

Abstract
Purpose:
Steroidogenic enzymes are essential for prostate cancer development. Enzymes inactivating potent androgens were not investigated thoroughly, which leads to limited interfere strategies for prostate cancer therapy. Here we characterized the clinical relevance, significance and regulation mechanism of enzyme HSD17B2 in prostate cancer development.
Experimental Design:
HSD17B2 expression was detected with patient specimens and prostate cancer cell lines. Function of HSD17B2 in steroidogenesis, AR signaling and tumor growth was investigated with prostate cancer cell lines and xenograft model. DNA methylation and mRNA alternative splicing were investigated to unveil the mechanisms of HSD17B2 regulation.
Results:
HSD17B2 expression was reduced as prostate cancer progresses. 17βHSD2 decreased potent androgen production by converting testosterone (T) or dihydrotestosterone (DHT) to their upstream precursors. HSD17B2 overexpression suppressed androgen-induced cell proliferation and xenograft growth. Multiple mechanisms were involved in HSD17B2 functional silencing including DNA methylation, androgen stimulation and mRNA alternative splicing. DNA methylation and T stimulation decreased HSD17B2 mRNA or protein level respectively. Two new catalytic-deficient isoforms, generated by alternative splicing, bound to wild type 17βHSD2 and promoted its degradation. Splicing factors SRSF1 and SRSF5 participated in the generation of new isoforms.
Conclusion:
Our findings provide evidence of the clinical relevance, significance and regulation of HSD17B2 in prostate cancer progression, which might provide new strategies for clinical management by targeting the functional silencing mechanisms of HSD17B2.
Introduction
Prostate cancer is the most common cancer in the US and the fastest increasing cancer in China in men (1; 2). Androgens activate androgen receptor (AR) signaling to promote prostate cancer progression (3). Thus steroidogenic enzymes involved in androgen metabolism are essential targets for prostate cancer treatment or biomarkers for disease diagnosis (4; 5).
Testosterone (T) produced by testis is the major androgen stimulating prostate cancer development until androgen deprivation therapy (ADT)(6). ADT resistance occurs and disease progresses into castration resistant prostate cancer (CRPC) by utilizing adrenal precursors, such as dehydroepiandrosterone (DHEA) or androstenedione (AD), to generate potent androgen dihydrotestosterone (DHT)(3; 7; 8). Steroidogenic enzymes which accelerate DHT synthesis have been investigated thoroughly in driving prostate cancer development and treatment resistance (9; 10). CYP17A is required for conversion of cholesterol to DHEA, providing androgen precursors to prostate cancer(7). Abiraterone, targeting CYP17A, is used for treatment of CRPC and castration-sensitive prostate cancer (11–14). 3βHSD1 is the rate-limiting enzyme for DHEA to DHT metabolism. A single nucleotide polymorphism (SNP) in 3βHSD1 increases its protein stability and leads to worse outcomes after ADT (15–19). Increased expression of AKR1C3 has been reported as a mechanism of drug resistance. AKR1C3 catalyzes AD or 5α-androstanedione (5α-dione) to T or DHT, respectively, by modifying the 17-keto group to 17β-OH which is essential for AR activation (20; 21). Increasing AKR1C3 leads to more efficient steroidogensis, which could subdue the response to abiraterone or enzalutamide (22; 23). Thus, steroidogenic enzymes promoting androgen synthesis obtain oncogenic function in disease development.
On the other hand, steroidogenic enzymes which inactivate androgens have not been investigated thoroughly. The regulation of androgen-inactivation enzymes is not well understood as is their role in tumor progression. The enzyme 17βHSD2 catalyzes the reverse reaction of AKR1C3. It catalyzes 17β-OH to 17-keto and leads to androgen inactivation (24–27). The significance and regulation of 17βHSD2 in prostate cancer remains elusive.
Here we show HSD17B2 expression and function is reduced in prostate cancer. Overexpression of HSD17B2 blocks potent androgen synthesis and thus suppresses AR signaling and cell growth. In vivo, 17βHSD2 inhibits xenograft proliferation as a tumor suppressor. The expression of HSD17B2 in prostate cancer is regulated by DNA methylation, androgen stimulation and mRNA alternative splicing. Our data demonstrate the tumor suppressor role of 17βHSD2 and its regulation mechanisms in prostate cancer, shedding light on disease interruption through HSD17B2.
Materials and methods
Cell lines
Cell lines LNCaP, PC3, Du145 and HEK293T cells were purchased from the American Type Culture Collection (Manassas, VA) and maintained in RPMI-1640 (LNCaP, PC3 and Du145) or DMEM (HEK293T) with 10% FBS. Dr. Charles Sawyers (Memorial Sloan Kettering Cancer Center, New York, NY) kindly provided LAPC4 cells, which were grown in Iscove’s Modified Dulbecco’s Medium with 10% FBS. VCaP was kindly provided by Dr. Jun Qin (SIBS, Shanghai, China). All experiments with LNCaP and VCaP were done in plates coated with poly-DL-ornithine (Sigma-Aldrich, St. Louis, MO). Cell lines was authenticated by Hybribio (Guangzhou, China) and determined to be mycoplasma free with primers 5’-GGGAGCAAACAGGATTAGATACCCT-3’ and 5’-TGCACCATCTGTCACTCTGTTAACCTC-3’.
Plasmids construction and transfection
Lentiviral vectors pCDH-puro or pLVX-tight-puro was used for HSD17B2 isoforms cloning. HSD17B2 shRNA and non-target shRNA control (pLKO.1 TRC, Mission RNAi) were constructed. The constructs were confirmed by DNA sequencing. The primer sequences are presented in supporting table 1.
Virus particles were harvested 48h after transfection in HEK-293T cells using PEI (Promega). Human prostate cancer cell lines were infected and selected with puromycin (Sigma Aldrich, St. Louis, Missouri, USA). The function of all vectors was validated by western blot.
Control gRNA (5’-ATCTGCCATGGCGTCCTGGC-3’) and HSD17B2 gRNAs (gA: 5’- ACTGTCCCACATAGTACTGT-3’, gB: 5’-TCAAGCCCCAAAAAGGGGAC-3’ and gD: 5’-TGTCCATTTGGAGCACCGAG-3’) were inserted into CRISPR plasmid backbone, lentiCRISPR v2, (a generous gift from Dr. Feng Zhang (Addgene Plasmid #52961)) according to the protocol they provided(28). Then they were used to generate the HSD17B2 knock out MDA-PCa-2b stable cell line by using a lentiviral system. 293T cells were cotransfected with 10 mg each of constructed plasmids (containing gRNAs), pMD2.G, and psPAX2 vector for 48 hours to package the virus. Then the virus was concentrated by using PEG-it Virus Precipitation Solution (System Biosciences) according to the provided protocol. Next, MDA-PCa-2b cells were infected with the concentrated virus for 24 hours with addition of polybrene (10 μg/ml), followed by selection with 2 μg/ml puromycin for ~2 weeks.
