

Abstract
The inner ear sensory epithelium harbors mechanosensory hair cells responsible for detecting sound and maintaining balance. This protocol describes a three-dimensional (3D) culture system which efficiently generates inner ear sensory epithelia from aggregates of mouse embryonic stem (mES) cells. By mimicking the activations and repressions of key signaling pathways during in vivo inner ear development, mES cell aggregates are sequentially treated with recombinant proteins and small molecule inhibitors for activating or inhibiting the Bmp, TGFβ, Fgf, and Wnt signaling pathways. These stepwise treatments promote mES cells to sequentially differentiate into epithelia representing the non-neural ectoderm, preplacodal ectoderm, otic placodal ectoderm, and ultimately, the hair cell-containing sensory epithelia. The derived hair cells are surrounded by a layer of supporting cells and are innervated by sensory neurons. This in vitro inner ear organoid culture system may serve as a valuable tool in developmental and physiological research, disease modeling, drug testing, and potential cell-based therapies.
Keywords: Inner ear, hair cells, sensory epithelium, vestibular, mouse pluripotent stem cells, organoid, three-dimensional culture
1. Introduction
Inner ear hair cells are mechanosensitive receptors that detect sound, gravity, and movement, and convert these signals into our sense of hearing and balance [1]. These sensory hair cells do not regenerate to any clinically relevant degree in mammalian inner ears, resulting in permanent hearing loss or vestibular impairments affecting millions of people worldwide [2].
Studies of mouse, chick, zebrafish, and Xenopus inner ear development have accumulated evidence that bone morphogenetic protein (Bmp) signaling, fibroblast growth factor (Fgf) signaling, and Wnt signaling are important in the development of the inner ear. During embryonic development in vivo, Bmp signaling activates a region of the definitive ectoderm, resulting in the formation of non-neural ectoderm [3–9]. Subsequent Bmp inhibition, along with Fgf signaling activation, induces the formation of the preplacodal region from the non-neural ectoderm [10,6,11–15]. The otic placode, along with other cranial placodes (e.g. olfactory, lens, epibranchial, etc.), is derived from the preplacodal region [3,16]. Wnt is known to be a signaling cue that promotes the formation of the otic placode [17,18]. Following induction, the otic placode invaginates to form the otic vesicle [19], which is the source of nearly all cell types of the mature inner ear, including the sensory hair cells [1] (Fig. 1).
Figure 1.

In vitro differentiation of inner ear sensory epithelium in 3D culture (top row) is achieved through manipulation of signaling pathways which are known to be essential in in vivo inner ear differentiation (bottom row).
We recently developed a method to derive inner ear sensory epithelia harboring functional hair cells from mouse embryonic stem cells (mESCs) in a three-dimensional (3D) culture, using stepwise treatment of signaling molecules, such as BMP-4 and FGF-2, that mimic those present in inner ear development in vivo [20–22] (Figs. 1, 2). Our method was built upon recent advances in cerebral and retinal tissue generation protocols in 3D culture [23–25]. In these culture systems, the definitive ectoderm, a common precursor for inner ear epithelia, retinal and cerebral tissues, was successfully generated. The key techniques for definitive ectoderm generation are to aggregate the dissociated pluripotent stem cells into spheroids in low-cell-adhesion U-bottom 96-well plates in a KnockOut serum replacement (KSR)-containing medium, followed by treatment with Matrigel that promotes the formation of a basement membrane. To guide the definitive ectoderm to develop into the non-neural ectoderm, we treat the mouse ES cells-derived aggregates with a recombinant human BMP-4 protein on differentiation day 3. To suppress undesirable mesoderm tissues from arising upon BMP-4 activation, the transforming growth factor-β (TGF-β) inhibitor SB-431542 [26] is added along with BMP-4. Following the induction of the non-neural ectoderm, Fgf signaling activation and Bmp inhibition are achieved through the addition of a recombinant human FGF-2 protein and the Bmp inhibitor LDN-193189 on differentiation day 4. The combined signaling cues result in the formation of the preplacodal region, which later develops into the otic placode between day 6 and day 8. On differentiation day 8, the aggregates are transferred to a minimum defined medium for a long-term culture. Similar to the in vivo morphogenesis events, cells of the otic placode invaginate and form the otic vesicles during day 9–12.
Figure 2.

Experimental procedures of 3D inner ear organoid culture.
Sensory epithelium harboring inner ear hair cells positive for vestibular hair cell markers, such as Myo7a, Brn3c, calretinin, Sox2, and Pax2, begin to arise on day 14 (Figs. 4B–D”). Like hair cells in vivo, hair cells generated in 3D organoids produce stereocilia bundles with a protruding kinocilium. Moreover, these in vitro derived hair cells are fully functional based on FM1–43 dye uptake assays and electrophysiology studies [20,27]. In addition to functional hair cells, a layer of Sox2-positive supporting cells as well as sensory neuron-like cells also arise in the differentiation culture [20].
Figure 4.
