

Abstract
Poly(ADP-ribose)polymerase-1 inhibitor (PARPi) AZD2461 was designed to be a weak P-glycoprotein (P-gp) analogue of FDA approved olaparib. With this chemical property in mind, we utilized the AZD2461 ligand architecture to develop a CNS penetrant and PARP-1 selective imaging probe, in order to investigate PARP-1 mediated neuroinflammation and neurodegenerative diseases, such as Alzheimer’s and Parkinson’s. Our work led to the identification of several high-affinity PARPi, including AZD2461 congener 9e (PARP-1 IC50 = 3.9±1.2 nM), which was further evaluated as a potential 18F-PET brain imaging probe. However, despite the similar molecular scaffolds of 9e and AZD2461, our studies revealed non-appreciable brain-uptake of [18F]9e in non-human primates, suggesting AZD2461 to be non-CNS penetrant.
Keywords: PARP-1 inhibitor, microPET imaging, P-glycoprotein, blood-brain barrier, AZD2461
1. Introduction
Poly(ADP-ribose) polymerase-1 is a nuclear protein that plays an integral role in numerous physiological cellular mechanisms including DNA damage repair, maintenance of genomic stability, and apoptosis.1 As such, this protein has become an attractive therapeutic target for cancer,2, 3 neuroinflamation and neurodegeneration,4 cardiovascular disease,5 and even drug addiction.6, 7 Moreover, PARP-1 mediated cell death, termed parthanatos, is the second most studied form of cell death and plays a key role in several neurodegenerative diseases.8 Consequently, a PARP-1 positron emission tomography (PET) neuroimaging probe would advance our limited understanding in the neurodegenerative pathways associated with PARP-1, and bring new insight into developing therapeutic strategies to combat these deadly neurological diseases. Unfortunately, all known PARP-1 imaging agents have been shown to be non-CNS penetrant.9
Despite the recent advances in PARP-1 inhibitor (PARPi) development,2 there have been limited studies illustrating P-gp status of PARPi.10 In 2008, olapraib (1) was shown to induce expression of the drug-efflux pump in mouse models,11 resulting in AstraZeneca to develop AZD2461 (2), a poor P-gp substrate, as a potential backup. Since then, 212, veliparib (3),10 and BGB-290 (4)13 are among the few PARP-1 inhibitors (PARPi) to be reported as weak P-gp substrates (Figure 1). With this chemical property in mind, we evaluated a structurally similar congener of AZD2461 as a potential blood-brain barrier (BBB) penetrable PET probe. Herein, we report the synthesis and PARP-1 binding profiles of AZD2461 analogues, as well as PARP-1 specificity and brain uptake of compound [18F]9e using microPET imaging studies and in vitro autoradiography, respectively.
Figure 1.

Chemical structures of reported CNS-penetrant PARP-1 inhibitors.
2. Results and discussion
2.1. Chemistry
Initial synthesis of AZD2461 fluoroethoxy analogues began with treating commercially available hydroxyl amines (5a-f) with NaH, followed by alkylating reagent 614 in DMF (Scheme 1). Boc-deprotection of the O-alkylated synthons was achieved with CF3COOH, affording intermediates 7a-f. Target compounds 9a-f were aquired by crude coupling of the desired fluoroethoxy free-amine (5a-f) with 8, using reagents HOBt and EDC in THF. These coupling conditions were also utilized to prepare 11a-b from synthons 8 and 10a-b.
Scheme 1. Synthesis of PARPi 9a-f, 11a-b, 14, and 17a.

aReagents and conditions: (i) Respective hydroxyl amine (5a-f), NaH, 6, DMF, rt, 2 h; (ii) TFA, DCM, rt, 3 h; (iii) 8, respective free-amine, HOBt hydrate, EDC HCl, TEA, THF, 60 °C, 12 h. (iv) 5a, PPh3, CBr4, THF, rt; (v) 15, (4-fluorophenyl)boronic acid, NiI2, trans-2-aminocyclohexanol hydrochloride, NaHMDS, 2-methyl-2-butanol 80 °C, 12 h.
Following our previously reported 20 minute C–N cross-coupling protocol,15 diazaspiro moiety 13 was obtained under aerobic conditions for development of 14. Synthesis of azaspiro motif 16 began with treating 5a with triphenylphosphine and carbon tetrabromide in THF to access intermediate 15. Coupling bromide synthon 15 and (4-fluorophenyl)boronic acid, using a modified Ni-catalyzed Suzuki reaction reported by Fu and co-workers16, allowed access to 16. Finally, 16 was reacted with penultimate compound 8 to afford 17 good yield.
2.2. Biological evaluation
2.2.1. PARP-1 radioligand binding
Binding profiles of compounds 2, 9a-f, 11a-b, 14, and 17 were obtained following our previously reported radioligand binding assay conducted in BRCA1 methylated ovarian cancer cells (OVCAR8).17 PARP-1 enzymatic inhibition and cLogP values of AZD2461 analogues containing O-alkylated fluoroethoxy nitrogen heterocycles were evaluated (9a-f) and outlined in Table 1. In terms of PARP-1 IC50, we identified compound 9e to be the most promising candidate in the fluoroethoxy series, with a comparable enzymatic affinity to AZD2461 parent compound 2. Next, we examined the binding properties of more rigid PARPi analogues (11a-b, 14, 17), each containing a fluorinated-aryl ring system. Compared to the IC50 profiles of 9a-f and AZD2461, we found these ligands to be more potent inhibitors of PARP-1 enzymatic activity. Compound 14, containing a 2,6-diazaspiro[3.3]heptane core, was found to be the most potent PARPi in this study, a property also observed in our recent report describing the unique pharmacological properties afforded when implementing this piperazine bioisostere in the olaparib architecture.18 However, for the purposes of this study, we furthered our investigation with 9e, due to the structural similarity of the compound and AZD2461, as well as the PARP-1 IC50 binding data (3.9 nM) and the favorable estimated cLogP value (~2.2).19
Table 1.
PARP-1 IC50 Values for 9a-f, 11a-b, 14, and 17a
| Compound | Coupled amine | PARP-1 IC50b | cLogPc |
|---|---|---|---|
| 2 | 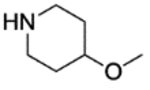 |
2.8±1.1 | 2.07 |
| 9a | 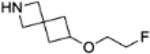 |
10.7±1.1 | 2.49 |
| 9b | 7.5±1.1 | 2.04 | |
|
9c (R) 9d (S) |
 |
10.1±1.1 6.8±1.1 |
2.15 2.15 |
| 9e | 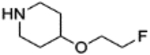 |
3.9±1.2 | 2.26 |
| 9f | 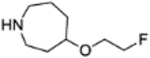 |
5.6±1.1 | 2.71 |
|
11a (Y = N) 11b (Y = CH) |
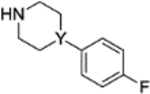 |
0.6±1.2 1.3±NA |
3.37 3.64 |
|
14 (Y = N) 17 (Y = CH) |
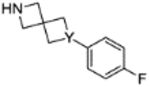 |
0.3±1.2 2.8±NA |
3.24 3.40 |
IC50 (nM) ± (SEM) values represent inhibition of PARP-1 enzymatic activity using our previously reported radioligand binding methodology (ref. 17).
Dose response curves were produced to calculate 50% maximum inhibition values (IC50) where n=3.