HPLC
Cells were seeded and incubated in 24-well plates with 0.2 million cells/well for ~24 h and then treated with indicated drugs and a mixture of radioactive ([3H]-labeled) and non-radioactive steroids (final concentration, 50 nM T and 10nM DHT; ~1,000,000 cpm/well; PerkinElmer, Waltham, MA) at 37°C. Aliquots of medium were collected at the indicated time and treated with 300 units of β-glucuronidase (Novoprotein Scientific Inc, China) at 37°C for 2h, extracted with 500 μL ethyl acetate:isooctane (1:1), dried under freeze dryer (Martin Christ Gefriertrocknungsanlagen, Germany).
High-performance liquid chromatography (HPLC) analysis was performed on a Waters Acquity ARC HPLC. Dried samples were reconstituted in 100 μl of 50% methanol and injected into the HPLC. Metabolites were separated on CORTECS C18 reverse-phase column 4.6×50 mm, 2.7 μM (Waters, Ireland) using a methanol/water gradient at 40°C. The column effluent was analyzed using β-RAM model 3 in-line radioactivity detector (LABLOGIC, USA). All HPLC studies were run in triplicate and repeated at least 3 times in independent experiments.
Gene expression and immunoblotting
Cells were starved for at least 48h with phenol red-free and serum free-medium and treated with the indicated androgens (100nM DHEA CAS#53–43-0, 10nM AD CAS#63–05-8, 10nM T CAS#58–22-0, 10nM 5a-dione CAS#846–46-8 and 1nM DHT CAS#521–18-6). Cell to cDNA Kit (EZBioscience) was used for cDNA synthesis directly from cells. Quantitative PCR (qPCR) experiment was conducted in Bio-Rad CFX96 (Bio-Rad) using EZBioscience 2× SYBR Green qPCR master mix (EZBioscience). The primers for qPCR were described in supplementary table 1.
Total protein was extracted from cells with RIPA buffer containing protease inhibitors (Piece, Prod#88666).The following primary antibodies were used: anti-17βHSD2 (1:1000, Santa Cruz), anti-FLAG (1:5000, Sigma), anti-β-actin (1:1000, Cell Signal Tech).
Pyrosequencing
DNA was extracted from cells with QIAamp DNA Mini Kit (QIAGEN) and pyrosequencing was performed by Genergy Biotechnology (Shanghai, China). Briefly, genomic DNA was treated with bisulfite conversion using Qiagen EpiTect® Bisulfite Kit (QIAGEN). Primers selectively amplified either methylated or unmethylated DNA were used. PCR products were sequenced on PyroMark Q96 ID (QIAGEN). The primers for PCR were described in supplementary table 1.
Cell proliferation assay
Cell proliferation assay was performed with cell counting kit-8 (Dojindo, Kumamoto, Japan) in accordance with the manufacturer’s instructions. Briefly, 10,000 cells/well dispensed in 100 μl aliquots were seeded in a 96-well plate and starved for 48h. Androgen was added as indicated. The viable cells were measured after 4 and 7 days according to the manufacturer’s protocol. The absorbance was read at 450 nm using a microplate reader (BioTeK, USA) to estimate the viable cells in each well. The growth curve was calculated by Graphpad Prism 5.0 software (San Diego, CA, USA).
Mouse xenograft studies
Male B-NDG® (B-NSG) mice (B:Biocytogen; N:NOD background; D:DNAPK (Prkdc) null; G:IL2rgknockout) mice, 4 to 6 weeks of age were obtained from Beijing Biocytogen. All mouse studies were conducted under a protocol approved by the Institutional Animal Care and Use Committee (SIBCB-S373–1802-006). 1×107 LNCaP-HSD17B2 stable cells were subcutaneously implanted into the right flank of the intact mice with Matrigel (Corning, #354234). Mice were randomly assigned into two groups when the xenografts reached approximately 50 to 100 mm3 (length × width × width × 0.5): sucrose control (5% sucrose, orally, n=10) and doxycycline (Sigma, dox, 2 mg/ml and 5% sucrose, orally, n=10). Sucrose and dox-containing water were replaced every two days. Tumor growth was observed every 2–3 days for 21 days by measuring the two-dimensional longest axis and shortest axis with a caliper. At the end of experiment, the animals were sacrificed, xenografts were collected for further analysis. The difference between treatment groups was assessed by Kaplan-Meier survival analysis using a log-rank test in SigmaStat 3.5.
Xenograft tissue metabolism
40–50 mg xenograft tissues were seeded and incubated in 24-well plates and then treated with a mixture of radioactive ([3H]-labeled) and non-radioactive steroids (final concentration, 50 nM T; ~1,000,000 cpm/well; PerkinElmer, Waltham, MA) at 37°C.
Immunohistochemistry (IHC)
Patient specimens were collected at Tongji hospital with patient consent under a hospital review board approved protocol. Consent was obtained from each patient or related guardian. Experiments were carried out in accordance with Declaration of Helsinki. Benign prostate tissues were collected from bladder cancer patients after radical cystectomy. Prostate cancer tissues were collected from patients with localized prostate cancer receiving only radical prostatectomy treatment. Xenograft samples or patient samples were fixed in 4% formaldehyde solution and embedded in paraffin. 5 μm thick sections were cut from paraffin-embedded tissue blocks, deparaffinized and rehydrated in ethanol, and then subjected to antigen retrieval by microwaving in sodium citrate (pH 6) for 15 minutes. Endogenous peroxidase activity was blocked using 3% hydrogen peroxide in PBS for 15 min. Sections were blocked with normal goat serum for 30 min at room temperature, followed by incubation with primary antibodies at 4°C overnight. The following primary antibodies were used: anti-17βHSD2 (1:25, Santa Cruz), anti-Ki67 (1:400, Cell Signal Tech). After washing with PBS three times on the second day, corresponding secondary antibodies were applied, and samples were further incubated at room temperature for 30min. Slides were visualized with DAB staining according the manufacturer’s instructions (GK500710, Genetech). Subsequently, they were counterstained with hematoxylin (G1080, Solarbio) and mounted in dimethyl benzene.
