

Abstract
Cancer cachexia is a multifactorial syndrome characterized by a progressive loss of skeletal muscle mass associated with significant functional impairment. Cachexia robs patients of their strength and capacity to perform daily tasks and live independently. Effective treatments are needed urgently. Here we investigated the therapeutic potential of activating the ‘alternative’ axis of the Renin-Angiotensin System (RAS), involving ACE2, angiotensin-(1–7), and the mitochondrial assembly receptor (MasR), for treating cancer cachexia. Plasmid overexpression of the MasR or pharmacological angiotensin-(1–7)/MasR activation did not affect healthy muscle fiber size in vitro or in vivo but attenuated atrophy induced by co-culture with cancer cells in vitro. In mice with cancer cachexia, the MasR agonist AVE 0991 slowed tumor development, reduced weight loss, improved locomotor activity, and attenuated muscle wasting, with the majority of these effects dependent on the orexigenic and not anti-tumor properties of AVE 0991. Proteomic profiling and immunohistochemistry revealed that mechanisms underlying AVE 0991 effects on skeletal muscle involved miR-23a-regulated preservation of the fast, glycolytic fibers. MasR activation is a novel regulator of muscle phenotype and AVE 0991 has orexigenic, anti-cachectic and anti-tumorigenic effects, identifying it as a promising adjunct therapy for cancer and other serious muscle wasting conditions.
Keywords: Renin-Angiotensin System, Mas receptor, cancer cachexia, anti-tumor
Introduction
Cancer cachexia is a complex, multifactorial syndrome characterized by a progressive loss of skeletal muscle mass that is associated with significant functional impairments (1). It affects 40–80% of all advanced cancer patients with the highest prevalence in those with pancreatic, gastric, oesophageal, colorectal and lung cancer, and in patients with advanced prostate, head/neck, liver, osteosarcoma, cervical, ovarian or breast cancer (2,3). The devastating consequences include profound weakness, impaired mobility and fatigue, reduced functional independence, and in the worst cases, compromised survival and death from metabolic, respiratory (diaphragm) or cardiac muscle (heart) failure (4). Cachexia is estimated to account for 20–30% of all cancer-related deaths (5) and weight loss and body mass index are predictors of survival in cancer patients (6,7). Unfortunately, effective treatment options for cancer cachexia are lacking and therefore identifying new therapeutic targets is critical.
A potential target that has received relatively little attention is the Renin-Angiotensin System (RAS). The ‘classical’ RAS axis, which involves conversion of angiotensin I (Ang I) to angiotensin II (Ang II) by the angiotensin converting enzyme (ACE) and signaling via the angiotensin type 1 (AT1) receptor, has been well-described as a negative regulator of skeletal muscle mass. Circulating Ang II levels are elevated in several muscle wasting conditions (8) and Ang II treatment induces muscle fiber atrophy in vitro and in vivo, effects associated with reduced Akt phosphorylation, increased myonuclear apoptosis and enhanced expression of the muscle-specific E3 ligases MuRF-1 and atrogin-1 (MAFb/x) (9–12). In contrast, the ‘alternative’ RAS axis counteracts signaling by the classical axis and so skeletal muscle size can be regulated by the balance between the two RAS axes. The alternative ACE2/Ang-(1–7)/MasR axis involves conversion of Ang I to angiotensin-(1–7) (Ang-(1–7) via one of two pathways: i) direct hydrolysis of Ang II to Ang-(1–7) via ACE2; or ii) indirect hydrolysis of Ang I to angiotensin-(1–9) (Ang-(1–9)) via ACE2 and subsequent conversion of Ang-(1–9) to Ang-(1–7) via ACE. ACE2 mediates production of Ang-(1–7) by two distinct pathways although the catalytic activity of ACE2 is ~400-fold higher with Ang II as a substrate than Ang I (13). Ang-(1–7) signals through the G-protein-coupled transmembrane mitochondrial assembly receptor (MasR) (14). Ang-(1–7) treatment counteracts muscle atrophy induced by Ang II administration in mice via mechanisms involving reduced TGF-β1 signaling, MuRF-1 and atrogin-1 expression and myonuclear apoptosis, and increased myosin heavy chain (MyHC) expression and Akt phosphorylation (9,10,15). Therefore, activation of the alternative RAS axis has therapeutic potential for muscle wasting conditions associated with increased Ang II/AT1 signaling. In this respect, infusion or administration of Ang-(1–7) in mice attenuated the muscle wasting and weakness associated with disuse (16) and endotoxemia (17), and reduced muscle fibrosis and enhanced strength in dystrophic mice (18,19). Recent studies reported increased plasma Ang II mRNA in cachectic but not non-cachectic cancer patients (20) and single nucleotide polymorphisms in the ACE gene resulting in increased ACE activity were associated with concurrent weight loss and low skeletal muscle index in cancer patients (21). The therapeutic potential of activating the alternative RAS axis for cancer cachexia has not been investigated. We tested the hypothesis that activation of the alternative ACE2/Ang-(1–7)/MasR axis attenuates muscle wasting in cancer and report for the first time that activating the alternative ACE2/Ang-(1–7)/MasR axis modulates muscle phenotype with the multiple benefits of having orexigenic, anti-cachectic and anti-tumorigenic effects.
Materials and Methods
Animals
All animal experiments were approved by the Animal Ethics Committee of The University of Melbourne and conducted in accordance with the Australian code of practice for the care and use of animals for scientific purposes as stipulated by the National Health and Medical Research Council (Australia). Mice were obtained from the Animal Resources Centre (Canning Vale, WA, Australia) and housed in the Biological Research Facility at The University of Melbourne under a 12:12-hr light-dark cycle. Water was available ad libitum and both water and standard laboratory chow was provided, changed and monitored daily. Methods for production and injection of AT1 and MasR recombinant adeno-associated virus (rAAV) vectors are described in the Supplemental Information and the primers with the restriction sites are shown in Supplemental Table S1.
In vivo AVE0991 study
The Colon-26 (C-26) mouse model of cancer cachexia is described in detail in the Supplemental Information. In the low dose AVE0991 study, CD2F1 mice (18–19-week-old) received a s.c. injection of C-26 cells (day 1), and three days later, began receiving the non-peptide, orally-active MasR agonist, AVE0991, via oral gavage at a dose of 1 mg/kg/day (n=8) or an equivalent volume of vehicle (sterile 0.9% NaCl containing 11.8% DMSO, n=8), given 10 times over a 13-day period (days 4–17). The volume given was 5 µl per gram body mass (i.e. 150 µl for a 30 g mouse). Seventeen days after C-26 injection (day 18), mice were anesthetized with sodium pentobarbitone (Nembutal, 60 mg.kg−1) via i.p. injection and the tibitalis anterior (TA), extensor digitorum longus (EDL), soleus, plantaris, gastrocnemius and quadriceps muscles as well as the epididymal fat, spleen, liver and heart, were surgically excised, blotted on filter paper and weighed on an analytical balance. The TA muscles were mounted in embedding medium, frozen in thawing isopentane and stored at −80ºC for subsequent analyses. The tumor was also excised and tumor length, width and depth assessed using digital calipers to facilitate determination of tumor volume. Mice were killed as a consequence of the cardiac excision.
In the high dose AVE0991 study, CD2F1 mice (10.5-month-old) received a s.c. injection of C-26 cells (day 1), and three days later, began receiving AVE0991 via oral gavage at a dose of 15 mg/kg/day (n=16) or an equivalent volume of vehicle (sterile 0.9% NaCl containing 11.8% DMSO, n=8), given 9 times over a 10-day period (days 4–14). The volume given was 6 µl per gram body mass (i.e. 180 µl for a 30 g mouse). Mice in the vehicle group and in one cohort of AVE0991 treated mice (n=8) were fed ad libitum and the second cohort of AVE0991 treated mice were pair-fed (PF) to the control group to account for effects on food intake (n=8). Water was available ad libitum for all groups. Sixteen days after C-26 injection (day 17), whole body metabolism and locomotor activity was assessed using the Promethion Metabolic Analyzer (Sable Systems International, North Las Vegas, NV 89032, USA). Mice were acclimated for 12-hours before data were collected every 5 min over a 12-hour light period and a 12-hour dark period. Oxygen consumption (VO2), energy expenditure, locomotor activity (Pedmeters, meters moved) and the number of beam breaks (sum of breaks in x, y and z beams) were recorded. During the entire data collection period, mice received drinking water ad libitum. Mice also received food ad libitum, with the exception of the PF animals that were only provided with the average amount of food consumed by the vehicle-treated mice over the 24-hour period. Eighteen days after C-26 injection (day 19), mice were anesthetized with sodium pentobarbitone (Nembutal, 60 mg.kg−1) via i.p. injection and dissections were performed as in the low dose study.