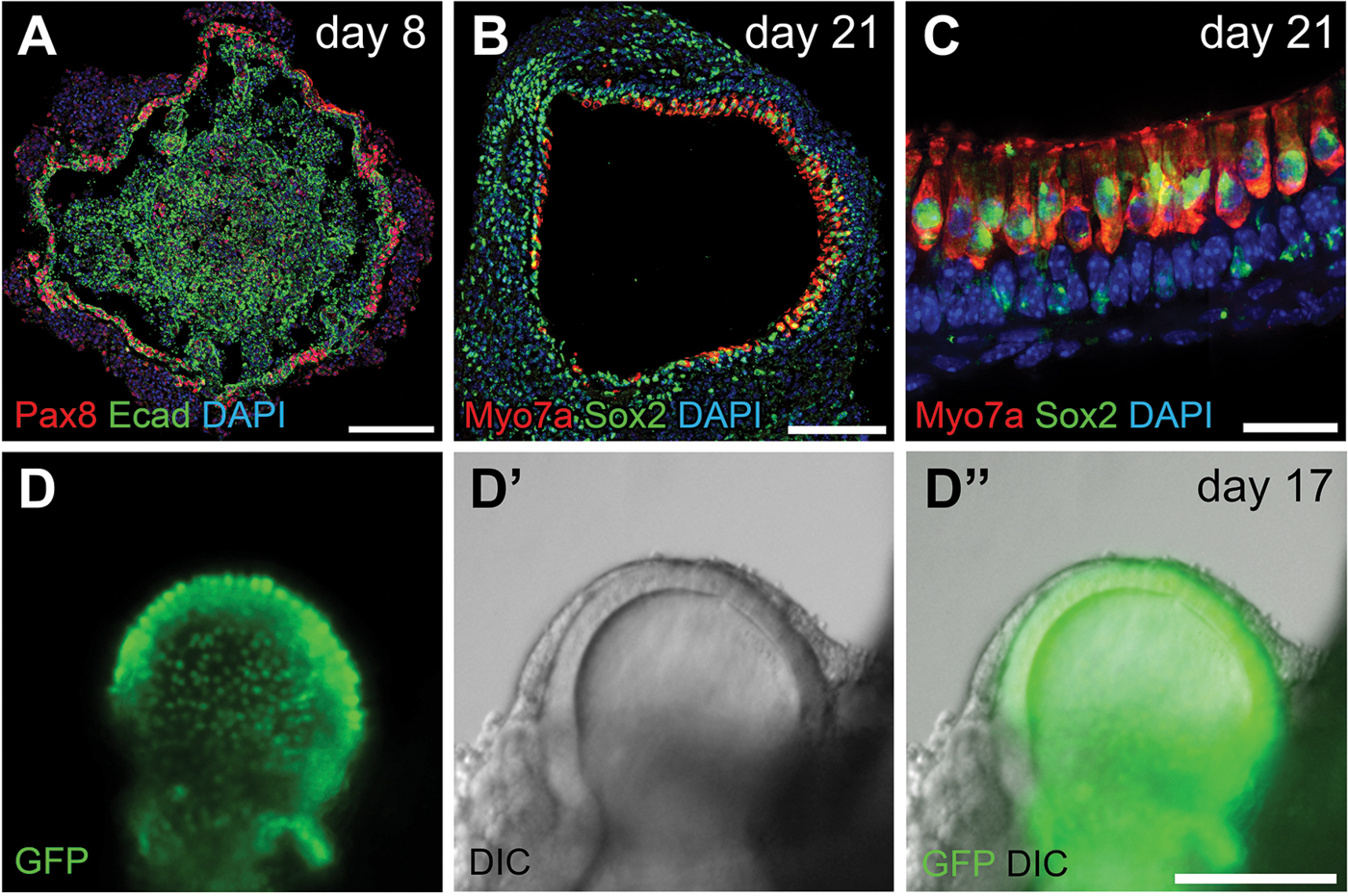
Preplacodal ectoderm on a day 8 aggregate and inner ear sensory epithelium on later staged aggregates. (A) Pax8 and E-cadherin (Ecad) label the preplacodal regions on a day 8 aggregate. (B–C) Inner ear hair cells expressing Myo7a and Sox2 tightly organized at the interior surface of vesicles on day 21. (D–D”) Live Imaging of a Atoh1-nGFP aggregate. GFP with nuclear localization signal (nGFP) is expressed under an Atoh1 promoter, thus marking the inner ear hair cells. GFP signals in (D) and (D”) are overlaid from two focal planes. Scale bars, 100 µm (A–B, D–D”); 20 µm (C).
In our previous studies, after the preplacodal region is derived through stepwise BMP-4/SB-431542/FGF-2/LDN-193189 treatment during the 3D differentiation culture, the aggregates undergo a self-guided development starting on day 8. We have demonstrated that the endogenous Wnt signaling is critical in otic placodal derivation from the preplacodal region during the self-guided development, as treatment with the Wnt inhibitor XAV939 during day 8–10 significantly reduced the formation of the otic vesicles [20]. In addition, in vivo studies in mice and zebrafish have also showed that Wnt signaling promotes the derivation of the otic placode from the preplacodal region at the expense of other placodal lineages [17,18]. In an effort to maximize the production of the otic hair cells for high-throughput assays, we have recently incorporated a treatment with the potent Wnt agonist CHIR-99021 [28,29] on day 8 of culture (Figs. 1, 2), and have found a significant increase in hair cell formation (E. Hashino and colleagues, unpublished).
The genetic context of the organoid cultures are no longer limited to wild-type or transgenic mice derivatives, thanks to the recent breakthroughs in genome engineering technologies. Specific mutants, reporter cell lines, and cell lines integrated with complex genetic modules can now be easily created in ES cells. This opens up exciting opportunities in applying the inner ear organoid culture system in research and therapies, such as elucidating mechanisms of inner ear development and physiology, disease modeling, high-throughput drug efficacy and toxicity testing, and cell-based therapeutics.
2. Materials
2.1. Reagents and Reagent setup
2.1.1. Reagents
Mouse ES cells acclimated to growth in LIF-2i medium [30] in feeder-free condition. We primarily use a R1 mES cell line (derived from R1 mice), R1/E mES cell line (ATCC), and Atoh1-nGFP mES cell line [31,32] (a generous gift from Dr. Stefan Heller, Stanford University) for differentiation culture, but this protocol has been tested with a mouse iPS cell line and several transgenic mES cell lines with comparable differentiation results.
0.1% Gelatin.
PBS (phosphate-buffered saline), pH 7.4.
Accutase cell dissociation reagent.
2.1.2. Solubilization and/or aliquoting of reagents (see Note 1)
10 mM PD-0325901 stock solution: Add 207 µL of DMSO (Dimethyl sulfoxide) to 1 mg of PD-0325901 powder and mix thoroughly. Store the resulting 10 mM PD-0325901 solution in 5.5 µL aliquots at −20 °C for up to 6 months.