The staining intensity was divided into four grades: no staining (0), weak (1), moderate (2) and strong (3). The final IHC scoring was performed from the multiplication between intensity and proportion scores of positive cells.
Chemical derivatization
Samples were derivatized by using Amplifex™ Keto Reagent (29; 30). Freeze-dried samples were reconstituted in 50 μL of Amplifex™ Keto working solution before diluted with 150 μL of 70% methanol. 200 μL of the final sample was injected onto the LC system.
Mass spectrometry
Samples were analyzed on a high-performance liquid chromatography station (Agilent, Germany) with a G4204A pumps, a G1367E auto-sampler, a G1316A column oven and a triple quadrupole 6490 (Agilent, Singapore) equipped with an ESI source operating in negative and positive ionization modes. The mobile phase for HPLC-MS/MS analyses was a mixture of water (A) and methanol (B) both containing 0.1% formic acid at 0.3 ml min−1 with a gradient elution: 0 min 45% B, 9 min 54% B, 9.51 min 90% B, 12.5 min 90% B, 12.51 min 45% B and kept at 45% B until the end of the run (15 min). Separation of drug metabolites was achieved using an Eclipse plus C18 RRHD analytical column 3.0 mm×50 mm, 1.8 μM (Agilent, USA) at 40°C. The injection volume was 10 μl, performed with auto-sampler. Androgen was ionized using electrospray ionization in positive ion mode (ESI). The temperature of the drying gas in the ionization source was 200 °C. The gas flow was 14l min−1, the nebulizer pressure was 20 psi and the capillary voltage was 3000 V (negative and positive). The analytes were quantified using multiple reaction monitoring (MRM) with the mass transitions and parameters for each compound as listed in supplementary table 2. Methanol and water were LC–MS grade and all were from Fisher Scientific.
Statistical analysis
All data were shown as mean ± standard deviation. Statistical analyses between groups were performed using Student’s t test and One-Way ANOVA. The correlation was determined by Pearson analysis. Survival was calculated by the Kaplan–Meier method and differences were analyzed by the log-rank test. p-value < 0.05 was considered statistically significant. All analyses were performed using Graphpad Prism 5.0 software.
Results
Expression of HSD17B2 declines with prostate cancer development.
To investigate its function in prostate cancer, we first detected the expression of HSD17B2 with patient specimens. 10 radical prostatectomy tissues from patients with localized prostate cancer were stained for 17βHSD2. 10 benign prostates from bladder cancer patients with radical cystectomy were used as benign control. Routine pathological examination methods such as hematoxylin and eosin (HE) staining were used to distinguish the tumor tissue. The immunohistochemistry (IHC) staining results indicated that prostate cancer had diminished expression of 17βHSD2 (Fig. 1 a and b, supplementary table 3). In the prostate cancer specimens, benign adjacent to tumor tissue had relatively higher expression of HSD17B2 compared with the related cancerous tissue (Fig. 1 c and d, supplementary table 3). Data mining with public data sets also indicated a decreasing level of HSD17B2 expression in prostate cancer compared with normal counterparts (Fig. 1 e & f)(31; 32). Consistently, HSD17B2 gene deletion was frequently found in both primary and metastatic prostate cancer, which may partially explain the low expression of HSD17B2 in prostate cancer (Fig. 1 g & h)(33; 34). Together all these data indicate the decreasing expression of HSD17B2 and its potential tumor suppressor role in prostate cancer.
Figure 1. HSD17B2 expression declines with prostate cancer progression.
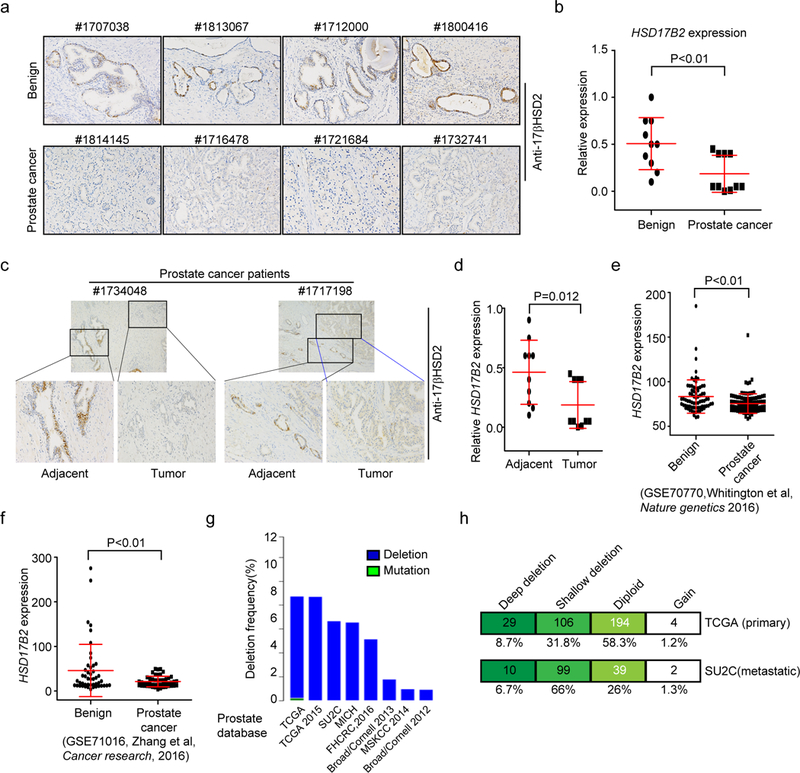
(a) IHC staining of 17βHSD2 with patient specimens. Specimens from radical cystectomy patients were selected as benign control and specimens from radical prostatectomy patients were taken as tumor tissue. (b) Relative expression level of HSD17B2 in benign and prostate cancer tissue. IHC scoring was performed from the multiplication between intensity and proportion scores of positive cells. P value was calculated with t-test. (c) and (d) HSD17B2 expression in benign adjacent to tumor and cancerous tissue. P value was calculated with paired t-test. (e) and (f) HSD17B2 expression in public data sets (GSE70770 and GSE71016). (g) Frequency of HSD17B2 gene deletion in different database. (h) HSD17B2 gene deletion in primary and metastatic prostate cancer. Deep deletion, 2 copies of HSD17B2 deleted; shallow deletion, 1 copy of HSD17B2 deleted.
17βHSD2 suppresses the conversion from testis and adrenal originated precursor to DHT.
