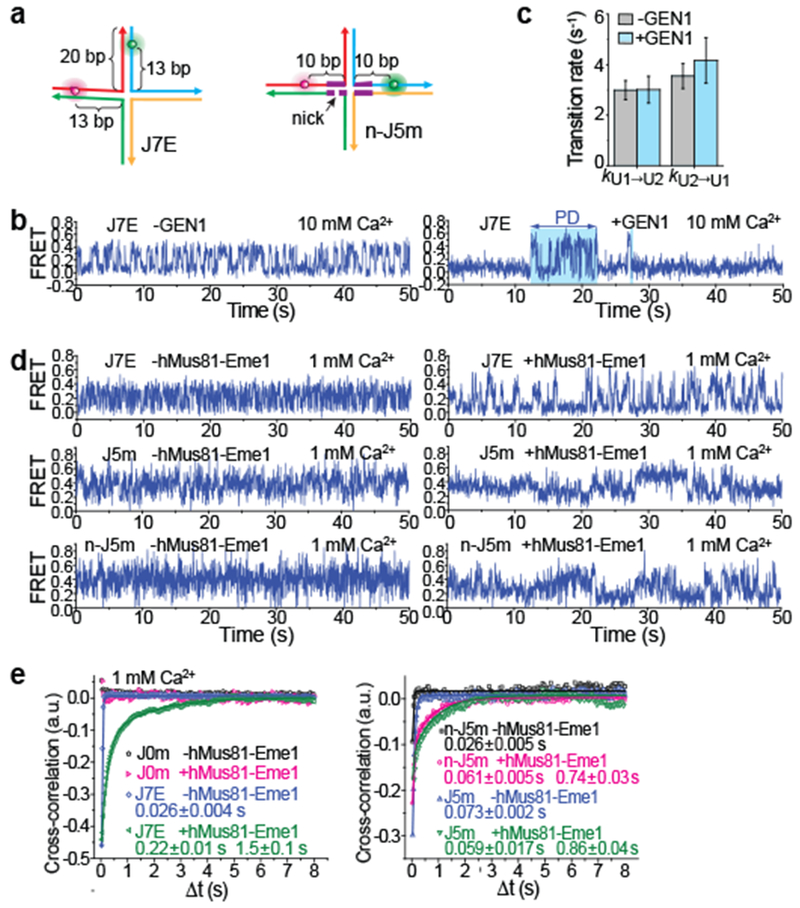
a, Schematics of J7E and n-J5m. b, smFRET- time traces of unbound and GEN1-bound J7E at 10 mM Ca2+. c, The conformer exchange rates between U1 and U2 for bare J7 and for GEN1-bound J7 within PD. Data are means ± s.e.m. of n = 1,000 HJ molecules. Error bars represent bootstrap estimates of s.e.m. d, smFRET-time traces of bare or hMus81-Eme1-bound J7E, J5m and n-J5m at 1 mM Ca2+. e, Cross-correlations of ID and IA for J7E, J0m, J5m and n-J5m, with and without hMus81-Eme1 bound, were fitted to single (for unbound HJs) or double (for hMus81-Eme1-bound HJs) exponential functions (see also Supplementary Table 1). Means ± s.e.m are indicated (n = 1,000 HJ molecules).
At 10 mM Ca2+, the unbound J7E exhibited transitions between U1 (E0.05) and U2 (E0.4) (Supplementary Fig. 15a). GEN1 binding induced a dominant peak at E0.1, representing the degenerate mixture of B1 and B2 (and U1), and a small peak at E0.4 (U2). Time traces showed that the bound J7E transited from the bound state (E0.1) to the PD mode exhibiting U1↔U2 transitions (Fig. 4b), similar to the behaviour of RuvC-bound J7. The PD lifetime (τPD) was 3.6 ± 0.3 s (Supplementary Fig. 15b) and kB→PD was 0.034 ± 0.003 s−1, both similar to those observed for RuvC-bound J7. Furthermore, the U1↔U2 transitions had similar rates to those obtained with protein-free J7E (Fig. 4c), indicating that the conformer exchange is unencumbered in PD of GEN1-bound junction.
