

Abstract
Both acute intoxication and longer-term cumulative ingestion of alcohol negatively impacts the metabolic phenotype of both skeletal and cardiac muscle, independent of overt protein calorie malnutrition, resulting in loss of skeletal muscle strength and cardiac contractility. In large part these alcohol-induced changes are mediated by a decrease in protein synthesis that in turn is governed by impaired activity of the protein kinase, the mechanistic target of rapamycin (mTOR). Herein, we summarize recent advances in mTOR signal transduction as well as similarities and differences between the effects of alcohol on this central metabolic controller between skeletal muscle and heart in response to acute versus chronic intake. While alcohol-induced alterations in global proteolysis via activation of the ubiquitin-proteasome pathway are equivocal, emerging data suggest alcohol increases autophagy in muscle. Further studies are necessary to define the relative contributions of these bidirectional changes in protein synthesis and autophagy in the etiology of alcoholic myopathy in skeletal muscle and heart.
Keywords: mTOR, translational control, protein synthesis, ubiquitin-proteasome pathway, autophagy, amino acids, alcoholic myopathy
1. INTRODUCTION
1.1. Scope of problem
The consumption of alcohol (i.e., ethanol) containing beverages has continued largely unabated for centuries because of its neurologic effects and its putative medicinal benefits. Today alcohol use and misuse represents a major global concern being directly or indirectly implicated in a range of disease conditions (21). Excessive consumption of alcohol is the fifth leading risk cause of disease and injury worldwide, although considerable variation exists between and within countries as to the amount and pattern of alcohol use. In the United States (US), the per capita consumption averages almost 9 L of pure ethanol per year and approximately 75,000 deaths annually are attributed to excessive alcohol consumption (108). These adverse health effects of alcohol result from an increased prevalence of gastrointestinal, hepatic, cardiovascular, infectious and neoplastic diseases (38).
While the effects of excessive alcohol on the brain and hepatobiliary system garner the greatest attention, the impact of alcohol on the structure and metabolic phenotype of striated muscle – both skeletal and cardiac – has been recognized for more than half a century and adversely affects morbidity and mortality. A significant loss of lean body and muscle mass is detected in 40–60% of individuals with prolonged heavy alcohol use, making it more prevalent than the inherited myopathies (89). This skeletal muscle myopathy ultimately leads to a loss of strength that is proportional to the lifetime alcohol consumption (74). Likewise, long duration of heavy alcohol consumption (> 80 g pure ethanol [≈8 standard drinks in the US] per day for > 10 years) often produces a spectrum of ultrastructural changes in the heart leading to the thinning of the ventricular wall, decreased ventricular function, and the development of alcoholic cardiomyopathy (83). The prevalence of this dilated cardiomyopathy varies widely (ranging between 4–40%) reflecting the criteria used for diagnosis, the diverse patient population studied as well as the amount, duration and pattern of alcohol misuse. Because of the difficulty in diagnosis, the prevalence of both skeletal and cardiac myopathy is believed to be underestimated. However, it is noteworthy, that many of alcohol’s effects on muscle protein metabolism and strength are not permanent and can be at least partially reversed with abstinence or controlled low-dose alcohol consumption (15, 118).
Early studies on the etiology of chronic alcoholic myopathy have largely eliminated disturbances in electrolytes, peripheral neuropathy, inactivity and a host of other factors as being causative (reviewed in (56, 84)). However, heavy alcohol consumption of long duration in humans can produce a secondary protein calorie malnutrition as well as deficiencies in vitamins and trace minerals (75) that may contribute to the development of alcoholic myopathy (119). While the relative importance of such nutritional effects are difficult to dissect in human studies, they can be largely excluded from tightly controlled animal studies (see Section 1.2). Hence, it is generally held that nutritional factors, while potentially contributory, are not essential for the development of either skeletal or cardiac muscle myopathy. In this regard, as described in subsequent sections, the effect of alcohol on striated muscle is characterized by its ability to alter metabolic processes critical for cellular protein homeostasis under basal conditions and often in response to a nutritional stimulus, such as refeeding or leucine. Thus, the current review focuses on recent advances that have improved our understanding of protein metabolism and the development of alcoholic myopathy. Throughout, we have highlighted gaps in our understanding and potential areas for future research.
1.2. Preclinical models
Although seminal epidemiological data pertaining to the effect of alcohol have been obtained in humans, there is a relative scarcity of human studies in which alcohol’s effect on muscle have been investigated and essentially no human studies on cellular mechanisms for the development of alcoholic myopathy. This paucity of human studies originates from the difficulty in quantitating and controlling the amount, type, pattern and duration of alcohol intake in addition to the inability to tightly control nutritional, genetic and environmental differences that may impact outcomes. Hence, the large majority of the available data in this area are derived from preclinical rodent models that appear to mimic the clinical condition. In general, chronic consumption is most frequently modeled in rats and mice by including alcohol either in the drinking water or as part of a nutritionally complete liquid diet. These models produce clinically relevant increases in the blood alcohol concentration (BAC) (39). While each of these methods has advantages and disadvantages, it is noteworthy that all chronic alcohol models include time-matched control animals that are pair-fed an isocaloric isonitrogenous non-alcohol containing diet. Thus, differences in the metabolic phenotype of muscle between alcohol-fed and control animals are most likely the result of alcohol and/or one of its oxidative metabolites (e.g., acetaldehyde). However, alcohol may also influence the digestion and absorption of select nutrients (66), and as this variable is not routinely monitored its contribution to the development myopathy in animals consuming an alcohol-containing diet for several months cannot be excluded.
Heavy episodic drinking (e.g., ≥ 5 standard drinks [≈14 g ethanol/drink] on an occasion for men and ≥ 4 standard drinks for women) is on the rise in the US (27). This binge drinking behavior is modeled in rodents by the bolus administration of alcohol either by oral gavage or intraperitoneal injection, with the latter approach being used most often despite its lack of clinical relevance. Although direct side-by-side comparisons are rare, the route of administration does not appear to greatly influence alterations in muscle protein metabolism and this may reflect the fact that the peak BAC and area under the BAC disappearance curve is remarkable similar regardless of the route of administration (60). These acute models of alcohol intoxication have provided valuable information related to possible early mechanisms or initiating causes for the metabolic disruption, but data from such models may not be indicative of changes observed with chronic consumption.
2. ALCOHOLIC MYOPATHY
While there are many similarities between the effects of alcohol (both acute and chronic) on protein homeostasis in skeletal and cardiac muscle, there are also clear tissue-specific differences that may be important in disease etiology and progression. In the following sections, we start by describing the effects of alcohol on skeletal muscle, as they are the most thoroughly investigated, and then compare and contrast these to alcohol-induced changes in heart. Table 1 provides a comparison of the key changes in protein hemostasis that have been reported for skeletal and cardiac muscle in response to acute and chronic alcohol intake, and these will be elaborated upon in subsequent sections.
Table 1.
Acute and chronic alcohol-induced changes in selected proteins regulating skeletal and cardiac muscle protein metabolism
| Skeletal muscle | Cardiac muscle | |||
|---|---|---|---|---|
| Acute | Chronic | Acute | Chronic | |
| Tissue weight/protein content | ― | ↓ | ― | |
| Protein synthesis | ↓ | ↓ | ↓ | ↓ |
| Translation efficiency | ↓ | ↓ | ↓ | ↓ |
| AKT phosphorylation (T308) | ― | ― | ||
| REDD1 total | ― | ― | ― | ↑ |
| AMPK phosphorylation (T172) | ― | ― | ↑ | ↑ |
| 4E-BP1 phosphorylation (T47/36) | ↓ | ↓ | ↓ | ↓ |
| eIF4G-eIF4E association | ↓ | ↓ | ↓ | ↓ |
| 4EBP1-eIF4E association | ↑ | ↑ | ↑ | ↑ |
| eIF4E phosphorylation (S209) | ― | ― | ND | |
| S6K1 phosphorylation (T389) | ↓ | |||
| S6 phosphorylation (S240/244) | ↓ | ↓ | ↓ | ND |
| eIF2B activity | ND | ↓ | ND | ― |
| eIF2Bε | ND | ― | ND | ― |
| eIF2α phosphorylation (T51) | ― | ― | ||
| eEF1A | ― | ↓ | ― | ↓ |
| eEF2 | ― | ― | ― | ↓ |
| eEF2 phosphorylation (T56) | ↓ | ― | ― | ― |
| Proteasome activity | ― | ― | ND | ― |
| Atrogin-1/MuRF1 | ↑ | ↑ | ― | ― |
| LC3B-II/I | ― | ↑ | ↑ | |
Directional changes in pathways and regulators of protein homeostasis in skeletal and cardiac muscle in response to acute alcohol intoxication (hours) and after chronic consumption (≥ 6 weeks) of an alcohol-containing diet. Original citations are contained in the text of the manuscript. Symbols: ↑, increased compared to time-matched control value; ↓ decreased compared to time-matched control value; ―, no change compared to time-matched control value; 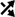 , reported changes are equivocal and not definitive.
, reported changes are equivocal and not definitive.
2.1. Muscle mass
Alcoholic skeletal muscle myopathy in humans is characterized by the loss of lean mass that can approach 20% of whole-body mass in severe conditions (110). This atrophic response to chronic excessive alcohol ingestion is often characterized by what is termed proximal wasting and evidenced by a reduction in the mass and cross-sectional area of muscles with a predominance of fast-twitch type II fibers as opposed to type I fibers (16, 26, 77, 85). Comparable changes have also been detected in rats and mice fed a nutritionally complete diet containing alcohol for at least 6 weeks (62, 87), providing reassurance on the fidelity of the preclinical model used.
