

Abstract
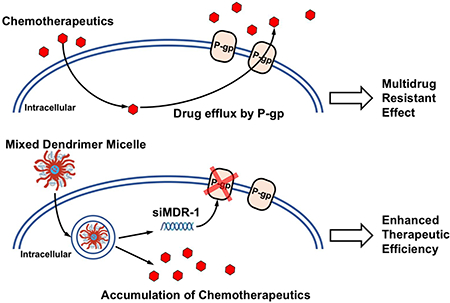
Multidrug resistance (MDR) significantly decreases the therapeutic efficiency of anti-cancer drugs. Its reversal could serve as a potential method to restore the chemotherapeutic efficiency. Downregulation of MDR-related proteins with a small interfering RNA (siRNA) is a promising way to reverse the MDR effect. Additionally, delivery of small molecule therapeutics simultaneously with siRNA can enhance the efficiency of chemotherapy by dual action in MDR cell lines. Here, we conjugated the dendrimer, generation 4 polyamidoamine (G4 PAMAM), with a polyethylene glycol (PEG)-phospholipid copolymer. The amphiphilic conjugates obtained spontaneously self-assembled into a micellar nanopreparation, which can be co-loaded with siRNA onto PAMAM moieties and sparingly water-soluble chemotherapeutics into the lipid hydrophobic core. This system was co-loaded with doxorubicin (DOX) and therapeutic siRNA (siMDR-1) and tested for cytotoxicity against MDR cancer cells: human ovarian carcinoma (A2780 ADR) and breast cancer (MCF7 ADR). The combination nanopreparation effectively downregulated P-gp in MDR cancer cells and reversed the resistance towards DOX.
Keywords: Multidrug resistance, polyamidoamine (PAMAM), mixed dendrimer micelles (MDM), combination delivery, micelle, siRNA delivery, chemotherapy
Graphical Abstract
1. Introduction:
Cancer has remained one of the leading causes of death for many decades. Although various chemotherapy options have been developed, many factors influence their therapeutic efficiency. In many cases, a chemotherapy showed efficacy in a patient but lost its efficacy on the same patient after several treatments because of the development of resistance to chemotherapy by the tumor [1]. Chemotherapy resistance hampers and limits the application of chemotherapy [2] due to increased drug efflux [3, 4], enhanced detoxification mechanisms [5, 6], malfunction in DNA repair systems [7–9] and inhibition of apoptotic pathways [10–12]. Effective reversal of resistance generated by the resistance-associated proteins in multidrug resistant (MDR) tumors is promising, but is still one of the most important unsolved problems in cancer treatment.
Clinically, reversing multidrug resistance by targeting P-gp has been extensively studied since P-gp is frequently found in solid tumors, hematologic cancers and cancer stem cells [13–15]. P-gp is a 170 kDa protein encoded by the MDR-1 (ABCB1) gene. Its physiological function in healthy tissues is to efflux large numbers of structurally unrelated hydrophobic compounds so as to protect the body from environmental toxins and xenobiotics. Clinical evidence has shown that overexpression of P-gp correlates with resistance towards chemotherapeutics including cisplatin, doxorubicin, methotrexate and topotecan [16–19], as well as a poor prognosis in patients with ovarian and breast carcinomas [20–23]. An association between MDR1/P-gp overexpression and resistance in lung cancer cell lines to several agents such as paclitaxel [24] and cisplatin [25] has been reported. Thus, knocking down P-gp expression would be expected to prevent or slow the efflux of drugs and help to restore intracellular drug levels required to induce apoptosis and cytotoxicity.
The silencing of MDR genes with small interfering RNA (siRNA) has been shown to be a powerful tool for restoring sensitivity to chemotherapy in MDR cancer cells [26–28]. Silencing the MDR-1/P-gp gene by siRNA can improve the effectiveness of anticancer drugs on MDR tumors [29–31]. Cationic G4 PAMAM dendrimers have been explored as non-viral delivery vectors for different oligonucleotides including siRNAs over the last two decades [32–36]. The dendriplexes formed between positively charged PAMAM and negatively charged plasmid DNA interacted with cells more efficiently [37]. Also, the G4 PAMAM dendrimer is expected to maintain the stability of siRNA, facilitate the cellular interaction, and assist in the endosomal escape of siRNA due to the “proton sponge effect” [38].
A nanoparticle drug delivery system that simultaneously delivers a chemotherapeutic drug and siRNA to the tumor has emerged as a promising treatment strategy for cancer [39–44]. In the current study, a unique co-delivery system based on mixed micelles composed of a lipid modified dendrimer copolymer and PEG5k-DOPE was used for co-delivery of siRNA and drugs to tumors via the enhanced permeability and retention (EPR) effect. This amphiphilic compound, G4-PAMAM-PEG2k-DOPE (Figure 1), forms micellar structures (or mixed micelles with PEG5k-DOPE) spontaneously. The cationic PAMAM structure plays the role of complexing siRNA, while the poorly soluble anticancer drugs are encapsulated in the hydrophobic cores of micelles. The positive charge of PAMAM promotes siRNA complexation as well as a cellular association and uptake by tumor cells. DOPE residues, together with tertiary amines among the PAMAM, facilitate endosomal escape and cytoplasmic delivery of active components [38]. In a previous study, such a system successfully delivered siRNA and DOX into the A549 cell line and knocked down green fluorescent protein (GFP) expression in a C166-GFP cell line [45]. Thus, downregulation of MDR transporters with siRNA-loaded nanocarriers using PAMAM-PEG2k-DOPE copolymer, along with co-administration of a traditional chemotherapeutic agent, represented a promising approach for inhibition of MDR tumors via a synergistic effect. Our goal was to examine the effect of a novel and versatile dendrimer-based nano-preparation combining siRNA and drugs active against MDR cancers.
Figure 1.

(Colored) Schematic diagram of MDM structure and PAMAM-PEG2k-DOPE.
2. Materials and Methods:
2.1. Materials
A PAMAM Dendrimer with an ethylenediamine core, generation 4, 10 % w/w solution in methanol (G(4)-D) was purchased from Sigma-Aldrich. 1,2-dioleoyl-sn-glycero-3- phosphoethanolamine-N-(methoxy(polyethylene glycol)-5000) (ammonium salt) (PEG5k-DOPE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and rhodamine-DOPE (Rh-PE) were purchased from Avanti Polar Lipids (AL, USA). NPC-PEG-2K-NPC (pNP-PEG2k-pNP) was purchased from Laysan Bio (AL, USA). Bovine serum albumin (BSA), methanol and dimethylformamide (DMF) were purchased from ThermoFisher Scientific (MA, USA). Lipofectamine RNAiMAX, FAM-labeled negative siRNA, Ambion™ siRNA targeting MDR-1 (siMDR-1): 5’-GGAAAAGAAACCAACUGUCdTdT-3’ (sense) [46], Ambion™ RNase III (1 U/μl) and E-Gel™ General Purpose Agarose Gels, 2% were also purchased from ThermoFisher Scientific (MA, USA). Nuclease-free water was purchased from Qiagen (MD, USA). Doxorubicin HCl was purchased from LC Laboratories (MA, USA). The CellTiter-Blue® Cell Viability Assay was purchased from Promega (WI, USA). The Adriamycin resistant human Caucasian ovarian carcinoma cell line (A2780 ADR) was purchased from Sigma-Aldrich. A2780 sensitive, MCF7 sensitive and MCF7 ADR (re-designated as NCI/ADR-RES Adriamycin resistant human ovarian carcinoma cell line) were purchased from the American Type Culture Collection (ATCC). Dulbecco’s modified Eagle’s media (DMEM), Roswell Park Memorial Institute medium (RPMI), fetal bovine serum (FBS) and Penicillin-streptomycin solution were obtained from CellGro (VA, USA). Hoechst 33342 was purchased from Molecular Probes Inc. (Eugene, OR). Para-formaldehyde was from Electron Microscopy Sciences (Hatfield, PA). Fluoromount-G was from Southern Biotech (Birmingham, AL). The Trypan blue solution was obtained from Hyclone (Logan, UT). Triethylamine (TEA), ethylenediaminetetraacetic acid (EDTA), Sodium Azide (NaN3), Sepharose® CL-4B, anhydrous chloroform (CHCl3), Molybdenum Blue spary reagent, Dragendorff reagent and all constitutions in Phosphate Buffered Saline (PBS) were purchased from Sigma-Aldrich (MO, USA). HEPES was purchased from MP Biomedicals (OH, USA) and D-(+)-Glucose, Anhydrous was purchased from Alfa Aesar (MA, USA). Heparin (1U/ml) was purchased from American Pharmaceutical Partners (IL, USA). Phycoerythrin labeled P-gp monoclonal antibody (UIC2 clone) was purchased from Abcam (MA, USA).