hMus81-Eme1 permits conformer exchange and branch migration
We next investigated the human heterodimeric endonuclease hMus81-Eme1. This enzyme acts cooperatively with other endonucleases by preferentially cleaving the junctions that have already been nicked by other endonucleases such as SLX1-SLX433–35. Because hMus81-Eme1 binding also requires junctions with longer arms36, we used J7E (Fig. 4a). EFRET histograms of hMus81-Eme1-bound J7E (Supplementary Fig. 15c) were similar to those of GEN1-bound J7E (Supplementary Fig. 15a) and RuvC-bound J7 (Fig. 3b). Time traces of unbound J7E exhibited EFRET fluctuations due to conformer exchange, and these fluctuations persisted in hMus81-Eme1 bound complexes (Fig. 4d and Supplementary Fig. 16), indicating that exchange occurs without full protein dissociation. EFRET fluctuations appear to involve more than two states, suggesting the existence of a PD intermediate between B1 and B2 (Fig. 4d). To test if hMus81-Eme1 binding still allows branch migration, we used J5m and a variant that contains a nick within the homologous region (n-J5m; Fig. 4a and Supplementary Fig. 15d). It has been shown that a nick does not prevent branch migration37. hMus81-Eme1 efficiently cleaved n-J5m but not J5m (Supplementary Fig. 1c), consistent with its role in junction resolution after the first unilateral cleavage33,36. hMus81-Eme1-bound n-J5m and J5m exhibited EFRET fluctuations with similar rates, but not the control J0m, indicating that hMus81-Eme1 permits branch migration (Fig. 4e and Supplementary Figs. 17 and 18).
Multivalent interactions between junctions and resolving enzymes
Since junctions in PD behave like their unbound counterparts in conformer exchange and branch migration, a significant amount of bonds at the DNA-protein interface must be broken. Because most resolving enzymes function as dimers, it is possible that one subunit within the dimer, or even more interactions at the binding interface, has been disengaged in PD, exposing the protein for competitive binding by additional DNA. Indeed, adding either unlabelled junction or duplex DNA as competitors to RuvC-bound J7 accelerated RuvC dissociation by at least 20-fold. (Supplementary Fig. 19). smFRET-time traces obtained immediately after adding DNA competitors typically exhibited an irreversible transition from the steady B1 state to a mode with rapid U1↔U2 transitions (Supplementary Fig. 19f). This is consistent with a model in which these molecules transit from the B state to the unbound state through PD, although it is impossible to identify the exact time point for the transition from PD to the unbound state. Junction and duplex DNA were equally competitive for Endo I and for Mus81-Mms4, the budding yeast homolog of hMus81-Eme138,39. Faster dissociation induced by competitor DNA suggests that in PD a significant fraction of the interactions between the resolving enzyme and the junction are lost, and the dissociated portion is available to interact with DNA competitors.
Discussion
Our study of four contrasting junction-resolving enzymes from bacteriophage, bacteria, fungi and humans suggests that they share similar properties whereby the dynamic processes of conformer exchange and branch migration can proceed without full dissociation. This is achieved via an intermediate termed PD in which the junction may freely sample U1, U2 and O states just like unbound junctions, and which serves as the intermediate for exchange between the B1 and B2 complexes, and for branch migration (Fig. 5a). Previous structural analyses could not detect PD due to its transient nature, and neither conformer exchange nor branch migration could occur within the constraints of a crystal lattice.
Figure 5: Proposed models for the coordination of junction resolution, conformer exchange and branch migration.