We further detected HSD17B2 expression in different cancer cell lines. PC3 and MDA-Pca-2b had the highest mRNA expression, while LNCaP, LAPC4 and VCaP had limited HSD17B2 (Fig. 2a). Thus, in PC3 and MDA-Pca-2b cells T or DHT was rapidly converted to AD or 5α-dione respectively but not in LNCaP or LAPC4 cells (Fig. 2 b & c, Supplementary Fig. 1). Stable cell lines with doxycycline (dox)-inducible HSD17B2 expression were generated in LNCaP and VCaP with lentivirus. The expression of HSD17B2 in LNCaP or VCaP inactivated T or DHT robustly (Fig. 2 d & e). We suspected that HSD17B2 elimination, which frequently occurs with disease progression, would promote adrenal precursors such as AD to be converted to potent androgens such as DHT. Two different CRISPR constructs were used to knock out HSD17B2 in MDA-Pca-2b (Supplementary Fig.2). More DHT was generated in the HSD17B2 knockout cell line, indicating a potential mechanism of androgen accumulation in CRPC (Fig. 2f). Together, the data demonstrate that down regulation of HSD17B2 facilitates DHT production in prostate cancer cell lines.
Figure 2. 17βHSD2 suppresses the conversion from testis or adrenal androgens to DHT.
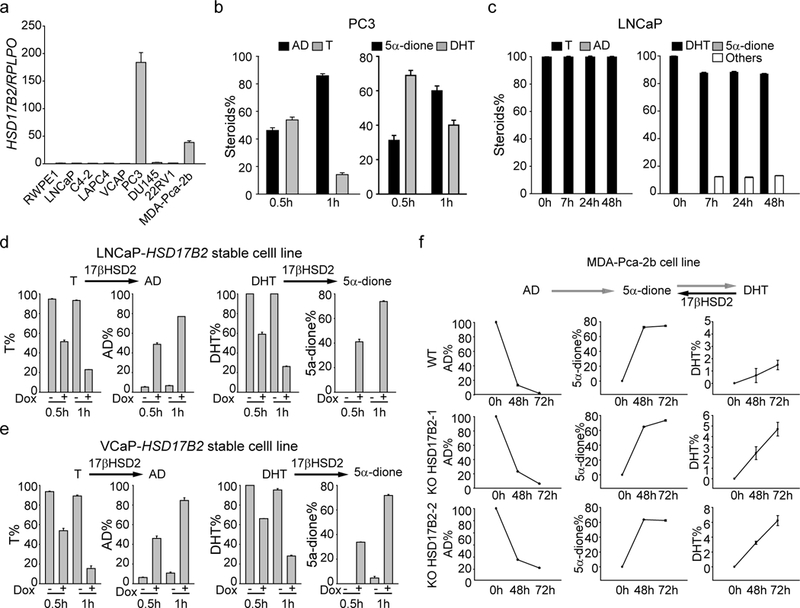
(a) Expression of HSD17B2 in different prostate cancer cell lines. (b) and (c) T and DHT metabolism in PC3 (high expression of HSD17B2) and LNCaP (low expression of HSD17B2). (d) and (e) Overexpression of HSD17B2 in LNCaP (d) or VCaP (e) led to inactivation of T and DHT. Doxycycline (Dox, 1 μg/ml) was used to induce the expression of HSD17B2 in the stable cell lines. (f) Knockout of HSD17B2 facilitated DHT accumulation in MDA-Pca-2b. Adrenal precursor AD was used to treat MDA-Pca-2b cell lines.
17βHSD2 inhibits AR signaling and tumor growth in vivo.
Due to its ability to inactivate androgens, the expression of HSD17B2 was likely to affect AR signaling and tumor growth. Induction of HSD17B2 expression in LNCaP and VCaP cells led to impaired T or DHT stimulated AR target gene expression (Fig.3 a & b). Its expression also suppressed testis or adrenal originated androgen induced cell proliferation (Fig. 3c). A dox-induced LNCaP-HSD17B2 stable cell line was used for xenograft experiments in intact NDG mice. When tumors reached ~100mm3, one group of mice was given dox plus sucrose in water and the control group had only sucrose without dox. The dox treated group had relative smaller tumors and took a longer time to reach ~500 mm3 (5 fold), indicating the tumor suppressor role of HSD17B2 in vivo (Fig. 3 d & e). Xenograft tumors were collected at the end of the mouse experiment. Significant higher mRNA and protein level of HSD17B2 were found in the dox treated group (Fig. 3 f & g). The IHC results also demonstrated that high level of 17βHSD2 correlated with low level of Ki-67, a cell proliferation marker, in the dox treated group (Fig. 3h and Supplementary Fig.3). Intratumoral T levels were also detected and less T was accumulated in the dox treated xenograft (Fig. 3i). Furthermore, fresh xenograft was treated with [3H] T and rapid conversion from T to AD was detected after dox treatment (Fig.3j). These data demonstrate the tumor suppressor role of HSD17B2 in prostate cancer in vivo.
Figure 3. 17βHSD2 inhibits tumor proliferation.