2.2. Conjugation of PAMAM-PEG2k-DOPE
G4 PAMAM-PEG2k-DOPE was prepared by conjugation between G4 PAMAM and p-nitrophenol (pNP)-PEG2k-DOPE according to a previously published method [45]. Briefly, to prepare pNP-PEG2k-DOPE, 500 mg (250 μmole) of pNP-PEG2k-pNP (MW 2000 Da) was dissolved in 6 ml of anhydrous CHCl3. Into the pNP-PEG2k-pNP solution, 38.7 mg (50 μmole) of DOPE in CHCl3 was added dropwise. This was followed by adding 23 μl of TEA, pNP-PEG2k-pNP and DOPE. The mixture was stirred overnight at room temperature. The reaction mixture was then evaporated to remove CHCl3 and freeze dried (Freezone 4.5, Labconco, MO, USA). Dry crude was dissolved with 2 ml of hydrochloric acid (0.1mM) and purified on a column packed with Sepharose® CL-4B using hydrochloric acid (0.1mM) as the mobile phase. All fractions were spotted on thin layer chromatography (TLC) and stained with Molybdenum Blue and Dragendorff reagents. The fractions stained with both stains were collected and lyophilized in a freeze dryer. Purified product was dissolved in anhydrous CHCl3 at 10 mg/ml and stored at −20°C for the next steps.
To conjugate PAMAM-PEG2k-DOPE (or D-PEG2k-DOPE), 0.48 ml of CHCl3 containing 4.8 mg (1.75 μmole) of pNP-PEG2k-DOPE was dried under nitrogen and reconstituted in 1 ml DMF. Methanol in G4 PAMAM (24.9 mg, 1.75 μmole) was also evaporated and re-dissolved in 2 ml DMF. The pNP-PEG2k-DOPE was added dropwise to 2 ml of G4 PAMAM in DMF, followed by 16 μl of TEA. The mixture was reacted at room temperature overnight. On the following day, DMF was removed under vacuum by rotary evaporation. The remaining product was freeze dried and hydrated with deionized water (diH2O). The reaction mixture was dialyzed against diH2O in a 100 kDa MWCO regenerated cellulose dialysis membrane (Spectrum Lab, CA, USA) for 48 hours. After dialysis, the product was lyophilized and dissolved in methanol at 10 mg/ml for future use.
2.3. Preparation of DOX free base loaded mixed dendrimer micelles
Doxorubicin free base (DOX) was prepared by reacting Doxorubicin HCl and a 10-fold mole excess of TEA in methanol for 48 hours. After DOX precipitated, methanol was removed by nitrogen air stream and DOX was reconstituted in chloroform. Mixed dendrimer micelles (MDM) were prepared by a thin-film hydration method. PAMAM-PEG2k-DOPE and PEG5k-DOPE were mixed at mole ratios of PAMAM-PEG2k-DOPE: PEG5k-DOPE=1:1, 1:10 and 1:20 (MDM 1:1, MDM 1:10, MDM 1:20, respectively) and made into a thin-film using nitrogen stream in a glass tube. After freeze-dried for 12 hours, the film was hydrated with RNase-free buffered HEPES with 5% w/v glucose (BHG) to form MDM at a micelle material concentration of 34 μM. To prepare DOX free base loaded mixed dendrimer micelles (MDM DOX), a mixture of PAMAM-PEG2k-DOPE, PEG5k-DOPE and DOX free base was made into a thin film with nitrogen evaporation in a glass tube and freeze-dried for 12 hours, followed by hydration with RNase-free BHG at a micelle material concentration of 34 μM. Non-encapsulated DOX free base was removed by filtering through a 0.22 μm sterile PES-membrane filter (ThermoFisher Scientific, MA, USA). Concentration of DOX loaded inside of the MDM DOX was quantified by spectrophotometer (UV mini 1240, Shimadzu, Kyoto, Japan) at 480 nm.
2.4. Particle size and zeta potential
The hydrodynamic diameter and zeta potential of MDMs were measured by a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). The size, PdI and zeta potential values were recorded at a 90-degree scattering angle in triplicate for each sample. All measurements were performed in BHG at a total micelle material concentration of 2 mg/ml.
2.5. Preparation of dendriplexes and complexation efficiency
Dendriplexes were prepared by mixing MDM formulations with siRNA in BHG at equal volumes and incubated at room temperature (RT) for 30 mins for full complexation.
To evaluate complexation efficiency, MDM 1:20 was prepared in BHG buffer and incubated with 750 ng of siRNA at various ratios between the number of cationic charges on PAMAM and number of anionic charges on siRNA (N/P ratios) to form dendriplexes. After 30 min incubation at room temperature, the dendriplexes were analyzed by gel electrophoresis using an E-Gel™ General Purpose Agarose Gels, 2% containing Ethidium Bromide according to the manufacture’s protocol (Invitrogen, MA, US). Free siRNA was used as control. Twenty microliter samples were loaded into wells and the gel was electroporated at 60 mV for 30 mins. siRNA bands were visualized under UV light.
2.6. RNase stability
RNase III (0.25 U/μl) was used to degrade any free siRNA. EDTA was used to terminate the activity of RNase. Heparin (1 U/μl) was used to dissociate siRNA from cationic nanoparticles. In our study, formulations were first complexed with siRNA at N/P ratios of 1 and 5, and incubated at room temperature for 30 mins. Then, the complex was mixed with RNase III and incubated at 37°C for 30 mins. After that, the heparin and EDTA mixture was incubated with the complex. After 30 mins incubation, 20 μl of the final reaction product was loaded into an E-Gel™ General Purpose Agarose Gels, 2% for electrophoresis (60 mV, 30 mins). Bands of siRNA were visualized under UV light.
2.7. Cell culture
A2780 ADR and A2780 sensitive cell lines were cultured in RPMI 1g/L glucose. MCF7 ADR was cultured in DMEM 1 g/L glucose and MCF7 sensitive was cultured in DMEM 4.5 g/L glucose. All media was supplemented with 10% FBS and antibiotics (100 IU/mL streptomycin). Cells were cultured at 37°C with 5% CO2. To maintain the MDR effect in A2780 ADR and MCF7 ADR, the cells were resuspended in media containing 100 nM doxorubicin HCl after each passage.