Culture of C2C12 cells
Murine C2C12 myoblasts (ATCC, Manassas, VA, USA) were plated in 6-or 12-well plates and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, Scoresby, VIC, Australia) supplemented with 10% (v/v) fetal calf serum (FBS, Life Technologies) and 1% L-glutamine (L-glut, Life Technologies) at 37ºC + 5% CO2. Upon confluency, the media was changed to DMEM containing 2% (v/v) horse serum (HS, Life Technologies) and 1% L-glut (DMEM/2% HS/1% L-glut) for 4 days (d) at 37ºC + 5% CO2 to induce differentiation during which time the media were changed every 48 h. Experiments were performed on day 4 of differentiation at which point healthy myotubes were observed.
For pharmacologic Ang II/AT1 activation, differentiated C2C12 myotubes were incubated in fresh DMEM/2% HS/1% L-glut without (control) or with Ang II (H-1705, 1 µM, Sigma-Aldrich) or the Ang-(1–7) inhibitor A779 (1 µM, Sigma-Aldrich) for 48 h at 37ºC + 5% CO2. Following incubation, myotubes were fixed for immunocytochemical analysis of cell diameter.
For serum starvation induced muscle wasting, differentiated C2C12 myotubes were washed once with 1 × PBS and incubated for 48 h at 37ºC + 5% CO2 in serum-free DMEM (Life Technologies), as described previously (22). Total protein synthesis was determined by a non-isotopic technique (SUnSET) which has been described in detail previously (23). Briefly, puromycin (Sigma-Aldrich, St. Louis, MO, USA) was added to the media at a final concentration of 1 µM exactly 30 min before cells were collected in ice-cold homogenizing buffer (10 mM TrisHCl [pH 7.4], 100 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton-X, 10% glycerol, 0.1% SDS, 20 mM Na4P2O7, 2 mM Na3VO4, 1 mM NaF, 0.5% sodium deoxycholate and 1 mM PMSF containing protease and phosphatase inhibitor cocktails) and analyzed by SDS-PAGE and western blot. Other cells were fixed and taken for immunocytochemical analysis of cell diameter or lysed for western blotting analysis of AT1 and MasR protein expression.
Methods for MasR overexpression and pharmacologic Ang-(1–7)/MasR activation in serum starved myotubes or in myotubes co-cultured with cancer cells are in Supplemental Information.
Proteomic profiling of AVE0991 treated serum starved myotubes
Proteins from vehicle (DMSO) and AVE0991 treated serum starved C2C12 myotubes were extracted using lysis buffer containing the non-ionic detergent Triton-X100 (1%). After tryptic digestion, a stable isotope dimethyl labeling was performed and a shotgun nano liquid chromatography-tandem mass spectrometry (LC-MS/MS) approach was used for the detection and quantification of the proteins, as described in detail in the Supplemental Information. The labeled mixtures of vehicle and AVE0991 treated serum starved samples were run three times and an average ratio for each protein was calculated based on the MS-Quant analysis. Proteins appearing in at least two technical replicates were considered for further analysis. Six experimental replicates were analyzed in this way.
Human cancer cachexia study
The human cancer cachexia study was approved by the University of Florida Institutional Review Board and conformed to the Declaration of Helsinki. Nine non-cancer controls (6 females/3 males) and twelve patients with pathologically diagnosed pancreatic ductal adenocarcinoma (PDAC) cancer (7 females/5 males) gave written informed consent for participation. Subject characteristics are detailed in Supplemental Table S2. The non-cancer controls were undergoing surgery for bile duct complications (n=7) and intraductal papillary mucinous neoplasm (IPMN, n=2) and had no weight loss in the previous 12 months. The PDAC patients were undergoing pancreatoduodenectomy and were classified as being cachectic (>5% weight loss in the previous 12 months, n=12). At the beginning of surgery, a sample of ~100 mg of rectus abdominus muscle was excised under aseptic conditions, immediately frozen in liquid nitrogen and stored at −80ºC for subsequent analyses.
Immunohistochemistry
Serial sections (5 µm) were cut transversely through the TA muscle using a refrigerated (−20ºC) cryostat (CTI Cryostat; IEC, Needham Heights, MA, USA). Sections were reacted with laminin (Sigma-Aldrich) for determination of myofiber cross-sectional area (CSA) and with SC-71 and BF-F3 (both developed by S. Schiaffino, University of Padova, obtained from the Developmental Studies Hybridoma Bank) to assess the percentage of myosin IIa and myosin IIb isoforms, respectively (24). We have previously shown that mouse TA muscles do not express detectable levels of type I fibers (25) and so the fibers not reacting with SC-71 or BF-F3 were assumed to be type IIx fibers. Digital images were obtained using an upright microscope with camera (Axio Imager D1, Carl Zeiss), controlled and quantified using AxioVision AC software (AxioVision AC release 4.8.2, Carl Zeiss).
Immunocytochemistry
Cells were washed for 2 × 5 min in PBS and fixed in 4% paraformaldehyde for 15 min. Cells were then washed in PBS (3 × 5 min), permeabilized with 0.1% Triton X-100 for 10 min, washed in PBS (3 × 5 min) and blocked in 3% (w/v) BSA in PBS for 1 h at RT. Cells were incubated overnight at 4ºC in anti-sarcomeric myosin (1:50 diluted in 3% BSA/PBS, MF 20, developed by D.A. Fischman, Weill Cornell Medical College, obtained from the Developmental Studies Hybridoma Bank, Iowa City, IA, USA). The following day, cells were washed in PBS (4 × 5 min) and incubated in goat-anti-mouse Alexa Fluor 555 secondary antibody (1:400, Life Technologies) and DAPI (1:1,000) for 2 h at RT. Cells were washed in PBS (4 × 5 min) and then imaged on a Zeiss Axiovert 40 CFL inverted microscope using a 20× objective to give a total magnification of 126×. Five images were taken in each well from pre-defined locations within each quadrant. Myotube diameter was quantified using AxioVision AC software (AxioVision AC release 4.8.2, Carl Zeiss, Wrek, Göttingen, Germany).
Statistics
All values are expressed as mean ± SEM unless stated otherwise. Groups were compared using an unpaired Student’s t-test, a one-way ANOVA or a two-way ANOVA, where appropriate. Bonferroni’s post hoc test was used to determine significant differences between individual groups. Correlations were performed using linear regression analysis. The level of significance was set at P<0.05 for all comparisons.
Further details about the Materials and Methods are provided in the Supplementary Information.
Results
Muscle-specific MasR overexpression does not alter muscle fiber size in healthy mice
While activation of the alternative RAS axis can counteract Ang II-induced muscle wasting (9,10,15), its effects on skeletal muscle size in healthy basal (in vivo) conditions was unknown. We therefore used recombinant adeno-associated viral (rAAV) vectors to directly increase MasR expression in skeletal muscle of healthy mice and examined its effect on muscle fiber size. This method achieves effective overexpression and is useful for such proof-of-principle studies. rAAV-mediated overexpression of the AT1 was used as a comparison as it would be anticipated to reduce skeletal muscle size. rAAV9 vectors expressing the MasR or AT1 were injected into the right tibialis anterior (TA) muscle and rAAV9 expressing an empty control vector was injected into the contralateral left TA muscle of healthy CD2F1 mice and analyzed 21 days (d) later. Intramuscular injection of rAAV9:MasR increased MasR protein abundance by ~5.3-fold compared with muscles injected with empty vector (P<0.001; Supplemental Fig. S1A), but had no effect on muscle fiber cross-sectional area (CSA, Supplemental Fig. S1B and C). In comparison, intramuscular injection of rAAV9:AT1 increased AT1 protein abundance by ~3.2-fold (P<0.001; Supplemental Fig. S1D) and reduced average muscle fiber CSA by 11% (P<0.05; Supplemental Fig. S1E), an effect attributed to an increased proportion of smaller fibers (P<0.05; Supplemental Fig. S1F). Supporting these results, pharmacologic activation of the ACE/Ang II/AT1 axis in healthy C2C12 cells using Ang II or A779 induced myotube atrophy (P<0.01, Supplemental Fig. S2A, B). These findings confirm that skeletal muscle fiber size in healthy basal conditions can be negatively regulated by increasing expression or activation of the AT1 but is not affected by direct MasR overexpression.
Genetic MasR overexpression and pharmacological Ang-(1–7)/MasR activation attenuates serum starvation-induced muscle atrophy in vitro
Since activation of the ACE2/Ang-(1–7)/MasR axis protects against muscle wasting in various conditions including disuse (16) and endotoxin-induced sepsis (17), we investigated the downstream mediators of this response in a non-targeted and non-biased manner to direct the focus of our subsequent in vivo analyses. We also wanted to avoid the complications of inflammatory and cachectic factors in media from cancer cells to better understand the mechanisms within skeletal muscle cells. To this end, we used a serum starvation model of muscle atrophy in vitro. C2C12 myotubes that had been differentiated for 4 d were incubated for a further 48 h in serum-free media (SS), causing a 40% reduction in myotube diameter (P<0.05; Supplemental Fig. S2C, D). The starvation-induced atrophy was associated with a decrease in protein synthesis (P<0.05; Supplemental Fig. S2E, F), 37% increase in AT1 protein abundance (P<0.05) but no change in MasR protein abundance (Supplemental Fig. S2G-I).