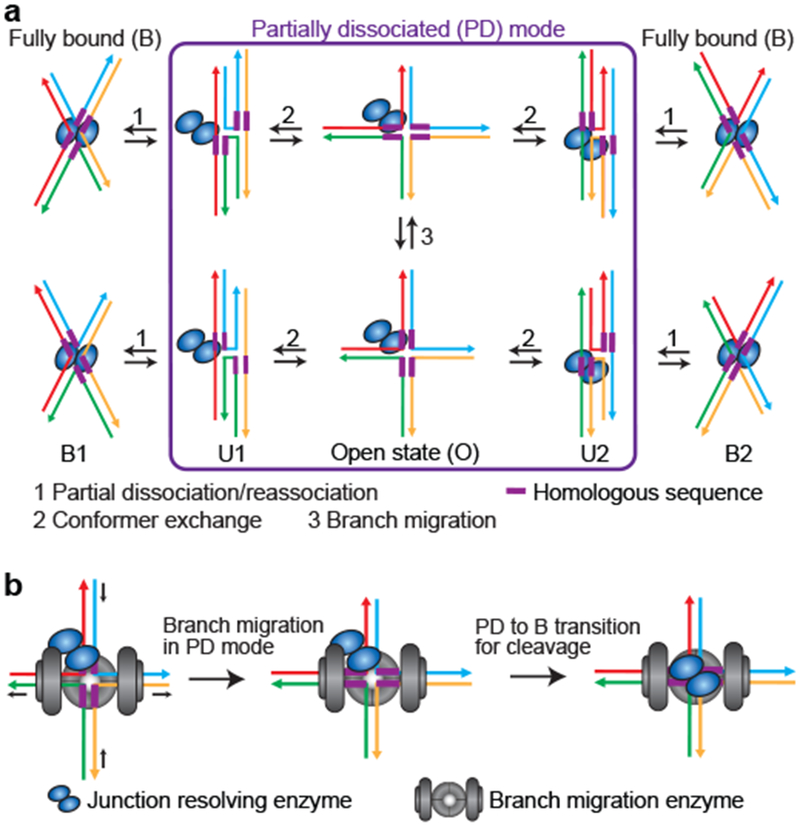
a, Schematic of a kinetic model deduced to describe the dynamics of resolving-enzyme-bound HJs. PD serves as an intermediate that allows a resolving-enzyme-bound junction to undergo both branch migration and conformer exchange. b, Schematic of the speculative ternary complex containing the branch migration-facilitating enzyme, the junction resolving enzyme and Holliday junction for the coordination of branch migration and junction resolution.
Several biochemical studies have shown that the enzymes facilitating branch migration interact specifically with their cognate resolving enzymes and stimulate their cleavage activity, implying possible coordination between junction branch migration and resolution 26,27,40–43. The PD mode discovered in this study provides a potential molecular mechanism for such coordination of branch migration and resolution. The branch migration enzyme and resolving enzyme bind together to the HJ to form a ternary complex, and the ternary complex formation may lock or bias the HJ in the PD mode to allow the branch migration enzyme to actively drive branch migration without full dissociation of the resolving enzyme; once the junction reaches its desired cleavage site, the resolving enzyme switches from the PD mode to the fully bound mode to achieve junction resolution (Fig. 5b). A structural model for such a ternary RuvABC-junction complex has been previously hypothesized where the HJ lies sandwiched between RuvA and RuvC, and the RuvA-RuvC-junction complex is flanked on two sides by RuvB27 although this RuvABC mode of action and its potential conservation in eukaryotes have not been demonstrated.
It should be noted that the rates of dynamics measured in vitro for the HJs bound by junction resolving enzymes can be slower than the actual rates in vivo as these rates may be affected by multiple factors: 1) The presence of the homologous sequence in the HJ core region could increase the frequency of PD. For example, the B→PD transition rates of J5m determined for all the four Endo I variants were 3-5 fold higher than those obtained for J0m. 2) Branch migration enzymes apply an active mechanical force (~25 pN for RuvAB44) to drive branch migration in one direction, which may significantly increase the tendency of visiting PD for the resolving enzyme-bound HJ. 3) The interaction strength between the resolving enzyme and HJ controls the frequency of PD, which differs among different resolving enzymes and at different ionic strengths. Both conformer exchange and branch migration for RuvC-bound and GEN1-bound HJs occur much faster than for Endo I-bound HJs. And the dynamics of hMus81-Eme1-bound HJs are even faster. In addition, high ionic strength (Na+ or divalent ion concentration) could increase the frequency of PD.