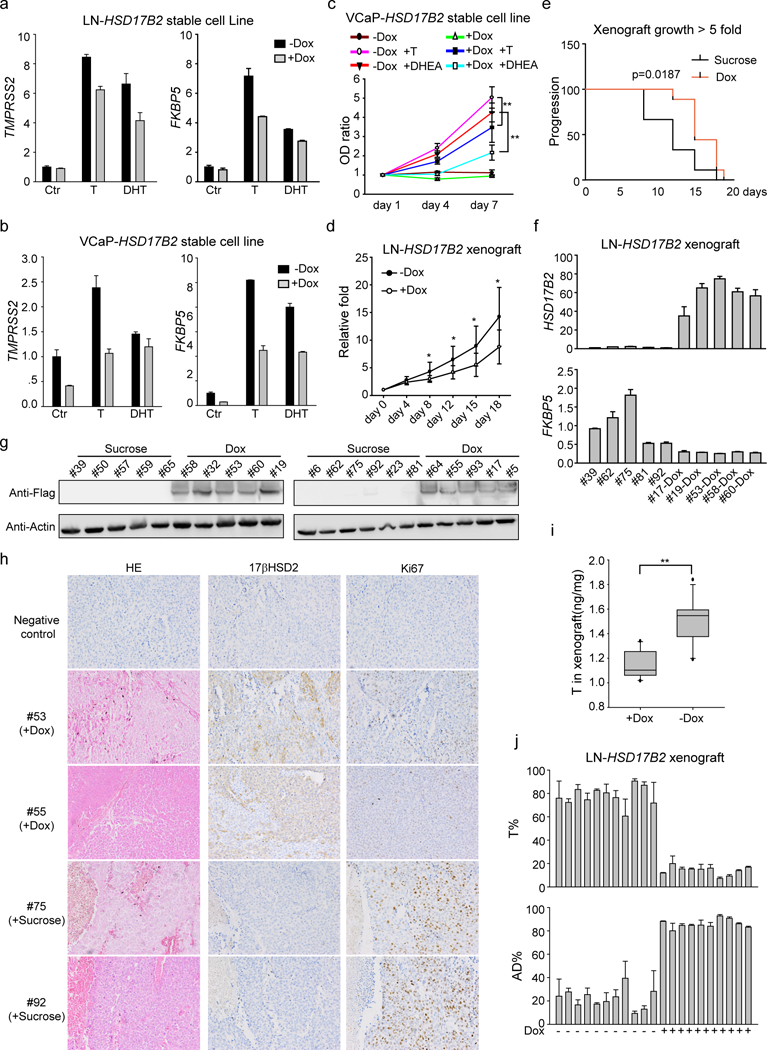
(a) and (b) Overexpression of HSD17B2 in LNCaP (a) and VCaP (b) inhibited androgen induced AR target genes expression. (c) 17βHSD2 inhibited T and DHEA induced cell proliferation. Dox (1 μg/ml) was used to induce HSD17B2 expression in VCaP. **, p<0.01. (d) and (e) HSD17B2 inhibited xenograft growth. Dox (2 mg/ml) was used to induce HSD17B2 expression. Growth curve (d) and Kaplan-Meier survival analysis (e, time for tumors reaching 5-fold size) were used to show the results of tumor growth. *, p <0.05. (f) and (g) HSD17B2 expression in xenograft. Both mRNA (f) and protein (g) level were detected in sucrose or dox treated xenograft. Fresh xenograft tumors were collected for immediate mRNA or protein detection. (h) Immunohistochemistry staining of xenograft. Ki67 was used as a proliferation marker. (i) Concentration of T in xenograft. **, p<0.01. (j) T metabolism in xenograft. Fresh xenograft was collected and treated with dox (xenografts from dox-treated group) or not (xenografts from sucrose-treated group). [3H]-T was used to treat xenograft tissues for metabolism assay.
Multiple mechanisms of HSD17B2 regulation in prostate cancer.
We further investigated the regulation mechanisms of HSD17B2 in prostate cancer. Gene deletion is one mechanism of silencing HSD17B2 expression (Fig. 1 g & h). We also examined the DNA methylation status of the HSD17B2 promoter. Limited CpG islands are located across the HSD17B2 promoter and two of them are in the binding site of SP1, a transcription factor facilitating HSD17B2 transcription (Fig. 4a)(35). It has been reported that SP1 has a higher affinity to the unmethylated DNA fragment, thus DNA methylation would diminish its binding to HSD17B2 promoter (36). The pyrosequencing results indicated that PC3 had the least DNA methylation in SP1 binding site, which correlated with its highest HSD17B2 expression (Fig. 4b). When treated with 5-azacytidine, an inhibitor of DNA methyltransferase, HSD17B2 expression in DU145 increased significantly but not in PC3, confirming that DNA methylation blocks HSD17B2 transcription in DU145 (Fig. 4c). To evaluate the effect of androgens on 17βHSD2, we treated PC3 with different androgens and found T, as the substrate of 17βHSD2, decreased but not increased its abundance (Fig. 4d). While AD, the product of 17βHSD2, increased its protein level slightly (Fig. 4d). The effect of DHEA was elusive and varied between different experiments, possibly due to its metabolism to downstream androgens. Also the effect of androgen stimulation was limited at the protein level only but not mRNA level (Supplementary Fig.4). In summary, DNA methylation and androgen stimulation would affect HSD17B2 expression in prostate cancer.
Figure 4. Multiple regulation mechanisms of HSD17B2 functional silencing.
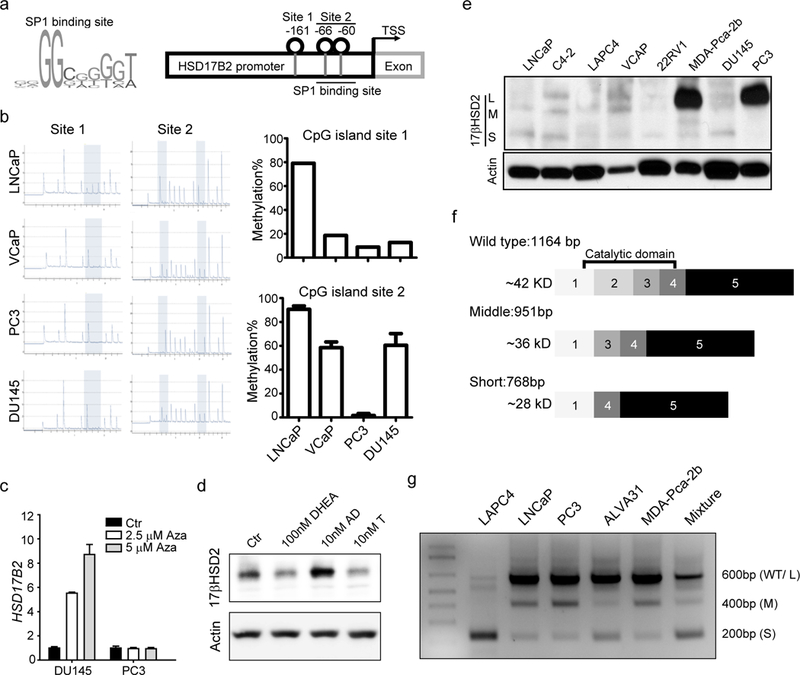
(a) Schema of SP1 binding site and HSD17B2 promoter. (b) DNA methylation of HSD17B2 promoter in prostate cancer cell lines. (c) HSD17B2 expression after 5-azacytidine treatment. HSD17B2 expression was normalized to RPLPO. Basal expression of HSD17B2 in each cell lines was taken as 1. (d) 17βHSD2 protein abundance after androgen stimulation. PC3 cells were treated with indicated androgens for 8h before collection for western blot. (e) Endogenous 17βHSD2 expression in different cell lines. (f) Schema of different HSD17B2 isoforms. (g) Amplicon of different isoforms in prostate cancer cell lines. Primers located in HSD17B2 exon 1 and exon 4 were used for PCR.