Calculated using ChemDraw Professional 15.1
2.2.2. P-gp assay
Compound 9e, and select compounds, were examined in a P-gp assay to determine if these PARPi act as stimulators or inhibitors of the plasma membrane protein (Figure 3). Luminescence generated from unmetabolized ATP was measured after incubating the compounds with recombinant human P-gp, an adenosine (ATP)-dependent drug efflux pump. Compounds acting as stimulators of P-gp ATPase, resulted in decreased luminescence due to the metabolized ATP. Similar luminescence signals were triggered with both positive control verapamil and PARPi olaparib, known substrates for P-gp.12 In contrast, increased luminescence was observed for known P-gp inhibitor Na3VO4 and 9e, 11b, 14, and 17 further indicating AZD2461 congener 9e to be a suitable brain imaging probe candidate.
Figure 3.

In vitro autoradiography blocking study conducted with [18F]9e and olaparib.
2.2.3. Radiochemistry
Radiolabeling precursors for [18F]9e were initially prepared by coupling commercially available compounds 8 with 2-(piperidin-4-yloxy)ethan-1-ol, in the presence of HOBt, EDC, TEA in THF, to obtain intermediate 18 in moderate yield (Scheme 2). Next, 18 was reacted with 4-toluenesulfonyl chlo-ride or methanesulfonyl chloride to afford precursors 19a and 19b, respectively. Access to [18F]9e was then achieved in a facile one-step radiolabeling strategy using precursor 19a. Although not optimized, this method afforded a radiochemical yield of 8–12% for [18F]9e, with a specific activity of 8,491 Ci/mmol (n = 2). The mesylate precursor 19b was also evaluated, however, low radiochemical yields were obtained (~1–3 %).
Scheme 2. Radiosynthesis of [18F]9ea.

aReagents and conditions: (i) 8, 2-(piperidin-4-yloxy)ethan-1-ol, HOBt hydrate, EDC HCl, TEA, THF, 60 °C, 12 h; (ii) TsCl or MsCl, TEA, DCM, 12 h; (iii) [18F]KF, K222, K2CO3, DMSO, 120 °C, 20 min.
2.2.4. In vitro autoradiography
In vitro autoradiography was performed with brain sections from Balb/c mice to verify binding specificity of radiolabeled compound [18F]9e (Figure 3). We found the distribution of [18F]9e to be mostly conserved in the cortex, hippocampus, and cerebellum. Olaparib was utilized as the blocking agent to screen any non-specific binding of the radioligand.
2.2.5. MicroPET imaging
Small animal PET imaging studies using rhesus macaques were conducted with compound [18F]9e and [18F]FTT (Figure 4), a PARP-1 PET tracer under clinical investigation20 known to be a Pg-p substrate. Despite possessing similar chemical structure and PARP-1 affinity as AZD2461, no appreciable brain uptake of [18F]9e was observed in any of the brain regions (Figure 5A). These images are similar to the those obtained and expected with [18F]FTT (Figure 5B), showing the PARPi to be non-BBB penetrable. Metabolic stability of [18F]9e was assessed using the monkey blood from each imaging study and found 42% and 45% of the parent compound intact at 40 min and 37 min, respectively (See SI).
Figure 4.

Chemical structure of [18F]FTT PET probe.
Figure 5.

Monkey PET images indicating the low brain uptake of [18F]9e (A) and [18F]FTT (B) indicated by SUV.
Rodent studies were then performed with [18F]9e, including control and P-gp knockout (k/o) mice models (data not shown), to validate the tracer as a non-P-gp substrate. However, analysis from select rodent studies showed [18F]9e to be non-metabolically stable, with over 80% decomposition of the parent compound in both mice blood and brain in just 5 min (See SI).
3. Conclusion
The goal of this study was to develop derivatives of AZD2461 for PARP-1 neuroimaging applications. We disclosed the synthesis and evaluation of AZD2461 analogues, including two novel PARPi containing spiro[3.3]heptane cores (14 and 17) with excellent PARP-1 affinity (0.3 nM and 2.8 nM, respectively). We identified AZD2461 congener 9e as a potent PARP-1 inhibitor (3.9 nM), and a weak substrate for P-gp. Radioligand [18F]9e was then examined as a potential PARP-1 PET neuroimaging probe, however, [18F]9e demonstrated no brain uptake in our monkey PET imaging studies. We further investigated [18F]9e in rodent models, including control and P-gp knockout (k/o) mice models, but found over 80% of the parent compound to be metabolized. Thus, it cannot be ruled out [18F]9e may indeed be a substrate for other clinically relevant MDR pumps,21 perhaps explaining the non-appreciable brain uptake observed in our non-human primate studies. Current evaluation is ongoing to identify a more metabolically stable PARPi that is a suitable candidate for PET neuroimaging.
4. Experimental
4.1. Chemistry
4.1.1. General experimental information
Chemical compounds 5a-f, 8, and 10a-b were purchased and used without further purification. Compound 8 can also be prepared following previously reported literature conditions.22, 23 Intermediate 614 and 1315 were synthesized following previously reported conditions. NMR spectra were taken on a Bruker DMX 500 MHz. Compound structures and identity were confirmed by 1H and 13C NMR, and mass spectroscopy. Compound purity greater than 95% was determined by LCMS analysis using a 2695 Alliance LCMS. All other commercial reagents were purchased and used without further purification. Purification of organic compounds were carried out on a Biotage Isolera One with a dual-wavelength UV-VIS detector. Chemical shifts (δ) in the NMR spectra (1H and 13C) were referenced by assigning the residual solvent peaks.
4.1.2. General synthetic procedure for compounds 7a-f
NaH 60% dispersion in mineral oil (3.3 mmol) was slowly added to a stirring solution of hydroxyl amine (5a-f) (3.0 mmol) in 6 mL of DMF at room temp, and stirred for 30 min. Compound 6 was then slowly added to reaction mixture, followed by an additional hour of stirring. A saturated NaHCO3 (aq) solution (40 mL) was then added to the crude reaction mixture and stirred at room temp for 1 h. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL) to afford the crude product. The subsequent residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with a 2:5 EtOAc/hexanes gradient, to afford the desired Boc-protected O-alkylated fluoroethoxy compounds, identified by LCMS. The intermediate was then dissolved in CH2Cl2 (2 mL), followed by dropwise addition of CF3COOH (2 mL), and stirred at room temperature for 3 h. Volatiles were then removed under reduced pressure and the crude product was neutralized with a saturated NaHCO3 (aq) solution (10 mL). The reaction mixture was extracted with CH2Cl2 (3 × 20 mL), and the organic layers were combined, dried, and concentrated to afford the free-amine intermediates (7a-f) as oils used in the next step without any further purification.
4.1.3. General synthetic procedure for compounds 9a-f, 11a-b, and 14
The desired free-amine intermediate (7a-f) (1.0 mmol), 8 (1.0 mmol), HOBt hydrate (1.0 mmol), EDC hydrochloride (1.0 mmol), and Et3N (2.0 mmol) were stirred in 5 mL of THF at 60 °C for 12 h. A saturated NaHCO3 (aq) solution (15 mL) was then added to the crude reaction mixture and stirred at room temp for 1 h. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL) to afford the crude product. The residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with 10% 7N NH3 in MeOH solution/CH2Cl2 to give the target compounds 9a−f. Compounds were analyzed for purity using LCMS, 1H and 13C NMR spectroscopy, and, if necessary, purified further using a Biotage SNAP flash purification cartridge, eluting with a 10% 7N NH3 in MeOH solution/EtOAc. It should be noted; the following yields of the target compounds were not optimized.
4.1.3.1. 4-(4-fluoro-3-(6-(2-fluoroethoxy)-2-azaspiro[3.3]heptane-2-carbonyl)benzyl)phthalazin-1(2H)-one (9a).