2.8. Cellular association assays
The cellular association of MDM and co-localization of DOX and FAM-siRNA delivered by MDM was evaluated using flow cytometry (Beckton Dickinson FACSCalibur™, NJ, USA). Cells (80,000) were seeded in each well of 12-well plates. After overnight incubation, cells were incubated with Rh-PE containing formulations (1 mole% out of the total moles in MDM) or MDM formulations loaded with DOX and FAM-siRNA for 4 hours in serum complete media. After that, cells were detached using trypsin and washed with PBS pH 7.4 three times and resuspended in 200 μl of PBS. The fluorescence signal was excited using a 488 nm laser and the emission was recorded using a 530/30 and 585/42 wavelength filter. A total of 10,000 gated living cell events were collected.
2.9. Cytotoxicity assays
Cytotoxicity of MDM formulations were evaluated in A2780 ADR, MCF7 ADR and MCF7 sensitive cell lines. Five thousand cells were seeded in each well of 96-well plates. Following overnight incubation, cells were treated with formulations in serum complete media at a lipid concentration ranging from 0.31–10 μM. Formulations were washed away after 4 hours incubation and replaced with fresh serum complete media. Cytotoxicity profiles were measured after another 44 hours incubation using a CellTiter-Blue® cell viability assay by following the manufacturer’s protocol (Promega, WI, USA).
2.10. Downregulation of membrane bound P-gp
Cells were seeded in 12-well plates at 80,000 cells per well or 6-well plates at 300,000 cells per well. After the cells were attached, the MDM 1:10 formulation complexing varying concentrations of siMDR-1 were incubated with cells for 4 hours in serum complete media. After the 4 hours incubation, media were replaced by fresh serum complete media. Cells were incubated at 37 °C with 5% CO2 for 44 hours before quantifying the P-gp expression and evaluating its function with flow cytometry.
2.11. Quantifying the amount of membrane bound P-gp
Phycoerythrin-labeled P-gp monoclonal antibody was used for membrane bound P-gp staining after downregulation of P-gp by MDM siMDR-1 treatments. In order to keep the membrane bound P-gp intact, A2780 ADR cells were harvested with enzyme-free cell dissociation buffer, while MCF7 ADR were harvested by gentle scraping. After harvesting, cells were counted and resuspended in blocking buffer containing 2% BSA (w/v) and 0.1% (w/v) NaN3 in PBS pH 7.4 at a concentration of 106 cells per 100 μl of cell suspension. After 30 mins, anti-P-gp antibody was added at 5 μl per 106 cells. Cells were incubated on ice for another 30 mins, followed by washing with PBS pH 7.4 three times before recording the fluorescence excited with a 488 nm blue laser using flow cytometry with a 585/42 wavelength filter. A total of 10,000 gated living cell events were collected.
2.12. Evaluating the function of membrane bound P-gp
A2780 ADR cells (80,000) were seeded in 12-well plates. Fourty-eight hours after treatments performed at a schedule of 4 hours incubation with formulation containing media and 44 hours incubation in serum complete media, media was replaced by 1 μg/ml of Rh-123 in serum complete media. Cells were incubated with Rh-123 for 2 hours before harvesting. After washing with PBS pH 7.4 three times, the fluorescent signal of Rh-123 trapped within the cells were excited with a 488 nm laser and detected with a 530/30 wavelength filter by flow cytometry. A total of 10,000 gated living cell events were collected.
2.13. Fluorescence imaging by confocal microscopy
Cells (80,000) were seeded on Fisherbrand® microscope cover glass in 12-well plates. After cells were attached, MDM formulations labeled with Rh-PE and complexed with FAM-siRNA were added to the medium and incubated with the cells for 4 hours. After 4 hours, cells were washed with fresh PBS pH 7.4 and fixed with PBS pH 7.4 containing 2% PFA for 30 mins at room temperature. Cells were then washed with PBS pH 7.4 three times and stained with 5 μg/ml Hoechst solubilized in PBS pH 7.4 for 15 mins. Cells were washed again with PBS pH 7.4 and mounted on Fisherbrand Superfrost® microscope slides with Fluoromount G® mounting buffer (SouthernBiotech, AL, USA) for analysis by confocal microscopy (Zeiss LSM 700, NY, USA) using a 63X lens.
2.14. Determination of CMC values of MDMs
Determination of the CMC values of MDM formulations was investigated using the pyrene method [47]. Briefly, 0.3 mg of pyrene in CHCl3 was taken in glass test tubes and dried. MDM, comprised of PAMAM-PEG2k-DOPE and PEG5k-DOPE at 1:1, 1:10 and 1:20 was added to different tubes at a lipid concentration range of 0–300 μM. Each tube was vortexed vigorously to re-suspend the pyrene powder. After overnight incubation at room temperature with agitation, the contents of each tube were filtered through 0.45 μm filters. A fluorescent spectrophotometer (F-2000, HITACHI, Tokyo, Japan) was used to detect the sharp increase of fluorescent signal of solutions at 339Ex/390Em.
2.15. HPLC analysis
The conjugation of PAMAM-PEG2k-DOPE was analyzed with the Hitachi HPLC system with a quaternary pump. PAMAM-PEG2k-DOPE and PAMAM were prepared in BHG buffer at 1 mg/ml for ultra-violet (UV) detection in a Protein KW804 size exclusion column (Shodex™, Showa Denka America, Inc., Tokyo, Japan) at 260 nm [48]. The column was eluted with 140 mM PBS pH 7.4 as the mobile phase. The injection volume was 30 μl and the flow rate was 1 ml/min.
2.16. Statistical analysis
All numerical in vitro data are expressed as mean ± SD, n=3. The data were analyzed for statistical significance using two-way ANOVA multiple comparisons with GraphPad prism 6 (GraphPad Software, Inc., San Diego, CA). Any P value less than 0.05 was considered statistically significant and was denoted as *, **, *** and **** for less than 0.05, 0.01, 0.001 and 0.0001, respectively.
3. Results:
3.1. Characterization of PAMAM-PEG2k-DOPE
The structure of PAMAM-PEG2k-DOPE was first characterized with high performance liquid chromatography (HPLC) using a size exclusion column. The 8.46-min retention time for PAMAM-PEG2k-DOPE indicated a larger size of the micellar structure compared to the peak of free PAMAM, which is around 11.69 min (Figure S1).