While the interactions of HJ and resolving enzymes have been extensively described in the literature, our results reveal their dynamic nature. Although their exact binding interface in PD awaits further investigation, a significant amount of protein-DNA interactions seen in the ground state of the complex, likely at least one of the two subunits, must have been lost to allow the observed HJ dynamics in PD with little to moderate levels of hindrance. Our data may have implications on other DNA-protein interactions that are multivalent and can be broken little by little or one at a time to facilitate the recruitment of other proteins to the same DNA molecule18.
Online Methods
Statistics and Reproducibility
All the experiments shown in this study were repeated at least three times independently with similar results.
DNA sequences and annealing procedures
DNA sequences for making the DNA constructs used in this study can be found in Supplementary Table 2. DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). J7, unlabeled J7 and J7E were annealed by mixing the four strands with the molar ratio 1:1:1:1 (final concentration ~10 μM each) in 10 mM Tris:HCl (pH 8.0) and 50 mM NaCl followed by slow cooling from 90°C to room temperature for ~ 2 hours. J3 was prepared by mixing equimolar amounts of the four component oligonucleotides in PNK buffer (New England Biolabs), labelling them with γ-32P-dATP (Perkin Elmer) and T7 polynucleotide kinase (New England Biolabs) followed by slow cooling from 90°C to room temperature for ~ 2 hours. 22-bp dsDNA was annealed by mixing the two strands with the molar ratio 1:1 (final concentration ~10 μM each) in 10 mM Tris:HCl (pH 8.0) and 50 mM NaCl followed by slow cooling from 90°C to room temperature for ~ 2 hours. J5m, n-J5m and J0m were constructed as previously described 7,37,45. When J5m undergoes spontaneous branch migration, there are six possible donor-acceptor separations of 10, 12, 14, 16, 18, 20 bp for different branch positions. The donor-acceptor separation for J0m is 16 bp.
Protein expression and purification
Wild-type Endo I and Endo I mutants were expressed and purified as previously described12,20,24,46. E. coli RuvC was purchased from Abcam (Cambridge, MA). C. thermophilum GEN1 was expressed and purified as previously described16. Full length human (h)Mus81-Eme1 was expressed and purified as previously described 36. All protein concentrations cited in the text refer to protein dimers. Purified proteins migrated as a single band on a polyacrylamide gel in the presence of SDS (Supplementary Fig. 20).
Single molecule imaging and data acquisition
All smFRET experiments were performed with a total internal reflection fluorescence (TIRF) microscope 47 at RT (22 ± 1°C) in imaging buffer composed of 20 mM Tris (pH 8.0 for Endo I and RuvC, pH7.5 for hMus81-Eme1 and GEN1), 10 mM NaCl, 0.1 mg/ml BSA, 1 mM DTT, oxygen scavenging system (0.5% wt/vol glucose, 3 mM Trolox, 165 U/ml glucose oxidase band 2170 U/ml catalase), with 5 mM EDTA or the desired concentrations of CaCl2/MgCl2. 5% (vol/vol) glycerol was included in the imaging buffer for hMus81-Eme1 and GEN1 measurements. 50-100 pM of Cy3-Cy5 labeled HJ molecules were immobilized on a quartz slide surface coated with polyethyleneglycol (mPEG-SC, Laysan Bio) in order to eliminate nonspecific surface adsorption of proteins 47,48. Surface immobilization was mediated by biotin-neutravidin binding between biotinylated HJs, neutravidin (Pierce), and biotinylated polymer (Bio-PEG-SC, Laysan Bio). After incubating HJ resolving enzymes (10 nM Endo I/K67A/EndoΔ/K67AΔ, 50 nM RuvC, 70 nM hMus81-Eme1, or 100 nM GEN1) with the surface-immobilized HJs for 5 min in imaging buffer containing 1 or 10 mM Ca2+, excess unbound proteins were flushed out of the sample chamber using five chamber volumes of imaging buffer and Cy3/Cy5 intensities from single HJs were recorded using an electron-multiplying CCD camera with time resolution of 0.03 s. These protein concentrations resulted in a bound fraction of almost 100%, and the protein binding was stable for a long period of time (for example, at least 1 hr for Endo I and RuvC) after flushing out the unbound proteins. EFRET histograms were generated by averaging for the time period of 0.15 s from ~10,000 HJ molecules each. smFRET data were excluded only if there was only Cy3 signal and lack of Cy5 signal for that single molecule, or if the intensity-time trace obtained for Cy3 or Cy5 exhibited multiple photo-bleaching steps.