Compound 9a can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 28%). 1H NMR (500 MHz, CDCl3) (reported as mixture of rotamers) δ 11.23 (m, 1H), 8.47–8.45 (m, 1H), 7.77–7.73 (m, 2H), 7.72–7.69 (m, 1H), 7.49–7.46 (m, 1H), 7.31–7.28 (m, 1H), 7.00–6.96 (m, 1H), 4.54–4.53 (t, J = 4.1 Hz, 1H), 4.45–4.43 (t, J = 4.0 Hz, 1H), 4.27 (s, 2H), 4.13 (s, 1H), 3.98 (s, 1H), 3.96–3.83 (m, 1H), 3.59–3.56 (m, 1H), 3.53–3.50 (m, 1H), 2.56–2.52 (m, 1H), 2.50–2.46 (m, 1H), 2.21–2.17 (m, 1H), 2.14–2.10 (m, 1H); 13C NMR (125 MHz, CDCl3) (reported as mixture of rotamers) δ 166.0 (2×C), 160.8, 157.0 (d, JC-F = 250.0 Hz), 156.9 (d, JC-F = 249.0 Hz), 145.7, 134.1, (d, JC-C-C-C-F = 3.2 Hz), 133.7, 132.3 (d, JC-C-C-F = 7.2 Hz), 130.8 (d, JC-C-C-F = 7.6 Hz), 131.6, 130.2 (d, JC-C-C-F = 3.4 Hz), 130.1 (d, JC-C-C-F = 3.5 Hz), 129.6, 128.4, 127.2, 125.2, 122.5 (d, JC-C-F = 17.6 Hz), 122.4 (d, JC-C-F = 17.0 Hz), 116.6 (d, JC-C-F = 22.5 Hz), 116.5 (d, JC-C-F = 22.7 Hz), 83.7 (d, JC–F = 169.4 Hz, CH2F), 83.6 (d, JC–F = 169.4 Hz, CH2F), 68.9, 68.8, 67.5 (d, JC–F = 19.7 Hz, O–CH2CH2F), 67.4 (d, JC–F = 19.7 Hz, O–CH2CH2F), 63.5, 63.4, 62.3, 62.2, 60.8, 59.7, 41.0, 37.8 (2×C), 30.7, 30.6; LC-MS (ESI) m/z: 440.09 [M+H]
4.1.3.2. 4-(4-fluoro-3-(3-(2-fluoroethoxy)azetidine-1-carbonyl)benzyl)phthalazin-1(2H)-one (9b).
Compound 9b can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 10%). 1H NMR (500 MHz, CDCl3) δ 11.42 (s, 1H), 8.47–8.45 (m, 1H), 7.77–7.70 (m, 3H), 7.51–7.50 (dd, J1 = 1.9 Hz, J2 = 4.2 Hz, 1H), 7.32–7.29 (m, 1H), 6.95 (t, J = 9.1 Hz, 1H), 4.58–4.56 (t, J = 4.0 Hz, 1H), 4.49–4.47 (t. J = 4.0 Hz, 1H), 4.38–4.33 (m, 2H), 4.27 (s, 2H), 4.19–4.16 (m, 1H), 4.07–4.06 (m, 1H), 4.00–3.98 (m, 1H), 3.71–3.56 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 166.2, 160.9, 156.9 (d, JC-F = 250.4 Hz), 145.7, 134.1 (d, JC-C-C-C-F = 3.4 Hz), 133.7, 132.4 (d, JC-C-C-F = 8.3 Hz), 131.6, 130.2 (d, JC-C-C-F = 3.4 Hz), 129.6, 128.3, 127.2, 125.0, 122.2 (d, JC-C-F = 16.9 Hz), 116.4 (d, JC-C-F = 22.7 Hz), 82.2 (d, JC–F = 169.9 Hz, CH2F), 68.6, 68.5 (d, JC–F = 19.7 Hz, O–CH2CH2F), 58.5, 58.4, 55.9, 37.8; LC-MS (ESI) m/z: 400.17 [M+H]
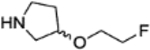
4.1.3.3. (R)-4-(4-fluoro-3-(3-(2-fluoroethoxy)pyrrolidine-1-carbonyl)benzyl)phthalazin-1(2H)-one (9c).
Compound 9c can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 12%). 1H NMR (500 MHz, CDCl3) (reported as mixture of rotamers) δ 11.81 (s, 1H), 8.45–8.43 (m, 1H), 7.74–7.70 (m, 3H), 7.38–7.35 (m, 1H), 7.27–7.25 (m, 1H), 6.99–6.96 (m, 1H), 4.57–4.39 (m, 2H), 4.26 (s, 2H), 4.19–4.06 (m, 1H), 3.76–3.70 (m, 2H), 3.69–3.65 (m, 1H), 3.63–3.41 (m, 2H), 3.32–3.27 (m, 1H); 2.08–2.02 (m, 1H), 2.01–1.92 (m, 1H); 13C NMR (125 MHz, CDCl3) (reported as mixture of rotamers) δ 165.1 (2×C), 161.1, 156.3 (2×C) (d, J(1)C-F = 248.0 Hz; J(2)C-F = 247.6 Hz), 145.7, 134.2 (d, JC-C-C-C-F = 3.7 Hz), 134.1 (d, JC-C-C-C-F = 3.7 Hz), 133.6, 131.5, 131.4 (2×C) (d, J(1)C-C-C-F = 8.6 Hz; J(2)C-C-C-F = 8.1 Hz), 129.6, 129.1 (d, JC-C-C-F = 3.8 Hz), 128.9 (d, JC-C-C-F = 4.0 Hz), 128.3, 127.0, 125.4 (d, JC-C-F = 17.7 Hz), 125.4 (d, JC-C-F = 18.2 Hz), 125.2 (2×C), 116.3 (2×C) (d, J(1)C-C-F = 22.0 Hz; J(1)C-C-F = 22.0 Hz), 83.7 (d, JC–F = 169.5 Hz, CH2F), 83.6 (d, JC–F = 169.7 Hz, CH2F), 78.5, 77.6, 68.2 (d, JC–F = 19.6 Hz, O–CH2CH2F), 52.8 (2×C), 51.2, 45.7 (2×C), 44.0, 37.8, 31.6, 29.9; LC-MS (ESI) m/z: 414.19 [M+H]
4.1.3.3. (S)-4-(4-fluoro-3-(3-(2-fluoroethoxy)pyrrolidine-1-carbonyl)benzyl)phthalazin-1(2H)-one (9d).
Compound 9d can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 14%). 1H NMR (500 MHz, CDCl3) (reported as mixture of rotamers) δ 11.65 (s, 1H), 8.45–8.44 (m, 1H), 7.72–7.71 (m, 3H), 7.39–7.36 (m, 1H), 7.28–7.26 (m, 1H), 7.00–6.96 (m, 1H), 4.57–4.40 (m, 2H), 4.27 (s, 2H), 4.19–4.08 (m, 1H), 3.77–3.71 (m, 2H), 3.69–3.64 (m, 1H), 3.59–3.42 (m, 2H), 3.32–3.28 (m, 1H), 2.09–1.93 (m, 2H); 13C NMR (125 MHz, CDCl3) (reported as mixture of rotamers) δ 165.1 (2×C), 161.0, 156.3 (2×C) (d, J(1)C-F = 247.6 Hz; J(2)C-F = 247.6 Hz), 145.7, 134.2 (d, JC-C-C-C-F = 3.2 Hz), 134.1 (d, JC-C-C-C-F = 3.0 Hz), 133.7, 131.5, 131.4 (2×C) (d, J(1)C-C-C-F = 8.1 Hz; J(2)C-C-C-F = 7.4 Hz), 129.6, 129.1 (d, JC-C-C-F = 3.8 Hz), 128.9 (d, JC-C-C-F = 3.7 Hz), 128.3, 127.1, 125.5 (d, JC-C-F = 18.5 Hz), 125.4 (d, JC-C-F = 18.3 Hz), 125.2 (2×C), 116.4 (2×C) (d, J(1)C-C-F = 22.0 Hz; J(1)C-C-F = 22.0 Hz), 83.7 (d, JC–F = 169.3 Hz, CH2F), 83.6 (d, JC–F = 169.8 Hz, CH2F), 78.5, 77.6, 68.2 (d, JC–F = 19.9 Hz, O–CH2CH2F), 52.8 (2×C), 51.2, 45.8 (2×C), 44.0, 37.8, 31.6, 29.9; LC-MS (ESI) m/z: 414.19 [M+H]
4.1.3.4. 4-(4-fluoro-3-(4-(2-fluoroethoxy)piperidine-1-carbonyl)benzyl)phthalazin-1(2H)-one (9e).
