

Abstract
Heart failure with preserved ejection fraction (HFpEF) is defined as clinical features of heart failure, ideally with biomarker evidence such as elevated plasma natriuretic peptide levels, in the setting of an ejection fraction (EF) greater than 50% and imaging evidence of diastolic left ventricular dysfunction (1,2). In the absence of cardiac imaging or invasive hemodynamics, this is a clinical syndrome that is often indistinguishable from heart failure with reduced ejection fraction (HFrEF). HFpEF and HFrEF present with a cadre of comparable signs and symptoms including jugular venous distention, pulmonary crackles on auscultation, breathlessness, orthopnea, exercise intolerance, exertional dyspnea, fatigue and peripheral edema. HFpEF accounts for at least half of all diagnoses of heart failure (1,2). Pulmonary hypertension (PH) is a common complication of HFpEF that is linked to worse disease morbidity and mortality. In fact, mortality has been linked to increases in the intrinsic pulmonary vascular resistance in the setting of increased left ventricular end diastolic pressure, characterized hemodynamically by rises in the transpulmonary pressure gradient, pulmonary vascular resistance or diastolic pressure gradient. Despite being the most common form of pulmonary hypertension, there are no approved therapies for the treatment of PH secondary to HFpEF. This review will summarize the hemodynamic classifications of PH in the setting of HFpEF, mechanisms of disease, the potential contribution of pulmonary vascular disease to poor outcomes in patients with HFpEF, and new approaches to therapy.
Hemodynamically Defining Pulmonary Vascular Disease related to HFpEF
The World Health Organization classifies pulmonary hypertension due to left heart disease (PH-LHD) as Group 2 pulmonary hypertension which incorporates HFpEF, HFrEF, as well as valvular disease (3). Overall, left heart disease is the most common etiology of pulmonary hypertension (4). By right heart catheterization (RHC), PH-LHD is defined by a mean pulmonary artery pressure (mPAP) ≥ 25 mmHg and a pulmonary artery wedge pressure (PAWP) > 15mmHg. PH-LHD is subsequently subdivided by ejection fraction into PH-HFpEF (ejection fraction > 50%) and PH- HFrEF (ejection fraction is <50%), although variable ejection fraction cut points have been used between 40-50%.
PH-HFpEF may be further classified based upon right heart catheterization hemodynamics (Figure 1) (5,6). Isolated post-capillary PH (Ipc-PH) refers to an increase in pulmonary arterial pressure (PAP) secondary to passive congestion from an elevated pulmonary arterial wedge pressure (PAWP), independent arterial vascular remodeling. The rise in PAP should be relative to the elevated PAWP and conversely, treatment that reduces the wedge pressure should proportionally reduce the mean pulmonary arterial pressure. In Ipc-PH, the transpulmonary gradient (TPG, defined as mPAP-PAWP) and the pulmonary vascular resistance (PVR, defined as TPG/Cardiac Output) are both normal, <12 mmHg and < 3 WU, respectively(3). Previously referred to as “mixed PH”, “reactive PH”, or “out of proportion PH”, the new term combined post- and pre-capillary (Cpc)-PH refers to the sub-phenotype of patients who have precapillary small vessel remodeling in addition to passive congestion. These patients have TPG>12 mm Hg, PVR >3 WU and/or diastolic pulmonary gradient (DPG, defined as diastolic PAP – PAWP) ≥7 mm Hg and, in contrast to patients with Ipc-PH, patients with Cpc-PH have a mean pulmonary artery pressure that remain elevated despite normalization of the PAWP (3).
Figure 1:

Subphenotypes of PH-HFpEF. Top Figure: Isolated post-capillary PH (Ipc-PH) refers to an elevated pulmonary arterial pressure (PAP) due to passive transmission of left sided pressures. This subphenotype can be defined by a transpulmonary gradient (TPG) less than or equal to 12, a diastolic pressure gradient (DPG) less than 7, or a pulmonary vascular resistance (PVR) less than or equal to 3 WU. Bottom Figure: Combined pre- and post-capillary PH (Cpc-PH) refers to an out of proportion elevation of PAP due to passive transmission of left sided pressures in addition to pulmonary vascular remodeling. This is defined by a TPG greater than 12, DPG greater than or equal to 7, or a PVR greater than 3.