For measuring the cleavage of Holliday junction resolving enzymes under single molecule conditions, 100 pM of Cy3/Cy5-labeled HJ (J5m or n-J5m) were immobilized on the PEG surface. Junction resolving enzymes (10 nM EndoΔ, 50 nM RuvC, or 70 nM hMus81-Eme1) were incubated with the surface-immobilized HJ for 5 min in 1 mM Ca2+ and the excess unbound proteins were flushed out using buffer containing 1 mM Ca2+. 1 mM Mg2+ was then introduced to the sample chamber to trigger the cleavage reaction at room temperature. For both of the two possible cleavage orientations, the cleavage released the part of HJ that contains Cy3. Therefore, the fraction of uncleaved J5m (or n-J5m) can be monitored by determining the mean Cy3 spot count per imaging area (~ 2,500 µm2) as a function of reaction time. For RuvC cleavage, RCUNC1 and J7E were also used following the same protocol except that 10 mM instead of 1 mM Mg2+ was introduced to the sample chamber to trigger the cleavage reaction and prism holder, and that sample stage, and objective were connected to a Thermo NESLAB RTE-7 circulating bath using custom parts to maintain sample temperature at 37 °C.
For the Ca2+-EDTA-Ca2+ buffer exchange experiments, 10 nM of wild type Endo I or Endo I mutant (K67A, EndoΔ, or K67AΔ) was incubated with the surface-immobilized HJs (J7, J0m or J5m) in 10 mM Ca2+ for 10 min. The first EFRET histogram was obtained 10 min after flushing out unbound proteins using buffer containing 10 mM Ca2+. The second EFRET histogram was obtained 10 min after the first buffer exchange to buffer containing 5 mM EDTA. Finally, the third EFRET histogram was obtained 5 min after the second buffer exchange to buffer containing 10 mM Ca2+. For the resolving enzyme cleavage assay, imaging buffer containing 1 mM Mg2+ was introduced to the sample chamber to trigger the cleavage reaction, after incubating and flushing HJ resolving enzymes in imaging buffer containing 1 mM Ca2+. Mean Cy3 spot count per image (each imaging area is ~2,500 μm2) was determined from images taken from 5-10 different slide regions at different time points after introducing the Mg2+ buffer. For the competition binding assay, unlabeled competitor DNA (J7 or 22-bp DNA duplex) was introduced to the sample chamber, after incubating and flushing RuvC proteins in buffer containing 10 mM Ca2+. EFRET histograms were obtained at different times after introducing competitor DNA.
Transition rate determination
The transition rate from the fully bound (B) state to the partially dissociated (PD) state, kPD→B, was determined from the single exponential fit to the histogram of the dwell times of the PD state. The reverse transition rate, kB→PD, was defined as the total number of B→PD transitions observed, divided by the total time that all the molecules spent in the B state. The total time here is the cumulative time not only from those molecules showing the B→PD transition, but also from the molecules that did not show such transitions but stayed in the B state throughout the observation time window, which is limited by the photobleaching life time of Cy dyes and typically ranges from 20 to 300 s under our experimental conditions. Because the B→PD transition occurs with a relatively low frequency, Cy3 or Cy5 can be photobleached before a B→PD transition can occur, making it not so meaningful to determine the percentage of molecules showing the B→PD transitions. Nonetheless, we determined such percentage values for Endo I-bound HJs, and found that these values vary a lot among the four Endo I variants and also depend on the DNA substrate (J0m or J5m): For J0m, it is 23% for Endo I, 42% for K67A, 45% for EndoΔ, and 91% for K67AΔ; For J5m, it is 51% for Endo I, 90% for K67A, 92% for EndoΔ, and 98% for K67AΔ. It is worth noting that the actual percentages of molecules that have the capability to show B→PD transitions are likely much more than those numbers have indicated due to the limited observation time window described above. The transition rates between U1 and U2 at 10 mM Ca2+ were determined using hidden Markov models as previously described 49. It is worth noting that when the U1 or U2 dwell times are shorter than our experimental time resolution (0.03 s), those dwell times would not show up in the time traces and hence were missed from our analysis. Given that U1/U2 dwell times have a single exponential distribution and the average U1 (or U2) dwell time is ~ 0.4 s, we can further estimate the portion of these undetectable U1 (or U2) dwell times to be ~ 7%. In the time traces showing initial binding of Endo I to J7 which locked the J7 molecules into B2 (Fig. 1f), we detected that 93% had been in U2, and only 7% had started from U1. Given that our detection would miss 7% of U2 dwell times, 93% could actually mean that almost 100% of J7 molecules that got locked in B2 had been in U2.