100 ng/µL human recombinant BMP-4 stock solution: Add 100 µL of 4 mM HCl to 10 µg of lyophilized BMP-4 and mix thoroughly. Store the resulting 100 ng/µL BMP-4 solution in 2 µL aliquots at −20 °C for up to 3 months, or at −80 °C for up to 6 months.
200 ng/µL human recombinant FGF-2 stock solution: Add 250 µL of sterile PBS with 0.1% BSA to 50 µg of lyophilized FGF-2 and mix thoroughly. Store the resulting 200 ng/µL FGF-2 solution in 2.5 µL aliquots at −80 °C for up to 3 months.
KnockOut serum replacement (KSR): Store in ~760 µL aliquots at −20 °C for up to 18 months. Protect from light.
10 mM CHIR-99021 stock solution: Store the 10 mM CHIR-99021 solution in 15.5 µL aliquots at −20 °C for up to 6 months.
10 mM SB-431542 stock solution: Store the 10 mM SB-431542 solution in 2 µL aliquots at − 20 °C for up to 6 months.
10 mM LDN-193189 stock solution: Store the 10 mM LDN-193189 solution in 2 µL aliquots at −20 °C for up to 6 months.
Matrigel: Aliquot 250 µL of ice-cold Matrigel into ice-cold microcentrifuge tubes sitting on ice using a pre-chilled pipet tip. Store aliquots at −20 °C for up to 2 years. Thaw aliquots overnight at 4 °C or for at least 2 h at 4 °C before use (see Note 2).
2.1.3. Preparation of culture media (see Note 1)
LIF-2i mES cell maintenance medium: For every 10 mL of LIF-2i medium, combine and mix the following reagents: 4.8 mL of Advanced DMEM/F12 medium, 4.8 mL of Neurobasal medium, 100 µL of GlutaMAX supplement, 50 µL of N-2 supplement, 100 µL of B-27 supplement minus vitamin A (see Note 3), 20 µL of Normocin (see Note 4), 10 µL of 1 × 106 units/mL LIF, 3 µL of 10 mM CHIR-99021, and 1 µL of 10 mM PD-0325901. Store the complete LIF-2i medium at 4 °C until use. It is best to finish using the complete medium within one week.
Ectodermal differentiation medium: For every 10 mL of ectodermal differentiation medium, combine and mix the following reagents: 9.65 mL of G-MEM, 100 µL of 100 mM sodium pyruvate, 100 µL of MEM non-essential amino acids solution, 150 µL of KnockOut serum replacement, 20 µL of Normocin, and 18 µL of 2-mercaptoethanol. Store the complete ectodermal differentiation medium at 4 °C until use. For every 96-well plate of differentiation culture, 25–30 mL of complete ectodermal differentiation medium should be made. It is best to finish using the complete medium within one week.
Maturation medium: For every 10 mL of maturation medium, combine and mix the following reagents: 9.8 mL of Advanced DMEM/F12, 100 µL of GlutaMAX supplement, 100 µL of N-2 supplement, and 20 µL of Normocin (see Note 4). Store the complete maturation medium at 4 °C until use. It is best to finish using the complete medium within two weeks.
Maturation medium with Matrigel and CHIR-99021: For every 10 mL of Matrigel (1% final concentration) and CHIR-99021 (3 µM final concentration) supplemented maturation medium, first add 100 µL of ice-cold Matrigel into 9.7 mL of ice-cold Advanced DMEM/F12 using a pre-chilled pipet tip. Immediately mix well by inverting the container (we usually use a 50 mL conical tube) several times (see Note 2). After the ice-cold Matrigel is diluted in the ice-cold Advanced DMEM/F12 and mixed well, the Matrigel will no longer gel at higher temperatures (RT or 37 °C) thus it is no longer necessary to keep the mixture ice-cold. Then continue to add the following reagents: 100 µL of GlutaMAX supplement, 100 µL of N-2 supplement, 20 µL of Normocin (see Note 4), and 3 µL of 10 mM CHIR-99021. It is best to make this Matrigel and CHIR-99021 supplemented maturation medium right before the day 8 treatment. At least 12.5 mL of this supplemented medium should be made for each 24-well plate to be seeded with aggregates on differentiation day 8.
2.2. Equipment
Cell culture dishes and plates: 35 mm, 60 mm, or 100 mm dishes, depending on how many 96-well plates of differentiation culture to start. At least two 96-well plates of differentiation culture can be started using ≥50% confluent mES cells cultured on a 35 mm dish. 6-well plates can also be used to culture mES cells, and the surface area of a well of a 6-well plate is equal to that of a 35 mm dish.
Low cell adhesion U-bottom 96-well plates: Nunclon Sphera coated (Thermo Fisher Scientific).
Low cell adhesion 24-well flat bottom plates: Nunclon Sphera coated (Thermo Fisher Scientific).
37 °C / 5% CO2 humidified incubator.
Biosafety cabinet.
Vacuum aspirator and disposable glass Pasteur pipets.
Water bath.
A centrifuge that is capable of centrifuging 2 mL microcentrifuge tubes, and a centrifuge that is capable of centrifuging 15 mL conical tubes and 96-well plates.
Automated cell counter or hemocytometer for manual cell counting.
Inverted microscope.
Pipet tips (20 µL, 200 µL, and 1 mL), microcentrifuge tubes (0.5 mL, 1.5 mL, and 2 mL), and conical tubes (15 mL and 50 mL).
Single channel (2 µL, 20 µL, 200 µL, and 1 mL) and multi-channel (200 µL) pipets.
Pipette basins.
Scissors for cutting the pipet tips to make wide-mouth pipet tips.
Sprayer filled with 70% ethanol.
3. Methods
3.1. Thawing cryopreserved mES cells and plating in a feeder-free condition
In a biosafety cabinet (see Note 1), coat a 60 mm cell culture dish (see Note 5) with 2 mL of 0.1% gelatin for at least 20 min in room temperature (RT).