In contrast, the development of alcoholic heart muscle disease often requires months of sustained alcohol intake (52). Divergent results exist regarding the effect of chronic alcohol-feeding on heart weight and protein content. Most animal studies report a decrease in ventricular weight or protein content in rodents (57, 116–118) or a thinning of the ventricular wall and septum is typically observed in animals and humans leading to ventricular dysfunction (52, 114). However, there are also now several studies reporting cardiac hypertrophy (i.e., increased heart-to-body weight ratio) in alcohol-fed mice (24, 64), but the mechanism underlying the difference from earlier reports remains unclear.
2.2. Protein synthesis
Early studies demonstrated a relatively specific decrease in in vivo-determined total mixed protein synthesis in fast- versus slow-twitch skeletal muscle of chronic alcohol-fed rats and mice, consistent with the presence of type I atrophy (62, 87). A decrease in global protein synthesis has also been reported in skeletal muscle from humans with chronic alcohol use (78). In rodent models, this inhibitory effect can be observed as early as 7–14 days after initiating feeding (86) and there is no refractoriness for up to at least 4 months (53, 117). Subsequently, alcohol was demonstrated to impair the synthetic rate of both sarcoplasmic and myofibrillar pools in skeletal muscle (62, 85, 88), and this may be causally related to the decrease in contractile proteins such as the Iβ, IIx and IIβ myosin isoforms (91), titin and nebulin (34).
A comparable decrease in skeletal muscle protein synthesis occurs in response to acute alcohol intoxication (85). This alcohol-induced decrease manifests rapidly within the first hour after intoxication and, depending on the initial dose administered, can be maintained for up to 24 hours, a time when the BAC was nondetectable (60, 90). The acute alcohol-induced decrease in synthesis is independent of the route of administration (oral versus intraperitoneal) as well as the sex and age of the rat (55, 60). Independent lines of investigation suggest that alcohol can directly affect skeletal muscle. For example, alcohol decreases protein synthesis in cultured myocytes, incubated whole muscle and the isolated perfused hindlimb (33, 60, 107). A direct action of alcohol on muscle is further supported by studies where animals were pretreated with a chemical inhibitor of alcohol dehydrogenase and the reduction in muscle protein synthesis was equivalent to that seen in vehicle-treated animals (55, 85). However, muscle protein synthesis can be inhibited by acetaldehyde, the key metabolite of alcohol oxidation, when present in high concentrations. For example, protein synthesis is reduced in myoblasts and myotubes cultured with acetaldehyde (33, 107) as well as in animals pretreated with a chemical inhibitor of acetaldehyde dehydrogenase (85).
Acute alcohol intoxication also decreases global protein synthesis in heart as well as the synthetic rate for cardiac myofibrillar, non-myofibrillar and mitochondrial proteins (50, 79, 97, 111). However, in contrast to skeletal muscle, animals must often ingest alcohol for extended periods of time (at least 12–14 weeks) before decreases in myocardial protein synthesis are detected (112, 116). In general, ribosomal number as estimated by total RNA is not altered by alcohol in either heart or skeletal muscle, suggesting that alcohol impairs protein synthesis by decreasing translational efficiency (50, 62).
2.3. Translational control
2.3.1. Regulation of mRNA translation
The translation of mRNA into protein occurs in three steps: initiation, elongation, and termination (Fig. 1). During initiation, the 40S ribosomal subunit binds to initiator methionyl-tRNAi (met-tRNAi) to form the 43S pre-initiation complex that subsequently binds to the mRNA, often at the 5’ m7GTP cap structure, to form the 48S pre-initiation complex (28). The complex then scans the 5’-untranslated region (5’-UTR) to locate the AUG start codon. The 60S ribosomal subunit joins the complex to form the active 80S ribosome that translates the open reading frame during the elongation step. Assembly of the 43S pre-initiation complex is mediated by a heterotrimeric complex referred to as eukaryotic initiation factor (eIF) 2. During formation of the 43S pre-initiation complex, eIF2 binds to GTP and met-tRNAi forming a ternary complex that then binds to the 40S ribosomal subunit. During the last step in initiation, the GTP bound to eIF2 is hydrolyzed and the eIF2•GDP complex is released from the complex, leaving behind the met-tRNAi. For eIF2 to re-bind met-tRNAi, the GDP bound to it must be exchanged for GTP, a process catalyzed by the guanine nucleotide exchange factor (GEF), eIF2B. The GEF activity of eIF2B is regulated indirectly through phosphorylation of the substrate, eIF2, whereby phosphorylation of eIF2 on its α-subunit converts the protein from a substrate into a competitive inhibitor of eIF2B.
Figure 1.
Key steps in the initiation phase of mRNA translation. The initiation step in translation involves the binding of the ternary complex, consisting of eukaryotic initiation factor 2 (eIF2) associated with GTP and the initiator form of methionyl-tRNA (met-tRNAi), to the 40S ribosomal subunit to form the 43S preinitiation complex. The eIF4F•eIF4B complex in association with mRNA subsequently binds to form the 48S preinitiation complex. The 40S ribosomal subunit then scans along the 5’-untranslated region of the mRNA and stops at an AUG start codon, triggering hydrolysis of GTP bound to eIF2 to GDP leading to release of the eIF2•GDP and eIF4F•eIF4B complexes. The GDP bound to eIF2 is exchanged for GTP by eIF2B and the eIF4F•eIF4B complex binds to another mRNA to re-start the process. GCN2 phosphorylates eIF2 on its α-subunit, converting it from a substrate of eIF2B into a competitive inhibitor, leading to decreased ternary complex formation. mTORC1 promotes assembly of the eIF4F complex through phosphorylation of the eIF4E binding proteins (4E-BPs) and programmed cell death 4 (PDCD4). Phosphorylation of 4E-BP by mTORC1 releases it from the eIF4E•4E-BP complex, allowing eIF4E to bind to eIF4G. Similarly, phosphorylation of PDCD4 by p70S6K1 (which is also activated by mTORC1) frees eIF4A from the eIF4A•PDCD4 complex, allowing it to bind to eIF4G. In addition, p70S6K1 phosphorylates eIF4B, thereby enhancing its stimulatory activity toward eIF4A.
Binding of mRNA to the 43S preinitiation complex is mediated by a complex of initiation factors referred to as eIF4F that is comprised of eIF4A, eIF4E, and eIF4G. eIF4E binds to both the m7GTP cap and eIF4G (Fig. 1). eIF4A is a RNA helicase whose activity is enhanced by eIF4B, and acts to unwind secondary structure in the 5’-UTR allowing the 40S ribosomal subunit to scan. In addition to binding to eIF4A and eIF4E, eIF4G binds to the 43S pre-initiation complex, thereby localizing it to the m7GTP cap. Assembly of the eIF4F complex is regulated through the interaction of eIF4A with programmed cell death 4 (PDCD4) and the interaction of eIF4E with the eIF4E binding proteins (4E-BP1–3). Binding of PDCD4 to eIF4A and the 4E-BPs to eIF4E prevents them from binding to eIF4G, leading to decreased cap-dependent binding of mRNA to the 43S preinitiation complex. The binding PDCD4 (programmed cell death 4) to eIF4A and 4E-BPs to eIF4E is regulated through phosphorylation of the binding proteins, whereby phosphorylation attenuates the binding of PDCD4 to eIF4A and the binding of 4E-BPs to eIF4E.
2.3.2. Regulation of mRNA translation by mTORC1
The mechanistic target of rapamycin (mTOR) complex 1 promotes the binding of mRNA to the 43S pre-initiation complex, in part, by phosphorylating the 4E-BPs, freeing eIF4E to bind to eIF4G 1 (81). Similarly, by activating the 70 kDa ribosomal protein S6 kinase 1 (p70S6K1), mTORC1 promotes phosphorylation of both PDCD4 and eIF4B thereby enhancing eIF4F assembly and function. Each of these phosphorylation events is necessary for maximal stimulation of protein synthesis by mTORC1 (12). As most mRNAs are translated in a cap-dependent manner, it is not surprising that inhibition of mTORC1, e.g. with Torin1, leads to a greater than 50% decrease in protein synthesis (109). However, inhibition of mTORC1 has a preferential effect on the translation of certain mRNAs, including those with a terminal oligopyrimidine (TOP) sequence at or near the 5’-cap (109) as well as those with highly-structured 5’ UTRs (63).