FRET efficiency calculation.
Apparent FRET efficiency (EFRET) was calculated from the fluorescence intensities of the donor (ID) and acceptor (IA) using the formula EFRET = IA / (IA + ID). The background and the cross-talk between the donor and acceptor were considered as previously described 47.
Cross-correlation analysis
The cross-correlation analysis was performed as previously described23,50. The cross-correlation functions were calculated between donor and acceptor time traces for each HJ molecule, and all the cross-correlations presented in figures are cross-correlations averaged among >200 HJ molecules. We found the cross-correlations for bare J7, J7E, J5m and n-J5m can be fit to a single exponential function, while those for resolving-enzyme-bound J7, J7E, J5m and n-J5m can be fit to a bi-exponential function, yielding one time component for bare HJs and two time components for resolving-enzyme-bound HJs.
Gel electrophoresis
To detect RuvC cleavage, 10 µl of each cleavage reaction was prepared as a mixture of 10 nM DNA substrate (J7E or RCUNC1), 10 nM RuvC, 50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 1 mM DTT, 100 µg/mL BSA. For control experiments, RuvC was not included in the mixture. Samples were incubated at RT or 37 °C for 15 min, then stopped by addition of 2 µl of 5× stop buffer (125 mM EDTA, 2.5% SDS, and 50% glycerol). Samples were then run on 10% polyacrylamide gel in 1× TBE buffer.
To determine the [Ca2+] dependence of Endo I binding to HJ, 0.2 nM 5’-32P-labelled four-way DNA junction J3 was incubated with serial two-fold dilutions of endonuclease I (from 1 µM to 15.3 pM) for 10 min at 22 °C in 20 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.1 mg/ml BSA and the specified concentration of either EDTA (1 mM) or CaCl2 (0.2, 1, 5, 10, 15 or 20 mM). These samples were then mixed with loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol FF and 2.5% Ficoll type 400), loaded onto 8% polyacrylamide gels and electrophoresed in TB buffer containing the specified concentration of either EDTA or CaCl2. Dried gels were exposed to storage phosphor screens (BAS-IP MP 2040), and quantified using a BAS- 1500 phosphorimager (Fuji) and Image Gauge V4.0 software. Data were analyzed as the fraction of DNA bound (fbound) versus the concentration of protein and were fit to a two-state model:
where total protein and DNA concentrations are P0 and D0 respectively and KD is the dissociation constant (i.e., the inverse of the binding affinity constant KA).
Code availability
All custom software and codes are available from T.H. (tjha@jhu.edu) or R.Z. (ruobozhou@fas.harvard.edu) upon request or can be downloaded from the Ha Research Group website at http://ha.med.jhmi.edu/resources/.
Data availability
The data that support the findings of this study are available from T.H. (tjha@jhu.edu) or R.Z. (ruobozhou@fas.harvard.edu) upon reasonable request. A Life Sciences Reporting Summary for this study is available.
Supplementary Material
Acknowledgements.
We acknowledge Ha lab members for experimental help and discussion. This work was supported by grants from the National Science Foundation (PHY-1430124) and the National Institutes of Health (GM 122569) to T.H., and grants from the Korean government (NRF 2018R1A2A1A190 to Y.C.). R.Z. is a Howard Hughes Medical Institute Fellow of the Life Sciences Research Foundation. T.H. is an employee of the Howard Hughes Medical Institute. Work in the Lilley lab is funded by Cancer Research UK program grant A18604.