To induce MasR overexpression in myotubes, we transfected C2C12 myoblasts with a control plasmid (rAAV-CMV-eGFP) or plasmid expressing the MasR (rAAV-CMV-MasR) and after 4 d differentiation, myotubes were incubated for 48 h in HS or SS. MasR overexpression had no effect on healthy myotube size but completed prevented starvation-induced atrophy (P<0.0001; Fig. 1A, B). To confirm that similar findings were seen when MasR overexpression was induced only in myotubes and not expressed at the myoblast stage, we transfected C2C12 myoblasts with a Tet-regulated lentivirus without (empty) or containing the MasR gene, differentiated for 4 d and then incubated myotubes for 48 h in HS or SS in the presence of doxycycline. MasR overexpression was confirmed by western blotting (Supplemental Fig. S3A). Inducing MasR overexpression in differentiated myotubes had no effect on healthy myotube size, but attenuated starvation-induced atrophy by 51% (P<0.02; Supplemental Fig. S3B, C). The similar findings between experiments inducing MasR overexpression in myotubes or myoblasts strongly support the protective effects of MasR overexpression for starvation-induced atrophy.
Figure 1.
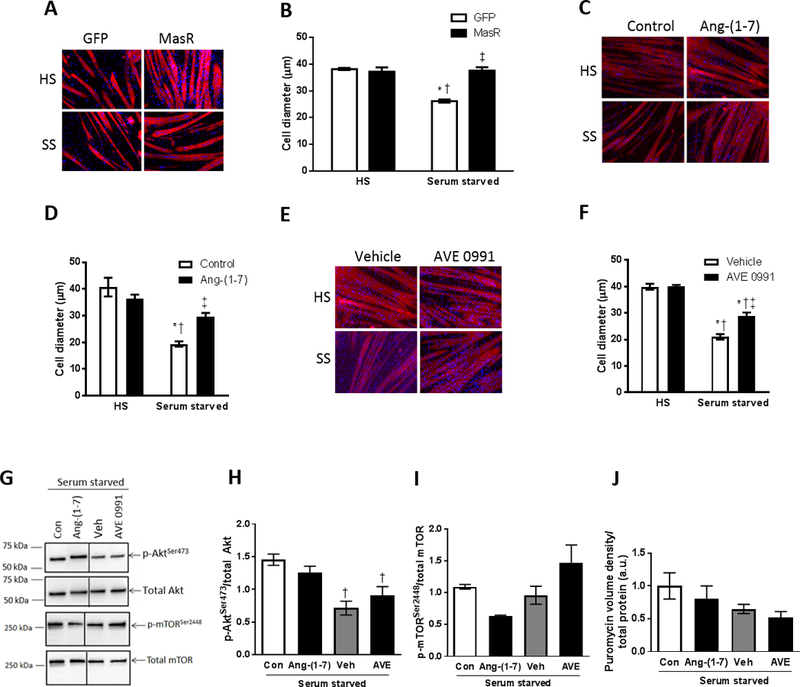
Genetic MasR overexpression and pharmacological Ang-(1–7)/MasR activation protects against serum starvation-induced muscle fiber atrophy in vitro. (A) C2C12 myoblasts were transfected with GFP control or MasR plasmid and after 4 days of differentiation into myotubes, were incubated in normal horse serum (HS) or serum-free media (serum starved, SS) and (B) cell diameter assessed after 48 h (two-way ANOVA; *P<0.001 vs. HS+GFP; †P<0.001 vs. HS+MasR; ‡P<0.001 vs. Serum starved+GFP; n=6). Pharmacological Ang-(1–7)/MasR activation was induced by incubating differentiated C2C12 myotubes for 48 h in HS or SS and treating at the same time: (C, D) with or without Ang-(1–7); or (E, F) with vehicle control (DMSO) or AVE0991 (MasR agonist) and cell diameter was assessed (D & F, two-way ANOVA; *P<0.05 vs. HS+Control/Vehicle; †P<0.05 vs. HS+Ang-(1–7)/AVE0991; ‡P<0.01 vs. Serum starved+Control/Vehicle; n=8). (G) Representative western blots and quantification of (H) phosphorylated total Akt, (I) phosphorylated and total mTOR, and (J) puromycin stain to assess protein synthesis in myotubes incubated for 48 h in serum free media and treated at the same time with or without Ang-(1–7), vehicle control or AVE0991 (one-way ANOVA; †P<0.05 vs. Serum starved+Control; n=3).
For pharmacological activation of the Ang-(1–7)/MasR axis, differentiated myotubes were incubated for 48 h in HS or SS without or with Ang-(1–7) or AVE0991 (MasR agonist). DMSO-treated myotubes were used as a vehicle control for the AVE0991 experiments. Neither Ang-(1–7) nor AVE0991 altered the size of healthy myotubes but both attenuated starvation-induced atrophy, by 64% and 41%, respectively (P<0.001, Fig. 1C-F). Neither Ang-(1–7) nor AVE0991 increased phosphorylation of Akt or mTOR (Fig. 1G-I) or enhanced protein synthesis (Fig. 1J, Supplemental Fig. S3D).
Next, we performed stable isotope dimethyl labeling and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify, in a non-targeted and non-biased manner, proteins and pathways altered by AVE0991 compared with vehicle treatment of serum starved C2C12 myotubes (Fig. 2A). AVE0991 was used because unlike Ang-(1–7) (H-1715), it is orally active and resistant to proteolytic enzymes (26) and hence more clinically relevant. A total of 911 proteins were identified and among them, 22 were increased (by a fold-change > 1.3) and 18 were decreased (by a fold-change ≤ 0.85) in AVE0991 compared with DMSO treated serum starved myotubes (Supplemental Tables S3, S4). The top-ranked downregulated proteins and the biological processes and molecular functions representing these proteins are shown in Supplemental Figs. S3E and F.
Figure 2.
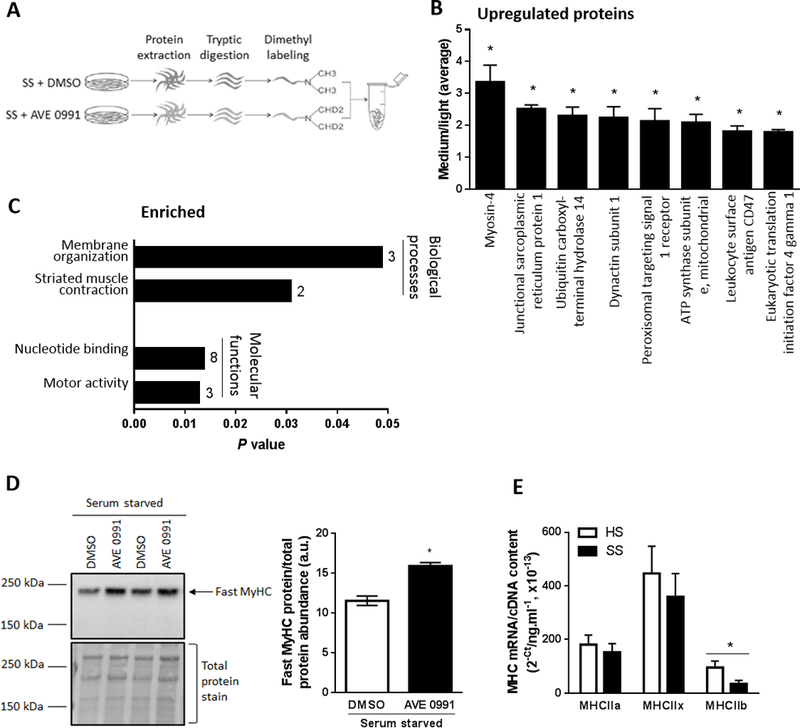
Proteomic profiling to identify proteins and pathways regulated by AVE0991 treatment of serum starved C2C12 myotubes. (A) Schematic of the experiment, with proteins from serum starved (SS) C2C12 myotubes treated with vehicle control (DMSO]) or AVE0991 ]extracted using lysis buffer containing 1% Triton-X 100, tryptic digested and purified using SPE cartridges. Peptides obtained from DMSO and AVE0991 treated serum starved myotubes were labeled with normal (light) or deuterated (medium) formaldehyde, respectively. The labelled peptides were mixed together and analysed by LC-MS/MS using an LTQ Orbitrap Elite mass spectrometer. (B) Upregulated proteins and (C) enriched biological processes and molecular functions representing upregulated proteins obtained from DAVID database in AVE0991 treated serum starved myotubes. Numbers next to solid bars in C indicates counts of proteins annotated to the indicated processes and functions (unpaired t test; *P<0.05 Serum starved+AVE0991 vs. Serum starved+DMSO; n=3). (D) Representative western blot and corresponding quantification of fast myosin heavy chain (MyHC) confirming upregulation of myosin-4, the top ranked upregulated protein, in AVE0991 treated serum starved myotubes (unpaired t test; *P<0.05; n=6). (E) Gene expression of the fast myosin heavy chain isoforms MHCIIa, MHCIIx and MHCIIb in C2C12 myotubes incubated for 48 h in HS or serum-free media (SS, unpaired t test; *P<0.05; n=6).