2.3.3. Regulation of mTOR by hormones and amino acids
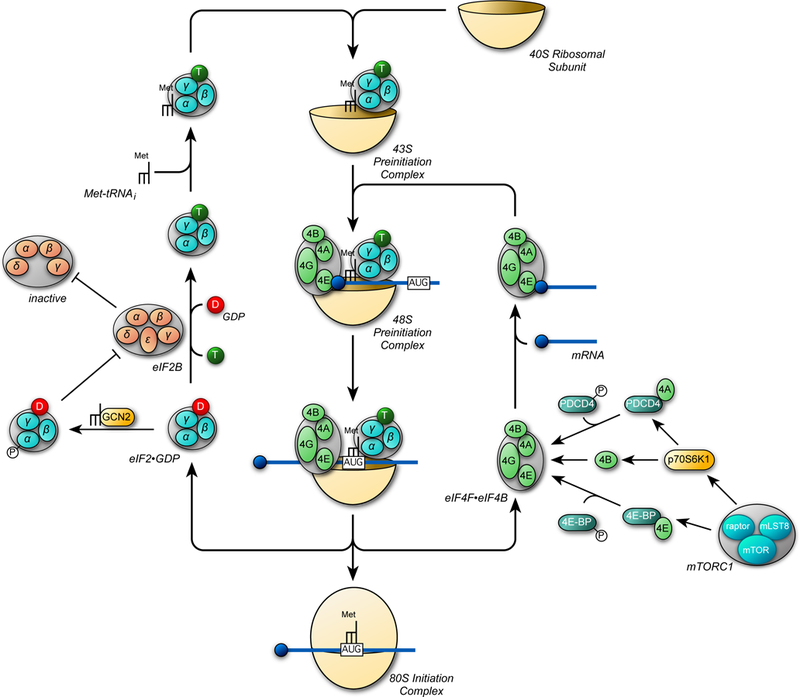
An obligate step in mTORC1 activation is its association with the small GTPase referred to as Ras homolog enriched in brain (Rheb, 7, 18, 35, 106) (Fig. 2). Rheb is farnesylated (9) and its localization to the cytoplasmic surface of late endosomal and/or lysosomal (LEL) membranes is necessary for it to activate mTORC1 (5). Although mTORC1 can bind to Rheb in either its GDP- or GTP-bound form, only binding to Rheb•GTP enhances the recruitment of substrates to mTORC1 in a Raptor-dependent manner (96). The GTP loading status of Rheb depends primarily on the activity of a GTPase activating protein (GAP) referred to as tuberous sclerosis complex 2 (TSC2, a.k.a. tuberin) (35, 122). TSC2 functions as part of a heterotrimeric complex to stimulate the GTPase activity of Rheb leading to decreased mTORC1 activation. The GAP activity of TSC2 is inhibited by phosphorylation by Akt (36). Akt is activated by phosphorylation on two key residues, Thr308 and Ser473 (69). Akt Thr308 is phosphorylated by phosphoinositide-dependent protein kinase 1 (PDK1) whereas Ser473 is phosphorylated by mTORC2. Although this might suggest that mTORC2 is an upstream regulator of mTORC1 activity, phosphorylation of Akt on Ser473 is dispensable for Akt-mediated TSC2 phosphorylation (23, 37), suggesting that mTORC1 is regulated by insulin and insulin-like growth factor I (IGF-I) in a PDK1-dependent but mTORC2-independent manner. In addition to Akt, the extracellular regulated kinase (ERK) and the 90 kDa ribosomal protein S6 kinase (p90RSK) also inactivate TSC2 GAP activity toward Rheb through phosphorylation on residues distinct from those phosphorylated by Akt (68, 94).
Figure 2.
Insulin/IGF-I signaling to mTORC1. Insulin and IGF-I both activate phosphatidylinositol 3-kinase (PI3K) leading to production of phosphatidylinositol-3,4,5-trisphosphate (PIP3) on the cytoplasmic face of the plasma membrane. PDK1, Akt, and mSIN1 have pleckstrin homology domains that interact with PIP3 resulting in their localization at the plasma membrane (67, 80). In addition, binding of mSIN1 to PIP3 results in activation of mTORC2 (67). Phosphorylation of Akt on Thr308 is sufficient for phosphorylation, and thus inactivation of TSC2 (23, 37). Phosphorylation of TSC2 by Akt leads to dissociation of the TSC complex from the late endosomal/lysosomal membrane, allowing Rheb to accumulate in the active GTP-bound form (6). Whether or not phosphorylation of TSC2 by either ERK- or p90RSK promotes TSC dissociation from the LEL membrane is unknown.
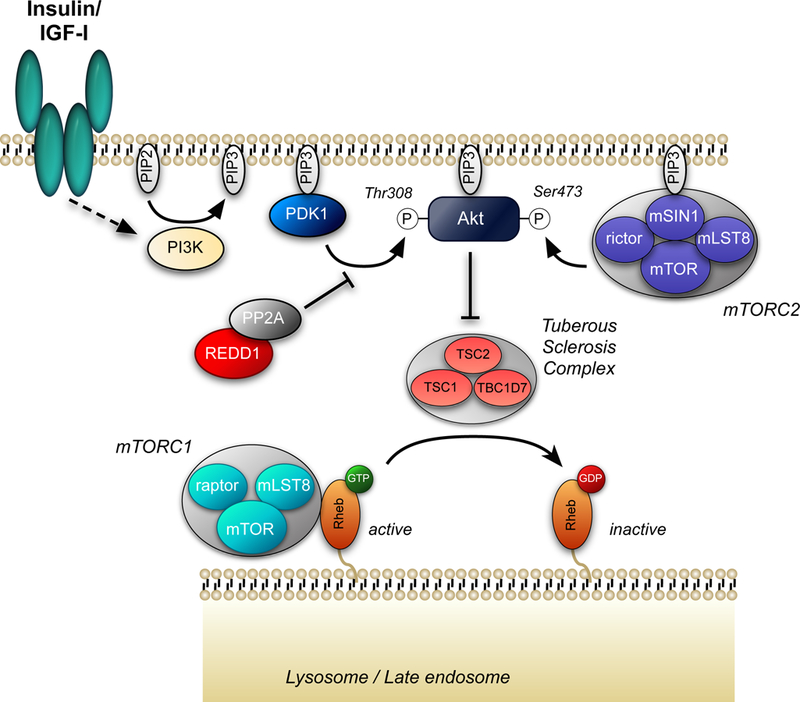
In contrast to insulin and IGF-I, amino acids activate mTORC1 in mouse embryo fibroblasts lacking TSC2 (93, 99). Moreover, whereas exogenous expression of a Rheb variant that constitutively binds GTP is sufficient to maintain mTORC1 in its active state in serum-deprived cells, it does not prevent the downregulation of mTORC1 activity caused by amino acid deprivation (11). Thus, most studies suggest that amino acids activate mTORC1 in a TSC2- and Rheb-independent manner. Instead, amino acids act through the Ras-related GTP-binding (Rag) proteins to activate mTORC1 (for recent reviews see: 4, 72, 121). The Rag proteins form a heterodimer consisting of either Rag A or B associated with either Rag C or D that binds to mTORC1, but not to mTORC2 (95). Interestingly, it is a complex of the GTP-bound form of Rag A or B and the GDP-bound form of Rag C or D that maximally activates mTORC1 (Fig. 3).
Figure 3.
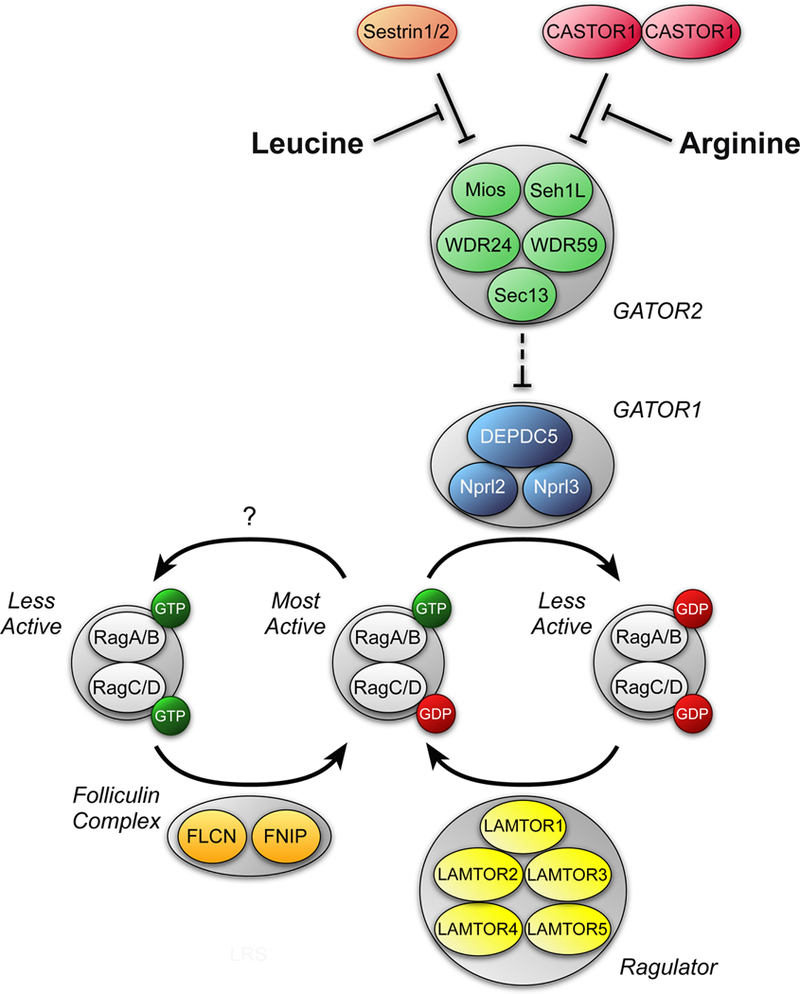
Regulation of mTORC1 by amino acids. Leucine and arginine activate mTORC1 in part by modulating the GTP loading status of Rags A and B. Leucine binds to Sestrins 1 and 2 and promotes their dissociation from GATOR2 (120), which consists of the proteins meiosis regulator for oocyte development (Mios), WD repeat-domain-containing proteins 24 and 59 (WDR24 and WDR59, respectively, Sec13-like protein (Sec13), and Sec13. Similarly, arginine binds to a protein referred to as cellular arginine sensor for mTORC1 (CASTOR) 1 and promotes its dissociation from GATOR2 (8). Binding of either Sestrin1/2 or CASTOR1 to GATOR2 represses the inhibitory effect of GATOR1 (consisting of DEP domain containing 5 (DEPDC5) and nitrogen permease regulator-like 2 and 3 (Nprl2 and Nprl3)) GAP activity toward Rags A and B. Ragulator acts to oppose the GAP activity of GATOR1 (comprised of LEL adaptor and MAPK and mTORC1 activators (LAMTOR) 1–5), and instead acts to promote exchange of GDP for GTP on Rags A and B. However, the mechanism through which amino acids act to regulate Ragulator GEF activity is unknown. The folliculin complex, composed of folliculin (FLCN) and the folliculin-interacting protein (FNIP), has have been reported to act as GAPs for Rags C and/or D. Neither the mechanism through which folliculin is regulated by amino acids nor its amino acid selectivity are known. Note that the RagA/B-GDP•RagC/D-GTP complex is not shown for simplicity, but would be expected to be less active than the RagA/G-GTP•RagC/D-GDP complex in activating mTORC1.