Warm at least 15 mL of LIF-2i medium in a 37°C water bath.
Take out a vial of cryopreserved mESCs from the liquid nitrogen tank.
Thaw the vial of mESCs by gentle agitation in a 37°C water bath. Remove the vial from the water bath when only a few ice crystals are remaining. To reduce the likelihood of contamination, keep the O-ring and cap out of the water.
Remove the vial from the water bath, and decontaminate the vial by spraying with 70% ethanol.
In a biosafety cabinet, transfer the cells from the cryogenic vial into a 15 mL conical tube by gentle pipetting.
Gently rinse the vial with an additional 1 mL of LIF-2i medium and then slowly transfer contents to the 15 mL tube.
Slowly add LIF-2i medium drop-wise to the 15 mL tube to bring the total volume to 10 mL.
Centrifuge the 15 mL tube at 180 × g for 10 min at RT.
Aspirate gelatin from the 60 mm dish. Leave the dish sitting open in the biosafety cabinet until dry.
When centrifuging is finished, carefully aspirate the supernatant from the 15 mL conical tube and resuspend the cell pellet in 3 mL of pre-warmed LIF-2i medium.
Add the cell suspension to the dried gelatin coated dish. It is recommended that the plating density should not exceed 50,000 cells/cm2.
Move the dish back and forth and side to side several times to evenly distribute cells across the cell culture dish. Incubate the dish in a humidified 37°C / 5% CO2 incubator.
3.2. mES cell maintenance and passaging in a feeder-free condition
1 day after plating, change medium by replacing spent medium with fresh pre-warmed LIF-2i to remove any resulting cell debris. Additional medium change is not required unless cells exceed 50% confluency or when the color of the culture medium is turning from red to orange or yellow.
Cells should be passaged when they are 50%–80% confluent. If the colonies are sparse but becoming too large, the cells should be passaged even when the overall confluency is less than 50%. mESCs are usually passaged twice a week, and the frequency of passaging can be controlled by adjusting the split ratio (usually between 1:10–1:50) during each passaging.
To passage mESCs, first coat a 60 mm cell culture dish (see Note 5) with 2 mL of 0.1% gelatin for at least 20 min in a biosafety cabinet at RT.
Warm a tube of LIF-2i medium in a 37°C water bath. Also warm an aliquot of accutase (400 µL or more) at RT.
Aspirate the spent LIF-2i medium from the culture dish.
Slowly add 400 µL of accutase and incubate the cells in the 37°C incubator for 1–3 min until cells start to detach from plate. Confirm the detachment of cells from dish under a microscope.
While cells are being dissociated in the incubator, aspirate gelatin from the 60 mm dish and leave the dish sitting open in the biosafety cabinet until dry.
Wash the dissociated cells off the dish by gently pipetting 1 mL of pre-warmed LIF-2i across the dish. Repeat the pipetting across the dish several times to completely wash off the cells and to break up the cell clumps. Avoid creating bubbles.
Collect cells into a 2 mL microcentrifuge tube. Gently pipet up and down several times using a p1000 tip to further break up cell clumps into single cells. Avoid creating bubbles.
Centrifuge cells for 2.5 min at 180 × g at RT.
Carefully remove supernatant completely by aspiration (see Note 6) and resuspend cell pellet with 1 mL of LIF-2i.
Add 20 µL (1:50 split ratio) – 100 µL (1:10 split ratio) of cells and 3 mL of LIF-2i to the dried gelatin-coated 60 mm dish. Select split ratio according to when you next wish to split the cells or to use the cells for a differentiation culture.
Move the dish back and forth and side to side a few times to evenly disperse cells across the surface of the dish. Incubate the dish in a humidified 37°C / 5% CO2 incubator.
3.3. Differentiation day 0: mESC dissociation and seeding in 96-well plate
Differentiation culture can be started when mESCs are ~50%–80% confluent.
Prepare a tube of ectodermal differentiation medium (25–30 mL for each 96-well plate culture), mix well, and warm medium in a 37°C water bath. Warm an aliquot of accutase (400 µL or more) at RT.
Aspirate spent LIF-2i medium from the mESC dish, and wash the cells three times with PBS (see Note 7).
To dissociate cells, slowly add 400 µL of accutase and incubate the dish in the 37°C incubator for 1–3 min until cells start to detach from plate. Confirm the detachment of cells from dish under a microscope.
Wash the dissociated cells off the dish by gently pipetting 1 mL of ectodermal differentiation medium across the plate. Repeat the pipetting across the dish several times to completely wash off the cells and to break up the cell clumps. Avoid creating bubbles.
Collet cells into a 2 mL microcentrifuge tube. Gently pipet up and down several times using a p1000 tip to further break up cell clumps into single cells (see Note 8). Avoid creating bubbles.
Centrifuge cells for 2.5 min at 180 × g at RT.
Carefully remove supernatant by aspiration (see Note 6). Resuspend cell pellet in 1 mL of ectodermal differentiation medium and mix well.
Determine the concentration of cells with an automatic cell counter or a hemocytometer.
Pipet appropriate volume of cells and mix with fresh pre-warmed ectodermal differentiation medium in a conical tube to acquire a final concentration of 30,000 cells per mL. The final volume should be 11 mL (i.e., 3.3 × 105 total cells) for each 96-well plate to be seeded (see Note 9).
Invert the tube several times to mix the cells. Pour cells into a multichannel pipet basin and gently pipet 100 µL of cells into each well (i.e., 3,000 cells/well) of a low-cell-adhesion U-bottom 96-well plate using a multichannel pipet.
Centrifuge the 96-well plate at 90 × g for 5 min to assist the aggregation of mESCs.
Incubate the plate in a humidified 37°C / 5% CO2 incubator for 24 h. Overnight the mESCs at the bottom of the low-cell-binding wells will aggregate to form spheres (Figs. 2; 3A–E).