Compound 9e can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 37%). 1H NMR (500 MHz, CDCl3) δ 11.84 (s, 1H), 8.45–8.43 (m, 1H), 7.73–7.67 (m, 3H), 7.30–7.28 (m, 1H), 7.27–7.24 (m, 1H), 6.98–6.95 (t, J = 8.8 Hz, 1H), 4.56–4.54 (t, J = 4.1 Hz, 1H), 4.46–4.44 (t, J = 4.1 Hz, 1H), 4.26 (s, 2H), 3.96 (bs, 1H), 3.73–3.68 (m, 1H), 3.67–3.63 (m, 1H), 3.62–3.60 (m, 1H), 3.55 (bs, 1H), 3.43 (bs, 1H), 3.08 (bs, 1H), 1.91–1.87 (m, 1H), 1.72–1.66 (m, 2H), 1.55 (bs, 1H); 13C NMR (125 MHz, CDCl3) δ 164.8, 161.0, 156.1 (d, JC-F = 247.2 Hz), 145.6, 134.2 (d, JC-C-C-C-F = 3.1 Hz), 133.6, 131.5, 131.1 (d, JC-C-C-F = 7.9 Hz), 129.5, 128.9 (d, JC-C-C-F = 3.7 Hz), 128.2, 127.0, 125.1, 124.5 (d, JC-C-F = 18.7 Hz), 116.1 (d, JC-C-F = 21.6 Hz), 83.8 (d, JC–F = 169.4 Hz, CH2F), 74.2, 68.2 (d, JC–F = 19.3 Hz, O–CH2CH2F), 44.1, 38.8, 37.7, 31.3, 30.3; LC-MS (ESI) m/z: 428.08 [M+H]
4.1.3.5. 4-(4-fluoro-3-(4-(2-fluoroethoxy)azepane-1-carbonyl)benzyl)phthalazin-1(2H)-one (9f).
Compound 9f can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 41%). 1H NMR (500 MHz, CDCl3) (reported as mixture of isomers and rotamers) δ 11.76–11.74 (m, 1H), 8.45–8.43 (m, 1H), 7.73–7.69 (m, 3H), 7.28–7.23 (m, 2H), 7.28–7.24 (m, 2H), 6.99–6.95 (m, 1H), 4.56–4.53 (m, 1H), 4.46–4.42 (m, 1H), 4.26 (s, 2H), 3.74 (bs, 1H), 3.69–3.65 (m, 1H), 3.62–3.56 (m, 2H), 3.55–3.51 (m, 1H), 3.39–3.20 (m, 2H), 2.04–1.91 (m, 2H), 1.87–1.77 (m, 2H), 1.74–1.68 (m, 2H), 1.55 (bs, 1H); 13C NMR (125 MHz, CDCl3) (reported as mixture of isomers and rotamers) δ 166.5, 166.3, 161.0 (2×C), 155.9 (d, JC-F = 247.4 Hz), 145.7, 134.2 (d, JC-C-C-C-F = 3.5 Hz), 134.1 (d, JC-C-C-C-F = 3.5 Hz), 133.6 (2×C), 131.5, 130.9 (d, JC-C-C-F = 7.5 Hz), 130.8 (d, JC-C-C-F = 7.6 Hz), 129.6, 128.5 (d, JC-C-C-F = 4.0 Hz), 128.2, 127.1, 125.5 (d, JC-C-F = 19.0 Hz), 125.4 (d, JC-C-F = 19.0 Hz), 116.3 (d, JC-C-F = 21.7 Hz), 116.2 (d, JC-C-F = 21.7 Hz), 84.0 (d, JC–F = 169.2 Hz, CH2F), 83.9 (d, JC–F = 168.9 Hz, CH2F), 77.5, 77.2, 67.7 (d, JC–F = 19.7 Hz, O–CH2CH2F), 67.6 (d, JC–F = 20.0 Hz, O–CH2CH2F), 48.7, 45.9, 43.8, 40.6, 37.8, 34.4, 32.9, 31.8, 30.1, 21.8, 21.3; LC-MS (ESI) m/z: 442.25 [M+H]
4.1.3.6. 4-(4-fluoro-3-(4-(4-fluorophenyl)piperazine-1-carbonyl)benzyl)phthalazin-1(2H)-one (11a).
Compound 11a can be prepared following the general synthetic procedure to afford a white crystalline solid (Yield 52%). 1H NMR (500 MHz, ((CD3)2SO) δ 12.60 (s, 1H), 8.27 (d, J = 7.7 Hz, 1H), 7.96 (d, J = 7.9 Hz, 1H), 7.88 (t, J = 7.2 Hz, 1H), 7.82 (t, J = 7.5 Hz, 1H), 7.45–7.43 (m, 1H), 7.38–7.37 (m, 1H), 7.24 (t, J = 9.1 Hz, 1H), 7.06 (t, J = 8.6 Hz, 2H), 6.96–6.93 (m, 2H), 4.33 (s, 2H), 3.75 (bs, 2H), 3.30 (bs, 2H), 3.12 (bs, 2H), 2.95 (bs, 2H). 13C NMR (125 MHz, ((CD3)2SO)) δ 163.8, 159.3, 155.4 (2×C) (d, J1C-F = 240.6 Hz, J2C-F = 239.1 Hz), 147.5, 144.8, 134.8 (d, JC-C-C-C-F = 3.8 Hz), 133.4, 131.6 (d, JC-C-C-F = 7.9 Hz), 131.5, 129.0, 128.8 (d, JC-C-C-F = 3.9 Hz), 127.9, 126.0, 125.4, 123.6 (d, JC-C-F = 18.3 Hz), 117.9 (d, JC-C-C-F = 7.8 Hz), 116.0 (d, JC-C-F = 21.8 Hz), 115.4 (d, JC-C-F = 21.9 Hz), 49.3, 49.2, 46.3, 41.2, 36.4; LC-MS (ESI) m/z: 461.06 [M+H]
4.1.3.7. 4-(4-fluoro-3-(4-(4-fluorophenyl)piperidine-1-carbonyl)benzyl)phthalazin-1(2H)-one (11b).