Based on data from a large retrospective cohort study by Vanderpool et al. including 10,023 patients who underwent right heart catheterization between 2005 and 2012 at the University of Pittsburgh, 48.9%, 34.2%, and 13.7% of patients with PH-HFpEF met criteria for pre-capillary involvement defined by TPG (>12 mm Hg), PVR (>3 Woods units), and DPG (≥7 mm Hg), respectively (Figure 2) (7). The prevalence of Cpc-PH in this cohort, as defined by the above TPG, PVR, and DPG thresholds are in line with previously published reports including a cohort from Johns Hopkins University of 1236 patients where 32% of patients met criteria for Cpc-PH by TPG>12 mm Hg and 10.6% by DPG ≥7 mm Hg and a cohort from Medical University of Vienna including 1094 patients with PH-LHD including 44% of patients with TPG>12 mm Hg and 16% of patients with DPG ≥7 mm Hg (8,9).
Figure 2:

Data obtained from the University of Pittsburgh and previously published with permission by Vanderpool et al representing the breakdown of patients with PH secondary to left heart disease (LHD) and PH-HFpEF by transpulmonary gradient (TPG), pulmonary vascular resistance (PVR), and diastolic pulmonary gradient (DPG)7.
PVR and TPG were traditionally used to differentiate Ipc-PH and Cpc-PH. However, both the TPG and the PVR have recently been criticized as the TPG is flow dependent and both PVR and TPG fail to recapitulate any underlying pulmonary vascular pathology (9,10). Therefore, there is an expanding breadth of evidence to support the use of the DPG as a more accurate indicator of pulmonary vascular remodeling to properly phenotype Ipc-PH and Cpc-PH (11). DPG, however, also has been disparaged for error in the measures of PAWP and diastolic PA pressures that could result from large v-waves, over- or under wedging, and/or catheter whip and catheter “ringing.” Such errors may yield relatively large differences in the DPG. Furthermore, DPG can be misread in an automated system based on timing of exhalation (12). Tampakakis et al. cautions against using DPG to define Cpc-PH because a proportion of patients in their cohort with right heart catheterization hemodynamics had a DPG ≥7 mm Hg but without actual pulmonary hypertension (mPAP <25 mm Hg). This indicates that DPG is a disease marker that may lack specificity for pulmonary vascular disease. Elevated DPG, for example, may be seen in acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), tachycardia, and in inotrope-dependent patients, independent underlying pulmonary vascular disease (8,13). Finally, studies conducted by Ghio et al. in patients with PH-HFrEF show a substantial improvement in cardiopulmonary hemodynamics in patients with elevations in DPG, PVR, and TPG when exposed to pulmonary vasodilators, suggesting that all three hemodynamic parameters may identify patients that respond to pulmonary vasodilation (14). Yet, despite the controversy surrounding DPG, the current European Society of Cardiology and the European Respiratory Society define Ipc-PH as a DPG < 7mmHg and/or PVR ≤ 3 WU while Cpc-PH is defined by a DPG ≥ 7 mm Hg and/or PVR > 3 WU (3). While these definitions remain debated, we would argue that Cpc-PH should be defined more liberally by TPG and PVR cut-off values, rather than by DPG cut-off values alone which eliminate a substantial number of patients based upon the prevalence noted above.
Applying the ESC/ERS guidelines indicated above to the University of Pittsburgh cohort, 13.7% of patients with PH-HFpEF met criteria for Cpc-PH. When these patients were re-classified based on TPG or PVR alone, 44.7% and 34.1% of patients with HFpEF met criteria for Cpc-PH, respectively, with significant overlap in patients who had both TPG >12 mm Hg and PVR ≥3 WU (7). Furthermore, TPG >12 mm Hg and PVR ≥ 3 WU were both independently associated with worse outcomes. Clinically, this suggests that we may underestimate the prevalence and risk of pre-capillary disease based on the current guidelines and perhaps underappreciating critical pathology that may lend itself to the development of specific therapies.
How do the cardiopulmonary hemodynamics of PH-HFpEF differ from PH-HFrEF?