The top-ranked upregulated protein was myosin-4 (fast myosin heavy chain, MyHC, IIb, Fig. 2B) and consistent with its upregulation, functional analyses using DAVID revealed the most prominent biological process and molecular function of upregulated proteins in AVE0991 treated serum starved myotubes involved striated muscle contraction and motor activity, respectively (Fig. 2C). Western blotting confirmed upregulation of fast MyHC in AVE0991 treated serum starved myotubes (P<0.05; Fig. 2D), indicating that the protective effect of AVE0991 in atrophied myotubes was associated with a shift towards a greater abundance of larger, fast type II muscle fibers. Quantitative PCR analysis confirmed that serum starvation was associated with a 64% decrease in MHCIIb mRNA expression (P<0.05, Fig. 2E).
Plasmid overexpression and pharmacological activation of the MasR attenuates cancer-induced muscle atrophy in vitro
We next examined whether the benefits of MasR activation extended to cancer-based muscle wasting. Because AT1 was elevated in serum starved myotubes, we first examined whether AT1 expression was similarly increased in humans (Supplemental Table S2) and mice with cancer cachexia. AT1 mRNA was ~100% higher in rectus abdominus muscle from cachectic (>5% weight loss) patients with pancreatic ductal adenocarcinoma (PDAC) cancer compared with non-cancer controls (P<0.05, Fig. 3A). However, the cachectic PDAC group were significantly older than controls (P<0.05, Supplemental Table S2) and so we examined the relationship between age and AT1 mRNA expression but found no significant correlation (r2=0.058; P=0.292, Fig. 3B). These findings indicate that the increase in AT1 mRNA in the cachectic PDAC group was not simply due to age. Similar to serum starved myotubes, MasR mRNA expression was not different between controls and PDAC patients (P=0.16; Fig. 3C) and was not significantly correlated with age (r2=0.038; P=0.399, Fig. 3D). There was no significant correlation between AT1 or MasR mRNA expression and the percentage weight loss in the previous 12 months in the PDAC patients (AT1, r2=0.18, P=0.18; MasR, r2=0.26, P=0.10; n=12).
Figure 3.
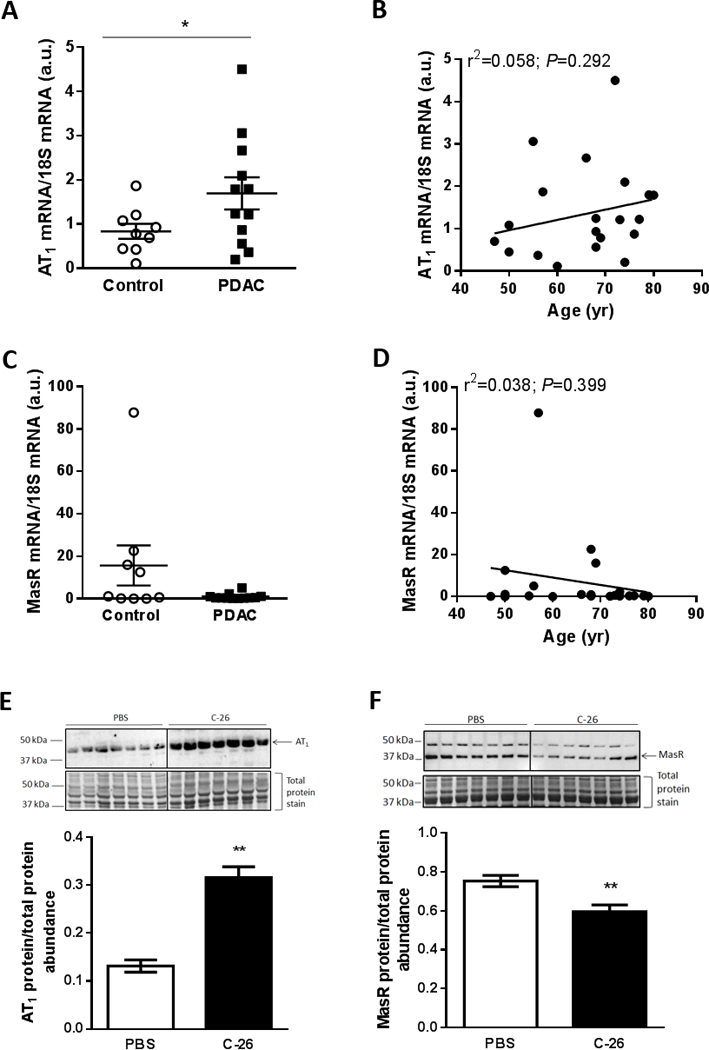
Muscle AT1 expression is elevated in humans and mice with cancer cachexia. (A) AT1 and (C) MasR mRNA expression in rectus abdominus muscle tissue from cachectic (>5% weight loss) patients with pancreatic ductal adenocarcinoma (PDAC) cancer compared with non-cancer controls (unpaired t test; *P<0.05; Control, n=9; PDAC, n=12). (B) AT1 and (D) MasR mRNA expression was not related to age (linear regression analysis; n=21). Representative western blot of (E) AT1 and (F) MasR protein expression and corresponding quantification in gastrocnemius muscles from PBS and C-26 tumor-bearing mice (unpaired t test; **P<0.01; n=7).
In gastrocnemius muscles from the Colon-26 (C-26) mouse model of cancer cachexia which had a 23% smaller mass than controls (PBS, 4.43±0.12; C-26, 3.42±0.13 mg.g−1 initial body mass, unpaired t test; P<0.01; n=7), AT1 protein expression was 240% higher (P<0.01; Fig. 3E) and MasR protein expression was 21% lower than controls (P<0.01; Fig. 3F).
As depicted in Fig. 4A, C2C12 myoblasts were transfected with GFP control or MasR plasmid and after 4 d differentiation, a transwell insert containing no cells (control) or Colon-26 (C-26) cancer cells was added and co-cultured for 48 h. Co-culture with C-26 cells induced a 45% reduction in myotube size (P<0.05, compare Con+GFP and C-26+GFP in Fig. 4B, C). However, MasR overexpression attenuated the atrophy from C-26 co-culture by 46% (P<0.05; Fig. 4B, C).
Figure 4.

Plasmid MasR overexpression and pharmacological Ang-(1–7)/MasR activation attenuates and reverses Colon-26 (C-26) cancer cell induced muscle fiber atrophy in vitro. (A) Schematic of the C2C12-C-26 co-culture system, with C2C12 myotubes incubated for 48 h with a transwell insert containing no cells (control) or C-26 cancer cells. (B, C) C2C12 myoblasts were transfected with GFP control or MasR plasmid and after 4 days of differentiation, were co-cultured with or without C-26 cancer cells and assessed 48 h later for myotube diameter (one-way ANOVA; *P<0.05 vs. Control+GFP; †P<0.05 vs. C-26+GFP; n=7–8). (D, E) C2C12 myotubes that had been differentiated for 4 days were treated with vehicle (DMSO), the AT1 antagonist telmisartan (Tel), the MasR agonist AVE0991 (AVE) or both telmisartan and AVE0991 (AVE+Tel) and at the same time were co-cultured with or without C-26 cancer cells and assessed 48 h later for myotube diameter (one-way ANOVA; *P<0.05 vs. Control+Vehicle; †P<0.05 vs. C-26+Vehicle; ‡P<0.05 vs. C-26+Telmisartan; n=8). (F, G) C2C12 myoblasts were transfected with GFP control or MasR plamid and after 4 days of differentiation, were treated with vehicle (DMSO) or AVE0991 and at the same time were co-cultured with or without C-26 cancer cells and assessed 48 h later for myotube diameter (one-way ANOVA; *P<0.05 vs. GFP+Vehicle; †P<0.01 vs. C-26+GFP+Vehicle; ‡P<0.001 vs. C-26+MasR+Vehicle; §P<0.0001 vs. C-26+GFP+AVE0991; n=6). Pharmacological Ang-(1–7)/MasR activation reverses Colon-26 (C-26) cancer cell induced muscle fiber atrophy in vitro. (H, I) C2C12 myotubes were co-cultured with or without C-26 cancer cells and assessed 24 h later for myotube diameter (unpaired t test; ****P<0.0001; n=6). (J, K) C2C12 myotubes were co-cultured with or without C-26 cancer cells and after 24 h, were treated with vehicle or AVE0991 and assessed for myotube diameter at 48 h after co-culture was initiated (one-way ANOVA; **P<0.01 vs. Control+Vehicle; †P<0.05 vs. C-26+Vehicle; n=4).
To investigate the effect of pharmacological MasR activation on C-26 induced muscle atrophy and to assess whether greater protection could be conferred by combined antagonism of the AT1, differentiated C2C12 myotubes were treated for 48 h with vehicle (Veh, DMSO), the AT1 antagonist telmisartan (Tel), AVE0991 (AVE) or both telmisartan and AVE0991 and at the same time, cultured with or without C-26 cancer cells (Fig. 4D, E). Treatment with AVE0991 alone or in combination with telmisartan attenuated the C-26 induced atrophy by 56% and 63%, respectively, indicating no additive effect of the two treatments (P<0.05, Fig. 4D, E). Treatment with telmisartan alone had no effect on myotube size (Fig. 4D, E).