Compound 11b can be prepared following the general synthetic procedure to afford a hard white solid (Yield 28%). 1H NMR (500 MHz, ((CD3)2SO) δ 12.60 (s, 1H), 8.25 (d, J = 7.6 Hz, 1H), 7.85–7.82 (m, 1H), 7.79–7.76 (m, 1H), 7.42–7.40 (m, 2H), 7.25–7.19 (m, 3H), 7.10 (t, J = 8.9 Hz, 1H), 4.64–4.61 (m, 1H), 4.33 (s, 2H), 3.40–3.38 (m, 1H), 3.10 (t, J = 12.4 Hz, 1H), 2.83–2.75 (m, 2H), 1.83–1.81 (m, 1H), 1.63–1.49 (m, 2H), 1.40 (bs, 1H). 13C NMR (125 MHz, ((CD3)2SO)) δ 163.6, 159.7 (d, JC-F = 243.0 Hz), 159.3, 155.3 (JC-F = 243.0 Hz), 144.8, 141.4 (d, JC-C-C-C-F = 2.6 Hz), 134.7 (d, JC-C-C-C-F = 2.9 Hz), 133.3, 131.4, 131.3 (d, JC-C-C-F = 7.8 Hz), 129.0, 128.5, 128.4 (d, JC-C-C-F = 7.8 Hz), 127.9, 126.0, 125.4, 124.3 (d, JC-C-F = 18.2 Hz), 115.9 (d, JC-C-F = 22.0 Hz), 114.9 (d, JC-C-F = 20.7 Hz), 59.7, 54.86, 46.9, 41.6, 40.7, 36.4, 32.6; LC-MS (ESI) m/z: 460.12 [M+H]
4.1.3.8. 4-(4-fluoro-3-(6-(4-fluorophenyl)-2,6-diazaspiro[3.3]heptane-2-carbonyl)benzyl)phthalazin-1(2H)-one (14).
Compound 14 can be prepared following the general synthetic procedure to afford a hard white solid (Yield 26%). 1H NMR (500 MHz, CDCl3) δ 11.83 (s, 1H), 8.25 (d, J = 7.7 Hz, 1H), 7.75–7.70 (m, 3H), 7.52–7.50 (dd, J1 = 2.0 Hz, J2 = 6.2 Hz, 1H), 7.34–7.31 (m, 1H), 7.00 (t, J = 9.2 Hz, 1H), 6.87 (t, J = 8.7 Hz, 1H), 6.34–6.32 (m, 2H), 4.31 (s, 2H), 4.28 (s, 2H), 4.20 (s, 2H), 3.94 (d, J = 7.6 Hz, 2H), 3.89 (d, J = 7.6 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 166.6, 161.1, 156.9 (d, JC-F = 250.2 Hz), 155.4 (JC-F = 236.0 Hz), 147.6, 145.6, 134.2 (d, JC-C-C-C-F = 3.0 Hz), 133.7, 132.6 (d, JC-C-C-F = 8.3 Hz), 131.5, 130.2 (d, JC-C-C-C-F = 3.3 Hz), 129.6, 128.2, 127.1, 125.1, 122.0 (d, JC-C-F = 16.8 Hz), 116.3 (d, JC-C-F = 22.5 Hz), 115.6 (d, JC-C-F = 22.3 Hz), 112.8 (d, JC-C-C-F = 7.5 Hz), 62.5, 61.2, 61.1, 58.6, 37.7, 33.7; LC-MS (ESI) m/z: 473.21 [M+H]
4.1.4. Synthetic procedure for compound 17
4.1.4.1. 4-(4-fluoro-3-(6-(4-fluorophenyl)-2-azaspiro[3.3]heptane-2-carbonyl)benzyl)phthalazin-1(2H)-one (17).
The synthesis of 16 was adapted from previously reported literature conditions.16, 24, 25 A solution of 5a (6.0 mmol) in THF (20 mL) at 0 °C was treated with triphenylphosphine (10 mmol), followed by CBr4 (10 mmol). After 1 h, the reaction mixture was warmed to room temp, and stirred for an additional 2 h. Next, the solvent was removed under reduced pressure to afford a crude oily residue. The subsequent residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with a 1:20 EtOAc/hexanes gradient, to afford 15 as a white solid. (Yield 26%) 1H NMR (500 MHz, CDCl3) δ 4.27–4.24 (quint, J = 7.3 Hz, 1H), 3.89 (s, 2H), 3.86 (s, 2H), 2.83–2.79 (m, 2H), 2.57–2.52 (m, 2H), 1.37 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 156.0, 79.4, 61.0, 60.7, 45.6, 36.4, 35.6, 28.3; LC-MS (ESI) m/z: 176.18 [M-Boc]. A 40 mL vial was charged with NiI2 (0.20 mmol), trans-2-aminocyclohexanol hydrochloride (0.20 mmol), NaHMDS (4.0 mmol), and 2-methyl-2-butanol (1.0 mL). The reaction was purged with N2 for 5 min, followed by the addition of 15 (2.0 mmol) in 2.5 mL of 2-methyl-2-butanol. The reaction mixture was heated to 80 °C and stirred vigorously for 12 h. After which, the crude reaction mixture was filtered and the solvent was removed under reduced pressure. The subsequent residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with a 1:5 EtOAc/hexanes gradient, to afford the Boc-protected precursor of 16 as a white solid. (Yield 74%). The intermediate was then dissolved in CH2Cl2 (2 mL), followed by dropwise addition of CF3COOH (2 mL), and stirred at room temperature for 3 h. Volatiles were then removed under reduced pressure and the crude product was neutralized with a saturated NaHCO3 (aq) solution (10 mL). The reaction mixture was extracted with CH2Cl2 (3 × 20 mL), and the organic layers were combined, dried, and concentrated to afford the free-amine intermediates (16) as light-tan solid. Finally, 16 (1.0 mmol), 16 (1. 0 mmol), HOBt hydrate (1.0 mmol), EDC hydrochloride (1.0 mmol), and Et3N (2.0 mmol) were stirred in 5 mL of THF at 60 °C for 12 h. A saturated NaHCO3 (aq) solution (15 mL) was then added to the crude reaction mixture and stirred at room temp for 1 h. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL) to afford the crude product. The residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with 10% 7N NH3 in MeOH solution/CH2Cl2 to give the target compounds 17 as a soft white solid. (Yield 18%) 1H NMR (500 MHz, CDCl3) (reported as mixture of rotamers) δ 11.38 (m, 1H), 8.48–8.46 (m, 1H), 7.77–7.71 (m, 3H), 7.52–7.49 (m, 1H), 7.34–7.29 (m, 1H), 7.10–7.06 (m, 2H), 7.01–6.93 (m, 3H), 4.29–4.28 (m, 3H), 4.16 (s, 1H), 4.08 (s, 1H), 3.94 (s, 1H), 3.42–3.25 (m, 1H), 2.64–2.60 (m, 1H), 2.58–2.54 (m, 1H), 2.31–2.27 (m, 1H), 2.24–2.19 (m, 1H). 13C NMR (125 MHz, CDCl3) (reported as mixture of rotamers) δ 166.1, 166.0, 160.9, 160.4 (2×C) (d, J1C-F = 244.7 Hz, J2C-F = 244.7 Hz), 157.0 (JC-F = 250.4 Hz), 145.7, 140.0 (d, JC-C-C-C-F = 2.8 Hz), 139.9 (d, JC-C-C-C-F = 2.7 Hz), 134.1, 134.0 (d, JC-C-C-F = 8.1 Hz), 133.7, 132.2 (d, JC-C-C-F = 8.2 Hz), 131.6, 130.2, 129.6, 128.4, 127.7 (d, JC-C-C-C-F = 3.2 Hz), 127.6 (d, JC-C-C-C-F = 2.8 Hz), 127.2, 125.2, 122.4 (d, JC-C-F = 17.2 Hz), 122.3 (d, JC-C-F = 17.2 Hz), 116.3 (d, JC-C-F = 22.7 Hz), 115.1 (d, JC-C-F = 21.2 Hz), 64.0, 63.9, 61.9, 61.8, 61.4, 59.3, 40.4, 37.8, 34.4, 34.2, 33.6, 33.3; LC-MS (ESI) m/z: 472.14 [M+H]
4.1.5. Synthetic procedure for compound 19a-b
4.1.5.1. 4-(4-fluoro-3-(4-(2-hydroxyethoxy)piperidine-1-carbonyl)benzyl)phthalazin-1(2H)-one (15).