The purified PAMAM-PEG2k-DOPE was further confirmed with 1H-NMR. The conjugate contains all components including PAMAM, PEG2k and DOPE (Figure S2). The characteristic peaks noted at different ppm values are as follows (where singlet, doublet, triplet, multiplet are noted as s, d, t, m respectively). For starting material pNP-PEG2k-pNP, 1H NMR (CDCl3, 400 MHZ): δ 8.29 (d, J=8.85 Hz, 2H), 7.40 (d, J=8.85 Hz, 2H), 4.46–4.45 (m,2H), 3.83–3.81 (m, 3H), 3.70–3.64 (m, 80H), 1.82 (bs, 4H). Two double peaks at δ ppm 8.29 and 7.40 are from the pNP group. Hydrogen on PEG chain give peaks at δ ppm 3.70–3.64. For DOPE, 1H NMR (CDCl3, 400 MHZ): δ 8.50 (bs, 3H), 5.38–5.30 (m, 3H), 5.25–5.18 (m, 1H), 4.45–4.35 (m, 1H), 4.25–4.05 (m, 3H), 4.00–3.90 (m, 2H), 3.18 (bs, 2H), 2.35–2.27 (m, 4H), 2.10–1.95 (m 8H), 1.59 (bs, 4H), 1.40–1.20 (m, 30H), 0.92–0.85 (m, 5H). The peak at δ ppm 5.38–5.30 was characterized as the –CH=CH– proton signal. For PAMAM, 1H NMR (DMSO-d6, 400 MHZ): δ 8.12–7.85 (m), 3.15–2.95 (m), 2.70–2.52 (m), 2.46–2.38 (m), 2.28–2.15 (m). For DOPE modified pNP-PEG-pNP, pNP-PEG2k-DOPE, 1H NMR (CDCl3, 400 MHZ): δ 8.29 (d, J=9.95 Hz, 2H), 7.40 (d, J=9.95 Hz, 2H), 5.37–5.34 (m,4H), 5.25–5.17 (m, 2H), 4.46–4.44 (m,2H), 4.39–4.36 (m, 1H), 4.25–4.20 (m, 2H), 4.18–4.14 (m, 1H), 4.05–3.80 (m, 3H), 3.83–3.81 (m, 2H), 3.75–3.60 (m, 93H), 3.48–3.46 (m, 1H), 3.42–3.37 (m, 3H), 2.33–2.27 (m, 5H), 2.18–2.13 (m, 8H), 2.05–1.95 (m, 8H), 1.62–1.51 (m, 5H), 1.35–1.25 (m, 33H), 0.90–0.75 (m, 9H). Existence of a proton signal on pNP at δ ppm 8.29 and 7.4 proved the availability of pNP in conjugation with PAMAM. The peak of a cis-double bond at δ ppm 5.37–5.34 also indicated successful conjugation of DOPE. For PAMAM-PEG2k-DOPE, 1H NMR (DMSO-d6, 400 MHZ): δ 8.02–7.92 (m), 5.33–5.30 (m), 4.04–4.02 (m), 3.69–3.66 (m), 3.58–3.56 (m), 3.28–3.01 (m), 2.68–2.55 (m), 2.42 (bs), 2.25–2.15 (m), 1.99–1.95 (m), 1.52–1.45 (m), 1.30–1.21 (m), 0.88–0.82 (m). All characteristic peaks, except for peaks on pNP, from starting materials were observed, demonstrating the successful conjugation of PAMAM-PEG2k-DOPE.
3.2. Size and zeta potential of MDM
As shown in Table 1, PAMAM-PEG2k-DOPE conjugate spontaneously formed micellar nano-preparations in an aqueous environment due to the hydrophobic moiety. However, there was a wide range of size distribution. Increasing the hydrophobic interactions and creating a core by addition of PEG5k-DOPE significantly decreased the polydispersity index (PdI). Sizes of MDM at all different mole ratios of PEG5k-DOPE were around 30 nm. Increasing the PEG5k-DOPE mole ratio helped to keep the PdI value at around 0.25, indicating a monodispersed size distribution. The surface charge of MDM decreased with the addition of PEG5k-DOPE. A slightly positive charge of MDM 1:10 is ideal for nucleic acid complexation and minimizes the cytotoxicity resulting from the high cationic charge. With an increase in PEG5k-PE content, MDM 1:20 showed a neutral zeta potential. MDM 1:10 formulation loaded with 8% (w/w) DOX has a homogeneous size peak of around 219 nm with a PdI around 0.187. The zeta potential value decreased after the complexation of siMDR-1 by 4 mV, demonstrating a successful complexation between formulation and siRNA.
Table 1.
Summary of size (Z-average), polydispersity index (PdI) and zeta potential of nano-preparations.
| Sample | Size* (nm) | PdI* | Zeta Potential* (mV) |
|---|---|---|---|
| PAMAM-PEG2k-PE | 41.06 ± 0.61 | 0.703 | 20.40 ± 3.39 |
| MDM 1:1 | 37.74 ± 2.20 | 0.464 | 17.80 ± 2.13 |
| MDM 1:10 | 30.44 ± 1.38 | 0.271 | 2.16 ± 0.64 |
| MDM 1:20 | 31.46 ± 2.07 | 0.238 | 0.13 ± 1.25 |
| MDM 1:10 DOX 4% w/w | 128.10 ± 1.55 | 0.322 | 2.14 ± 0.98 |
| MDM 1:10 DOX 8% w/w | 219.40 ± 5.71 | 0.187 | 8.39 ± 0.06 |
| MDM 1:10 DOX 8% w/w siMDR | 175.80 ± 1.04 | 0.204 | 4.55 ± 0.25 |
Results indicate average mean ± SD, n=3.
3.3. siRNA complexation and protection against RNases
Among the MDMs, MDM 1:20 had the lowest amount of dendrimer conjugate. Thus, it was the least likely group for siRNA complexation. By evaluating the siRNA complexation ability of MDM 1:20, it is possible to postulate the siRNA complexation ability in MDM 1:1 and MDM 1:10. As shown in Figure 2, 750 ng of siRNA was used in each lane. Full complexation of siRNA was reached at a N/P ratio higher than 1 in both PAMAM-PEG2k-DOPE and MDM 1:20 groups. This suggested that PAMAM-PEG2k-DOPE and MDM have the ability to complex siRNA. Also, the presence of PEG5k-DOPE, even at 1:20, did not impair the complexation between siRNA and PAMAM.
Figure 2.

Gel electrophoresis image (2% Agarose gel at 60 mV for 30 mins) indicating the ability of PAMAM-PEG2k-DOPE and MDM 1:20 in complexing siRNA. Full complexations were reached at N/P 2. 750 ng of siRNA was used in each lane.
The ability of MDM 1:10 to protect siRNA from RNase mediated degradation was investigated. Since the PAMAM-PEG2k-DOPE was responsible for complexing and delivering siRNA in our formulations, it was also evaluated by itself. RNase digestion was terminated by EDTA and any remaining siRNA was expected to be released from the dendriplex by using heparin. The free siRNA was visualized using gel electrophoresis. A N/P ratio of 5 was used to assure the full complexation of siRNA for both PAMAM-PEG2k-DOPE and MDM (Figure 3a). Following exposure to RNase, siRNA bands were still present when complexed with PAMAM-PEG2k-DOPE or MDM. However, the siRNA band was barely visible when free siRNA was treated with both RNase and EDTA/heparin. This result indicates the successful protection of the siRNA by PAMAM-PEG2k-DOPE and MDM. Thus, we revealed the efficacy of PAMAM-PEG2k-DOPE conjugate as well as the MDM formulation based on this novel conjugate in protecting siRNA.
Figure 3.

a, Investigation of siRNA protection by PAMAM-PEG2k-DOPE (D-PEG-PE) and MDM using gel electrophoresis. 750 ng of siRNA was used in each lane. b, Cellular association of the conjugate and MDM formulations was checked by flow cytometry using A2780 ADR cell line. Results indicate mean ± SD, n=3. ****p ≤ 0.0001, ***p ≤ 0.001.
3.4. Cellular association of PAMAM-PEG2k-PE and MDM
As shown in Table 1, siRNA complexation slightly decreased the zeta potential of the formulation. There was a possibility that siRNA complexation might impair the cellular association of MDM formulations because positive charge-motivated interaction plays a major role in cellular association. Thus, the cellular association abilities of MDM and PAMAM-PEG2k-DOPE with or without complexed siRNA were quantified in serum complete media by measuring the fluorescent signal from Rh-PE incorporated inside the micellar nano-preparations. To understand the major driving force for cellular association, PAMAM-PEG2k-DOPE was used either at the same total concentration as the MDM (2.5 μM) or at a concentration that was used in the MDM formulation (Figure 3b). PAMAM-PEG2k-DOPE at 2.5 μM showed an 8-fold increase in association compared to the control group. The fluorescent signal decreased significantly under the shielding effect of PEG5k-DOPE.
The cellular association of the MDM carrying siRNA was also investigated using another MDR cell line, the MCF7 ADR cell line. Two different concentrations of siRNA were used to investigate whether the cellular association is affected by the amount of complexed siRNA (Figure 4a). In both cell lines treated with Rh-PE containing formulations, MDM alone or complexed with 100 and 500 nM siRNA, achieved same fluorescence increase, which was around 3-fold, indicating that the siRNA complexation did not exceed the ability of association with the cells.