The combination of plasmid-mediated MasR overexpression and AVE0991 treatment was also investigated and compared with either intervention alone (MasR overexpression, +34%; AVE0991, +25%). Combination treatment conferred the greatest increase in the size of myotubes co-cultured with C-26 cells, with effects being additive (+66%, P<0.0001, Fig. 4F, G).
These findings demonstrate that genetic overexpression or pharmacologic activation of the MasR can attenuate cancer-induced wasting when treatment is given prior to or at the same time as co-culture is initiated. However, whether it could reverse atrophy was not known. We therefore co-cultured C2C12 myotubes with C-26 cells for 24 h, which induced a 37% decrease in myotube size (P<0.0001, Fig. 4H, I), before adding vehicle or AVE0991 and assessed myotube diameter after another 24 h. AVE0991 treatment initiated 24 h after co-culture completely reversed the C-26 induced atrophy, with myotube size not significantly different from controls (Fig. 4J, K).
Co-culture with C-26 cells for 48 h induced a 72% decrease in MHCIIb mRNA expression (P<0.02, Fig. 5A). We used Ingenuity Pathway Analysis (IPA) to identify potential interacting pathways between MasR and Myh4 (MHCIIb). This identified microRNA-23a (miR-23a-3p) as a mediator of MasR regulation of Myh4, with MasR signaling inhibiting miR-23a expression, and Myh4 being a direct target of miR-23a-3p (Fig. 5B). The 3’UTR of My4 contains a binding site for the seed sequence of miR-23a-3p (Fig. 5B). We therefore investigated whether increasing miR-23a-3p expression attenuated the protective effects of AVE0991 for cancer-induced myotube wasting. C2C12 myoblasts were transfected with a miR-23a-3p mimic or mimic negative control and after 4 days of differentiation, myotubes were co-cultured for 48 h with C-26 cells and treated without (DMSO) or with AVE0991. The miR-23a-3p mimic attenuated the AVE0991-induced increase in myotube size by 19% (P<0.01, Fig. 5C, D. By identifying relevant pathways, these findings provide further insight into the mechanism of fiber protection by MasR signaling.
Figure 5.
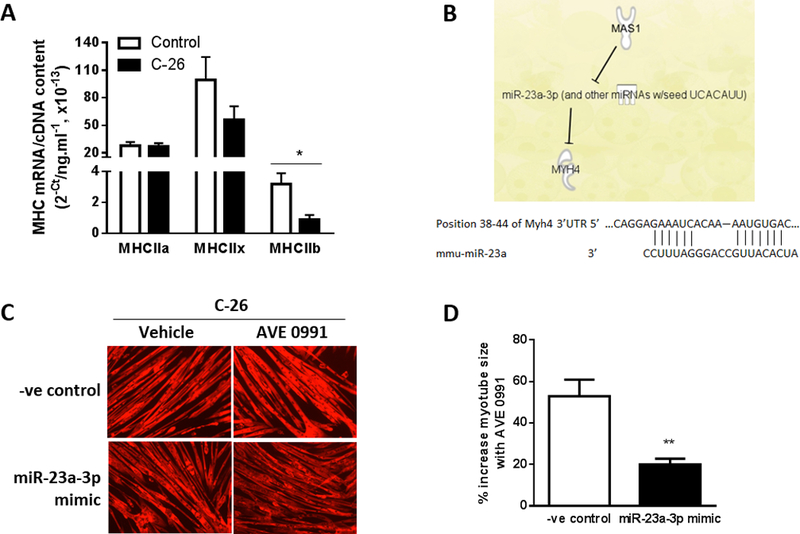
miR-23a-3p mediates the effects of MasR activation of myosin-4. (A). MHCIIa, MHCIIx and MHCIIb mRNA expression in C2C12 myotubes co-cultured for 48 h with or without C-26 cancer cells (unpaired t test; *P<0.02; n=6). (B) Ingenuity Pathway Analysis (IPA) revealed microRNA-23a (miR-23a-3p) as a mediator of MasR regulation of Myh4. The binding site on the 3’UTR of Myh4 mRNA for the seed sequence of miR-23a. (C, D) C2C12 myoblasts were transfected with a miRNA mimic negative control (-ve control) or miR-23a-3p mimic and after 4 days of differentiation into myotubes, were co-cultured with C-26 cancer cells and treated with vehicle or AVE0991 and assessed 48 h later for myotube diameter. Overexpression of miR-23a-3p using a mimic attenuated the AVE0991 induced increase in size of myotubes co-cultured with C-26 cells (unpaired t test; **P<0.01; n=6).
Pharmacological MasR activation slows tumor growth and attenuates muscle wasting in mice with cancer cachexia by inducing an oxidative-to-glycolytic muscle fiber transition
To test the clinical relevance of our in vitro findings, we next examined whether MasR activation could attenuate cancer-induced muscle wasting in vivo. To this end, we used oral gavage of AVE0991 rather than rAAV-mediated MasR overexpression since oral administration is more likely to be implemented in the clinic and have greater patient compliance, and because gene therapy approaches still need to be refined for clinical use. Although whole body effects of AVE0991 may complicate findings within the muscle, additional systemic benefits would further strengthen its therapeutic potential and interest to patients and clinicians. C-26 tumor-bearing mice were treated with 1 mg/kg AVE0991 via daily oral gavage from days 4–17 and on day 18, skeletal muscle mass and fiber size were assessed (Supplemental Fig. S4A). AVE0991 induced small but significant improvements in cumulative food and water intake, which are indicators of quality of life (Supplemental Fig. S4B, C). It also attenuated loss of body mass without altering tumor size (Supplemental Fig. S4D, E). Despite low dose AVE0991 having no effect on the percentage change in tumor-free body mass, the mass of various hindlimb muscles or other tissues (Supplemental Fig. S4F-H), or on average muscle fiber CSA (Supplemental Fig. S5A, B), it did induce a shift in fiber proportions from oxidative type IIa fibers towards the higher force-producing fast, glycolytic type IIb fibers (Supplemental Fig. S5A, C). Furthermore, AVE0991 decreased mRNA expression of the slow isoforms of both troponin I and troponin C and reduced Smad3 mRNA (−44%, P<0.01; Supplemental Fig. S5D, E).
Since AVE0991 had previously been administered to rodents over doses ranging from 1 to 20 mg/kg (27–30), we next investigated whether a higher dose of AVE0991 (15 mg/kg) would confer greater benefits in mice with severe cancer cachexia (Fig. 6A). In addition, an AVE0991 treated group that was pair-fed to the vehicle control, was included to examine the relationship between observed benefits and food intake (Fig. 6A). In mice fed ad libitum, high dose AVE0991 increased food intake from day 15 (Fig. 6B), water intake from day 13 (Supplemental Fig. S6A) and attenuated loss of body mass from day 16 (Fig. 6C). As expected, pair-fed AVE0991 treated mice had a similar food intake (Fig. 6B), water intake (Supplemental Fig. S6A) and loss of relative body mass to vehicle controls (Fig. 6C). In mice fed ad libitum but not in pair-fed mice, high dose AVE0991 decreased tumor area from day 12 (Fig. 6D) and at the end of the experimental period, reduced tumor volume by 30% (C-26+Vehicle, 2743±269 mm3; C-26+AVE0991, 1917±186 mm3; C-26+AVE0991 PF, 2331±394 mm3; one-way ANOVA; P<0.05 C-26+AVE0991 vs. C-26+Vehicle; n=8–16). Whole body metabolism and locomotor activity were assessed 16 days after C-26 injection and there was a main effect for AVE0991-treated mice either fed ad libitum or pair-fed to controls to have a lower oxygen consumption (VO2) than vehicle controls (P<0.05, Fig. 6E). Energy expenditure was not different between groups (Fig. 6F) but AVE0991 treated mice fed ad libitum had greater motor activity (Pedmeters, P<0.05 treatment main effect, Fig. 6G) and movement as assessed by the number of beam breaks (P<0.05 treatment main effect, Supplemental Fig. S6B) compared with both vehicle control and pair-fed mice. Energy expenditure normalized to locomotor activity was lower in AVE0991 treated mice fed ad libitum compared with vehicle control and pair-fed mice (P<0.05, Fig. 6H). Furthermore, AVE0991 treated mice fed ad libitum but not pair-fed had an attenuated loss of tumor-free body mass (−9%, P<0.01), and greater plantaris (+22%, P<0.05), tibialis anterior (TA, +9%) and quadriceps muscle mass (+14%, P<0.05) compared with vehicle controls (Fig. 6I, J). In AVE0991 treated mice, heart and liver mass were both reduced by 10% in pair-fed mice compared with those fed ad libitum (P<0.05, Fig. 6K).
Figure 6.