Adir et al. directly compared pulmonary hemodynamics in patients with PH-HFpEF and PH-HFrEF. An ejection fraction above and below 50% was used to discriminate between HFrEF and HFpEF. Pulmonary hypertension with left heart disease was defined as mPAP>25 mmHg by right heart catheterization and PAWP >15 mmHg. A DPG <7 mmHg was used to define isolated post capillary PH (Ipc-PH) whereas DPG ≥ 7 mmHg defined combined pre and post capillary PH (Cpc-PH). Patients with PH-HFpEF were more likely to be older, female, non-smokers, with a higher BMI, compared with patients with PH-HFrEF. They had less history of coronary disease but significantly more diabetes, hypertension, and atrial fibrillation. When adjusted for age, gender, and pulmonary artery wedge pressure, there was a significant difference in DPG and mitral regurgitation when comparing cardiopulmonary hemodynamics. All other parameters were not significantly different between patients with HFpEF and HFrEF with PH. The similarity in hemodynamic profiles between the two groups supports the accepted underlying common pathology of elevated left sided filling pressures causing secondary remodeling of the pulmonary arteries, veins and intermediate vessels, irrespective of ejection fraction (15,16).
Are provocation maneuvers important for the diagnosis of PH-LHD?
Prior to undergoing right heart catheterization, patients are typically aggressively diuresed and fasted pre-procedurally. Case series have suggested that this relative state of intravascular dehydration may actually mask PH secondary to left heart disease, and result in the misdiagnosis of pulmonary arterial hypertension (pre-capillary PH) (17). Robbins et al. confirmed that more than 20% of patients initially diagnosed as pulmonary arterial hypertension (PAH) by right heart catheterization hemodynamics are re-classified as pulmonary hypertension secondary to left heat disease (PH-LHD) with a PAWP >15 mm Hg after receiving a 500mL rapid fluid challenge resulting in an acute rise in PAWP with post-fluid hemodynamics akin to patients with an initial diagnosis of PH-LHD (18). The effects of fluid loading are more pronounced in patients with HFpEF compared to healthy controls and are most significant in elderly women (17). More recent work has suggested a more stringent PAWP >18 mm Hg as threshold for the diagnosis of occult post-capillary PH in the setting of a fluid challenge (19). In contrast to the findings of Robbins, this more conservative PAWP threshold results in the reclassification of 6% of patients with previously diagnosed pre-capillary PH and 8% of patients previously diagnosed as no PH were re-diagnosed as occult post-capillary PH (20). In a prospective study conducted by Anderson et al., exercise and acute volume loading as provocation maneuvers were directly compared in healthy patients and patients with HFpEF. Compared to acute volume loading, Anderson et al. found exercise to be a more sensitive and specific provocation maneuver to detect occult PH-LHD manifesting as a greater than two-fold increase in PAWP with exercise (21). Other proposed definitions for PH-LHD are a PAWP > 20 or 25 mm Hg during upright and supine exercise, respectively, with a consensus definition yet to be reached (22,23). Recently, the use of indexed values has been considered to define occult PH secondary to LHD. The mPAP/CO slope of ≥ 3 mm Hg/L/min could reduce the false positive diagnosis of PH-LHD, which occur due to physiologic elevations of PAWP even in healthy patients. Work by Herve et al., suggests that both a mPAP >30 mm Hg at maximal exercise and a total pulmonary resistance (TPR) > 3 WU at maximal exercise have high sensitivity and specificity in identifying patients with PH-LHD in a population of patients previously undiagnosed with PH based on resting mean pulmonary artery pressure < 20 mm Hg (24). Based on these findings, exercise induced PH is now defined according to the European Respiratory Society by a resting mPAP < 25 mm Hg and exercise mPAP > 30 mm Hg with a TPR> 3 WU (25). Therefore, provocation maneuvers, both via exercise and acute fluid challenge, can be useful to diagnose occult PH-LHD. Transpulmonary resistance and mean pulmonary artery pressure at maximal exercise possess the highest diagnostic yield to discriminate patients with occult PH-LHD compared to controls, however neither have been incorporated into diagnostic guidelines for PH-LHD, owing to a lack of consensus on standardized exercise or fluid challenge protocols.
Epidemiology of HFpEF and PH associated with HFpEF
Heart failure has been estimated to afflict nearly 10% of the western population (26). The most recent estimates, based on data collected from NHANES 2011-2014, suggest that 6.5 million Americans > 20 years of age carry a diagnosis of heart failure with more than 50% of those patients having HFpEF (27). The prevalence of HFpEF is rising at a rate of 1% per year (2). In the United States, this accounts for 30.7 billion dollars in healthcare costs (28). The burgeoning epidemic of HFpEF is largely related to the concomitant increased prevalence of diabetes, hypertension, hyperlipidemia, and obesity, all known risk factors for HFpEF (29).