Compounds 8 (1.0 mmol), 2-(piperidin-4-yloxy)ethan-1-ol (1. 0 mmol), HOBt hydrate (1.0 mmol), EDC hydrochloride (1.0 mmol), and Et3N (2.0 mmol) were stirred in 5 mL of THF at 60 °C for 12 h. A saturated NaHCO3 (aq) solution (15 mL) was then added to the crude reaction mixture and stirred at room temp for 1 h. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL) to afford the crude product. The residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with 10% 7N NH3 in MeOH solution/CH2Cl2 to give the target compounds 15 as a white crystalline solid. (Yield 38%) 1H NMR (500 MHz, CDCl3) δ 11.78 (s, 1H), 8.42–8.40 (m, 1H), 7.69–7.67 (m, 3H), 7.28–7.26 (m, 1H), 7.25–7.26 (m, 1H), 6.97–6.93 (m, 1H), 4.24 (m, 2H), 4.01 (bs, 1H), 3.69–3.68 (m, 2H), 3.57–3.50 (m, 3H), 3.43 (bs, 2H), 3.05 (bs, 1H), 2.81 (bs, 1H), 1.91–1.88 (m, 1H), 1.73 (bs, 1H), 1.68–1.62 (m, 1H), 1.52 (bs, 1H); 13C NMR (125 MHz, CDCl3) δ 164.8, 160.9, 156.0 (d, JC-F = 246.9 Hz), 145.7, 134.2 (d, JC-C-C-C-F = 3.1 Hz), 133.6, 131.5, 131.1 (d, JC-C-C-F = 7.8 Hz), 129.5, 128.9 (d, JC-C-C-F = 3.5 Hz), 128.2, 127.0, 125.1, 124.5 (d, JC-C-F = 18.6 Hz), 116.1 (d, JC-C-F = 22.0 Hz), 74.2, 69.4, 61.8, 44.3, 39.0, 37.7, 31.3, 30.5; LC-MS (ESI) m/z: 426.06 [M+H]
4.1.5.2. 2-((1-(2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperidin-4-yl)oxy)ethyl 4-methylbenzenesulfonate (19a).
Reagent p-toulenesulfonyl chloride (1.3 mmol) was slowly added to a stirring solution of 18 (1.0 mmol) and TEA (2.1 mmol) in 20 mL of DCM at 0 °C. The reaction mixture was allowed to warm to room temp, and stirred for 12 h. Then, the reaction mixture was filtered and solvent was removed under reduced pressure to afford a crude oily reside. The subsequent residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with a 1:20 MeOH/EtOAc gradient, to afford 19a as a white crystalline solid. (Yield 75%) 1H NMR (500 MHz, CDCl3) δ 11.61 (s, 1H), 8.45–8.43 (m, 1H), 7.76–7.74 (m, 3H), 7.73–7.69 (m, 2H), 7.31–7.29 (m, 3H), 7.28–7.25 (m, 1H), 6.99–6.96 (t, J = 8.7 Hz, 1H), 4.26 (s, 2H), 4.13–4.11 (t, J = 4.7 Hz, 2H), 3.79 (bs, 1H), 3.66–3.58 (m, 3H), 3.52 (bs, 1H), 3.35 (bs, 1H), 3.05 (bs, 1H), 2.40 (s, 3H), 1.80–1.76 (m, 1H), 1.65 (bs, 1H), 1.58–1.56 (m, 1H), 1.47 (bs, 1H); 13C NMR (125 MHz, CDCl3) δ 164.8, 160.9, 156.0 (d, JC-F = 247.6 Hz), 145.7, 144.9, 134.2 (d, JC-C-C-C-F = 3.3 Hz), 133.6, 133.0, 131.5, 131.2 (d, JC-C-C-F = 7.9 Hz), 129.8, 129.6, 128.9 (d, JC-C-C-F = 3.4 Hz), 128.3, 127.9, 127.1, 124.5, 125.1, 124.5 (d, JC-C-F = 18.4 Hz), 116.1 (d, JC-C-F = 22.0 Hz), 74.1, 69.5, 65.7, 43.9, 38.6, 37.7, 31.3, 30.5, 21.6; LC-MS (ESI) m/z: 426.06 [M+H]; LC-MS (ESI) m/z: 580.20 [M+H]
4.1.5.3. 2-((1-(2-fluoro-5-((4-oxo-3,4-dihydrophthalazin-1-yl)methyl)benzoyl)piperidin-4-yl)oxy)ethyl methanesulfonate (19b).
Reagent methane sulfonyl chloride (1.3 mmol) was slowly added to a stirring solution of 18 (1.0 mmol) and TEA (2.1 mmol) in 20 mL of DCM at 0 °C. The reaction mixture was allowed to warm to room temp, and stirred for 12 h. Then, the reaction mixture was filtered and solvent was removed under reduced pressure to afford a crude oily reside. The subsequent residue was loaded onto a Biotage SNAP flash purification cartridge, eluting with a 1:20 MeOH/EtOAc gradient, to afford 19b as a white crystalline solid. (Yield 80%) 1H NMR (500 MHz, CDCl3) δ 11.33 (s, 1H), 8.46–8.44 (m, 1H), 7.75–7.71 (m, 3H), 7.30–7.26 (m, 2H), 7.01–6.97 (t, J = 8.8 Hz, 1H), 4.34–4.33 (t, J = 4.5 Hz, 2H), 4.27 (s, 2H), 3.98 (bs, 1H), 3.76–3.68 (m, 2H), 3.61 (bs, 1H), 3.53 (bs, 1H), 3.45–3.43 (m, 1H), 3.11 (bs, 1H), 3.03 (s, 3H), 1.92–1.88 (m, 1H), 1.75 (bs, 1H), 1.71–1.66 (m, 1H), 1.54 (bs, 1H); 13C NMR (125 MHz, CDCl3) δ 164.9, 160.8, 156.1 (d, JC-F = 246.7 Hz), 145.7, 134.2 (d, JC-C-C-C-F = 3.2 Hz), 133.7, 133.0, 131.6, 131.2 (d, JC-C-C-F = 7.7 Hz), 129.6, 129.0 (d, JC-C-C-F = 3.7 Hz), 128.3, 127.1, 125.1, 124.6 (d, JC-C-F = 18.5 Hz), 116.2 (d, JC-C-F = 21.8 Hz), 74.5, 69.3, 66.0, 44.1, 38.9, 37.8, 37.7, 31.3, 30.4; LC-MS (ESI) m/z: 504.86 [M+H]
4.2. Biological evaluation
4.2.3. PARP-1 radioligand binding
The affinity of each compound for the PARP-1 enzyme was evaluated using a radioligand binding method previously reported by our group.17 Briefly, OVCAR8 ovarian cancer cells were seeded in 96-well Stripwell plates at a density of 40,000 cells/well 24 hrs prior to the study. On the day of study, compounds were diluted in RPMI to concentrations of 100 μM - 0.064 nM. Next, [125I]KX1, a known PARP-1 specific radioligand, was added to the plate followed by each compound dilution. Reactions were allowed to equilibrate for 1 hr and were then washed with 200 μL of PBS. Wells were separated and counted on an automatic Wizard gamma counter (Perkin Elmer, Waltham MA). Dose response curves were produced to calculate 50% maximum inhibition values (IC50) using non-linear fit sigmoidal dose response curves in GraphPad 7.0 (Prism, La Jolla CA). Experiments were repeated three times with adjusting dose concentrations to increase accuracy of IC50 values.