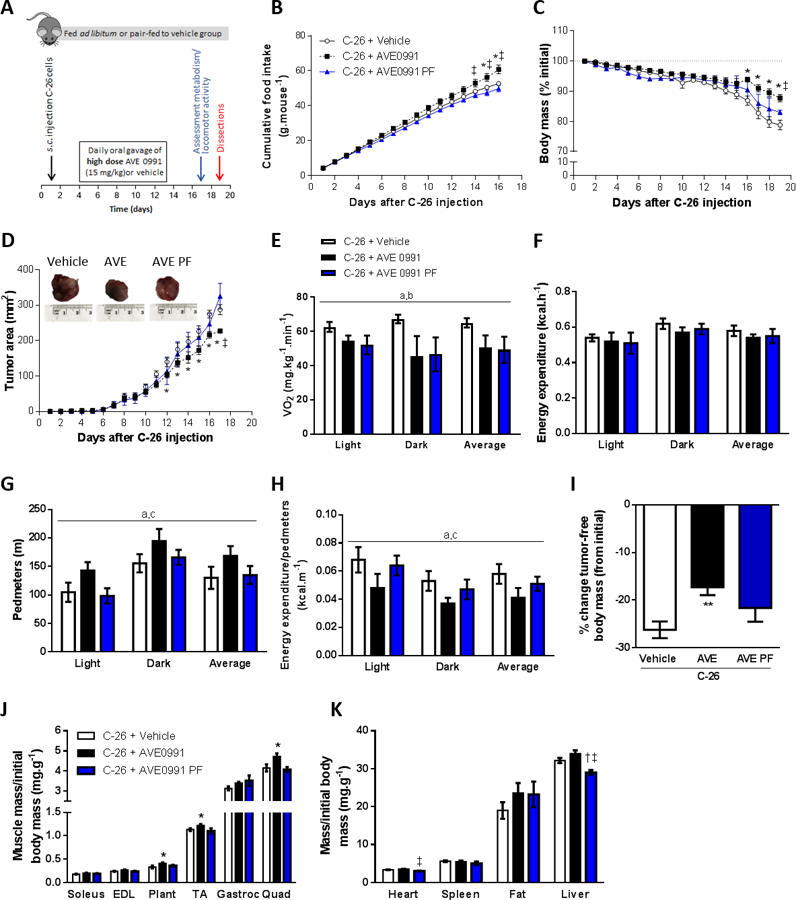
High dose AVE0991 (15 mg/kg) attenuates wasting and slows tumor growth in severely cachectic C-26 tumor-bearing mice. (A) Experimental timeline involving treatment of C-26 tumor-bearing mice treated with high dose AVE0991 (15 mg/kg) or vehicle control. (B) Daily cumulative food intake, (C) relative body mass and (D) tumor area were improved in AVE0991 treated mice fed ad libitum (two-way ANOVA; *P<0.05 C-26+AVE0991 vs. C-26+Vehicle; †P<0.05 C-26+AVE0991 PF vs. C-26+Vehicle; ‡P<0.05 C-26+AVE0991 PF vs. C-26+AVE0991; n=4–16). On day 17, mice were placed in a Promethion metabolic analyzer for 24 h and average oxygen consumption (VO2, E) energy expenditure (F), locomotor activity (Pedmeters, G) and energy expenditure relative to locomotor activity (H) were assessed for each of the 12-h light and dark cycles, and for the average of both cycles (24-h period) (two-way ANOVA; aP<0.05 Treatment main effect C-26+AVE0991 vs. C-26+Vehicle; bP<0.05 Treatment main effect C-26+AVE0991 PF vs. C-26+Vehicle; CP<0.05 Treatment main effect C-26+AVE0991 PF vs. C-26+AVE0991; n=5–6). At the end of treatment (day 19), the tumor was excised, weighed and measured for size and mass (representative tumors shown in inset in D) and (I) the percentage change in tumor-free body mass from initial (day 1) was calculated (one-way ANOVA; **P<0.01 vs. C-26+Vehicle; n=8–16). Selected tissues were also excised and weighed: (J) skeletal muscles soleus, extensor digitorum longus (EDL), plantaris (Plant), tibialis anterior (TA), gastrocnemius (Gastroc), quadriceps (Quad); (K) heart, spleen, epididymal fat and liver (one-way ANOVA; *P<0.05 C-26+AVE0991 vs. C-26+Vehicle; †P<0.05 C-26+AVE0991 PF vs. C-26+Vehicle; ‡P<0.05 C-26+AVE0991 PF vs. C-26+AVE0991; n=8–16).
AVE0991 treatment of mice fed ad libitum but not pair-fed to controls enhanced average TA muscle fiber CSA by 16% (P<0.05) due to increased CSA of the type IIx (+20%, P<0.05) and type IIb fibers (+18%, P<0.05, Fig. 7A, B). In mice fed ad libitum, AVE0991 also induced a shift in fiber type proportions away from type IIx fibers towards glycolytic type IIb fibers (P<0.05; Fig. 7A, C). Moreover, AVE0991 reduced mRNA expression of the slow isoforms of troponin I and C (P<0.05), with the decrease in slow troponin C being independent of food intake (Fig. 7D). There was no significant difference between groups in mRNA expression of MuRF-1, atrogin-1, TGF-β1, smad3 or caspase 3 (Fig. 7E).
Figure 7.
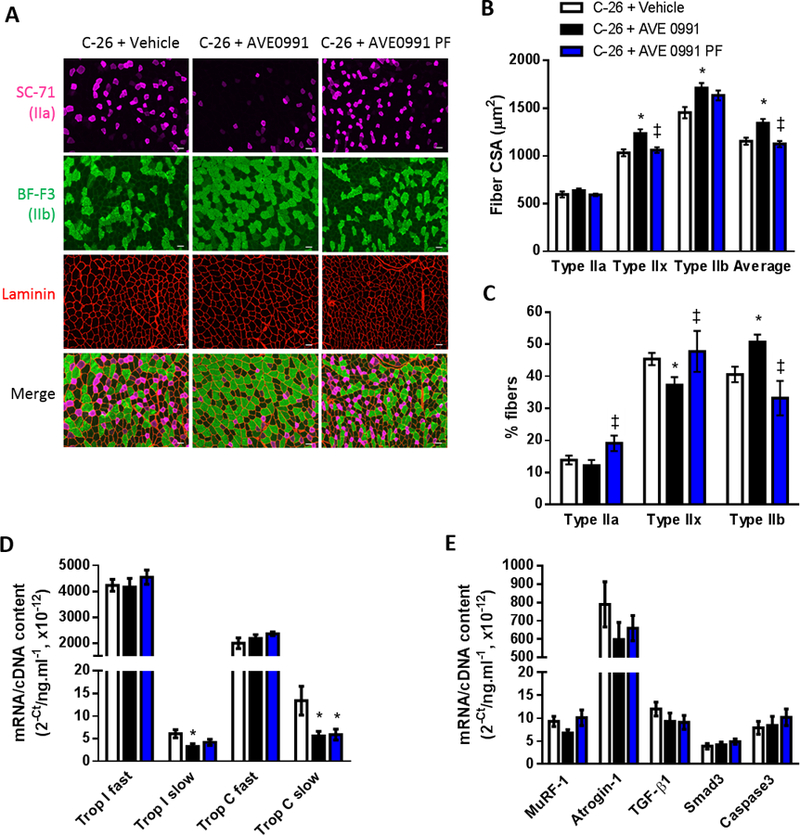
High dose AVE0991 (15 mg/kg) induces TA muscle fiber hypertrophy in severely cachectic C-26 tumor-bearing mice fed ad libitum. (A) Representative images of TA muscle cross-sections reacted for laminin (red) to indicate all muscle fibers and myosin IIa (SC-71, pink) and myosin IIb (BF-F3, green) to indicate type IIa and type IIb fibers, respectively. Since mouse TA muscles do not express detectable levels of type I fibers, those fibers not reacting with SC-71 or BF-F3 (i.e. black fibers in the merge) were assumed to be type IIx fibers. Quantification of SC-71, BF-F3 and laminin based on reaction intensity facilitated determination of: (B) average fiber CSA and CSA of the type IIa, type IIx (non-reacting with SC-71 or BF-F3) and type IIb fibers; and (C) the proportion of type IIa, type IIx and type IIb fibers (one-way ANOVA; *P<0.05 vs. C-26+Vehicle; ‡P<0.05 vs. C-26+AVE0991; C-26+Vehicle, n=16; C-26+AVE0991, n=15; C-26+AVE0991 PF, n=7). Gene expression of (D) the fast and slow isoforms of troponin I and troponin C, (E) the ubiquitin ligases MuRF-1 and atrogin-1, TGF-β1, Smad3 and caspase3 in TA muscles from vehicle or AVE0991 treated C-26 tumor-bearing mice (one-way ANOVA; *P<0.05 vs. C-26+Vehicle; n=7–8. Using a 20× objective, total magnification for images in Fig. 7A is 126×. Scale bar = 50 µm.