Thaw an aliquot of Matrigel from a −20°C freezer (one 250 µL aliquot for each 96-well plate differentiation culture) by placing it in a 4°C fridge overnight.
Figure 3.
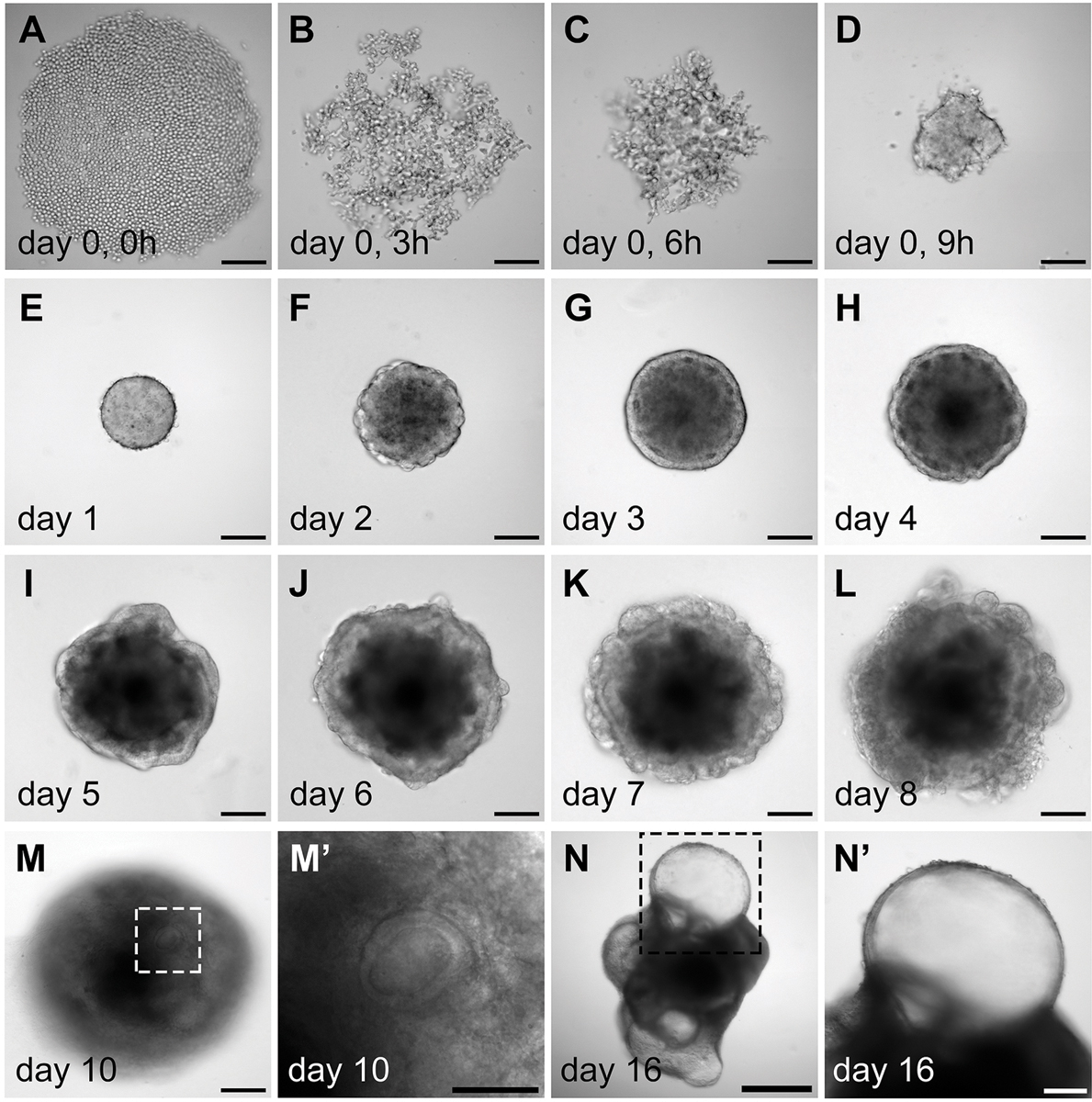
Morphology of the aggregates during in vitro differentiation. (A–D) Aggregation of the mouse ES cells during day 0, which is likely mediated by cell surface adhesion proteins. (E–L) Morphological changes of the aggregates during day 1 to day 8 culture. Note that after Matrigel addition on day 1, an epithelium develops on the surface of the aggregates on day 3 (G). In a self-guided manner, some of the interior cell mass breaches out during day 6 to day 8. Concurrently, the epithelium is rearranged from the outer surface to the interior of the aggregates (J–L). (M– M’) Vesicles embedded inside the aggregates become apparent on day 10. (N–N’) A day 16 aggregate with multiple protruding vesicles. (M’) and (N’) are higher magnification images of (M) and (N), respectively. Scale bars, 100 µm (A–M, N’); 50 µm (M’); 500 µm (N).
3.4. Differentiation day 1: Addition of Matrigel
Add 220 µL of cold Matrigel to 5.28 mL of ice-cold ectodermal differentiation medium in a cold 15 mL conical tube using a pre-chilled pipet tip to make 5.5mL of complete medium (4% Matrigel concentration). Immediately mix by inverting the tube several times (see Note 2).
Warm the complete medium to 37°C in a water bath.
By holding a multichannel pipet at an angle, carefully remove 50 µL of medium from each well of the 96-well plate and make sure the aggregates at the bottom of the wells are not disturbed.
Add 50 µL of the complete medium (4% Matrigel in ectodermal differentiation medium) to each well using a multichannel pipet. Carefully pipet up and down several times to mix, while making sure that the aggregates are not sucked into the pipet tips as this will damage the aggregates. The final concentration of Matrigel is now 2%.
Incubate the plate in a humidified 37°C / 5% CO2 incubator for 48 h (Figs. 2; 3E).