The GTP loading status of Rag A and B depends in part on the GTPase stimulating activity of a protein complex referred to as GAP activity towards Rags (GATOR) 1. By promoting GTP hydrolysis by Rag A and B, GATOR1 acts to repress mTORC1 activity. In contrast, an independent complex, GATOR2, opposes GATOR1 function leading to mTORC1 activation. Recent studies have identified three GATOR2-interacting proteins that act as amino acid sensors to transduce the signal from leucine, arginine, and methionine to GATOR2, and subsequently to mTORC1. Specifically, in cells deprived of leucine, arginine, or methionine, the Sestrin proteins 1, 2, and 3 (82, 120), the cellular arginine sensor for mTORC1 (CASTOR1) (8), and S-adenosylmethionine sensor upstream of mTORC1 (SAMTOR) (22) respectively, bind to and repress the function of GATOR2. Adding back the deprived amino acid results in dissociation of the repressor protein from GATOR2 leading to mTORC1 activation.
2.3.4. mTOR regulation by alcohol
There is considerable inconsistency in the published data pertaining to alcohol-induced changes in signaling upstream of mTORC1 in skeletal muscle (Table 1). For example, while there are minor or no changes in the phosphorylation state of the insulin receptor, IGF-I receptor, and insulin-like substrate-1 in response to alcohol (46, 73), Thr308-phosphorylated Akt has been reported to be decreased (47, 73, 104) or unchanged (46, 47, 105) by either acute or chronic alcohol intake. A dose-dependent effect of alcohol on Akt phosphorylation may account for some of this discrepancy (17). Where assessed, there is no change in the relative phosphorylation of the Akt substrate PRAS40 on Thr246 between alcohol-fed and pair-fed control rats (46). Furthermore, no alcohol-induced change in TSC2 phosphorylation or the association of TSC1•TSC2 in skeletal muscle has been detected (61). As a result of these equivocal changes, many studies have focused on unraveling alcohol-induced changes that negatively impact the activity of mTORC1 and protein-protein interactions within mTORC1 that are illustrated in Figure 1.
As highlighted above, mTORC1 is a central regulator of mRNA translation and there is considerable information pertaining to the role of this kinase in the development of alcoholic myopathy. A decrease in mTORC1 activity is often evidenced in vivo by decreased phosphorylation of 4E-BP1 and S6K1 – two authentic downstream substrates for this Ser/Thr kinase (Fig. 1). In this regard, there is a consistent decrease in 4E-BP1 phosphorylation in skeletal muscle in response to either acute exposure or chronic ingestion of alcohol. However, a coordinate decrease in S6K1 phosphorylation has only been seen in some (45, 73) but not other (49, 54, 60, 61, 100) studies, and the mechanism for alcohol’s preferential inhibitory effect on 4E-BP1 phosphorylation remains to be elucidated. The alcohol-induced decrease in 4E-BP1 phosphorylation is physiologically significant, as this change was associated with a relative increase in the binding of the translational repressor molecule 4E-BP1 with eIF4E, and a reciprocal decrease in the formation of the active eIF4E•eIF4G complex (46, 49, 54). Such changes would be expected to decrease cap-dependent translation. Despite the variable effect of alcohol on S6K1 phosphorylation, acute and chronic alcohol has consistently been shown to decrease the phosphorylation of several downstream target proteins, such as S6 (Ser240/244), eIF4G (Ser1108) and eIF4B (Ser422), but not the phosphorylation of PDCD4 (49, 54, 61). All of these changes in protein phosphorylation are independent of changes in the relative amount of the total protein. By placing the alcohol-fed rats on a diet without alcohol for 3 days, all of the above mentioned alcohol-induced changes in mTOR signaling and protein synthesis in skeletal muscle could be reversed (118). Finally, the abundance and phosphorylation of eIF4E, which can enhance cap-dependent translation when increased, do not differ in skeletal muscle of control and alcohol-fed rats (62). A direct inhibitory effect of alcohol on in vitro-determined mTORC1 kinase activity has also been reported in cultured myocytes as have the reduction in the phosphorylation state of downstream target proteins (30).
The suppressive effects of alcohol on mTORC1 activity cannot be attributed to alterations in the total cellular content of the various core and associated proteins (e.g., mTOR, Raptor, mLST8, Deptor, and PRAS40) in the complex, either under in vivo (59, 61) or in vitro (30, 31) conditions. However, alcohol feeding markedly alters various protein-protein interactions within this complex that are internally consistent with the reduction in muscle protein synthesis (59). Specifically, alcohol increases the molecular interaction of the scaffolding protein Raptor with the negative regulatory protein Deptor, while simultaneously decreasing the association of Raptor with 4E-BP1 (45). While the total amount of RagA and RagC in muscle of alcohol-fed rats is unchanged, there is a marked decrease in the binding of Rag A to Raptor that might be expected to impair nutrient-stimulation of protein synthesis (45). Finally, although unexpected, it has been repeatedly demonstrated that the extent of mTOR binding to Raptor is increased by alcohol. Such findings may indicate alcohol promotes a “closed conformation” rendering it less active. Chronic alcohol also increases Ser792-phosphorylated Raptor (45, 59). As AMP-activated protein kinase (AMPK) phosphorylates this reside on Raptor, and thereby inhibits mTORC1 activity, it was therefore unexpected that there is no concomitant increase in AMPK activity in skeletal muscle of chronic alcohol-fed rats. The reason for this disconnect between AMPK activity and Raptor phosphorylation is unknown. Finally, the relative abundance of the inhibitory protein REDD1 (regulated in development and DNA damage responses 1) in skeletal muscle is also unchanged by acute and chronic alcohol consumption (45, 54, 103).
For cardiac muscle, there is consensus that the phosphorylation of both 4E-BP1 and S6K1 is decreased by acute alcohol intoxication (50, 111) and chronic alcohol ingestion (114, 116, 118). It is noteworthy that chronic alcohol feeding has only been reported to decrease protein synthesis and mTORC1 in male, but not female, animals (57, 114). When observed, the decrease in myocardial mTORC1 appears governed in part by the generation of acetaldehyde as overexpression of mitochondrial aldehyde dehydrogenase prevents the alcohol-induced decrease in 4E-BP1 and S6K1 phosphorylation (20). Additionally, while the relative abundance of all proteins in mTORC1 were unchanged in alcohol-fed animals, there was an increased binding of mTOR with Raptor similar to that observed in skeletal muscle (61).
In contrast to skeletal muscle, there is general agreement that acute and chronic alcohol increases AMPK phosphorylation (activity) in heart (25, 40, 57). Results pertaining to the abundance of another negative regulator of mTORC1, REDD1, indicate either an increase (57) or no change (115) in heart in response to alcohol feeding. As these studies were performed in rats and mice, respectively, a species-specific response cannot be excluded. Furthermore, the bulk of the data suggest the phosphorylation of T308- or S473-Akt, a positive effector of mTORC1 activity, is not altered by either acute (14, 47, 58, 111) or chronic alcohol (115, 116). As myocardial mTORC1 activity is simultaneously determined to be decreased in all of the above mentioned studies, collectively these data suggest alterations in the phosphorylation state of AMPK and Akt are not essential for the alcohol-induced decrease in cardiac protein synthesis.
2.3.5. 43S Pre-initiation complex
As described in Section 2.3.1, protein synthesis may in part be mediated through the formation of the 43S preinitiation complex (Figures 4 and 5A). In this regard, chronic alcohol decreased eIF2B activity that is required to regenerate the active eIF2•GTP complex to which the met-tRNAimet binds (62). The underlying mechanism for this change has not been pursued since the original observation, but appears independent of alcohol-induced changes in the expression of the catalytic subunit eIF2Bε as well as the amount of total and phosphorylated eIF2α (62). Furthermore, decreased eIF2B activity is not detected in skeletal muscle after acute alcohol intoxication (51), suggesting a more sustained exposure is necessary to evoke the change. Counter to what is seen in skeletal muscle, eIF2B activity in heart does not differ between alcohol- and pair-fed control rats (62). Furthermore, there are no alcohol-induced changes in the abundance of eIF2Bε or eIF2α (62, 64). There are no reports on the effects of acute alcohol intoxication on the eIF2/2B system and 43S preinitiation complex formation in heart. These acute and chronic effects of alcohol in skeletal and cardiac muscle are summarized in Table 1.
2.3.6. Translation elongation and termination
Chronic alcohol consumption decreases the RNA content of free nonpolysome-associated 40S and 60S subunits in fast-twitch, but not slow-twitch, skeletal muscle suggesting alcohol also impairs the elongation-termination phase of translation (62). Consistent with this observation, the protein abundance of eukaryotic elongation factor (eEF)1A is decreased in skeletal muscle from alcohol-fed rats (117). Such a reduction would be consistent with impaired translocation of aminoacyl-tRNA to the A-site of the ribosome. Contrary to expectations, the amount of total and Thr56-phosphorylated eEF2, which directs the movement of tRNA to the P-site, is not different in muscle from alcohol-fed and control rats (117). However, acute alcohol intoxication in mice decreases eEF2 phosphorylation, which would be expected to enhance elongation, but does not alter total or Ser1108 phosphorylation of eEF1A (117). In contrast, eEF2 phosphorylation is increased in myocytes incubated with alcohol (29). More research is needed to directly measure the impact of alcohol on elongation per se, and to explore the mechanism underlying these differences between acute and chronic alcohol on translation elongation in skeletal muscle.