Figure 4.

a. Cellular association assay of MDM 1:10 complexed with 0 nM, 100 nM and 500 nM siMDR-1 in A2780-ADR and MCF7-ADR. MDM Rh-PE are MDMs containing 1% mole of Rh-PE. Results indicate mean ± SD, n=3. ****p ≤ 0.0001. b. Analysis of the co-localization of DOX and FAM-siRNA in MCF7 ADR cell line treated with Lipofectamine FAM-siRNA, MDM 1:10 loaded with DOX and MDM 1:10 co-loaded with DOX and FAM-siRNA using flow cytometry. FL1-H indicates the fluorescence signal at 530 nm. FL2-H indicate the fluorescence signal at 585 nm.
3.5. Localization of DOX and FAM-siRNA delivered by MDM
PAMAM-PEG2k-DOPE, MDM 1:1, MDM 1:10 and MDM 1:20 were investigated for their siRNA delivery efficiency using confocal microscopy in the MCF7 ADR cell line (Figure 5, S5). The same mole amount of PAMAM-PEG2k-DOPE was used in PAMAM-PEG2k-DOPE, MDM 1:1, MDM 1:10 and MDM 1:20 groups. As shown in Figure 5, FAM-labelled siRNA and DOX were successfully co-delivered into the cytoplasm in all groups. PAMAM-PEG2k-DOPE reached the highest fluorescence signal from FAM-siRNA than other MDM formulations because of the absence of PEG shielding. However, PAMAM-PEG2k-DOPE itself has limited delivery efficiency for DOX. With the increasing amount of PEG from MDM 1:1 to MDM 1:20, the signal from FAM-sRNA delivered into cells was decreasing (Figure S5) and the signal from DOX inside of cells also got attenuated (Figure 5). In the merged image of MDM 1:10 loaded with DOX and FAM-siRNA, the yellow signal indicated the co-localization of the DOX and FAM-siRNA. Same result was also observed in Figure 4b. Among the population sorted with flow cytometry, 97.54% of cell population exhibited overexpression of in both FL-1H and FL-2H, indicating the successful delivery of both DOX and FAM-siRNA in the same cell. In the merged image of MCF7 ADR treated with MDM loaded with Rh-PE and FAM-siRNA (Figure S5), there were also individual green and red spots suggesting the escape of free FAM-siRNA from the formulation into the cytoplasm.
Figure 5.

(Colored) Confocal images of MCF7 ADR cell line treated with PAMAM-PEG2k-DOPE and different MDM formulations loaded DOX and complexed with FAM-labeled siRNA. Blue: Hoechst 33342; Green: FAM-siRNA; Red: DOX. Magnitude: 63X.
3.6. Downregulation of the membrane bound P-gp
As shown in Figure 6a, both Adriamycin-resistant cell lines, A2780 ADR and MCF7 ADR, expressed significantly higher P-gp compared to their sensitive counterparts, confirming the selection of these cell lines as appropriate models.
Figure 6.

a, Characterization of the membrane bound P-gp expression on MCF-7, A2780 cell lines and their Adriamycin resistant counter cell lines using flow cytometry. b, siRNA-mediated P-gp downregulation by MDM 1:10 complexed with siMDR-1 or siRNA control (siNEG) and c, inhibition of P-gp function by MDM 1:10 complexed with siMDR-1 or siRNA Control (siNEG) in the A2780 ADR cell line. Results indicate mean ± SD, n=3. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
siRNA-mediated P-gp downregulation by MDM formulations was investigated in the A2780 ADR cell line due to their high P-gp levels. Cells were harvested with enzyme free cell dissociation buffer to maintain the integrity of membrane bound P-gp, to determine the ability of cells to pump out chemotherapeutic agents. As shown in Figure 6b, MDM 1:10 complexed with 100 nM and 500 nM siMDR-1 successfully downregulated P-gp by around 40 % at 48 hours after treatment. MDM 1:10 complexed with 500 nM negative siRNA control didn’t show any difference between MDM 1:10 alone in P-gp downregulation.
3.7. Downregulation of membrane bound P-gp function
Rhodamine-123 (Rh-123) was used as a known substrate of P-gp to investigate the cell’s efflux function [49]. If the amount of P-gp was downregulated low enough to enhance drug accumulation, more Rh-123 signal was expected inside of cells. Results showed that in cells treated with MDM 1:10/siMDR-1, the fluorescence signal inside of cells increased by 75% compared to the control treated with free Rh-123, and 35 % compared to the group treated with MDM without siMDR-1 (Figure 6c). While cells treated with MDM 1:10 complexed with negative siRNA control didn’t show any fluorescence increase than control group. This result demonstrated that downregulation of P-gp impaired the efflux effect resulting from P-gp. More Rh-123 was trapped inside A2780 ADR cells due to the decreased membrane bound P-gp. Also, MDM complexed with 100 nM or 500 nM siMDR-1 showed a similar fluorescence increase, which was in agreement with the membrane bound P-gp downregulation result (Figure 6b). Both of these results indicate that the membrane bound P-gp was decreased, and as a result, impaired the function of P-gp in pumping out chemotherapeutics.
3.8. Cytotoxicity of MDM 1:10 loaded with both DOX and siMDR-1
Having demonstrated an effective approach for delivery of siMDR-1, MDM was further explored as a co-delivery system for both siRNA and hydrophobic small molecules. Both A2780 ADR and MCF7 ADR were used to assess the cytotoxicity resulting from MDM 1:10 co-loaded with DOX and siMDR-1. Cells were incubated with various formulations for 4 hours. Cell viability was investigated using CellTiter-Blue® assay after 44 hours incubation in serum-complete media. As shown in Figures 7a, b, in both cell lines, MDM loaded with both DOX and siMDR-1 led to a significantly higher cytotoxicity compared to all other groups, including MDM, MDM DOX and free DOX. MDM complexed with negative siRNA control showed a similar cytotoxic effect as MDM loaded with DOX and MDM complexed with siMDR-1 didn’t shown any therapeutic benefits than MDM alone (Figure S4b). These results indicated the combinatorial effect of DOX and siMDR-1 in MCF7 ADR and A2780 ADR cell lines due to siMDR-1-induced downregulation of the P-gp efflux pump and increased amount of DOX inside cells.
Figure 7.

Cytotoxicity assay of MDM 1:10 formulations loaded with DOX or DOX/siRNA against a, MCF7 ADR cells and b, A2780 ADR cells. Abscissa labelled “3.67/500/1.7” indicates 3.67 μM nano-preparation loaded with 500 nM siRNA and 1.7 μM DOX. c, Cytotoxicity assay of MCF7 sensitive cell line and MCF7 ADR cell line treated with free DOX 3.4 μM, free DOX 3.4 μM with 200 nM of siMDR-1, MDM 1:10 loaded with 3.4 μM DOX and MDM 1:10 loaded with 3.4 μM DOX and 200 nM siMDR-1. d, Cytotoxicity assay of A2780 ADR cells treated with MDM 1:10 or MDM 1:10 siMDR-1 together with free DOX HCl 3.4 μM. Abscissa labelled “3.68/200” indicates 3.68 μM nano-preparation loaded with 200 nM siRNA. All results indicate mean ± SD, n=3. **P<0.01, ***P<0.001****P<0.0001.