3.5. Differentiation day 3: Addition of BMP-4 and SB-431542
In a 15 mL conical tube, add 1.5 µL of 100 ng/µL BMP-4 and 1.5 µL of 10 mM SB-431542 to 3 mL of ectodermal differentiation medium. Mix by inverting the tube several times.
Warm the complete medium to 37°C in a water bath.
Pour the complete medium into a multichannel pipet basin.
Pipet 25 µL of the BMP-4/SB-431542 containing ectodermal differentiation medium to each well of the 96-well plate using a multichannel pipet. Carefully pipet up and down several times to mix well, while making sure that the aggregates are not sucked into the pipet tips. The final concentration of BMP-4 is now 10 ng/mL, and that of SB-431542 is 1 µM.
Incubate the plate in a humidified 37°C / 5% CO2 incubator for 24 h (Figs. 2; 3G).
3.6. Differentiation day 4: Addition of FGF-2 and LDN-193189 (see Note 10)
In a 15 mL conical tube, add 2.25 µL of 200 ng/µL FGF-2 and 1.8 µL of 10 mM LDN-193189 to 3 mL of ectodermal differentiation medium. Mix by inverting the tube several times.
Warm the complete medium to 37°C in a water bath.
Pipet 25 µL of the FGF-2/LDN-193189 containing ectodermal differentiation medium to each well of the 96-well plate using a multichannel pipet. Carefully pipet up and down several times to mix well, while making sure that the aggregates are not sucked into the pipet tips. The final concentration of FGF-2 is now 25 ng/mL, and that of LDN-193189 is 1 µM.
Incubate the plate in a humidified 37°C / 5% CO2 incubator for 4 days (Figs. 2; 3H).
3.7. Differentiation day 8: Transfer aggregates to Matrigel and CHIR-99021 supplemented maturation medium for long-term culture
On day 7, thaw an aliquot of Matrigel from a −20°C freezer by placing it in a 4°C fridge overnight.
On day 8, make Matrigel (1% final concentration) and CHIR-99021 (3 µM final concentration) supplemented maturation medium (see Note 2). The volume of the complete medium prepared should be at least 12.5 mL for each 24-well plate to be seeded. Up to four 24-well plates can be seeded with aggregates from each 96-well plate.
Warm the Matrigel and CHIR-99021 supplemented maturation medium to 37°C in a water bath.
Using a wide-mouth pipet tip on a 200 µL single channel pipet, carefully transfer individual aggregates from the 96-well plate to a 15 mL conical tube (see Note 11).
After all aggregates are settled at the bottom of the conical tube, carefully remove the spent medium and wash three times with at least 5 mL of PBS. Carefully remove excess PBS after the washes (see Note 12).
Add a few mL of the pre-warmed Matrigel and CHIR-99021 supplemented maturation medium to the 15 mL conical tube, and quickly pour the medium along with the aggregates to a 60 mm dish. If there are remaining aggregates in the conical tube, repeat this step to transfer the remaining aggregates to the 60 mm dish.
At this stage the aggregates are big enough to be visible by eyes. Using a wide-mouth pipet tip on a 200 µL single channel pipet, carefully transfer each aggregates along with 50 µL of medium from the 60 mm dish to each well of a low-cell-adhesion 24-well flat bottom plate. Aggregates in one 96-well plate can be transferred to up to four 24-well plates.
Gently add 450 µL of the Matrigel and CHIR-99021 supplemented maturation medium to each well of the 24-well plate to make a final volume of 500 µL of medium in each well.
Incubate the plate in a humidified 37°C / 5% CO2 incubator for 48 h (Figs. 2; 3L; 4A).
3.8. Differentiation day 10 and later: Long-term culture in maturation medium
Perform a half medium change every other day starting on day 10 (i.e., days 10, 12, 14 and so on), by removing 250 µL of spent medium and gently adding 250 µL of pre-warmed maturation medium (without Matrigel or CHIR-99021).
Return the plate in a humidified 37°C / 5% CO2 incubator. Aggregates can be cultured for up to 30 days under these conditions. For longer culture durations, different medium compositions or culture formats are likely to be required. Vesicles can be observed starting from day 9 or day 10 (Figs. 3M, M’), and inner ear hair cells should first arise within otic vesicle epithelia between day 14 and day 16 (Figs. 3N, N’; 4B–D”). Aggregates can be used for analysis/experiments (e.g. immunofluorescence, electrophysiology recording, flow cytometry, qPCR, Western blot, etc.) at any point during the differentiation culture depending on the experimental needs.
Acknowledgements
This work was supported by a National Institutes of Health grant R01 DC013294 (to E.H.).
Footnotes
Notes
All reagents and culture media coming in contact with living cells or aggregates must be prepared in a biosafety cabinet with sterile tubes and pipet tips, etc. All experiments/treatments on living cells or aggregates must be carried out in a biosafety cabinet, except for viewing/imaging under a microscope, centrifuging, thawing cells in a water bath, and fixing aggregates for immunofluorescence staining.
It is extremely important that Matrigel and all pipet tips, conical tubes, and medium coming in contact with Matrigel should be ice-cold, since Matrigel starts to gel above 10°C. After the ice-cold Matrigel is diluted in culture media and well mixed, the Matrigel will no longer gel at higher temperatures (RT or 37 °C) thus it is no longer necessary to keep it cold.
It is important to use B-27 supplement without vitamin A rather than using the regular B-27 supplement in the LIF-2i medium for mES cell culture. Vitamin A (retinol) can be converted to retinoic acid in culture, which can induce spontaneous neuronal differentiation of the mES cells [33–35].
Normocin is used as an anti-microbial reagent in all culture and differentiation media in this protocol. It is especially important to not use aminoglycosides-containing anti-microbial reagents like the widely used penicillin-streptomycin in the maturation media, as aminoglycosides are known to be toxic to inner ear hair cells [36].