There are few data on the effects of alcohol on translation elongation in heart. In contrast to skeletal muscle, chronic alcohol feeding decreases both eEF1A and eEF2 in heart (112, 117, 118). However, similar to muscle, no inhibitory effect is seen in heart in response to acute alcohol intoxication (117). The relative importance of chronic alcohol-induced decreases in elongation versus initiation remain to be determined and represent a gap in our knowledge.
2.4. Leucine resistance
Branched-chain amino acids in general and leucine in particular are fundamental nutrient signals regulating the translational control of muscle protein synthesis (see Section 2.3.3 and Fig.3). In this regard, alcohol markedly impairs the typical stimulatory effects of orally administered leucine. For example, the ability of a maximally stimulating dose of leucine to enhance protein synthesis and increase 4E-BP1 phosphorylation as well as increase the formation of the active eIF4E•eIF4G complex in skeletal muscle is attenuated by acute alcohol intoxication (49). Furthermore, acute alcohol completely blocks leucine’s ability to increase the phosphorylation of S6K1 (Thr389 and Thr421/424) and S6. Similarly, alcohol inhibits mTORC1 activity stimulated by an intravenous infusion of a complete amino acid mixture (100). This alcohol-induced leucine resistance could not be attributed to differences in the prevailing plasma concentration of leucine or insulin (e.g., leucine is an insulin secretagogue) after oral leucine (49). Similarly, culture of myocytes with alcohol for 24 hours antagonizes the ability of leucine to promote phosphorylation of mTOR, S6K1, S6 and 4E-BP1 as well as blunts the interaction between mTOR and Rheb (31). The available literature is consistent with the role of leucine resistance as a contributor to the development of alcoholic skeletal muscle myopathy. In contrast, acute alcohol intoxication does not impair the ability of oral leucine to increase cardiac protein synthesis, the formation of the active eIF4E•eIF4G complex, or the phosphorylation of 4E-BP1 and mTOR (111). No data are currently available on whether chronic alcohol consumption alters the anabolic response of the heart to either leucine or refeeding. Hence, alcohol appears to produce a tissue-specific leucine resistance. Finally, there are large gaps in knowledge related to alcohol’s effect on the various regulator protein complexes (e.g., GATOR, CASTOR, Sestrin, Ragulator) under either basal or after nutrient stimulation.
2.5. Circulating hormones and substrates
Alcohol-induced decreases in muscle protein synthesis could be mediated by a decreased concentration of anabolic hormones (e.g., insulin, growth factors and testosterone) or an increased concentration of catabolic hormones (glucocorticoids). Similarly, a change in the prevailing concentration of total or branched-chain amino acids may also govern proportional changes in muscle protein synthesis. The overwhelming number of studies indicate that acute and chronic alcohol either does not significantly alter or slightly increases the plasma insulin concentration in humans and rodents (as reviewed in (102). Likewise, under well-controlled experimental conditions, inhibitor studies have demonstrated that the suppressive effects of alcohol are not due to elevations in the plasma corticosterone concentration (49, 55). Finally, there are no differences in the concentration of total amino acids, branched-chain amino acids or leucine between control and alcohol-fed rats (3, 59). Hence, the consensus from the available literature suggests that changes in the concentration of selected hormones and protein substrates are not causally related to the reduction in muscle protein synthesis.
The exception to this generalization is the decreased concentration of the anabolic hormone IGF-I in blood and muscle of chronic alcohol-fed animals that has been consistently reported by independent laboratories using several preclinical models (48, 101). The alcohol-induced decrease in the plasma IGF-I concentration or the IGF-I mRNA/protein content in skeletal muscle is directly correlated with a reduction in protein synthesis and the formation of the active eIF4E•eIF4G complex (56). Moreover, protein synthesis can be increased back to pair-fed control levels in alcohol-fed rats injected with IGF-I formulated to prolong its circulating half-life (53). In contrast, plasma IGF-I levels are not routinely decreased by acute alcohol intoxication (59) and therefore this potential mechanism does not appear operational under this circumstance. However, acute alcohol intoxication does impair the normal stimulatory actions of IGF-I (and insulin) on S6K1 and S6 phosphorylation in skeletal muscle (46, 59) providing evidence for the presence of an anabolic resistant state in response to short-term alcohol exposure.
2.6. Protein degradation
2.6.1. Ubiquitin-proteasome pathway
The contribution of protein breakdown to the alcohol-induced type II atrophy remains controversial. Studies have reported the urinary excretion of 3-methylhistidine, a biomarker for myofibrillar degradation, is either unchanged (92), decreased (70) or increased (75). Estimates of global muscle proteolysis have also been reported to be unchanged in chronic alcohol infused monkeys (71) and chronic alcohol-fed rats (70). Finally, there is no change detected in global proteolysis of whole muscles or cultured myocytes incubated acutely (4–24 hours) with alcohol (33) or in the isolated perfused hindlimb or incubated epitrochlearis treated with alcohol (113).
Because of its central regulatory role in the degradation of myofibrillar proteins, the ubiquitin-proteasome pathway has also been examined (10). While chronic alcohol consumption has been reported to decrease in vitro-determined 20S proteasome activity in skeletal muscle (44), the majority of studies show no alcohol-induced change (45, 107, 113) in vitro-determined 20S proteasome activity in skeletal muscle. In contrast, there are now several reports that chronic alcohol feeding increases the mRNA content for two muscle-specific E3 ubiquitin ligases, muscle atrophy F-box (MAFbx or atrogin-1) and muscle RING finger-1 (MuRF1) (45, 76, 77), with acute alcohol intoxication increasing both ligases in a dose- and time-dependent manner (113). While these E3 ligases are causally related to muscle atrophy in other catabolic conditions based on data from gene knockout mice, there is also increasing appreciation that elevations in atrogin-1 and MuRF1 mRNA are not consistently associated with enhanced proteasome activity (1). Moreover, treatment of alcohol-fed rats with the antioxidant procysteine attenuated the type II fiber atrophy but exaggerated the elevation in atrogin-1 and MuRF1, thereby questioning their role in the loss of muscle mass (77). Finally, the activity of calpain 1 and 2, that are necessary for the initial cleavage of myofibrillar proteins, as well as their inhibitor calpastatin do not differ in skeletal muscle of control and alcohol-fed rats (44). Hence, there is currently a lack of compelling data to support the hypothesis that alcohol-induced skeletal muscle wasting results from an increase in global proteolysis and activation of the ubiquitin-proteasome pathway. Similarly, there is a paucity of data supporting the activation of this pathway in the etiology of alcoholic cardiomyopathy (57, 113).
2.6.2. Autophagy
Autophagy is an evolutionarily conserved process whereby intracellular macromolecules and organelles are engulfed by a double membrane that forms a structure referred to as the autophagosome (2). Subsequent fusion of the autophagosome with a lysosome results in acidification and digestion of the luminal contents by lysosomal enzymes. The digestion products, e.g. amino acids, can be metabolized to generate ATP or used in the liver for gluconeogenesis (41), thereby helping the organism to survive various stresses. For example, in wild type neonatal mice, a drop in blood amino acid concentrations shortly after birth leads to repression of mTORC1 activity, leading to activation of autophagy in skeletal muscle (13). The amino acids produced by autophagy are used in the liver for production of glucose to prevent perinatal hypoglycemia. In contrast, in mice in which the wild type Rag A gene is replaced with a constitutively active mutant, mTORC1 is not repressed after birth even though blood amino acids fall to a similar extent as in wild type mice, and activation of autophagy and induction of hepatic gluconeogenesis is impaired, resulting in hypoglycemia and death unless glucose is exogenously supplied. mTORC1 inhibits autophagy by phosphorylating Unc-51 like autophagy activating kinase 1 (ULK1) and UV Radiation Resistance Associated Gene (UVRAG) (42, 43). ULK1 is a protein kinase that activates an early step in autophagy and phosphorylation by mTORC1 represses its protein kinase activity (42). In contrast, UVRAG is part of a complex containing Vps34 that plays a role in autophagosome maturation and fusion with the lysosome (65). Phosphorylation of UVRAG leads to impairment of this step in autophagy (43).
As a second major mechanism central to the degradation and recycling of intracellular proteins and organelles, autophagy has been the focus of recently studies in the development of alcoholic myopathy. Lysosomes are central to this process as they contain a family of proteases, the cathepsins. However, neither chronic alcohol intake nor acute intoxication alters the activity of cathepsins B, D, H and L in skeletal muscle (44). In contrast, emerging evidence, although not entirely consistent, suggests alcohol may increase skeletal muscle autophagy. Specifically, autophagy-related gene (Atg)7, Beclin 1 and the abundance of the lipidated LC3B protein (i.e., LC3B-II) are all increased, while p62 (which is consumed during autophagy) is decreased in skeletal muscle of male alcohol-fed mice (107). Moreover, an increase in LC3B-II is also observed in muscle of alcoholic patients (107). In contrast, in alcohol-fed female mice, LC3B-II, p62 and ULK1 phosphorylation are all unchanged compared to control animals (103). Likewise, little or no change is detected in skeletal muscle from female mice after acute alcohol intoxication (104). As the alcohol-feeding model was similar in the above studies, it is possible that the discrepancies in these studies represent a sexual dimorphic response. However, there is also no apparent increase in autophagy in myoblasts isolated from chronic binge alcohol consuming male rhesus macaques (98).