Footnotes
Competing financial interests. The authors declare no competing financial interests.
References
- 1.Liu Y & West SC Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol 5, 937–944 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Lilley DM Structures of helical junctions in nucleic acids. Q Rev Biophys 33, 109–159 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Lilley DM & White MF The junction-resolving enzymes. Nat Rev Mol Cell Biol 2, 433–443 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Declais AC & Lilley DM New insight into the recognition of branched DNA structure by junction-resolving enzymes. Curr Opin Struct Biol 18, 86–95 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Wyatt HD & West SC Holliday junction resolvases. Cold Spring Harb Perspect Biol 6, a023192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarbajna S & West SC Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci 39, 409–419 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Karymov M, Daniel D, Sankey OF & Lyubchenko YL Holliday junction dynamics and branch migration: single-molecule analysis. Proc Natl Acad Sci U S A 102, 8186–8191 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinney SA, Freeman AD, Lilley DM & Ha T Observing spontaneous branch migration of Holliday junctions one step at a time. Proc Natl Acad Sci U S A 102, 5715–5720 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKinney SA, Declais AC, Lilley DMJ & Ha T Structural dynamics of individual Holliday junctions. Nat Struct Biol 10, 93–97 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Joo C, McKinney SA, Lilley DMJ & Ha T Exploring rare conformational species and ionic effects in DNA Holliday junctions using single-molecule spectroscopy. Journal of Molecular Biology 341, 739–751 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Hohng S et al. Fluorescence-force spectroscopy maps two-dimensional reaction landscape of the holliday junction. Science 318, 279–283 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadden JM, Declais AC, Carr SB, Lilley DM & Phillips SE The structural basis of Holliday junction resolution by T7 endonuclease I. Nature 449, 621–624 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Biertumpfel C, Yang W & Suck D Crystal structure of T4 endonuclease VII resolving a Holliday junction. Nature 449, 616–U614 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Declais AC et al. The complex between a four-way DNA junction and T7 endonuclease I. EMBO J 22, 1398–1409 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorecka KM, Komorowska W & Nowotny M Crystal structure of RuvC resolvase in complex with Holliday junction substrate. Nucleic Acids Res 41, 9945–9955 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y et al. Crystal Structure of a Eukaryotic GEN1 Resolving Enzyme Bound to DNA. Cell Rep 13, 2565–2575 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha T et al. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci U S A 93, 6264–6268 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ha T Single-molecule approaches embrace molecular cohorts. Cell 154, 723–726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duckett DR et al. The structure of the Holliday junction, and its resolution. Cell 55, 79–89 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Declais AC, Hadden J, Phillips SE & Lilley DM The active site of the junction-resolving enzyme T7 endonuclease I. J Mol Biol 307, 1145–1158 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Duckett DR, Panis MJ & Lilley DM Binding of the junction-resolving enzyme bacteriophage T7 endonuclease I to DNA: separation of binding and catalysis by mutation. J Mol Biol 246, 95–107 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Declais AC & Lilley DM Mechanistic aspects of the DNA junction-resolving enzyme T7 endonuclease I. Biochemistry 45, 3934–3942 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Kim HD et al. Mg2+-dependent conformational change of RNA studied by fluorescence correlation and FRET on immobilized single molecules. Proc Natl Acad Sci U S A 99, 4284–4289 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freeman AD, Declais AC & Lilley DM The importance of the N-terminus of T7 endonuclease I in the interaction with DNA junctions. J Mol Biol 425, 395–410 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Bennett RJ, Dunderdale HJ & West SC Resolution of Holliday junctions by RuvC resolvase: cleavage specificity and DNA distortion. Cell 74, 1021–1031 (1993). [DOI] [PubMed] [Google Scholar]