This protocol uses 60 mm dishes (Surface area = 2,827 mm2) to culture mES cells. To reduce the amount of LIF-2i culture medium and reagents (e.g. gelatin, accutase, etc.) being used, mES cells can also be cultured in 35 mm dishes or 6-well plates (Surface area = 962 mm2 per dish/well). If using the latter two types of culture dishes/plates, use 1 mL of gelatin to coat the dish/well, use 300 µL accutase for cell dissociation, use 1 mL LIF-2i to wash the accutase dissociated cells from the dish/well, and use 2 mL of LIF-2i medium to feed the cells. Though the surface area of a 35 mm dish or a well of a 6-well plate is ~70% smaller than that of a 60 mm dish, mES cells cultured in one dish/well are still enough to start at least two 96-well plates in a differentiation experiment when the cells are more than 50% confluent.
To completely remove the medium without the risk of aspirating away the cell pellet, put the glass Pasteur pipet tip against the interior lateral wall of the round-bottom 2 mL microcentrifuge tube near the opening area of the tube. Slowly tilt the 2 mL tube from an up-right position to a horizontal position, which allows the liquid to be slowly aspirated at the opening area of the tube. The cell pellet should stay at the bottom of the 2 mL tube. This technique only works with the round-bottom 2 mL tubes but not the 1.5 mL or 0.5 mL microcentrifuge tubes or the 15 mL conical tubes, as in the latter three types of tubes the bottoms are narrow, thus some liquid will remain at the bottom when the tubes are tilted to horizontal or even to up-side-down positions. It is essential to use this technique to remove as much liquid from the cell pellet as possible, as excessive reagents in the remaining liquid may negatively affect the downstream experiments.
It is important to thoroughly wash the ES cells to completely remove LIF-2i, as excessive residual inhibitory components in the LIF-2i medium (i.e., LIF, CHIR-99021, and PD-0325901) may repress the differentiation of the ES cells in the 3D differentiation culture.
After the 1–3 min of accutase digestion followed by washing and pipetting, nearly all the cells should be dissociated into single cells. This can be confirmed using an automated cell counter or under the microscope on a hemocytometer during cell counting. Therefore, there is no need to forcefully pipet cells through a cell strainer tube to ensure single cell dissociation.
For example, if the concentration of the cell suspension is 1 × 106 cells/mL and one 96-well plate is to be seeded, add 330 µL of cell suspension (3.3 × 105 cells ÷ 1 × 106 cells/mL = 0.33 mL) in 10.67 mL of ectodermal differentiation medium to make 11 mL of cells with a 30,000 cells/mL concentration.
The timing of FGF-2/LDN-193189 treatment may need to be optimized depending on the mES cell line being used. Day 4 appears optimal for most cell lines, including R1 and R1/E. However, treatment between day 3.5 and 4.5 may be more suitable for other cell lines.
Make wide-mouth pipet tips by cutting the end of the pipet tip using a sterile scissor in a biosafety cabinet. Make sure the resulting wide-mouth opening of the pipet tip is bigger than the aggregates to be transferred.
Thoroughly wash the aggregates to avoid small molecules and recombinant proteins from previous treatments (i.e. BMP-4, SB-431542, FGF-2, and LDN-193189) from affecting aggregates in the maturation stages.
References
- 1.Whitfield TT (2015) Development of the inner ear. Curr Opin Genet Dev 32:112–118. doi: 10.1016/j.gde.2015.02.006 [DOI] [PubMed] [Google Scholar]
- 2.Warchol ME (2011) Sensory regeneration in the vertebrate inner ear: Differences at the levels of cells and species. Hearing Res 273 (1–2):72–79. doi: 10.1016/j.heares.2010.05.004 [DOI] [PubMed] [Google Scholar]
- 3.Grocott T, Tambalo M, Streit A (2012) The peripheral sensory nervous system in the vertebrate head: A gene regulatory perspective. Dev Biol 370 (1):3–23. doi: 10.1016/j.ydbio.2012.06.028 [DOI] [PubMed] [Google Scholar]
- 4.Wilson PA, Hemmatibrivanlou A (1995) Induction of Epidermis and Inhibition of Neural Fate by Bmp-4. Nature 376 (6538):331–333. 10.1038/376331a0 [DOI] [PubMed] [Google Scholar]
- 5.Wilson PA, Lagna G, Suzuki A, HemmatiBrivanlou A (1997) Concentration-dependent patterning of the Xenopus ectoderm by BMP4 and its signal transducer smad1. Development 124 (16):3177–3184 [DOI] [PubMed] [Google Scholar]
- 6.Kwon HJ, Bhat N, Sweet EM, Cornell RA, Riley BB (2010) Identification of Early Requirements for Preplacodal Ectoderm and Sensory Organ Development. Plos Genet 6 (9). doi:ARTN e1001133 10.1371/journal.pgen.1001133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barth KA, Kishimoto Y, Rohr KB, Seydler C, Schulte-Merker S, Wilson SW (1999) Bmp activity establishes a gradient of positional information throughout the entire neural plate. Development 126 (22):4977–4987 [DOI] [PubMed] [Google Scholar]
- 8.Neave B, Holder N, Patient R (1997) A graded response to BMP-4 spatially coordinates patterning of the mesoderm and ectoderm in the zebrafish. Mech Develop 62 (2):183–195. 10.1016/S0925-4773(97)00659-X [DOI] [PubMed] [Google Scholar]
- 9.Harvey NT, Hughes JN, Lonic A, Yap C, Long C, Rathjen PD, Rathjen J (2010) Response to BMP4 signalling during ES cell differentiation defines intermediates of the ectoderm lineage. J Cell Sci 123 (10):1796–1804. doi: 10.1242/jcs.047530 [DOI] [PubMed] [Google Scholar]
- 10.Pieper M, Ahrens K, Rink E, Peter A, Schlosser G (2012) Differential distribution of competence for panplacodal and neural crest induction to non-neural and neural ectoderm. Development 139 (6):1175–1187. doi: 10.1242/dev.074468 [DOI] [PubMed] [Google Scholar]