Despite the uncertain nature of the above mentioned in vivo results from alcohol-treated animals, several studies have provided definitive data that relatively short-term incubation (6 to 24 hours) of C2C12 myotubes and myoblasts in vitro with alcohol increases autophagy (32, 107). For example, alcohol increases autophagy in myotubes as assessed by GFP-LC3B vesicle accumulation and the direct measurement of autophagic flux (107). Moreover, knockdown of Atg7 prevents alcohol-induced increases in autophagy and the decrease in myocyte cross sectional area. Further, chemical inhibitor studies indicate the acute in vitro stimulatory effect of alcohol on autophagy is largely dependent on the generation of acetaldehyde rather than the direct effects of alcohol. This general conclusion has been independently confirmed in alcohol-treated myoblasts where alcohol increases LC3B-II and Atg7, and decreases p62; changes that appear to be regulated by a FoxO1-ULK1-mediated increase in the binding of Atg14 with the BECN1•VPS34 complex (32). Additional studies are required to confirm whether alcohol increases autophagy in vivo in skeletal muscle and under what experimental conditions.
There is a more consistent literature pertaining to the effect of alcohol on cardiac autophagy. Collectively, the data indicate that both acute and chronic alcohol intake increase autophagy in heart as evidenced by an increased LC3B-II/I ratio and Atg7, and a decrease in Ser757-phosphorylated ULK-1 (19, 40). This increase in autophagy is mediated by elevated levels of acetaldehyde in the heart (19), and appears AMPK-dependent as it is largely prevented in alcohol-treated AMPK knockout mice (40). Similarly, short-term incubation of freshly isolated cardiomyocytes or H9c2 cells with alcohol also increases LC3B-II abundance, an effect that is prevented by pretreatment with the AMPK inhibitor Compound C or the knockdown of ULK-1 (19, 25, 40). Given the leucine resistance produced by alcohol, at least in skeletal muscle, additional studies should be directed to determine whether alcohol also antagonizes the ability of leucine or refeeding to suppress autophagy.
3. CONCLUSIONS AND DIRECTIONS FOR FUTURE DIRECTIONS
To summarize, acute intoxication and long-term heavy alcohol decrease the activity of the key regulator of protein homeostasis mTORC1 in skeletal and cardiac muscle. As a result, a host of downstream protein targets governing translation initiation and elongation are hypo-phosphorylated impairing both sarcoplasmic and myofibrillar protein synthesis. Emerging data indicate that, at least for skeletal muscle, alcohol also produces leucine resistance that minimizes the normal protein anabolic response to refeeding. The data also suggest that despite the similar decrease in mTORC1 and protein synthesis, the underlying mechanisms, as they related to protein-protein interactions with mTORC1 as well as upstream regulators, may differ between heart and skeletal muscle as well as between acute intoxication and chronic consumption. Because of the rapid recent advances in understanding basic nutrient signaling via mTORC1, there is a considerable knowledge gap in how alcohol affects these newly discovered regulatory elements. Finally, there is a paucity of data from humans that is essential to determine the translational nature of the mechanistic data generated from preclinical models and cell culture.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R37 AA11290 (CHL)) and (DK15658 (SRK)). The authors also wish to acknowledge the many collaborators they have had the pleasure of working with over the years and apologize to those authors whose work we failed to cite because of space limitations.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding or financial holdings that might be perceived as affecting the objectivity of this review.
4. REFERENCES
- 1.Atherton PJ, Greenhaff PL, Phillips SM, Bodine SC, Adams CM, Lang CH. 2016. Control of skeletal muscle atrophy in response to disuse: clinical/preclinical contentions and fallacies of evidence. Am J Physiol Endocrinol Metab 311: E594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, et al. 2016. Mammalian autophagy: How does It work? Annu Rev Biochem 85: 685–713 [DOI] [PubMed] [Google Scholar]
- 3.Bernal CA, Vazquez JA, Adibi SA. 1993. Leucine metabolism during chronic ethanol consumption. Metabolism: clinical and experimental 42: 1084–6 [DOI] [PubMed] [Google Scholar]
- 4.Broer S, Broer A. 2017. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J 474: 1935–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buerger C, DeVries B, Stambolic V. 2006. Localization of Rheb to the endomembrane is critical for its signaling function. Biochemical and Biophysical Research Communications 344: 869–80 [DOI] [PubMed] [Google Scholar]
- 6.Cai S-L, Tee AR, Short JD, Bergeron JM, Kim J, et al. 2006. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. Journal of Cell Biology 173: 279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. 2003. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. The Journal of Biological Chemistry 278: 32493–96 [DOI] [PubMed] [Google Scholar]
- 8.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, et al. 2016. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 165: 153–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark GJ, Kinch MS, Rogers-Graham K, Sebti SM, Hamilton AD, Der CJ. 1997. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J Biol Chem 272: 10608–15 [DOI] [PubMed] [Google Scholar]
- 10.Collins GA, Goldberg AL. 2017. The logic of the 26S proteasome. Cell 169: 792–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dennis MD, Baum JI, Kimball SR, Jefferson LS. 2011. Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J Biol Chem 286: 8287–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dennis MD, Jefferson LS, Kimball SR. 2012. Role of p70S6K1-mediated phosphorylation of eIF4B and PDCD4 proteins in the regulation of protein synthesis. Journal of Biological Chemistry 287: 42890–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, et al. 2013. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493: 679–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Mas MM, Abdel-Rahman AA. 2015. Estrogen modulation of the ethanol-evoked myocardial oxidative stress and dysfunction via DAPK3/Akt/ERK activation in male rats. Toxicology and Applied Pharmacology 287: 284–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Estruch R, Sacanella E, Fernandez-Sola J, Nicolas JM, Rubin E, Urbano-Marquez A. 1998. Natural history of alcoholic myopathy: a 5-year study. Alcoholism, Clinical and Experimental Research 22: 2023–8 [PubMed] [Google Scholar]
- 16.Fernandez-Sola J, Sacanella E, Estruch R, Nicolas JM, Grau JM, Urbano-Marquez A. 1995. Significance of type II fiber atrophy in chronic alcoholic myopathy. Journal of the Neurological Sciences 130: 69–76 [DOI] [PubMed] [Google Scholar]
- 17.Gao L, Zhang X, Wang FR, Cao MF, Zhang XJ, et al. 2010. Chronic ethanol consumption up-regulates protein-tyrosine phosphatase-1B (PTP1B) expression in rat skeletal muscle. Acta Pharmacologica Sinica 31: 1576–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garami A, Zwartkruis FJT, Nobukuni T, Joaquin M, Roccio M, et al. 2003. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP1 signaling, is inhibited by TSC1 and 2. Molecular Cell 11: 1457–66 [DOI] [PubMed] [Google Scholar]
- 19.Ge W, Guo R, Ren J. 2011. AMP-dependent kinase and autophagic flux are involved in aldehyde dehydrogenase-2-induced protection against cardiac toxicity of ethanol. Free Radical Biology & Medicine 51: 1736–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge W, Li Q, Turdi S, Wang XM, Ren J. 2011. Deficiency of insulin-like growth factor 1 reduces vulnerability to chronic alcohol intake-induced cardiomyocyte mechanical dysfunction: role of AMPK. Journal of Cellular and Molecular Medicine 15: 1737–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilmore W, Chikritzhs T, Stockwell T, Jernigan D, Naimi T, Gilmore I. 2016. Alcohol: taking a population perspective. Nature reviews. Gastroenterology & Hepatology 13: 426–34 [DOI] [PubMed] [Google Scholar]
- 22.Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, et al. 2017. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358: 813–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, et al. 2006. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 Is required for signaling to Akt-FOXO and PKCα, but not S6K1. Developmental Cell 11: 859–71 [DOI] [PubMed] [Google Scholar]
- 24.Guo R, Hu N, Kandadi MR, Ren J. 2012. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy 8: 593–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo R, Ren J. 2012. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovascular Research 94: 480–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanid A, Slavin G, Mair W, Sowter C, Ward P, et al. 1981. Fibre type changes in striated muscle of alcoholics. Journal of Clinical Pathology 34: 991–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hingson RW, Zha W, White AM. 2017. Drinking beyond the binge threshold: Predictors, consequences, and changes in the U.S. American journal of preventive medicine 52: 717–27 [DOI] [PubMed] [Google Scholar]
- 28.Hinnebusch AG. 2017. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem Sci [DOI] [PubMed]
- 29.Hong-Brown LQ, Brown CR, Huber DS, Lang CH. 2007. Alcohol regulates eukaryotic elongation factor 2 phosphorylation via an AMP-activated protein kinase-dependent mechanism in C2C12 skeletal myocytes. The Journal of biological chemistry 282: 3702–12 [DOI] [PubMed] [Google Scholar]