When we detected 17βHSD2 protein by immunoblotting, extra bands in C4–2 and other cell lines were found, indicating a potential regulatory mechanism related with alternative splicing (Fig.4e). We used a mixed cDNA library from various prostate cancer cell lines to clone HSD17B2. Three different isoforms were identified in prostate cancer cells. Compared with the wild type (L isoform), the middle-length (M) isoform of HSD17B2 was lack of exon 2 (213 base pair) and the short-length (S) isoform was lack of exon 2 and 3 (186 base pair) (Fig. 4f). The missing of exons did not interrupt the protein translation. A primer pair located in exon 1 and 4 respectively was used to confirm the existence of these isoforms in different prostate cancer cell lines. Three different PCR products confirmed the existence of the isoforms, consistent with the cloning constructs and immunoblotting results (Fig. 4g).
We further investigated the function of the new isoforms. Overexpression of the M or S isoforms showed no effect on T metabolism, unlike the wild type one (Fig. 5a). It was possibly because loss of exon 2 or 3 abolished the catalytic domain. It has been reported that 17βHSD2 forms a dimer to execute its function, so the interaction between different isoforms was examined (37). The results indicated that every isoform could interact with itself or the other two (Fig. 5b). Besides, the M or S isoforms were not as stable as the wild type one. When treated with cycloheximide (CHX), an inhibitor of protein biosynthesis, the protein level of M and S isoform, especially S isoform, disappeared quickly (Fig. 5c). The protease inhibitor, MG132 facilitated M and S isoform accumulation but not the wild type (Fig. 5d). Thus we hypothesized that M and S isoform could promote wild type 17βHSD2 degradation by binding L isoform as a heterodimer. When co-expressed with M or S isoform in 293T cell lines, the L isoform had a shorter retention time (Fig. 5e). A dox induced M or S expression stable cell line was generated in PC3. The induction of M or S isoform expression would decrease endogenous wild type 17βHSD2 protein level (Fig. 5f). Furthermore, the induction of M or S isoform expression would rescue the suppression of AR signaling caused by wild type 17βHSD2 overexpression in LNCaP (Fig. 5g). The splicing factors participated into the generation of these isoforms were investigated. By overexpressing different splicing factors in PC3, M and S isoforms could only be detected when cells were transfected with SRSF1 or SRSF5 (Fig. 5h). Also higher expression of SRSF1 or SRSF5 correlated with low expression of HSD17B2 from different public data sets (Fig. 5 i & j)(38; 39). Taken together, these data demonstrate that mRNA alternative splicing produces two new catalytic-deficient isoforms degrades wild type 17βHSD2, resulting in functional silencing of HSD17B2 in both mRNA level and protein level.
Figure 5. Function of new isoforms of 17βHSD2.
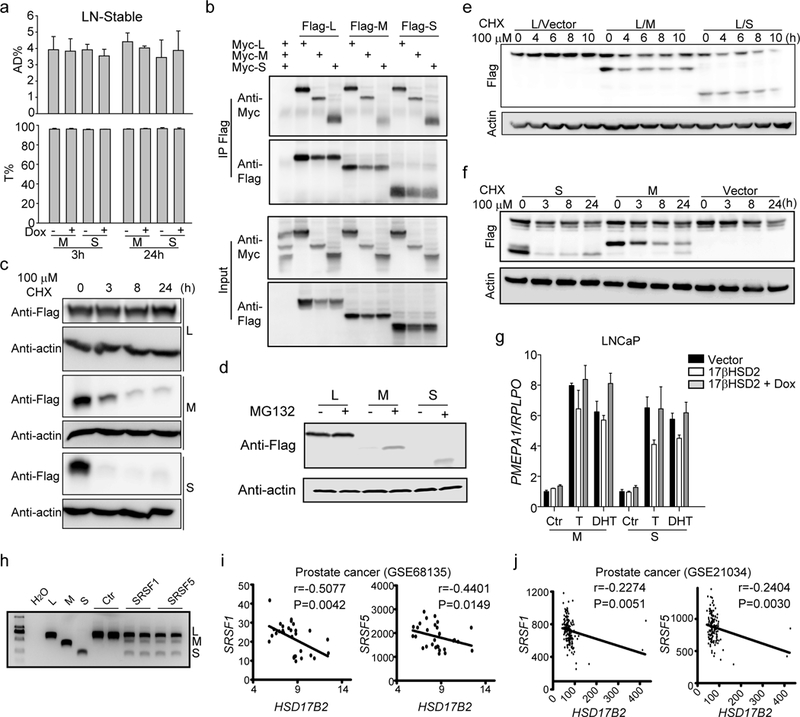
(a) M and S isoforms do not inactivate T. Dox was used to induce M or S expression in LNCaP stable cell line. (b) Isoforms of 17βHSD2 interact with one another. (c) Half-life of different 17βHSD2 isoforms. Plasmids were transfected into 293T cells and cycloheximide (CHX) was added 18h after transfection. (d) MG132 increased M and S isoform stability. Plasmids were transfected into the 293T cell line and MG132 was added 24h after transfection. (e) and (f) M and S isoforms promoted wild type 17βHSD2 degradation. Transient overexpression of M or S in 293T (e) or stable-expression in PC3 (f) promoted wild type 17βHSD2 degradation. (g) M or S isoforms suppressed wild type 17βHSD2 function in target gene regulation. LNCaP stable cell lines expressing dox-induced M or S isoform were transfected with wild type HSD17B2 and treated with or without dox as indicated. T, 10 nM; DHT, 1 nM. (h) SRSF1 and SRSF5 overexpression produced more M and S mRNA. Primers located in HSD17B2 exon 1 and exon 4 were used for PCR. (i) and (j) Correlation between HSD17B2 and SRSF1/5 in different public data sets (GSE 21034, GSE 68135).
Discussion
Steroidogenesis plays a critical role in the development and progression of prostate cancer (40; 41). In benign prostate tissue and throughout various stages of prostate cancer, steroidogenic enzymes convert T of gonadal origin or androgens of adrenal origin to DHT, which potently activates AR signaling to sustain tissue development or cancer progression (3). Steroidogenic enzymes which promote DHT synthesis, such as CYP17A, 3βHSD1 and AKR1C3, are involved in therapy resistance. However enzymes which inactivate DHT and T have not received much attention.