When compared to patients with HFrEF, patients with HFpEF are typically older (30). Multiple studies have reported that patients with HFpEF are more likely to be female when compared to patients with HFrEF. The significance of this epidemiological observation has been scrutinized as a result of the skewed sex distribution of ageing individuals and the failure of many of these studies to include appropriately age-matched cohorts. Pooled cohorts from large clinical trials were used to develop predictive models for HFpEF and HFrEF. Among age-matched individuals with similar risk factors for heart failure, female sex did not increase the risk for HFpEF (1,30). Based on population based studies and clinical registries, the burden of cardiovascular risk factors including coronary artery disease and hyperlipidemia are highly variable in patients with HFpEF (20-77% and 16-77%, respectively) (26). Other risk factors include obesity, smoking status, atrial fibrillation, and chronic kidney disease (30,31).
Although studies have cited a wide range to describe the prevalence of pulmonary hypertension in patients with HFpEF, we feel the prevalence is approximately 35% based upon our data at the University of Pittsburgh which is in line with that of Tampakakis and Gerges (6–8). When compared to aged-matched patients with HFpEF without pulmonary vascular disease, patients with PH-HFpEF are more often female with a worse functional class. There is a similar occurrence of comorbid conditions such as hypertension, diabetes, obesity, and coronary artery disease between patients with HFpEF with and without accompanying PH (32). Compared to pulmonary arterial hypertension (PAH), patients with PH-HFpEF are more likely to be older, have co-occurring hypertension, diabetes, obesity, coronary artery disease, and renal dysfunction. Additionally, patients with PH-HFpEF are more apt to have biatrial enlargement and left ventricular hypertrophy. Robbins and colleagues found that two or more features of the metabolic syndrome were present in 94% of patients with pulmonary vascular hypertension associated with left heart disease (Figure 3) (33).
Figure 3:
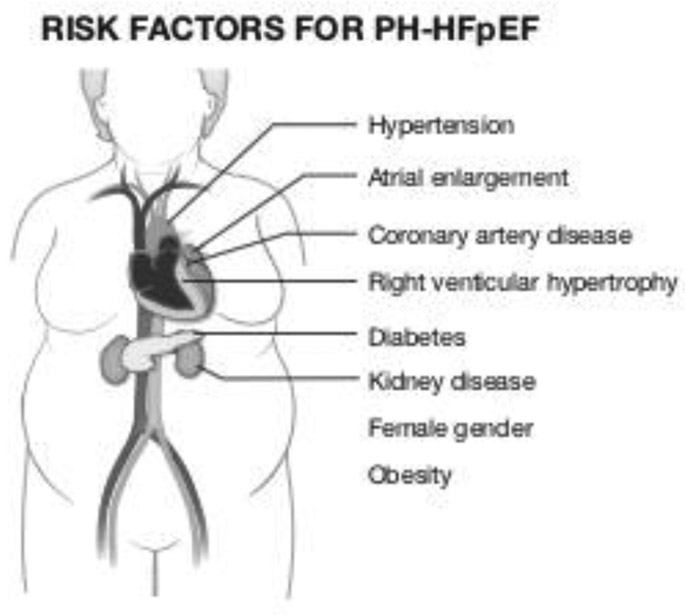
Risk factors for the development of PH-HFpEF include female gender, atrial enlargement, coronary artery disease, right ventricular hypertrophy and features of the metabolic syndrome including diabetes, hypertension, and obesity.
Impact of Pulmonary Vascular Remodeling on Mortality
In the University of Pittsburgh cohort of 10,023 subjects who received a right heart catheterization over a 7-year period, 4621 patients were diagnosed with PH-LHD, allowing for evaluation of the impact of different definitions of PH-LHD on outcomes. During a median 1383 day follow up period there were 1406 deaths. Overall, the survival of patients with PH-HFpEF was similar to Group 1 pulmonary arterial hypertension but with more cardiac hospitalizations, while patients with PH-HFrEF had the highest mortality and hospitalization rates. In the subset of patients with PH-HFpEF, age, RA pressure, mPAP, PAWP, PA pulse pressure, PA compliance, TPG, PVR, and DPG were significantly associated with higher mortality. Regardless of the parameter used to establish pre-capillary involvement of PH-LHD (PVR ≥ 3 WU, TPG >12 mmHg, DPG ≥ 7 mmHg), the risk of death was greater with pre-capillary disease. PVR ≥ 3 WU had the highest risk for mortality compared to TPG and DPG cut-off values. The mortality rate for PH-HFpEF was 19.3% at one year and 43.5% at five years(7). In a survival analysis performed by Gerges et al, the median overall survival of patients with HFpEF and the Cpc-PH subphenotype (DPG ≥ 7 mmHg) was 54 months, significantly shorter than the 102 months survival of patients with Ipc-PH subphenotype (6).