- 30.Hong-Brown LQ, Brown CR, Kazi AA, Huber DS, Pruznak AM, Lang CH. 2010. Alcohol and PRAS40 knockdown decrease mTOR activity and protein synthesis via AMPK signaling and changes in mTORC1 interaction. Journal of cellular biochemistry 109: 1172–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong-Brown LQ, Brown CR, Kazi AA, Navaratnarajah M, Lang CH. 2012. Rag GTPases and AMPK/TSC2/Rheb mediate the differential regulation of mTORC1 signaling in response to alcohol and leucine. American journal of physiology. Cell physiology 302: C1557–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong-Brown LQ, Brown CR, Navaratnarajah M, Lang CH. 2017. FoxO1-AMPK-ULK1 regulates ethanol-induced autophagy in muscle by enhanced ATG14 association with the BECN1-PIK3C3 complex. Alcoholism, Clinical and Experimental Research 41: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong-Brown LQ, Frost RA, Lang CH. 2001. Alcohol impairs protein synthesis and degradation in cultured skeletal muscle cells. Alcoholism, Clinical and Experimental Research 25: 1373–82 [PubMed] [Google Scholar]
- 34.Hunter RJ, Neagoe C, Jarvelainen HA, Martin CR, Lindros KO, et al. 2003. Alcohol affects the skeletal muscle proteins, titin and nebulin in male and female rats. The Journal of Nutrition 133: 1154–7 [DOI] [PubMed] [Google Scholar]
- 35.Inoki K, Li Y, Xu T, Guan K-L. 2003. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes and Development 17: 1829–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoki K, Li Y, Zhu T, Wu J, Guan K-L. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nature Cell Biology 4: 648–57 [DOI] [PubMed] [Google Scholar]
- 37.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, et al. 2006. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125–37 [DOI] [PubMed] [Google Scholar]
- 38.Jayasekara H, English DR, Room R, MacInnis RJ. 2014. Alcohol consumption over time and risk of death: a systematic review and meta-analysis. American journal of epidemiology 179: 1049–59 [DOI] [PubMed] [Google Scholar]
- 39.Jelic P, Shih MF, Taberner PV. 1998. Diurnal variation in plasma ethanol levels of TO and CBA mice on chronic ethanol drinking or ethanol liquid diet schedules. Psychopharmacology (Berl) 138: 143–50 [DOI] [PubMed] [Google Scholar]
- 40.Kandadi MR, Hu N, Ren J. 2013. ULK1 plays a critical role in AMPK-mediated myocardial autophagy and contractile dysfunction following acute alcohol challenge. Current pharmaceutical design 19: 4874–87 [DOI] [PubMed] [Google Scholar]
- 41.Kaur J, Debnath J. 2015. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: 461–72 [DOI] [PubMed] [Google Scholar]
- 42.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim YM, Jung CH, Seo M, Kim EK, Park JM, et al. 2015. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell 57: 207–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koll M, Ahmed S, Mantle D, Donohue TM, Palmer TN, et al. 2002. Effect of acute and chronic alcohol treatment and their superimposition on lysosomal, cytoplasmic, and proteosomal protease activities in rat skeletal muscle in vivo. Metabolism 51: 97–104 [DOI] [PubMed] [Google Scholar]
- 45.Korzick DH, Sharda DR, Pruznak AM, Lang CH. 2013. Aging accentuates alcohol-induced decrease in protein synthesis in gastrocnemius. American journal of physiology. Regulatory, integrative and comparative physiology 304: R887–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar V, Frost RA, Lang CH. 2002. Alcohol impairs insulin and IGF-I stimulation of S6K1 but not 4E-BP1 in skeletal muscle. American journal of physiology. Endocrinology and metabolism 283: E917–28 [DOI] [PubMed] [Google Scholar]
- 47.Lang CH, Derdak Z, Wands JR. 2014. Strain-dependent differences for suppression of insulin-stimulated glucose uptake in skeletal and cardiac muscle by ethanol. Alcoholism, Clinical and Experimental Research 38: 897–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lang CH, Fan J, Lipton BP, Potter BJ, McDonough KH. 1998. Modulation of the insulin-like growth factor system by chronic alcohol feeding. Alcoholism, Clinical and Experimental Research 22: 823–9 [PubMed] [Google Scholar]
- 49.Lang CH, Frost RA, Deshpande N, Kumar V, Vary TC, et al. 2003. Alcohol impairs leucine-mediated phosphorylation of 4E-BP1, S6K1, eIF4G, and mTOR in skeletal muscle. American journal of physiology. Endocrinology and metabolism 285: E1205–15 [DOI] [PubMed] [Google Scholar]
- 50.Lang CH, Frost RA, Kumar V, Vary TC. 2000. Impaired myocardial protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4F. American journal of physiology. Endocrinology and metabolism 279: E1029–38 [DOI] [PubMed] [Google Scholar]
- 51.Lang CH, Frost RA, Kumar V, Wu D, Vary TC. 2000. Impaired protein synthesis induced by acute alcohol intoxication is associated with changes in eIF4E in muscle and eIF2B in liver. Alcoholism, Clinical and Experimental Research 24: 322–31 [PubMed] [Google Scholar]
- 52.Lang CH, Frost RA, Summer AD, Vary TC. 2005. Molecular mechanisms responsible for alcohol-induced myopathy in skeletal muscle and heart. The international journal of biochemistry & cell biology 37: 2180–95 [DOI] [PubMed] [Google Scholar]
- 53.Lang CH, Frost RA, Svanberg E, Vary TC. 2004. IGF-I/IGFBP-3 ameliorates alterations in protein synthesis, eIF4E availability, and myostatin in alcohol-fed rats. American journal of physiology. Endocrinology and metabolism 286: E916–26 [DOI] [PubMed] [Google Scholar]
- 54.Lang CH, Frost RA, Vary TC. 2007. Skeletal muscle protein synthesis and degradation exhibit sexual dimorphism after chronic alcohol consumption but not acute intoxication. American journal of physiology. Endocrinology and metabolism 292: E1497–506 [DOI] [PubMed] [Google Scholar]
- 55.Lang CH, Frost RA, Vary TC. 2008. Acute alcohol intoxication increases REDD1 in skeletal muscle. Alcoholism, Clinical and Experimental Research 32: 796–805 [DOI] [PubMed] [Google Scholar]
- 56.Lang CH, Kimball SR, Frost RA, Vary TC. 2001. Alcohol myopathy: impairment of protein synthesis and translation initiation. The international journal of biochemistry & cell biology 33: 457–73 [DOI] [PubMed] [Google Scholar]
- 57.Lang CH, Korzick DH. 2014. Chronic alcohol consumption disrupts myocardial protein balance and function in aged, but not adult, female F344 rats. American journal of physiology. Regulatory, integrative and comparative physiology 306: R23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lang CH, Kumar V, Liu X, Frost RA, Vary TC. 2003. IGF-I induced phosphorylation of S6K1 and 4E-BP1 in heart is impaired by acute alcohol intoxication. Alcoholism, Clinical and Experimental Research 27: 485–94 [DOI] [PubMed] [Google Scholar]
- 59.Lang CH, Lynch CJ, Vary TC. 2010. Alcohol-induced IGF-I resistance is ameliorated in mice deficient for mitochondrial branched-chain aminotransferase. The Journal of Nutrition 140: 932–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lang CH, Pruznak AM, Deshpande N, Palopoli MM, Frost RA, Vary TC. 2004. Alcohol intoxication impairs phosphorylation of S6K1 and S6 in skeletal muscle independently of ethanol metabolism. Alcoholism, Clinical and Experimental Research 28: 1758–67 [DOI] [PubMed] [Google Scholar]
- 61.Lang CH, Pruznak AM, Nystrom GJ, Vary TC. 2009. Alcohol-induced decrease in muscle protein synthesis associated with increased binding of mTOR and raptor: Comparable effects in young and mature rats. Nutrition & metabolism 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lang CH, Wu D, Frost RA, Jefferson LS, Kimball SR, Vary TC. 1999. Inhibition of muscle protein synthesis by alcohol is associated with modulation of eIF2B and eIF4E. The American journal of physiology 277: E268–76 [DOI] [PubMed] [Google Scholar]
- 63.Larsson O, Morita M, Topisirovic I, Alain T, Blouin MJ, et al. 2012. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proceedings of the National Academy of Sciences 109: 8977–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li SY, Gilbert SA, Li Q, Ren J. 2009. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. Journal of molecular and cellular cardiology 47: 247–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang C, Lee JS, Inn KS, Gack MU, Li Q, et al. 2008. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10: 776–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieber CS. 2000. ALCOHOL: its metabolism and interaction with nutrients. Annual review of nutrition 20: 395–430 [DOI] [PubMed] [Google Scholar]
- 67.Liu P, Gan W, Chin YR, Ogura K, Guo J, et al. 2015. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer discovery 5: 1194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma L, Teruya-Feldstein J, Bonner P, Bernardi R, Franz DN, et al. 2007. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Research 67: 7106–12 [DOI] [PubMed] [Google Scholar]
- 69.Manning BD, Toker A. 2017. AKT/PKB signaling: Navigating the network. Cell 169: 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin FC, Peters TJ. 1985. Assessment in vitro and in vivo of muscle degradation in chronic skeletal muscle myopathy of alcoholism. Clinical science 68: 693–700 [DOI] [PubMed] [Google Scholar]
- 71.Molina PE, McNurlan M, Rathmacher J, Lang CH, Zambell KL, et al. 2006. Chronic alcohol accentuates nutritional, metabolic, and immune alterations during asymptomatic simian immunodeficiency virus infection. Alcoholism, Clinical and Experimental Research 30: 2065–78 [DOI] [PubMed] [Google Scholar]
- 72.Moro T, Ebert SM, Adams CM, Rasmussen BB. 2016. Amino acid sensing in skeletal muscle. Trends Endocrinol Metab 27: 796–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen VA, Le T, Tong M, Silbermann E, Gundogan F, de la Monte SM. 2012. Impaired insulin/IGF signaling in experimental alcohol-related myopathy. Nutrients 4: 1058–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nicolas JM, Garcia G, Fatjo F, Sacanella E, Tobias E, et al. 2003. Influence of nutritional status on alcoholic myopathy. The American journal of clinical nutrition 78: 326–33 [DOI] [PubMed] [Google Scholar]
- 75.Nunez NP, Carter PA, Meadows GG. 2002. Alcohol consumption promotes body weight loss in melanoma-bearing mice. Alcoholism, Clinical and Experimental Research 26: 617–26 [DOI] [PubMed] [Google Scholar]