To assess whether the observed beneficial effects of AVE0991 were simply due to inhibition of tumor growth, a subset of mice with similar tumor burden were analyzed. As shown in Supplemental Fig. S6C-F, comparisons were made between mice in each group that had similar tumor burden (tumor mass as a percentage of total end body mass) of: 2.05%, 2.28% and 2.28% (low tumor burden) and 4.96%, 5.09% and 5.06% (high tumor burden). Since only one mouse per group was assessed, statistical analyses were not performed. The attenuated loss of tumor-free body mass and increase in quadriceps muscle mass with AVE0991 remained in mice fed ad libitum with both low and high tumor burden (Supplemental Fig. S6C, D). The increase in plantaris and TA muscle mass with AVE0991 was only maintained in mice fed ad libitum with low tumor burden (Supplemental Fig. S6D). Also maintained in AVE0991 treated mice fed ad libitum with low and high tumor burden was: the increase in average CSA and CSA of type IIx and IIb fibers in mice; and the shift in fiber type proportions away from type IIx fibers towards type IIb fibers (Supplemental Fig. S6E, F). Thus, the majority of AVE0991 effects were still evident when mice of similar tumor burden were compared.
Relevance to other cancers
We investigated the therapeutic potential of AVE0991 for attenuating wasting induced by other cancer cells and found that 48 h co-culture with Lewis Lung Carcinoma (LLC) cells induced a 23% (P<0.05) decrease in C2C12 myotube size, which was attenuated by 18% with AVE0991 treatment (Supplemental Fig. S7A, B).
Discussion
The two regulatory axes of the Renin-Angiotensin System (RAS) exert opposing effects in several tissues including skeletal muscle (31,32), with the ‘classical’ ACE/Ang II/AT1 axis inducing muscle wasting which can be counteracted by the ‘alternative’ ACE2/Ang-(1–7)/MasR axis (9,10,15). Activation of the alternative RAS axis therefore has therapeutic potential for muscle wasting conditions associated with hyperactivity of the classical axis. Here, we show that AT1 expression is elevated in preclinical and clinical cancer cachexia and that pharmacological activation of the alternative axis attenuates cancer cell induced atrophy in vitro, and improves locomotor activity, enhances muscle mass and fiber size, reduces weight loss and slows tumor growth in mice with cancer cachexia. These effects were dependent on the orexigenic properties of the MasR agonist AVE0991. Thus the multifactorial benefits of AVE0991 having orexigenic, anti-cachectic and anti-tumorigenic effects identify this strategy as a novel and promising adjunct therapy for cancer.
Systemic activation of the ACE2/Ang-(1–7)/MasR axis reduces many of the deleterious effects of Ang II infusion in mouse skeletal muscle (9,10,15) and infusion of Ang-(1–7) in mice attenuates muscle wasting in several conditions, including the muscular dystrophies (18,33), hindlimb immobilization (16) and endotoxin-induced sepsis (17). However, its role in the regulation of skeletal muscle size in healthy, basal conditions had not previously been examined. We demonstrate for the first time that increasing ACE2/Ang-(1–7)/MasR signaling has no direct effect on the regulation of healthy skeletal muscle size in vivo, with muscle-specific MasR overexpression not altering muscle fiber size in healthy mice. The lack of effect was not due to an absence of endogenous ligand as serum Ang-(1–7) levels are higher than Ang II in the healthy condition (34). Furthermore, genetic MasR overexpression or pharmacologic Ang-(1–7)/MasR activation had no effect on the size of healthy myotubes in vitro. Conversely, we demonstrated that muscle-specific AT1 overexpression or pharmacological AT1 activation induced fiber atrophy in healthy mice. These experiments were done in conjunction with those involving intramuscular injection of rAAV9:MasR and together, confirm that the size of healthy skeletal muscle can be regulated by overexpression of the AT1 but not the MasR. In contrast, MasR overexpression or pharmacologic activation of the ACE2/Ang-(1–7)/MasR axis attenuated myotube atrophy induced with serum starvation or co-culture with cancer in vitro. The latter findings were not specific to C-26 induced wasting, with AVE0991 also attenuating wasting induced by co-culture with LLC cells, indicating efficacy across multiple cancer types. Since the combination of increasing MasR levels and enhancing receptor activity conferred greater protection against cancer-induced atrophy in vitro, with effects being additive of the individual interventions, it is unlikely that the addition of ligands will induce further improvements in AVE0991 activation. The findings also support the notion that the eventual best treatment may come from combining AVE0991 with receptor overexpression when gene therapy approaches become suitable for clinical use.
Since cancer cachexia is one of the most prevalent muscle wasting diseases affecting 40–80% of all advanced cancer patients (2,3,35) and estimated to account for up to 30% of all cancer-related deaths (5), we investigated whether ACE2/Ang-(1–7)/MasR axis activation conferred protective effects against cancer-based wasting in vivo. This was demonstrated through low-dose (1 mg/kg) AVE0991 treatment improving food and water intake and weight loss, with even greater benefits with higher dose (15 mg/kg) AVE0991 administration which also improved locomotor activity, enhanced muscle mass and fiber size, attenuated the loss of tumor-free body mass and reduced tumor development in the C-26 mouse model of cancer cachexia. Although anti-cancer properties of Ang-(1–7) have been reported (36), this is one of the first reports of the clinically applicable non-peptide MasR agonist AVE0991 having anti-tumor effects. Most of the benefits of AVE0991 remained when mice of similar tumor burden were compared but were diminished or lost in pair-fed mice, indicating that the anti-cachectic effects were at least in part mediated by the orexigenic properties of AVE0991 and were not simply due to slowed tumor growth. However, our findings in the cancer-free serum starvation cell culture model confirmed that AVE0991 also has direct effects on skeletal muscle cells. In this regard, the identification of AVE0991 as a regulator of skeletal muscle phenotype by preserving the large, fast type IIb fibers (myosin-4, fast myosin heavy chain) reveals a novel role for MasR activation and highlights it as a potential application for conditions associated with specific atrophy of fast, glycolytic type II skeletal muscle fibers (37), including cancer cachexia (38). Our investigation demonstrated that these effects were mediated by miR-23a-3p and add to the growing body of evidence implicating components of the RAS in miRNA regulation, with an AT1-regulated miRNA fingerprint conserved in vascular smooth muscle cells of humans and rodents (39). Specifically, the miR-23a∼27a∼24–2 cluster is one of 24 known miRNA clusters in the human genome that respond to AT1 stimulation (39) and our data indicate that it may also be responsive to MasR modulation. Taken together, these findings provide further insight into the mechanism of muscle fiber protection by MasR signaling.
High levels of circulating Ang II have been reported in several muscle wasting conditions (8), including patients with cancer cachexia (20) and we extended these findings by revealing increased AT1 mRNA in muscles from cachectic pancreatic cancer patients and in muscles from cachectic C-26 tumor-bearing mice. We were unable to localize the AT1 using immunofluorescence in these samples due to the lack of a specific antibody, but we generated an AT1 antibody for western blotting that has much greater specificity and thus, validity, than those available commercially (Supplemental Fig. S8A, B). Furthermore, although we were unable to identify the cell type(s) contributing to the elevated AT1 in cachectic muscles, our finding of increased AT1 in serum starved myotubes support a local production of AT1 in muscle fibers during conditions of wasting. Overexpression and pharmacologic activation of the MasR was protective against muscle wasting in each of the conditions in which AT1 was elevated, suggesting that increased muscle AT1 may be a useful indicator for the therapeutic efficacy of MasR activation.
In conclusion, activation of the alternative ACE2/Ang-(1–7)/MasR axis attenuates skeletal muscle wasting in conditions associated with increased muscle AT1 expression. We have identified the MasR agonist AVE0991 as a novel regulator of muscle phenotype, with therapeutic potential for conditions where skeletal muscle atrophy and weakness are indicated. AVE0991 has anti-tumor and anti-cachectic effects that are dependent on its orexigenic properties, with clinical relevance as an adjunct therapy for cancer patients and other conditions associated with anorexia and cachexia.
Supplementary Material
Statement of significance:
Findings demonstrate that MasR activation has multiple benefits of being orexigenic, anti-cachectic and anti-tumorigenic, revealing it as a potential adjunct therapy for cancer.
Acknowledgments
The authors thank: Prof. Jeffrey S. Chamberlain (Department of Neurology, The University of Washington, Seattle, USA) for provision of the rAAV-CMV-MCS and rAAV-CMV-eGFP expression vectors; Prof. Martha Belury (Department of Human Nutrition, The Ohio State University) for donating the C-26 cells and Prof. Donna McCarthy (College of Nursing, The Ohio State University) for arranging the shipment of these cells; and Dr. Audrey Chan (Centre for Muscle Research, Department of Physiology, The University of Melbourne) for expert technical assistance.
The MF 20 monoclonal antibody, developed by D.A. Fischman (Weill Cornell Medical College), and the SC-71 and BF-F3 monoclonal antibodies, developed by S. Schiaffino (University of Padova), were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Grant Support
Supported by project grants from the National Health and Medical Research Council (NHMRC, Australia, 1041865, to G.S. Lynch and K.T. Murphy), Victorian Cancer Agency (16852, to K.T. Murphy) and National Institute of Arthritis, Musculoskeletal and Skin Diseases (R01AR060209, to A.R. Judge). K.T. Murphy was supported by a Career Development Fellowship from the NHMRC (1023178).