Effect of P-gp downregulation in non-resistant cell line was also investigated. In Figure 7c, both MCF7 and MCF7 ADR cell lines didn’t show any increased cytotoxicity coming from siMDR-1 when treated free siMDR-1 in addition to free DOX. MCF7 sensitive cells didn’t shown any decreased cell viability from MDM loaded with DOX and siMDR-1 (46.3%) compared to MDM DOX (43.3%) either. However, MCF7 ADR treated with MDM loaded with DOX and siMDR-1 (45.8%) resulted in a much lower cell viability than cells treated with MDM DOX (57.0%). Given that the resistance in MCF7 ADR increase the cell viability treated with DOX formulations, similar cytotoxic effect in MCF7 sensitive (46.3%) and MCF7 ADR (45.8%) cell lines treated with MDM loaded with DOX and siMDR-1 suggested the successful reversal of MDR effect.
To prove that a co-delivery system for DOX and siMDR-1 is necessary, A2780 ADR cells were first treated with MDM 1:10 or MDM 1:10 complexed with siMDR-1, followed with treatment of free DOX HCl. As shown in Figure 7d, cells treated with MDM 1:10 siMDR-1 didn’t show a significantly higher cytotoxicity than MDM 1:10 when DOX was delivered separately.
4. Discussion:
Reversal of the MDR effect has continued to attract great interest over the last two decades [11]. The co-delivery of both nucleic acids and a chemotherapy together can cause a combinatorial effect and ensure that cells affected are exposed to both therapeutic agents, thereby, increasing the therapeutic efficiency. In this project, a tri-block copolymer PAMAM-PEG2K-DOPE based on G4 PAMAM was conjugated. The structure of PAMAM-PEG2K-DOPE was characterized by both HPLC (Figure S1) and 1H-NMR (Figure S2). The conjugate has a significantly shorter retention time than PAMAM alone measured by size exclusion chromatography using HPLC, although the single molecular weight is only 3 kDa different. Due to the addition of a hydrophobic moiety on the conjugate, PAMAM-PEG2K-DOPE spontaneously forms a nano-structure, with a particle size much larger than PAMAM alone.
The new copolymer self-assembles into a micellar structure together with PEG5k-DOPE, forming a mixed dendrimer micelle, which contains a siRNA complexing region and a hydrophobic core for hydrophobic chemotherapies (Figures 1, S3). The necessity of a PEG5k-DOPE shield to increase the stability of nanocarriers and enable a long-term circulation in vivo has been well-discussed [50, 51]. Addition of PEG5k-DOPE reduces the interaction between cell membrane and the cationic charges in PAMAM, decreasing the PdI value, the zeta potential of nano-preparation and the cytotoxic effect. As shown in Figure S4a, with an increased amount of PEG5k-DOPE in MDM, cytotoxicity coming from MDM decreases. MDM 1:10 and MDM 1:20 have a much lower cytotoxic effect than PAMAM-PEG2K-DOPE alone and MDM 1:1 because the high amount of PEG5k chains stabilized the micellar structure, shielding positive charge on the PAMAM moiety. Considering the structure and cytotoxicity aspects, it is necessary to include PEG5k-DOPE in MDM. Also, as showed in Figure 2, even at the highest PEG concentration, MDM 1:20 showed a similar ability to complex siRNA. The long PEG chains decreased the overall charge of MDM, but they didn’t influence the electrostatic interaction between PAMAM and siRNA.
It was also argued that due to the high chemical stability of PEG, the PEGylated nanocarriers would be resistant to cellular internalization in vivo [52, 53]. Thus, a very high ratio of PEG5k-DOPE would jeopardize the performance of MDM by decreasing the cellular interaction. As shown in Figure S5, MDM 1:1 has a strong cellular association, while the fluorescence signal decreased with an increasing density of the PEG5k-DOPE compartment. The MDM 1:20 formulation resulted in significantly lower cell association and internalization in the MCF7 ADR cell line. Taking structural stability, cellular association, siRNA complexation and cytotoxicity into consideration, MDM 1:10, with its relatively lower cytotoxicity and higher cell internalization, represented an ideal formulation to be used in following experiments.
One of the advantages of MDM is the encapsulation of hydrophobic chemotherapeutics in its hydrophobic core. As a model, DOX free-base was loaded into the MDM by hydrating the mixed film of co-polymer together with PEG5k-PE and DOX base. It was shown in Table 1 that loading of DOX base into the core of the nano-preparation promoted a size increase of MDM. With 4% w/w DOX loaded in MDM, the PdI value was above 0.3 with one peak around 30 nm and the other around 220 nm, which referred to empty MDM and MDM DOX, respectively. When 8% w/w of DOX was loaded, the resultant size of MDM DOX was a single peak of 220 nm with a PdI value of 0.187, indicating a homogenous size distribution. It was reported that hydrophobic DOX had a property to form precipitates as DOX fibers due to its planar molecular structure [54]. Thus, the hydrophobic DOX in MDM formulations might act as a core of the MDM, which inserted hydrophobic tails of lipid moieties into DOX. Similar size increase in DOX loaded formulations was also observed by Y Zhou et al., who reported that the size of DOX loaded micelle increased from 53 nm to 145 nm with the increasing feed of DOX base [55].
The positive charge of MDM 1:10 decreased from 8.39 mV to 4.55 mV after it complexed with siRNA. Though cellular interaction of MDM is driven by surface cationic charges, such a decrease in charge does not affect the cellular interaction but significantly enhances the cellular association as observed in Figure 3b. We suggest that complexation of siRNA favors the cellular association by causing some structural change of MDM or decreasing the interaction between MDM and some serum proteins. Usually, the serum proteins form a protein corona around nanoparticles, impeding active targeting and cell association [56]. Most importantly, vesicles for siRNA delivery have to protect the siRNA from degradation and ensure the endosomal escape of siRNA. We have demonstrated that the complexation between MDM and siRNA protects siRNA from RNase-mediated degradation and confirmed the successful delivery co-localization of siRNA and DOX by the MDM delivery system with confocal microscopy and flow cytometry.
MCF7 ADR was used in these experiments because of the size of cells was much larger than A2780 ADR, where visualizing under confocal microscopy was challenging. In Figure 5, though there is a strong signal coming from the internalized FAM-siRNA, the DOX signal in PAMAM-PEG2k-DOPE group is not obvious. This could be a result of the formation of PAMAM-PEG2k-DOPE nanoaggregates instead of micellar structure, indicated by the PdI value around 0.7, as seen in Table 1. The lack of hydrophobic cores limits the encapsulation of DOX in PAMAM-PEG2k-DOPE, thus resulting in the low DOX signal.
We have also demonstrated that the delivery of siMDR-1 with MDM 1:10 successfully downregulated the P-gp on cell surface by 40 % (Figure 6b). However, the amount of P-gp that can be downregulated is limited because proteins that have already been expressed on cell surface will not be affected by RNA interference. Also, cells proliferated during the 48 hours’ treatment before staining of P-gp. Thus, reversal of multidrug resistance requires a long-term treatment. In order to reach higher efficacy, multiple treatments are required to achieve a stronger therapeutic effect. Another concern also arises. Although the amount of P-gp on the cell membrane is downregulated, the remaining P-gp may still be enough to pump out the majority of chemotherapeutic agents, resulting in MDR. Thus, evaluating the function of the remaining P-gp in pumping out small molecules is necessary. We have successfully assessed the function of P-gp by using Rh-123 as the substrate (Figure 6c). Increased fluorescent signal in cells demonstrated that more Rh-123 molecules were trapped inside of the A2780 ADR cells due to the decreased membrane bound P-gp. Not only the amount of membrane bound P-gp, but also the MDR-related drug efflux effect, was decreased.