Numerous studies have now associated pulmonary vascular remodeling in the setting of LHD (HFpEF or HFrEF) with a higher patient morbidity or mortality. For example, elevations in PVR and TPG are associated with increased mortality (34–36). DPG has also recently been rigorously evaluated as prognostic marker with conflicting results. In an analysis of 1389 patients with PH-LHD (HFrEF and HFpEF), Gerges et al. demonstrated that patients with pre-capillary disease defined by an elevated TPG (>12 mm Hg) who were further stratified by DPG ≥ 7 mm Hg had a worse prognosis when compared to patients with a DPG < 7 mm Hg (unadjusted analysis) and more vascular pathology including medial hypertrophy, intimal and adventitial fibrosis, and more occluded vessels (9). Vanderpool and colleagues similarly demonstrated that DPG ≥ 7 mm Hg, TPG >12 mm Hg, and PVR ≥ 3 WU are all significant predictors of mortality in HFpEF (adjusted analysis) (7). In contrast, a study performed on patients receiving orthotopic heart transplants with an elevated TPG demonstrates no impact on 5-year post-transplant survival with a concomitant elevation in DPG, questioning the implication that DPG is reflective of underlying pulmonary vascular disease (37). Work by Assad et al., exploring RHC data of 2,817 patients with PH-LHD (both HFrEF and HFpEF) at Vanderbilt University demonstrated no mortality difference in patients with Cpc-PH versus Ipc-PH, as defined by DPG (adjusted analysis) (38). In a retrospective cohort study which enrolled over 1200 patients, Tampakakis et al. was unable to demonstrate any association between DPG and mortality in patients with PH-LHD (both HFrEF and HFpEF) but did confirm that PVR and TPG were significant predictors of mortality in this cohort (8). These findings were recapitulated in a recent study by Tampakakis utilizing three cohorts of patients with PH-HFpEF of PH-HFrEF from academic institutions where DPG alone again, failed to predict mortality. DPG ≥ 7 mmHg and PVR > 3 WU did, however, portend a worse mortality (39). Perhaps DPG fails to predict mortality because it fails to recapitulate the degree of RV dysfunction that occurs in patients with PH-HFpEF. As described by Brittain et al, the right heart does not pump against a pressure gradient but rather against a pressure. Also, the calculation of the DPG, unlike the systemic vascular resistance or the pulmonary arterial capacitance, does not incorporate any measures of right ventricular function, such as stroke volume (40).
Pulmonary arterial capacitance (PAC, defined as the stroke volume/pulmonary pulse pressure), a parameter reflective of pulsatile RV afterload and irreversible remodeling of the pulmonary vasculature, is an increasingly utilized marker of mortality. Al-Naamani specifically compared pulmonary arterial capacitance to TPG, DPG, and SVR in 73 patients with PH-HFpEF and found the capacitance to be the strongest predictor of mortality and to be a superior predictor of mortality than TPG, DPG, or pulmonary vascular resistance. Findings from Tampakakis using a combined cohort of patients with PH-HFpEF and PH-HFrEF substantiates the value of pulmonary arterial capacitance and arterial elastance as better predictors of mortality when compared to PVR or TPG (39). Furthermore, pulmonary arterial capacitance improvement with vasodilators portends improved survival (41). Pulmonary arterial capacitance, therefore, should be considered a burgeoning marker of pulmonary vascular remodeling in PH-HFpEF that is highly correlated with survival.
Non-Invasive Measures of PH-HFpEF
Echocardiography centering on markers of diastolic dysfunction has a critical role in diagnosing HFpEF. According to the American Society of Echocardiography guidelines for the use of echocardiography to assess diastolic dysfunction, four diagnostic parameters have been identified: 1) annular e’ velocity; 2) average E/e’; 3) LA volume index; 4) and peak tricuspid regurgitation velocity(42). Chronically elevated LV filling pressures due to diastolic dysfunction results in an increase in the PA pressure that can be estimated by echocardiogram using continuous wave Doppler to measure the tricuspid regurgitate peak systolic velocity. Tricuspid regurgitant velocity >2.5 m/second is considered abnormal and correlates to a systolic PA pressure of 30-35 mm Hg (42–44). In patients who underwent echocardiography followed by invasive measurement of left ventricular end diastolic pressure there was only a modest relationship between non-invasive classification of diastolic dysfunction and invasive parameters to confirm diastolic dysfunction, further reinforcing the need for invasive RHC to diagnose PH-HFpEF (45).