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL , et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–95. [DOI] [PubMed] [Google Scholar]
- 2.Sun L, Quan XQ, Yu S. An Epidemiological Survey of Cachexia in Advanced Cancer Patients and Analysis on Its Diagnostic and Treatment Status. Nutr Cancer 2015;67:1056–62. [DOI] [PubMed] [Google Scholar]
- 3.Blum D, Stene GB, Solheim TS, Fayers P, Hjermstad MJ, Baracos VE , et al. Validation of the Consensus-Definition for Cancer Cachexia and evaluation of a classification model--a study based on data from an international multicentre project (EPCRC-CSA). Ann Oncol 2014;25:1635–42. [DOI] [PubMed] [Google Scholar]
- 4.Powers SK, Lynch GS, Murphy KT, Reid MB, Zijdewind I. Disease-induced skeletal muscle atrophy and fatigue. Med Sci Sports Exerc 2016;48:2307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer 2002;2:862–71. [DOI] [PubMed] [Google Scholar]
- 6.Martin L, Senesse P, Gioulbasanis I, Antoun S, Bozzetti F, Deans C , et al. Diagnostic criteria for the classification of cancer-associated weight loss. J Clin Oncol 2015;33:90–9. [DOI] [PubMed] [Google Scholar]
- 7.Renfro LA, Loupakis F, Adams RA, Seymour MT, Heinemann V, Schmoll HJ , et al. Body Mass Index Is Prognostic in Metastatic Colorectal Cancer: Pooled Analysis of Patients From First-Line Clinical Trials in the ARCAD Database. J Clin Oncol 2016;34:144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida T, Delafontaine P. Mechanisms of Cachexia in Chronic Disease States. Am J Med Sci 2015;350:250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cisternas F, Morales MG, Meneses C, Simon F, Brandan E, Abrigo J , et al. Angiotensin-(1–7) decreases skeletal muscle atrophy induced by angiotensin II through a Mas receptor-dependent mechanism. Clin Sci (Lond) 2015;128:307–19. [DOI] [PubMed] [Google Scholar]
- 10.Meneses C, Morales MG, Abrigo J, Simon F, Brandan E, Cabello-Verrugio C. The angiotensin-(1–7)/Mas axis reduces myonuclear apoptosis during recovery from angiotensin II-induced skeletal muscle atrophy in mice. Pflügers Arch 2015;467:1975–84. [DOI] [PubMed] [Google Scholar]
- 11.Song YH, Li Y, Du J, Mitch WE, Rosenthal N, Delafontaine P. Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest 2005;115:451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rezk BM, Yoshida T, Semprun-Prieto L, Higashi Y, Sukhanov S, Delafontaine P. Angiotensin II infusion induces marked diaphragmatic skeletal muscle atrophy. PLoS One 2012;7:e30276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1–7)-Mas receptor axis. Hypertens Res 2009;32:533–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I , et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A 2003;100:8258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morales MG, Abrigo J, Meneses C, Simon F, Cisternas F, Rivera JC , et al. The Ang-(1–7)/Mas-1 axis attenuates the expression and signalling of TGF-β1 induced by AngII in mouse skeletal muscle. Clin Sci (Lond) 2014;127:251–64. [DOI] [PubMed] [Google Scholar]
- 16.Morales MG, Abrigo J, Acuna MJ, Santos RA, Bader M, Brandan E , et al. Angiotensin-(1–7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis Model Mech 2016;9:441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morales MG, Olguin H, Di Capua G, Brandan E, Simon F, Cabello-Verrugio C. Endotoxin-induced skeletal muscle wasting is prevented by angiotensin-(1–7) through a p38 MAPK-dependent mechanism. Clin Sci (Lond) 2015;129:461–76. [DOI] [PubMed] [Google Scholar]
- 18.Acuña MJ, Pessina P, Olguin H, Cabrera D, Vio CP, Bader M , et al. Restoration of muscle strength in dystrophic muscle by angiotensin-1–7 through inhibition of TGF-β signalling. Hum Mol Genet 2014;23:1237–49. [DOI] [PubMed] [Google Scholar]
- 19.Riquelme C, Acuna MJ, Torrejon J, Rebolledo D, Cabrera D, Santos RA , et al. ACE2 is augmented in dystrophic skeletal muscle and plays a role in decreasing associated fibrosis. PLoS One 2014;9:e93449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Penafuerte CA, Gagnon B, Sirois J, Murphy J, MacDonald N, Tremblay ML. Identification of neutrophil-derived proteases and angiotensin II as biomarkers of cancer cachexia. Br J Cancer 2016;114:680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johns N, Stretch C, Tan BH, Solheim TS, Sorhaug S, Stephens NA , et al. New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J Cachexia Sarcopenia Muscle 2017;8:122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ham DJ, Caldow MK, Lynch GS, Koopman R. Arginine protects muscle cells from wasting in vitro in an mTORC1-dependent and NO-independent manner. Amino Acids 2014;46:2643–52. [DOI] [PubMed] [Google Scholar]
- 23.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P , et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 2011;25:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elf K, Shevchenko G, Nygren I, Larsson L, Bergquist J, Askmark H , et al. Alterations in muscle proteome of patients diagnosed with amyotrophic lateral sclerosis. Journal of proteomics 2014;108:55–64. [DOI] [PubMed] [Google Scholar]
- 25.Murphy KT, Chee A, Gleeson BG, Naim T, Swiderski K, Koopman R , et al. Antibody-directed myostatin inhibition enhances muscle mass and function in tumor-bearing mice. Am J Physiol 2011;301:R716–R26. [DOI] [PubMed] [Google Scholar]
- 26.Santos RA, Ferreira AJ. Pharmacological effects of AVE 0991, a nonpeptide angiotensin-(1–7) receptor agonist. Cardiovasc Drug Rev 2006;24:239–46. [DOI] [PubMed] [Google Scholar]
- 27.Ebermann L, Spillmann F, Sidiropoulos M, Escher F, Heringer-Walther S, Schultheiss HP , et al. The angiotensin-(1–7) receptor agonist AVE0991 is cardioprotective in diabetic rats. Eur J Pharmacol 2008;590:276–80. [DOI] [PubMed] [Google Scholar]
- 28.Ma Y, Huang H, Jiang J, Wu L, Lin C, Tang A , et al. AVE 0991 attenuates cardiac hypertrophy through reducing oxidative stress. Biochem Biophys Res Commun 2016;474:621–5. [DOI] [PubMed] [Google Scholar]
- 29.Cunha TM, Lima WG, Silva ME, Souza Santos RA, Campagnole-Santos MJ, Alzamora AC. The nonpeptide ANG-(1–7) mimic AVE 0991 attenuates cardiac remodeling and improves baroreflex sensitivity in renovascular hypertensive rats. Life Sci 2013;92:266–75. [DOI] [PubMed] [Google Scholar]
- 30.Rodrigues-Machado MG, Magalhaes GS, Cardoso JA, Kangussu LM, Murari A, Caliari MV , et al. AVE 0991, a non-peptide mimic of angiotensin-(1–7) effects, attenuates pulmonary remodelling in a model of chronic asthma. Br J Pharmacol 2013;170:835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cabello-Verrugio C, Morales MG, Rivera JC, Cabrera D, Simon F. Renin-angiotensin system: an old player with novel functions in skeletal muscle. Med Res Rev 2015;35:437–63. [DOI] [PubMed] [Google Scholar]
- 32.Cabello-Verrugio C, Rivera JC, Garcia D. Skeletal muscle wasting: new role of nonclassical renin-angiotensin system. Curr Opin Clin Nutr Metab Care 2017;20:158–63. [DOI] [PubMed] [Google Scholar]
- 33.Sabharwal R, Cicha MZ, Sinisterra RD, De Sousa FB, Santos RA, Chapleau MW. Chronic oral administration of Ang-(1–7) improves skeletal muscle, autonomic and locomotor phenotypes in muscular dystrophy. Clin Sci (Lond) 2014;127:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pignone A, Rosso AD, Brosnihan KB, Perfetto F, Livi R, Fiori G , et al. Reduced circulating levels of angiotensin-(1−−7) in systemic sclerosis: a new pathway in the dysregulation of endothelial-dependent vascular tone control. Ann Rheum Dis 2007;66:1305–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fearon KC, Voss AC, Hustead DS. Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr 2006;83:1345–50. [DOI] [PubMed] [Google Scholar]
- 36.Gallagher PE, Arter AL, Deng G, Tallant EA. Angiotensin-(1–7): a peptide hormone with anti-cancer activity. Current medicinal chemistry 2014;21:2417–23. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Pessin JE. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care 2013;16:243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M , et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer cell 2005;8:421–32. [DOI] [PubMed] [Google Scholar]
- 39.Kemp JR, Unal H, Desnoyer R, Yue H, Bhatnagar A, Karnik SS. Angiotensin II-regulated microRNA 483–3p directly targets multiple components of the renin-angiotensin system. Journal of molecular and cellular cardiology 2014;75:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.