4.2.4. Pgp-Glo™ assay
P-gp activity of each compound was measured using Promega Pgp-Glo™ Assay Systems. 25μg of diluted recombinant human P-gp membranes were added to untreated white opaque multiwell plates along with Pgp-Glo™ Assay Buffer, a non-limiting concentration of ATP (5mM) and 20μl of each test compound (20μM) for 1 hr at 37°C. Untreated and Na3VO4-treated control samples were also tested in addition to Verapamil-treated samples (positive control). After incubation, 50μl of ATP Detection Reagent was added to all wells to stop the P-gp reaction. Samples were mixed briefly on a plate shaker then incubated plate at room temperature for 20 minutes to allow luminescent signal to develop. Luminescence was read on a plate-reading luminometer. This luciferase-based detection reaction provides a linear response to ATP concentration in each sample. Thus any changes in signal directly reflect changes in ATP concentration.
4.2.5. Radiochemistry
The radiosynthesis of [18F]9e was accomplished on a Synthera synthesis module with full automation. Briefly, [18F]fluoride (800 mCi) was produced by proton irradiation of enriched H218O via the reaction of 18O (p, n) 18F by the Cyclone cyclotron (IBA). The [18F]fluoride in a H218O solution is delivered to the hotcell, trapped on a preactivated Sep-Pak Light QMA Carb cartridge (Waters), and eluted to the reaction vial with 1 mL of eluent containing 2 mg of potassium carbonate and 7 mg of Kryptofix in a mixture of 0.85 mL of acetonitrile and 0.15 mL of water. The residual water was evaporated azotropically with 1 mL of acetonitrile at 100 °C under a stream of nitrogen gas and vacuum. A solution of 4 mg of tosylate precursor 19a in 0.7 mL of methyl sulfide (DMSO) was added to the reaction vial for a 20 min reaction. The crude product was diluted with 3 mL of the mobile phase and passed through an Alumina N Light cartridge (Waters) and a 0.45 μm nylon filter to the HPLC loop for high-performance chromatography (HPLC). A Phenomenex Luna 5 μm C18 100 Å LC Column 250 × 10 mm semipreparative column with a mobile phase of acetonitrile and water (40:60 by volume) was used for HPLC purification. At a flow rate of 4 mL/min, the product was eluted at 11 min and diluted with water to a volume of 50 mL. The diluted product solution was passed through a Sep-Pak Plus C18 cartridge (Waters). The trapped product was rinsed with water to waste and then eluted with ethanol (0.6 mL, with 0.1% ascorbic acid) followed by 8 mL of normal saline to the final production vial through a 0.2 μm nylon filter. After being shaken well, the final product was ready for quality control (QC) and animal studies. The yield ranged from 8 to 11% (decay corrected to the start of synthesis) in an average time of 65 min from receipt of [18F]fluoride in a H218O solution from the cyclotron.
4.2.6. In vitro autoradiography
The normal Balb/c mouse (male, 10–12 weeks-old) frozen brain sections (10-μm thickness) were prepared using a cryostat microtome (CM1900, Leica, Germany) one day before and kept in −80℃ freezer. The sections were first thawed at room temperature for 20 min and rehydrated with ice-cold PBS buffer (pH 7.4) for 5 min. Then all sections were incubated with 4 nM [18F]9e with, or without, 10 μM olaparib at room temperature for 1 h to define the control and nonspecific binding groups. After the radioligand incubation, all of them were washed three times with ice-cold PBS for 3 min, dried up with a fan, and exposed to a BAS-SR 2040 imaging plate (20×40 cm, Fujifilm, Japan) for 2 hr. In the end the plate were scanned with a Typhoon 7000 phosphorimager (GE Healthcare) in a condition of 500 V and a resolution of 25 μm.
4.2.7. MicroPET imaging
All animal studies were performed under protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Male rhesus macaques (13–21 year old) were sedated with ketamine/dexdomidor and anesthesia maintained for imaging with 1% isoflurane. Temperature was maintained with a recirculating water warm pad and vital signs such as blood pressure, pulse oximetry, and EKG were monitored continuously. Three rhesus macaques were scanned on the G-PET, a high sensitivity, high resolution PET scanner using gadolinium orthosilicate crystals incorporated into an Anger-logic detector developed for brain imaging. Data was acquired for up to 50 min (6 × 10 sec, 3 × 60 sec, 3 × 120 sec, 3 × 180 sec, 4 × 300 sec, and 1 × 600 sec) in list mode after an intravenous injection of 101.6 MBq [18F]FTT to one of the rhesus macaques, and 91.4–128.8 MBq [18F]9e to two of the rhesus macaques. A venous blood sample was drawn into a heparinized syringe at 37 min post [18F]9 injection for metabolite analysis. The acquired data were sorted into sinograms and reconstructed using the fully 3D LOR-RAMLA iterative reconstruction algorithm. Six volumes of interest (VOIs) including striatum, thalamus, frontal cortex, occipital cortex, whole brain, and cerebellum were manually delineated with PMOD image analysis software (version 3.7, PMOD Technologies LLC). Time activity curves were extracted from all the VOIs and performed as percentage injection dose per c.c. (%ID/c.c.) and standard uptake value (SUV).
4.2.8. Metabolite analysis
The blood samples were centrifuged at 3000 G for 10 min to separate plasma and red blood cells. 2 mL acetonitrile was added to the sample of plasma (1 mL). The plasma solution was vortexed, followed by centrifugation at 3000 G for 10 min. After the supernatant had been separated from the pellet, each portion was counted with a gamma counter (PerkinElmer Wizard 2480) to determine the extraction efficiency. Each supernatant was diluted with water and passed through a 0.45 um nylon filter for HPLC injection. With Agilent 1200 series, 200 uL solution was injected onto Agilent SB-C18 column (250 × 10 mm) for analysis. The mobile phase was 34% acetonitrile in water (volume) and the flow rate was 1 mL/min. The HPLC eluent was collected 1 tube/min and 16 tubes were collected for each injection. The collected HPLC fractions were measured by Gamma Counter for further data analysis. Meanwhile the cold reference was also injected and monitored at 254 nm UV wavelength.
Supplementary Material
Figure 2.

Luminescence generated with 9e, 11b, 14, and 17, olaparib, Verapamil, and Na3VO4. Verapamil, a substrate for P-gp that stimulates P-gp ATPase activity resulting in decreased luminescence, was utilized as a positive control. Increase in luminescence results from the light-generating reaction from luciferase and unmetabolized ATP. Unconsumed ATP indicates a decrease in P-gp ATPase stimulation, rendering the compound as a P-gp inhibitor. Data is normalized to known P-gp inhibitor Na3VO4.
ACKNOWLEDGMENT
David Livingston (Harvard Medical School) for the OVCAR8 cancer cells.
Funding Sources
SWR and LNP conducted this research through the support of training grants 5T32DA028874–07 and T32GM008076, respectively.