Cytotoxicity profiles of MDM in both A2780/A2780 ADR and MCF7/MCF7 ADR cell lines have further revealed that the enhanced therapeutic efficiency of DOX was achieved by the dual functioning of siMDR-1 and the drug (Figure 7). To maximize the effect of a co-delivery system, it is crucial to elucidate the ideal combinations of the two cargos. The effect of DOX might mask the effect of nucleic acid therapy and induce cell death before P-gp is downregulated. Thus, the combination and physiological mechanism of two cargos in this platform may be further optimized. Under consideration of the bio-distribution in vivo, concentrations of siRNA ranging from 100–500 nM were investigated in cytotoxicity assays. These results not only exhibit the capacity of MDM in loading siRNA, but also will provide information for in vivo studies to achieve a sufficient delivery of siRNA in tumor tissues. Especially at this stage when little is known about how different the mechanisms of action of the anticancer drug and the siRNA will impact on their biodistribution and efficacy from in vitro to in vivo.
Additionally, ligand-targeting properties are a promising approach for inclusion in the formulation for specific delivery of a drug combination into tumor cells, thereby, enhancing the therapeutic efficiency and reducing the systemic toxicity. PEG moieties provide flexibility in substitution for ligand-conjugated PEG [57]. Under such circumstance, long PEG chains may compete and overcome the targeting effect of ligands. Thus, it might not be realistic to attach targeting moieties to an even longer PEG chain and expect to achieve a higher internalization efficiency. In such a case, instead of using PEG5k-DOPE, PEG3k-DOPE can be used as a substitution in the formulation, while targeting moieties can be attached to PEG5k-DOPE.
Finally, the dendrimer-based nano-preparation provides an ideal co-delivery platform for both chemotherapy and nucleic acid. In addition to the combination of P-gp and DOX, other RNA interfering agents in combination with hydrophobic molecules can be used in this platform to reverse multidrug resistance, including siBcl-2 and siSurvivin. Moreover, the abundance of cationic charges on PAMAM provides the possibility for delivery of large nucleic acid molecules, such as mRNA or DNA. The combination effect of both nucleic acid and chemotherapy is expected to enhance the therapeutic efficiency of chemotherapy.
5. Conclusions:
Reversal of the MDR effect is an urgent task when validating existing chemotherapies as well as when developing new drugs. Downregulation of the multidrug resistance-related protein, P-gp, using its corresponding siMDR-1 is a promising approach. Based on G4 PAMAM, a co-delivery system for siRNA and chemotherapeutic agents has been developed. By conjugating PAMAM with PEG2k-DOPE, a triblock co-polymer with both a hydrophobic moiety and siRNA complexing ability makes it suitable for co-delivery of hydrophobic chemotherapy and nucleic acid molecules. PAMAM dendrimer plays important roles in complexing siRNA, facilitating cellular interaction and assisting endosomal escape. PEG moieties shield the cationic charge and homogenize the structure of the nano-preparation. A balance between cellular interaction and cytotoxicity is a crucial factor in terms of maximizing the therapeutic efficiency.
We have demonstrated that MDM 1:10 is the most promising platform for co-delivery of nucleic acid with hydrophobic chemotherapy. When complexed with siMDR-1, MDM 1:10 downregulated the amount of membrane bound P-gp as well as the function of P-gp, resulting in reversal of the MDR effect. In addition to successful delivery of siMDR-1 into MDR cell lines, a synergistic anti-cancer effect has been achieved by the co-delivery of DOX base and siMDR-1 with our dendrimer-based nano-preparation. Evaluation of such systems in vivo after systemic administration using different dosage schedules would be a priority task for future advances.
Supplementary Material
6. Acknowledgement:
This work was supported by the National Institute of Health (NIH) [1R01CA200844]
Footnotes
Declaration of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7. References:
- 1.Housman G, et al. , Drug resistance in cancer: an overview. Cancers (Basel), 2014. 6(3): p. 1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarisozen C, et al. , Polymers in the co-delivery of siRNA and anticancer drugs to treat multidrug-resistant tumors. Journal of Pharmaceutical Investigation, 2017. 47(1): p. 37–49. [Google Scholar]
- 3.Rodriguez-Antona C and Ingelman-Sundberg M, Cytochrome P450 pharmacogenetics and cancer. Oncogene, 2006. 25(11): p. 1679–91. [DOI] [PubMed] [Google Scholar]
- 4.Shen H, et al. , Comparative metabolic capabilities and inhibitory profiles of CYP2D6.1, CYP2D6.10, and CYP2D6.17. Drug Metab Dispos, 2007. 35(8): p. 1292–300. [DOI] [PubMed] [Google Scholar]
- 5.Townsend DM and Tew KD, The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene, 2003. 22(47): p. 7369–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gagnon JF, et al. , Irinotecan inactivation is modulated by epigenetic silencing of UGT1A1 in colon cancer. Clin Cancer Res, 2006. 12(6): p. 1850–8. [DOI] [PubMed] [Google Scholar]
- 7.Kohn EC, et al. , Dose-intense taxol: high response rate in patients with platinum-resistant recurrent ovarian cancer. J Natl Cancer Inst, 1994. 86(1): p. 18–24. [DOI] [PubMed] [Google Scholar]
- 8.Selvakumaran M, et al. , Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res, 2003. 63(6): p. 1311–6. [PubMed] [Google Scholar]
- 9.Fink D, Aebi S, and Howell SB, The role of DNA mismatch repair in drug resistance. Clin Cancer Res, 1998. 4(1): p. 1–6. [PubMed] [Google Scholar]
- 10.Altieri DC, Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene, 2003. 22(53): p. 8581–9. [DOI] [PubMed] [Google Scholar]
- 11.Gillet JP and Gottesman MM, Mechanisms of multidrug resistance in cancer. Methods Mol Biol, 2010. 596: p. 47–76. [DOI] [PubMed] [Google Scholar]
- 12.Lima RT, et al. , Specific downregulation of bcl-2 and xIAP by RNAi enhances the effects of chemotherapeutic agents in MCF-7 human breast cancer cells. Cancer Gene Ther, 2004. 11(5): p. 309–16. [DOI] [PubMed] [Google Scholar]
- 13.Fojo AT, et al. , Expression of a multidrug-resistance gene in human tumors and tissues. Proc Natl Acad Sci U S A, 1987. 84(1): p. 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldstein LJ, et al. , Expression of a multidrug resistance gene in human cancers. J Natl Cancer Inst, 1989. 81(2): p. 116–24. [DOI] [PubMed] [Google Scholar]
- 15.Shervington A and Lu C, Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Invest, 2008. 26(5): p. 535–42. [DOI] [PubMed] [Google Scholar]
- 16.Goren D, et al. , Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin Cancer Res, 2000. 6(5): p. 1949–57. [PubMed] [Google Scholar]
- 17.Maliepaard M, et al. , Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res, 1999. 59(18): p. 4559–63. [PubMed] [Google Scholar]
- 18.Trippett T, et al. , Defective transport as a mechanism of acquired resistance to methotrexate in patients with acute lymphocytic leukemia. Blood, 1992. 80(5): p. 1158–62. [PubMed] [Google Scholar]
- 19.Veneroni S, et al. , Expression of P-glycoprotein and in vitro or in vivo resistance to doxorubicin and cisplatin in breast and ovarian cancers. Eur J Cancer, 1994. 30A(7): p. 1002–7. [DOI] [PubMed] [Google Scholar]