17βHSD2 catalyzes the conversion from T or DHT to AD or 5a-dione, respectively by modifying the 17β-OH moiety to 17-keto (24). Its expression is gradually reduced as disease progresses, indicating its tumor suppressor function. Our data demonstrate that 17βHSD2 inhibits gonadal and adrenal androgen conversion to DHT, indicating its important role in both localized prostate cancer and metastatic CRPC. Overexpression of HSD17B2 in prostate cancer cell lines diminishes AR signaling and suppresses androgen-induced cell proliferation and xenograft growth. Thus, it might also serve as a prostate cancer prognostic biomarker.
Investigation of the regulatory mechanisms of HSD17B2 in prostate cancer will shed light on novel strategies for prostate cancer treatment. DNA methylation in the HSD17B2 promoter blocks the binding of SP1 and restrains HSD17B2 expression, showing the importance of epigenetic regulation in prostate cancer. Surprisingly, T as the substrate of 17βHSD2 reduces its abundance, indicating a positive-feedback loop to stimulate prostate cancer development. As more potent androgen accumulated in prostate cancer, it will not only sustain the tumor proliferation but also suppress the androgen inactivation machinery by reducing 17βHSD2 protein level.
Another regulation mechanism is mRNA alternative splicing. The new isoforms generated by mRNA alternative splicing have no enzymatic activity due to the destruction of the catalytic domain. The new isoforms also bind to wild type 17βHSD2 and promote its degradation, thus enhancing androgen-induced AR signaling in prostate cancer. The significance of the alternative splicing is not only cutting down the mRNA level of the wild type HSD17B2 but only decreasing its protein abundance. SRSF1 and SRSF5 are involved in HSD17B2 mRNA alternative splicing and might serve as novel therapeutic targets or biomarkers.
In summary, we have found HSD17B2 expression decreases with prostate cancer progression. 17βHSD2 inactivates the potent androgens, T and DHT, to restrain tumor growth. Functional silencing of HSD17B2 in prostate cancer is achieved through multiple mechanisms including gene deletion, DNA methylation, circulating androgen stimulation and mRNA alternative splicing (Fig. 6). Two new isoforms generated by splicing factors SRSF1 and SRSF5 bind to wild type 17βHSD2 to promote its degradation. Our work unveils the essential role of HSD17B2 in prostate cancer progression and the novel mechanisms of its regulation, which might provide new strategies for clinical management.
Figure 6. Schema of HSD17B2 function and regulation.
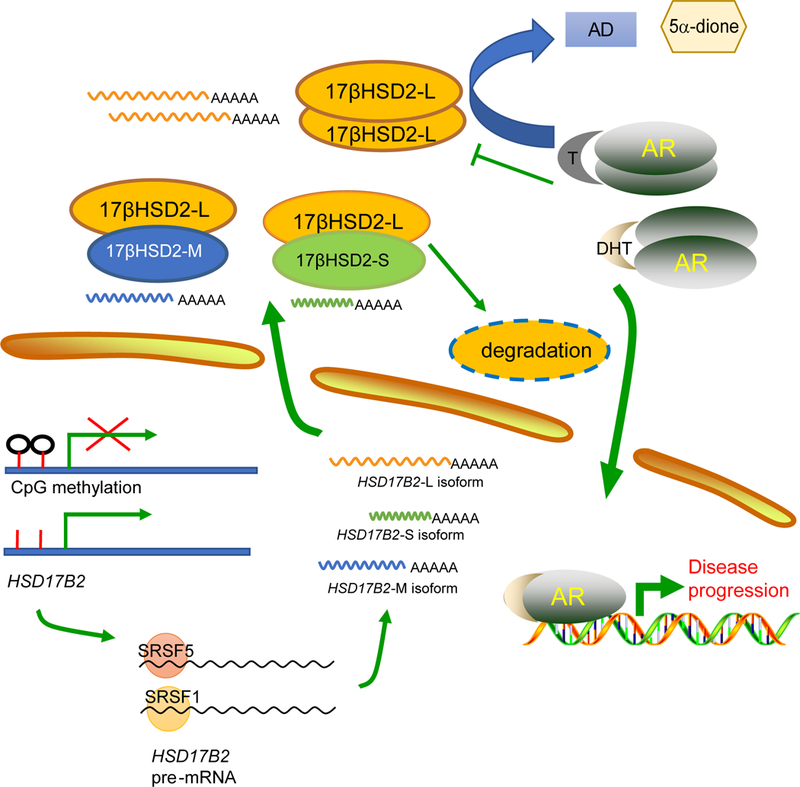
17βHSD2 inactivates T and DHT to prevent disease progression. DNA methylation, androgen stimulation and alternative splicing were involved in HSD17B2 functional silencing.
Supplementary Material
Translational Relevance.
Androgens sustain prostate cancer development. Steroidogenic enzymes promoting androgen synthesis are well established therapeutic targets and predictive biomarkers, while enzymes inactivating androgens have not been investigated thoroughly. Here we show the clinical relevance and significance of HSD17B2 in prostate cancer, providing potential biomarker for disease diagnosis. Mechanisms of HSD17B2 functional silencing were further investigated to provide potential novel strategies for disease intervention.
Acknowledgements
We thank the staff members of Mass Spectrometry at National Facility for Protein in Shanghai (NFPS), Zhangjiang Lab, China for providing technical support and assistance in data collection and analysis. This work has been supported in part by funding from National Key R&D program of China (2018YFA0508200 to Z.L.), Strategic Priority Research Program of Chinese Academy of Sciences, Grant No. XDB19000000 (to Z.L.), the National Natural Science Foundation of China (81722033 and 31771575 to Z.L., 81672526 to D.W., 81872075 to J.T. ), Prostate Cancer Foundation Young Investigator Award (#15YOUN11 to Z.L.), Youth Innovation Promotion Association (Chinese Academy of Sciences, to J.T.), a Prostate Cancer Foundation Challenge Award (to N.S.), and, grants from the National Cancer Institute (R01CA168899, R01CA172382, and R01CA190289; to N.S.). The authors claim no conflict of interest.