- 26.van Gool AJ, Shah R, Mezard C & West SC Functional interactions between the holliday junction resolvase and the branch migration motor of Escherichia coli. EMBO J 17, 1838–1845 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zerbib D, Mezard C, George H & West SC Coordinated actions of RuvABC in Holliday junction processing. J Mol Biol 281, 621–630 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Lilley DM & White MF Resolving the relationships of resolving enzymes. Proc Natl Acad Sci U S A 97, 9351–9353 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fogg JM, Schofield MJ, White MF & Lilley DM Sequence and functional-group specificity for cleavage of DNA junctions by RuvC of Escherichia coli. Biochemistry 38, 11349–11358 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Bennett RJ & West SC Structural analysis of the RuvC-Holliday junction complex reveals an unfolded junction. J Mol Biol 252, 213–226 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Shah R, Bennett RJ & West SC Genetic recombination in E. coli: RuvC protein cleaves Holliday junctions at resolution hotspots in vitro. Cell 79, 853–864 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Fogg JM & Lilley DM Ensuring productive resolution by the junction-resolving enzyme RuvC: large enhancement of the second-strand cleavage rate. Biochemistry 39, 16125–16134 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Wyatt HD, Sarbajna S, Matos J & West SC Coordinated Actions of SLX1-SLX4 and MUS81-EME1 for Holliday Junction Resolution in Human Cells. Mol Cell 52, 234–247 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Castor D et al. Cooperative Control of Holliday Junction Resolution and DNA Repair by the SLX1 and MUS81-EME1 Nucleases. Mol Cell 52, 221–233 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gwon GH et al. Crystal structures of the structure-selective nuclease Mus81-Eme1 bound to flap DNA substrates. Embo Journal 33, 1061–1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang JH, Kim JJ, Choi JM, Lee JH & Cho Y Crystal structure of the Mus81-Eme1 complex. Genes Dev 22, 1093–1106 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palets D, Lushnikov AY, Karymov MA & Lyubchenko YL Effect of single-strand break on branch migration and folding dynamics of Holliday junctions. Biophys J 99, 1916–1924 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parsons CA & West SC Specificity of binding to four-way junctions in DNA by bacteriophage T7 endonuclease I. Nucleic Acids Res 18, 4377–4384 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaskell LJ, Osman F, Gilbert RJ & Whitby MC Mus81 cleavage of Holliday junctions: a failsafe for processing meiotic recombination intermediates? EMBO J 26, 1891–1901 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matulova P et al. Cooperativity of Mus81.Mms4 with Rad54 in the resolution of recombination and replication intermediates. J Biol Chem 284, 7733–7745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Mazina OM & Mazin AV Human Rad54 protein stimulates human Mus81-Eme1 endonuclease. Proc Natl Acad Sci U S A 105, 18249–18254 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Constantinou A, Davies AA & West SC Branch migration and Holliday junction resolution catalyzed by activities from mammalian cells. Cell 104, 259–268 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Zhang R et al. BLM helicase facilitates Mus81 endonuclease activity in human cells. Cancer Res 65, 2526–2531 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Amit R, Gileadi O & Stavans J Direct observation of RuvAB-catalyzed branch migration of single Holliday junctions. Proc Natl Acad Sci U S A 101, 11605–11610 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 45.Karymov MA, Bogdanov A & Lyubchenko YL Single molecule fluorescence analysis of branch migration of holliday junctions: effect of DNA sequence. Biophys J 95, 1239–1247 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hadden JM, Convery MA, Declais AC, Lilley DM & Phillips SE Crystal structure of the Holliday junction resolving enzyme T7 endonuclease I. Nat Struct Biol 8, 62–67 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Roy R, Hohng S & Ha T A practical guide to single-molecule FRET. Nat Methods 5, 507–516 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ha T et al. Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 419, 638–641 (2002). [DOI] [PubMed] [Google Scholar]
- 49.McKinney SA, Joo C & Ha T Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophysical Journal 91, 1941–1951 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou R et al. SSB Functions as a Sliding Platform that Migrates on DNA via Reptation (vol 146, pg 222, 2011). Cell 146, 485–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from T.H. (tjha@jhu.edu) or R.Z. (ruobozhou@fas.harvard.edu) upon reasonable request. A Life Sciences Reporting Summary for this study is available.