- 20.Bell DR, et al. , Detection of P-glycoprotein in ovarian cancer: a molecular marker associated with multidrug resistance. J Clin Oncol, 1985. 3(3): p. 311–5. [DOI] [PubMed] [Google Scholar]
- 21.Green H, et al. , mdr-1 single nucleotide polymorphisms in ovarian cancer tissue: G2677T/A correlates with response to paclitaxel chemotherapy. Clin Cancer Res, 2006. 12(3 Pt 1): p. 854–9. [DOI] [PubMed] [Google Scholar]
- 22.Linn SC, et al. , Prognostic relevance of P-glycoprotein expression in breast cancer. Ann Oncol, 1995. 6(7): p. 679–85. [DOI] [PubMed] [Google Scholar]
- 23.Baekelandt MM, et al. , P-glycoprotein expression is a marker for chemotherapy resistance and prognosis in advanced ovarian cancer. Anticancer Res, 2000. 20(2B): p. 1061–7. [PubMed] [Google Scholar]
- 24.Yabuki N, et al. , Gene amplification and expression in lung cancer cells with acquired paclitaxel resistance. Cancer Genet Cytogenet, 2007. 173(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 25.Amiri-Kordestani L, et al. , Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist Updat, 2012. 15(1–2): p. 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lage H, MDR1/P-glycoprotein (ABCB1) as target for RNA interference-mediated reversal of multidrug resistance. Curr Drug Targets, 2006. 7(7): p. 813–21. [DOI] [PubMed] [Google Scholar]
- 27.Stege A, Kruhn A, and Lage H, Overcoming multidrug resistance by RNA interference. Methods Mol Biol, 2010. 596: p. 447–65. [DOI] [PubMed] [Google Scholar]
- 28.Yin Q, et al. , Overcoming multidrug resistance by co-delivery of Mdr-1 and survivin -targeting RNA with reduction-responsible cationic poly(beta-amino esters). Biomaterials, 2012. 33(27): p. 6495–506. [DOI] [PubMed] [Google Scholar]
- 29.Liu C, et al. , Novel biodegradable lipid nano complex for siRNA delivery significantly improving the chemosensitivity of human colon cancer stem cells to paclitaxel. J Control Release, 2009. 140(3): p. 277–83. [DOI] [PubMed] [Google Scholar]
- 30.Abbasi M, et al. , Cationic polymer-mediated small interfering RNA delivery for P-glycoprotein downregulation in tumor cells. Cancer, 2010. 116(23): p. 5544–54. [DOI] [PubMed] [Google Scholar]
- 31.Yadav S, et al. , Evaluations of combination MDR-1 gene silencing and paclitaxel administration in biodegradable polymeric nanoparticle formulations to overcome multidrug resistance in cancer cells. Cancer Chemother Pharmacol, 2009. 63(4): p. 711–22. [DOI] [PubMed] [Google Scholar]
- 32.Taratula O, et al. , Surface-engineered targeted PPI dendrimer for efficient intracellular and intratumoral siRNA delivery. J Control Release, 2009. 140(3): p. 284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Zhou J, and Luo Y, SiRNA delivery systems based on neutral cross-linked dendrimers. Bioconjugate Chemistry, 2012. 23(2): p. 174–83. [DOI] [PubMed] [Google Scholar]
- 34.Waite CL and Roth CM, PAMAM-RGD conjugates enhance siRNA delivery through a multicellular spheroid model of malignant glioma. Bioconjugate Chemistry, 2009. 20(10): p. 1908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi Y, et al. , Potential Use of Lactosylated Dendrimer (G3)/alpha-Cyclodextrin Conjugates as Hepatocyte-Specific siRNA Carriers for the Treatment of Familial Amyloidotic Polyneuropathy. Molecular Pharmaceutics, 2012. 9(6): p. 1645–1653. [DOI] [PubMed] [Google Scholar]
- 36.Tang Y, et al. , Efficient in vitro siRNA delivery and intramuscular gene silencing using PEG-modified PAMAM dendrimers. Molecular Pharmaceutics, 2012. 9(6): p. 1812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo D, et al. , Poly(ethylene glycol)-Conjugated PAMAM Dendrimer for Biocompatible, High-Efficiency DNA Delivery. Macromolecules, 2002. 35(9): p. 3456–3462. [Google Scholar]
- 38.Patil ML, et al. , Internally cationic polyamidoamine PAMAM-OH dendrimers for siRNA delivery: effect of the degree of quaternization and cancer targeting. Biomacromolecules, 2009. 10(2): p. 258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patil YB, et al. , The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials, 2010. 31(2): p. 358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, et al. , Co-delivery of drugs and DNA from cationic core-shell nanoparticles self-assembled from a biodegradable copolymer. Nat Mater, 2006. 5(10): p. 791–6. [DOI] [PubMed] [Google Scholar]
- 41.Chen Y, et al. , Multifunctional nanoparticles delivering small interfering RNA and doxorubicin overcome drug resistance in cancer. Journal of Biological Chemistry, 2010. 285(29): p. 22639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen AM, et al. , Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small, 2009. 5(23): p. 2673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun TM, et al. , Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS Nano, 2011. 5(2): p. 1483–94. [DOI] [PubMed] [Google Scholar]
- 44.Xiong X-B and Lavasanifar A, Traceable Multifunctional Micellar Nanocarriers for Cancer-Targeted Co-delivery of MDR-1 siRNA and Doxorubicin. ACS Nano, 2011. 5(6): p. 5202–5213. [DOI] [PubMed] [Google Scholar]
- 45.Biswas S, et al. , Lipid modified triblock PAMAM-based nanocarriers for siRNA drug co-delivery. Biomaterials, 2013. 34(4): p. 1289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Navarro G, et al. , P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine (Lond), 2012. 7(1): p. 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goddard ED, et al. , Fluorescence probes for critical micelle concentration determination. Langmuir, 1985. 1(3): p. 352–5. [DOI] [PubMed] [Google Scholar]
- 48.Pande S and Crooks RM, Analysis of poly(amidoamine) dendrimer structure by UV-vis spectroscopy. Langmuir, 2011. 27(15): p. 9609–13. [DOI] [PubMed] [Google Scholar]
- 49.Altenberg GA, et al. , Unidirectional fluxes of rhodamine 123 in multidrug-resistant cells: evidence against direct drug extrusion from the plasma membrane. Proc Natl Acad Sci U S A, 1994. 91(11): p. 4654–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lankveld DP, et al. , Blood clearance and tissue distribution of PEGylated and non-PEGylated gold nanorods after intravenous administration in rats. Nanomedicine (Lond), 2011. 6(2): p. 339–49. [DOI] [PubMed] [Google Scholar]
- 51.Wang H, et al. , Folate-PEG coated cationic modified chitosan--cholesterol liposomes for tumor-targeted drug delivery. Biomaterials, 2010. 31(14): p. 4129–38. [DOI] [PubMed] [Google Scholar]
- 52.Maeda T and Fujimoto K, A reduction-triggered delivery by a liposomal carrier possessing membrane-permeable ligands and a detachable coating. Colloids Surf B Biointerfaces, 2006. 49(1): p. 15–21. [DOI] [PubMed] [Google Scholar]
- 53.Romberg B, Hennink WE, and Storm G, Sheddable coatings for long-circulating nanoparticles. Pharm Res, 2008. 25(1): p. 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X, et al. , Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim Biophys Acta, 1998. 1415(1): p. 23–40. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, et al. , Doxorubicin-loaded redox-responsive micelles based on dextran and indomethacin for resistant breast cancer. Int J Nanomedicine, 2017. 12: p. 6153–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen D, et al. , Plasma protein adsorption and biological identity of systemically administered nanoparticles. Nanomedicine (Lond), 2017. 12(17): p. 2113–2135. [DOI] [PubMed] [Google Scholar]
- 57.Torchilin VP, et al. , p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim Biophys Acta, 2001. 1511(2): p. 397–411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.