- 76.Otis JS, Brown LA, Guidot DM. 2007. Oxidant-induced atrogin-1 and transforming growth factor-beta1 precede alcohol-related myopathy in rats. Muscle & nerve 36: 842–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Otis JS, Guidot DM. 2009. Procysteine stimulates expression of key anabolic factors and reduces plantaris atrophy in alcohol-fed rats. Alcoholism, Clinical and Experimental Research 33: 1450–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pacy PJ, Preedy VR, Peters TJ, Read M, Halliday D. 1991. The effect of chronic alcohol ingestion on whole body and muscle protein synthesis--a stable isotope study. Alcohol and alcoholism 26: 505–13 [DOI] [PubMed] [Google Scholar]
- 79.Patel VB, Siddiq T, Richardson PJ, Preedy VR. 2000. Alcohol-induced reductions in cardiac protein synthesis in vivo are not ameliorated by treatment with the dihydropyridine calcium channel blocker amlodipine. Alcoholism, Clinical and Experimental Research 24: 727–32 [PubMed] [Google Scholar]
- 80.Pearce LR, Komander D, Alessi DR. 2010. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11: 9–22 [DOI] [PubMed] [Google Scholar]
- 81.Pelletier J, Graff J, Ruggero D, Sonenberg N. 2015. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res 75: 250–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peng M, Yin N, Li MO. 2014. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell 159: 122–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Piano MR, Phillips SA. 2014. Alcoholic cardiomyopathy: pathophysiologic insights. Cardiovascular toxicology 14: 291–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Preedy VR, Adachi J, Asano M, Koll M, Mantle D, et al. 2002. Free radicals in alcoholic myopathy: indices of damage and preventive studies. Free Radical Biology & Medicine 32: 683–7 [DOI] [PubMed] [Google Scholar]
- 85.Preedy VR, Keating JW, Peters TJ. 1992. The acute effects of ethanol and acetaldehyde on rates of protein synthesis in type I and type II fibre-rich skeletal muscles of the rat. Alcohol and alcoholism 27: 241–51 [PubMed] [Google Scholar]
- 86.Preedy VR, Macallan DC, Griffin GE, Cook EB, Palmer TN, Peters TJ. 1997. Total contractile protein contents and gene expression in skeletal muscle in response to chronic ethanol consumption in the rat. Alcohol 14: 545–9 [DOI] [PubMed] [Google Scholar]
- 87.Preedy VR, Peters TJ. 1988. The effect of chronic ethanol feeding on body and plasma composition and rates of skeletal muscle protein turnover in the rat. Alcohol and alcoholism 23: 217–24 [PubMed] [Google Scholar]
- 88.Preedy VR, Peters TJ. 1990. Changes in protein, RNA and DNA and rates of protein synthesis in muscle-containing tissues of the mature rat in response to ethanol feeding: a comparative study of heart, small intestine and gastrocnemius muscle. Alcohol and alcoholism 25: 489–98 [PubMed] [Google Scholar]
- 89.Preedy VR, Salisbury JR, Peters TJ. 1994. Alcoholic muscle disease: features and mechanisms. The Journal of pathology 173: 309–15 [DOI] [PubMed] [Google Scholar]
- 90.Reilly ME, Mantle D, Richardson PJ, Salisbury J, Jones J, et al. 1997. Studies on the time-course of ethanol’s acute effects on skeletal muscle protein synthesis: comparison with acute changes in proteolytic activity. Alcoholism, Clinical and Experimental Research 21: 792–8 [PubMed] [Google Scholar]
- 91.Reilly ME, McKoy G, Mantle D, Peters TJ, Goldspink G, Preedy VR. 2000. Protein and mRNA levels of the myosin heavy chain isoforms Ibeta, IIa, IIx and IIb in type I and type II fibre-predominant rat skeletal muscles in response to chronic alcohol feeding. Journal of muscle research and cell motility 21: 763–73 [DOI] [PubMed] [Google Scholar]
- 92.Reinus JF, Heymsfield SB, Wiskind R, Casper K, Galambos JT. 1989. Ethanol: relative fuel value and metabolic effects in vivo. Metabolism 38: 125–35 [DOI] [PubMed] [Google Scholar]
- 93.Roccio M, Bos JL, Zwartkruis FJT. 2005. Regulation of the small GTPase Rheb by amino acids. Oncogene 25: 657–64 [DOI] [PubMed] [Google Scholar]
- 94.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proceedings of the National Academy of Sciences USA 101: 13489–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, et al. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato T, Nakashima A, Guo L, Tamanoi F. 2009. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. Journal of Biological Chemistry 284: 12783–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Siddiq T, Salisbury JR, Richardson PJ, Preedy VR. 1993. Synthesis of ventricular mitochondrial proteins in vivo: effect of acute ethanol toxicity. Alcoholism, Clinical and Experimental Research 17: 894–9 [DOI] [PubMed] [Google Scholar]
- 98.Simon L, LeCapitaine N, Berner P, Vande Stouwe C, Mussell JC, et al. 2014. Chronic binge alcohol consumption alters myogenic gene expression and reduces in vitro myogenic differentiation potential of myoblasts from rhesus macaques. American journal of physiology. Regulatory, integrative and comparative physiology 306: R837–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. 2005. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. The Journal of Biological Chemistry 280: 18717–27 [DOI] [PubMed] [Google Scholar]
- 100.Sneddon AA, Koll M, Wallace MC, Jones J, Miell JP, et al. 2003. Acute alcohol administration inhibits the refeeding response after starvation in rat skeletal muscle. American journal of physiology. Endocrinology and metabolism 284: E874–82 [DOI] [PubMed] [Google Scholar]
- 101.Sonntag WE, Boyd RL. 1989. Diminished insulin-like growth factor-1 levels after chronic ethanol: relationship to pulsatile growth hormone release. Alcoholism, Clinical and Experimental Research 13: 3–7 [DOI] [PubMed] [Google Scholar]
- 102.Steiner JL, Crowell KT, Lang CH. 2015. Impact of alcohol on glycemic control and insulin action. Biomolecules 5: 2223–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Steiner JL, Gordon BS, Lang CH. 2015. Moderate alcohol consumption does not impair overload-induced muscle hypertrophy and protein synthesis. In Physiological reports [DOI] [PMC free article] [PubMed]
- 104.Steiner JL, Kimball SR, Lang CH. 2016. Acute alcohol-induced decrease in muscle protein synthesis in female mice is REDD-1 and mTOR-independent. Alcohol and alcoholism 51: 242–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steiner JL, Lang CH. 2014. Alcohol impairs skeletal muscle protein synthesis and mTOR signaling in a time-dependent manner following electrically stimulated muscle contraction. Journal of applied physiology 117: 1170–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. 2003. Tuberous sclerosis complex gene products, tuberin and hamartin, control mTOR by acting as a GTPase-activating protein complex toward Rheb. Current Biology 13: 1259–68 [DOI] [PubMed] [Google Scholar]
- 107.Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, et al. 2014. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 10: 677–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thavorncharoensap M, Teerawattananon Y, Yothasamut J, Lertpitakpong C, Chaikledkaew U. 2009. The economic impact of alcohol consumption: a systematic review. Substance abuse treatment, prevention, and policy 4: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485: 109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Urbano-Marquez A, Estruch R, Fernandez-Sola J, Nicolas JM, Pare JC, Rubin E. 1995. The greater risk of alcoholic cardiomyopathy and myopathy in women compared with men. Journal of the American Medical Association 274: 149–54 [DOI] [PubMed] [Google Scholar]
- 111.Vary T 2009. Oral leucine enhances myocardial protein synthesis in rats acutely administered ethanol. The Journal of Nutrition 139: 1439–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vary TC, Deiter G. 2005. Long-term alcohol administration inhibits synthesis of both myofibrillar and sarcoplasmic proteins in heart. Metabolism 54: 212–9 [DOI] [PubMed] [Google Scholar]
- 113.Vary TC, Frost RA, Lang CH. 2008. Acute alcohol intoxication increases atrogin-1 and MuRF1 mRNA without increasing proteolysis in skeletal muscle. American journal of physiology. Regulatory, integrative and comparative physiology 294: R1777–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vary TC, Kimball SR, Sumner A. 2007. Sex-dependent differences in the regulation of myocardial protein synthesis following long-term ethanol consumption. American journal of physiology. Regulatory, integrative and comparative physiology 292: R778–87 [DOI] [PubMed] [Google Scholar]
- 115.Vary TC, Lang CH. 2008. Differential phosphorylation of translation initiation regulators 4EBP1, S6k1, and Erk 1/2 following inhibition of alcohol metabolism in mouse heart. Cardiovascular toxicology 8: 23–32 [DOI] [PubMed] [Google Scholar]
- 116.Vary TC, Lynch CJ, Lang CH. 2001. Effects of chronic alcohol consumption on regulation of myocardial protein synthesis. American journal of physiology. Heart and circulatory physiology 281: H1242–51 [DOI] [PubMed] [Google Scholar]
- 117.Vary TC, Nairn AC, Deiter G, Lang CH. 2002. Differential effects of alcohol consumption on eukaryotic elongation factors in heart, skeletal muscle, and liver. Alcoholism, Clinical and Experimental Research 26: 1794–802 [PubMed] [Google Scholar]
- 118.Vary TC, Nairn AC, Lang CH. 2004. Restoration of protein synthesis in heart and skeletal muscle after withdrawal of alcohol. Alcoholism, Clinical and Experimental Research 28: 517–25 [DOI] [PubMed] [Google Scholar]
- 119.Wilkens Knudsen A, Jensen JE, Nordgaard-Lassen I, Almdal T, Kondrup J, Becker U. 2014. Nutritional intake and status in persons with alcohol dependency: data from an outpatient treatment programme. European journal of nutrition 53: 1483–92 [DOI] [PubMed] [Google Scholar]
- 120.Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, et al. 2015. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351: 43–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolfson RL, Sabatini DM. 2017. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab 26: 301–09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. 2003. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nature Cell Biology 5: 578–81 [DOI] [PubMed] [Google Scholar]