- 11.Kwon HJ, Riley BB (2009) Mesendodermal Signals Required for Otic Induction: Bmp-Antagonists Cooperate With Fgf and Can Facilitate Formation of Ectopic Otic Tissue. Dev Dynam 238 (6):1582–1594. doi: 10.1002/dvdy.21955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichert S, Randall RA, Hill CS (2013) A BMP regulatory network controls ectodermal cell fate decisions at the neural plate border. Development 140 (21):4435–4444. doi: 10.1242/dev.098707 [DOI] [PubMed] [Google Scholar]
- 13.Ahrens K, Schlosser G (2005) Tissues and signals involved in the induction of placodal Six1 expression in Xenopus laevis. Dev Biol 288 (1):40–59. doi: 10.1016/j.ydbio.2005.07.022 [DOI] [PubMed] [Google Scholar]
- 14.Brugmann SA, Pandur PD, Kenyon KL, Pignoni F, Moody SA (2004) Six1 promotes a placodal fate within the lateral neurogenic ectoderm by functioning as both a transcriptional activator and repressor. Development 131 (23):5871–5881. doi: 10.1242/dev01516 [DOI] [PubMed] [Google Scholar]
- 15.Litsiou A, Hanson S, Streit A (2005) A balance of FGF, BMP and WNT signalling positions the future placode territory in the head (vol 132, pg 4051, 2005). Development 132 (21):4895–4895 [DOI] [PubMed] [Google Scholar]
- 16.Schlosser G (2006) Induction and specification of cranial placodes. Dev Biol 294 (2):303–351. doi: 10.1016/j.ydbio.2006.03.009 [DOI] [PubMed] [Google Scholar]
- 17.McCarroll MN, Lewis ZR, Culbertson MD, Martin BL, Kimelman D, Nechiporuk AV (2012) Graded levels of Pax2a and Pax8 regulate cell differentiation during sensory placode formation. Development 139 (15):2740–2750. doi: 10.1242/dev.076075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohyama T, Mohamed OA, Taketo MM, Dufort D, Groves AK (2006) Wnt signals mediate a fate decision between otic placode and epidermis. Development 133 (5):865–875. doi: 10.1242/dev.02271 [DOI] [PubMed] [Google Scholar]
- 19.Sai XR, Yonemura S, Ladher RK (2014) Junctionally restricted RhoA activity is necessary for apical constriction during phase 2 inner ear placode invagination. Dev Biol 394 (2):206–216. doi: 10.1016/j.ydbio.2014.08.022 [DOI] [PubMed] [Google Scholar]
- 20.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E (2013) Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500 (7461):217–+. doi: 10.1038/nature12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koehler KR, Hashino E (2014) 3D mouse embryonic stem cell culture for generating inner ear organoids. Nat Protoc 9 (6):1229–1244. doi: 10.1038/nprot.2014.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Longworth-Mills E, Koehler KR, Hashino E (2016) Generating Inner Ear Organoids from Mouse Embryonic Stem Cells. Methods Mol Biol 1341:391–406. doi: 10.1007/7651_2015_215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y (2011) Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472 (7341):51–U73. doi: 10.1038/nature09941 [DOI] [PubMed] [Google Scholar]
- 24.Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasail Y (2008) Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell stem cell 3 (5):519–532. doi: 10.1016/j.stem.2008.09.002 [DOI] [PubMed] [Google Scholar]
- 25.Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M, Sasai Y (2012) Self-Formation of Optic Cups and Storable Stratified Neural Retina from Human ESCs. Cell stem cell 10 (6):771–785. doi: 10.1016/j.stem.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 26.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling (vol 27, pg 275, 2009). Nat Biotechnol 27 (5):485–485. doi: 10.1038/nbt0509-485a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu XP, Koehler KR, Mikosz AM, Hashino E, Holt JR (2016) Functional development of mechanosensitive hair cells in stem cell-derived organoids parallels native vestibular hair cells. Nat Commun 7. doi:ARTN 11508 10.1038/ncomms11508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kriks S, Shim JW, Piao JH, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang LC, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480 (7378):547–U177. doi: 10.1038/nature10648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chambers SM, Qi YC, Mica Y, Lee G, Zhang XJ, Niu L, Bilsland J, Cao LS, Stevens E, Whiting P, Shi SH, Studer L (2012) Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol 30 (7):715–+. doi: 10.1038/nbt.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self-renewal. Nature 453 (7194):519–U515. doi: 10.1038/nature06968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oshima K, Shin K, Diensthuber M, Peng AW, Ricci AJ, Heller S (2010) Mechanosensitive Hair Cell-like Cells from Embryonic and Induced Pluripotent Stem Cells. Cell 141 (4):704–716. doi: 10.1016/j.cell.2010.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lumpkin EA, Collisson T, Parab P, Moer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, Johnson JE (2003) Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns 3 (4):389–395. 10.1016/S1567-133x(03)00089-9 [DOI] [PubMed] [Google Scholar]
- 33.Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa S, Sasai Y (2000) Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 28 (1):31–40. 10.1016/S0896-6273(00)00083-0 [DOI] [PubMed] [Google Scholar]
- 34.Wichterle H, Lieberam I, Porter JA, Jessell TM (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110 (3):385–397. 10.1016/S0092-8674(02)00835-8 [DOI] [PubMed] [Google Scholar]
- 35.Okada Y, Shimazaki T, Sobue G, Okano H (2004) Retinoic-acid-concentration-dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Dev Biol 275 (1):124–142 doi: 10.1016/j.ydbio.2004.07.038 [DOI] [PubMed] [Google Scholar]
- 36.Forge A, Schacht J (2000) Aminoglycoside antibiotics. Audiol Neuro-Otol 5 (1):3–22. 10.1159/000013861 [DOI] [PubMed] [Google Scholar]