Abbreviations list:
- SNP
single nucleotide polymorphism
- T
testosterone
- DHT
dihydrotestosterone
- AR
androgen receptor
- 5α
dione: androstenedione
- DHEA
dehydroepiandrosterone
- AD
androstenedione
- HE
hematoxylin and eosin
- IHC
immunohistochemistry
- Dox
doxycycline
Footnotes
Conflict of interest:
The authors declare no potential conflict of interest.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians 66:115–32 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. 2018. Cancer statistics, 2018. CA: a cancer journal for clinicians 68:7–30 [DOI] [PubMed] [Google Scholar]
- 3.Sharifi N, Auchus RJ. 2012. Steroid biosynthesis and prostate cancer. Steroids 77:719–26 [DOI] [PubMed] [Google Scholar]
- 4.Armstrong CM, Gao AC. 2015. Drug resistance in castration resistant prostate cancer: resistance mechanisms and emerging treatment strategies. American journal of clinical and experimental urology 3:64–76 [PMC free article] [PubMed] [Google Scholar]
- 5.Penning TM, Byrns MC. 2009. Steroid hormone transforming aldo-keto reductases and cancer. Ann N Y Acad Sci 1155:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huggins C, Hodges CV. 1972. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA: a cancer journal for clinicians 22:232–40 [DOI] [PubMed] [Google Scholar]
- 7.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, et al. 2008. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer research 68:4447–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, et al. 2007. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer research 67:5033–41 [DOI] [PubMed] [Google Scholar]
- 9.Penning TM. 2015. Mechanisms of drug resistance that target the androgen axis in castration resistant prostate cancer (CRPC). The Journal of steroid biochemistry and molecular biology 153:105–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, et al. 2011. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer research 71:6503–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, et al. 2011. Abiraterone and increased survival in metastatic prostate cancer. The New England journal of medicine 364:1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. 2013. Abiraterone in metastatic prostate cancer without previous chemotherapy. The New England journal of medicine 368:138–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, et al. 2017. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. The New England journal of medicine 377:338–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, et al. 2017. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. The New England journal of medicine 377:352–60 [DOI] [PubMed] [Google Scholar]
- 15.Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, et al. 2013. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 154:1074–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hearn JW, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, et al. 2016. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. The Lancet. Oncology 17:1435–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hettel D, Sharifi N. 2018. HSD3B1 status as a biomarker of androgen deprivation resistance and implications for prostate cancer. Nature reviews. Urology 15:191–6 [DOI] [PubMed] [Google Scholar]
- 18.Hearn JWD, Xie W, Nakabayashi M, Almassi N, Reichard CA, et al. 2018. Association of HSD3B1 Genotype With Response to Androgen-Deprivation Therapy for Biochemical Recurrence After Radiotherapy for Localized Prostate Cancer. JAMA oncology 4:558–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almassi N, Reichard C, Li J, Russell C, Perry J, et al. 2018. HSD3B1 and Response to a Nonsteroidal CYP17A1 Inhibitor in Castration-Resistant Prostate Cancer. JAMA oncology 4:554–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adeniji AO, Chen M, Penning TM. 2013. AKR1C3 as a target in castrate resistant prostate cancer. The Journal of steroid biochemistry and molecular biology 137:136–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchais-Oberwinkler S, Henn C, Moller G, Klein T, Negri M, et al. 2011. 17beta-Hydroxysteroid dehydrogenases (17beta-HSDs) as therapeutic targets: protein structures, functions, and recent progress in inhibitor development. The Journal of steroid biochemistry and molecular biology 125:66–82 [DOI] [PubMed] [Google Scholar]
- 22.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. 2017. Inhibition of AKR1C3 Activation Overcomes Resistance to Abiraterone in Advanced Prostate Cancer. Molecular cancer therapeutics 16:35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, et al. 2015. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer research 75:1413–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu ML, Huang YW, Lin SX. 2002. Purification, reconstitution, and steady-state kinetics of the trans-membrane 17 beta-hydroxysteroid dehydrogenase 2. The Journal of biological chemistry 277:22123–30 [DOI] [PubMed] [Google Scholar]
- 25.Wu L, Einstein M, Geissler WM, Chan HK, Elliston KO, Andersson S. 1993. Expression cloning and characterization of human 17 beta-hydroxysteroid dehydrogenase type 2, a microsomal enzyme possessing 20 alpha-hydroxysteroid dehydrogenase activity. The Journal of biological chemistry 268:12964–9 [PubMed] [Google Scholar]
- 26.Casey ML, MacDonald PC, Andersson S. 1994. 17 beta-Hydroxysteroid dehydrogenase type 2: chromosomal assignment and progestin regulation of gene expression in human endometrium. The Journal of clinical investigation 94:2135–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castagnetta LA, Carruba G, Traina A, Granata OM, Markus M, et al. 1997. Expression of different 17beta-hydroxysteroid dehydrogenase types and their activities in human prostate cancer cells. Endocrinology 138:4876–82 [DOI] [PubMed] [Google Scholar]
- 28.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin W, Jarvis M, Star-Weinstock M, Altemus M. 2013. A sensitive and selective LC-differential mobility-mass spectrometric analysis of allopregnanolone and pregnanolone in human plasma. Analytical and bioanalytical chemistry 405:9497–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bussy U, Huertas M, Chung-Davidson YW, Li K, Li W. 2016. Chemical derivatization of neurosteroids for their trace determination in sea lamprey by UPLC-MS/MS. Talanta 149:326–34 [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, Wang J, Wang Y, Zhang Y, Castro P, et al. 2016. MNX1 Is Oncogenically Upregulated in African-American Prostate Cancer. Cancer research 76:6290–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitington T, Gao P, Song W, Ross-Adams H, Lamb AD, et al. 2016. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nature genetics 48:387–97 [DOI] [PubMed] [Google Scholar]
- 33.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng YH, Imir A, Suzuki T, Fenkci V, Yilmaz B, et al. 2006. SP1 and SP3 mediate progesterone-dependent induction of the 17beta hydroxysteroid dehydrogenase type 2 gene in human endometrium. Biology of reproduction 75:605–14 [DOI] [PubMed] [Google Scholar]
- 36.Zhu WG, Srinivasan K, Dai Z, Duan W, Druhan LJ, et al. 2003. Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Molecular and cellular biology 23:4056–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin SX, Yang F, Jin JZ, Breton R, Zhu DW, et al. 1992. Subunit identity of the dimeric 17 beta-hydroxysteroid dehydrogenase from human placenta. The Journal of biological chemistry 267:16182–7 [PubMed] [Google Scholar]
- 38.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, et al. 2010. Integrative genomic profiling of human prostate cancer. Cancer cell 18:11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prueitt RL, Wallace TA, Glynn SA, Yi M, Tang W, et al. 2016. An Immune-Inflammation Gene Expression Signature in Prostate Tumors of Smokers. Cancer research 76:1055–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mostaghel EA, Montgomery B, Nelson PS. 2009. Castration-resistant prostate cancer: targeting androgen metabolic pathways in recurrent disease. Urologic oncology 27:251–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai C, Heemers H., Sharifi N. 2017. Androgen signaling in prostate cancer. Cold Spring Harb Perspect Med [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.