ABBREVIATIONS
- ATP
adenosine triphosphate
- BBB
blood-brain barrier
- EDC HCl
ethylcarbodiimide hydrochloride
- HOBt
1-hydroxybenzotriazole
- MDR
multidrug resistance
- P-gp
permeability glycoprotein
- TEA
triethylamine
- THF
tetrahydrofuran
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Monkey blood, and mice brain and blood HPLC metabolite analysis of [18F]9e (PDF)
1H and 13C NMR spectra of compounds 9a-f, 11a-b, 14, 17, and 19a (PDF)
REFERENCES
- 1.Bai P Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y-Q; Wang P-Y; Wang Y-T; Yang G-F; Zhang A; Miao Z-H An Update on Poly(ADP-ribose)Polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. J. Med. Chem 2016, 59, 9575–9598. [DOI] [PubMed] [Google Scholar]
- 3.Weaver AN; Yang ES Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol 2013, 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martire S; Mosca L; d’Erme M PARP-1 Involvement in Neurodegeneration: A Focus on Alzheimer’s and Parkinson’s Diseases. Mech. Ageing Dev 2015, 146.-, 53–64. [DOI] [PubMed] [Google Scholar]
- 5.Pacher P; Szabó C Role of Poly(ADP‐ribose) Polymerase 1 (PARP‐1) in Cardiovascular Diseases: The Therapeutic Potential of PARP Inhibitors. Cardiovasc. Drug Rev. 2007, 25, 235–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scobie N, Poly K (ADP)-ribose Polymerase-1 Inhibitors as a Potential Treatment for Cocaine Addiction. CNS Neurol. Disord. Drug Targets 2015, 14, 727–730. [DOI] [PubMed] [Google Scholar]
- 7.Scobie KN; Damez-Werno D; Sun H; Shao N; Gancarz A; Panganiban CH; Dias C; Koo J; Caiafa P; Kaufman L; Neve RL; Dietz DM; Shen L; Nestler EJ Essential Role of Poly(ADP-ribosyl)ation in Cocaine Action. Proc. Natl. Acad. Sci. USA 2014, 111, 2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan J; Dawson TM; Dawson VL, Cell Death Mechanisms of Neurodegeneration In Neurodegenerative Diseases: Pathology, Mechanisms, and Potential Therapeutic Targets, Beart P; Robinson M; Rattray M; Maragakis NJ, Eds. Springer International Publishing: Cham, 2017; pp 403–425. [Google Scholar]
- 9.Carney B; Kossatz S; Reiner T Molecular Imaging of PARP. J. Nucl. Med 2017, 58, 1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denise L; Patricia M; Steven B; Julien T; J., O. L. J.; T., H. B.; Britta S PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci 2014, 103, 1913–1920. [DOI] [PubMed] [Google Scholar]
- 11.Rottenberg S; Jaspers JE; Kersbergen A; van der Burg E; Nygren AOH; Zander SAL; Derksen PWB; de Bruin M; Zevenhoven J; Lau A; Boulter R; Cranston A; O’Connor MJ; Martin NMB; Borst P; Jonkers J High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oplustil O’Connor L; Rulten SL; Cranston AN; Odedra R; Brown H; Jaspers JE; Jones L; Knights C; Evers B; Ting A; Bradbury RH; Pajic M; Rottenberg S; Jonkers J; Rudge D; Martin NM; Caldecott KW; Lau A; O’Connor MJ The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, 6084–6094. [DOI] [PubMed] [Google Scholar]
- 13.Wang L; Tang Z; Luo L; Wei M; Li K; Song J; Zhang T; Wang H; Ren B; Zhou C Treatment Cancers Using Combination Comprising PARP Inhibitors. WO2018059437A1, 2018. [Google Scholar]
- 14.Henrottin J; Zervosen A; Lemaire C; Sapunaric F; Laurent S; Van den Eynde B; Goldman S; Plenevaux A; Luxen A N1-Fluoroalkyltryptophan Analogues: Synthesis and in vitro Study as Potential Substrates for Indoleamine 2,3-Dioxygenase. ACS Med. Chem. Lett 2015, 6, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reilly SW; Bryan NW; Mach RH Pd-Catalyzed Arylation of Linear and Angular Spirodiamine Salts Under Aerobic Conditions. Tetrahedron Lett. 2017, 58, 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.González-Bobes F; Fu GC Amino Alcohols as Ligands for Nickel-Catalyzed Suzuki Reactions of Unactivated Alkyl Halides, Including Secondary Alkyl Chlorides, with Arylboronic Acids. J. Am. Chem. Soc 2006, 128, 5360–5361. [DOI] [PubMed] [Google Scholar]
- 17.Makvandi M; Xu K; Lieberman BP; Anderson RC; Effron SS; Winters HD; Zeng C; McDonald ES; Pryma DA; Greenberg RA; Mach RH A Radiotracer Strategy to Quantify PARP-1 Expression In Vivo Provides a Biomarker That Can Enable Patient Selection for PARP Inhibitor Therapy. Cancer Res. 2016, 76, 4516–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reilly SW; Puentes LN; Wilson K; Hsieh C-J; Weng C-C; Makvandi M; Mach RH Examination of Diazaspiro Cores as Piperazine Bioisosteres in the Olaparib Framework Shows Reduced DNA Damage and Cytotoxicity. J. Med. Chem 2018, 61, 5367–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes JD; Blagg J; Price DA; Bailey S; DeCrescenzo GA; Devraj RV; Ellsworth E; Fobian YM; Gibbs ME; Gilles RW; Greene N; Huang E; Krieger-Burke T; Loesel J; Wager T; Whiteley L; Zhang Y Physiochemical Drug Properties Associated with In Vivo Toxicological Outcomes. Bioorg. Med. Chem. Lett 2008, 18, 4872–4875. [DOI] [PubMed] [Google Scholar]
- 20.Michel LS; Dyroff S; Brooks FJ; Spayd KJ; Lim S; Engle JT; Phillips S; Tan B; Wang-Gillam A; Bognar C; Chu W; Zhou D; Mach RH; Laforest R; Chen DL PET of Poly (ADP-Ribose) Polymerase Activity in Cancer: Preclinical Assessment and First In-Human Studies. Radiology 2017, 282, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eckford PDW; Sharom FJ ABC Efflux Pump-Based Resistance to Chemotherapy Drugs. Chem. Rev 2009, 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- 22.Zmuda F; Malviya G; Blair A; Boyd M; Chalmers AJ; Sutherland A; Pimlott SL Synthesis and Evaluation of a Radioiodinated Tracer with Specificity for Poly(ADP-ribose) Polymerase-1 (PARP-1) in Vivo. J. Med. Chem 2015, 58, 8683–8693. [DOI] [PubMed] [Google Scholar]
- 23.Menear KA; Adcock C; Boulter R; Cockcroft X.-l.; Copsey L; Cranston A; Dillon KJ; Drzewiecki J; Garman S; Gomez S; Javaid H; Kerrigan F; Knights C; Lau A; Loh VM; Matthews ITW; Moore S; O’Connor MJ; Smith GCM; Martin NMB 4-[3-(4-Cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A Novel Bioavailable Inhibitor of Poly(ADP-ribose) Polymerase-1. J. Med. Chem 2008, 51, 6581–6591. [DOI] [PubMed] [Google Scholar]
- 24.Meyers MJ; Long SA; Pelc MJ; Wang JL; Bowen SJ; Schweitzer BA; Wilcox MV; McDonald J; Smith SE; Foltin S; Rumsey J; Yang Y-S; Walker MC; Kamtekar S; Beidler D; Thorarensen A Discovery of novel spirocyclic inhibitors of fatty acid amide hydrolase (FAAH). Part 2. Discovery of 7-azaspiro[3.5]nonane urea PF-04862853, an orally efficacious inhibitor of fatty acid amide hydrolase (FAAH) for pain. Bioorg. Med. Chem. Lett 2011, 21, 6545–6553. [DOI] [PubMed] [Google Scholar]
- 25.Long SA; Meyers MJ; Pelc MJ; Schweitzer BA; Thorarensen A; Wang LJ Preparation of 7-azaspiro[3.5]nonane-7-carboxamides as Inhibitors of Fatty Acid Amide Hydrolase (FAAH). WO2010049841A1, 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.