Cardiac MRI is an alternative, non-invasive modality to diagnose diastolic dysfunction and is the gold standard for the diagnosis of left ventricle and left atrium volume and left ventricle mass. Cardiac MRI may be better suited to measure left atrial volume index, which is useful to help classify the severity of the diastolic dysfunction and also to differentiate PH-HFpEF from PAH (46). Furthermore, cardiac MRI can effectively quantify myocardial fibrosis in non-infarcted myocardium by measuring extracellular volume fraction that is associated, in a dose dependent manner, with poor outcomes from heart failure including hospitalizations and all-cause mortality. This remains true independent of heart failure etiology or ejection fraction (47).
Right heart catheterization is considered the gold standard to diagnose PH secondary to HFpEF. Several studies have failed to find an echocardiographic profile in patients with HFpEF that is diagnostic of PH-HFpEF. Studies by Raeisi-Giglou and colleagues indicate that a mere 41% of patients with PH-HFpEF by RHC had LV diastolic dysfunction on echocardiography (48). Thus, while echocardiography may be sufficient to diagnose diastolic dysfunction, it is often insufficient to diagnose PH in the setting of HFpEF and an invasive RHC remains a necessary diagnostic tool.
Pathobiology of PH-HFpEF
The Fifth World Symposium on Pulmonary Hypertension in Nice, France proposed a model to explain the pathology of PH-HFpEF that continues to evolve. According to this model, underlying systolic or diastolic left ventricular heart disease increases left ventricular end-diastolic pressure causing left atrial enlargement, left atrial interstitial fibrosis, and reduced compliance (49). Left atrium changes, coupled with left ventricular diastolic dysfunction and exercise induced mitral regurgitation, result in a passive backward transmission of elevated leftsided filling pressures resulting in an elevated PAWP and mPAP (50). Acute elevations in left atrial pressure contribute to ‘alveolar-capillary stress failure’, resulting in an acute hydrostatic insult to the microvasculature and a disruption to the micro vessel permeability. This allows erythrocytes, protein, and fluid to leak into the alveolar lumen that ultimately impairs alveolar gas exchange. Alveolar-capillary stress failure manifests, clinically, as pulmonary edema and hemodynamically as an increase in PAWP and parallel rise in mPAP. Chronic pulmonary edema and elevated left sided filling pressures may result in a dynamic interplay of hormonal and inflammatory mediators, decreased nitric oxide (NO) availability, increased endothelin-1 expression, and a diminished natriuretic peptide vasodilation response. Additionally, hypoxia induces vasoconstriction and structural remodeling of the small pulmonary arteries. Histopathological changes identified include thickening of the alveolar-capillary membrane due to collagen deposition, arterial hypertrophy, fibrosis and luminal occlusion yielding pre- and post-capillary pulmonary hypertension and resulting in “out of proportion pulmonary hypertension” or CpC-PH (49,51). This model suggests that remodeling of the pulmonary arteries occurs as an aftermath of chronic pulmonary vascular congestion.
As outlined by Ter Maaten and colleagues, renal dysfunction and HFpEF are increasingly seen as interdependent. Patients with HFpEF typically have a higher central venous pressure due to RV dysfunction and often co-occurring pulmonary hypertension which results in decreased renal blood flow and renal perfusion pressure. This activates the renin-angiotensin-aldosterone system (RAAS) and reduces GFR. Additionally, reduced systolic filling and elevated end diastolic pressure reduces stroke volume and, in combination with chronotropic incompetence, reduces cardiac output that diminishes renal blood flow. A proposed systemic reduction in bioavailable nitric oxide (NO) in HFpEF and PH related to HFpEF further limits renal blood flow and sodium excretion. Chronic kidney disease is also an independent risk factor for the development of HFpEF. Aberrancies in phosphorous, parathyroid hormone, fibroblast growth factor 23 (FGF23), and Vitamin D may contribute to ventricular hypertrophy, fibrosis, and microvascular dysfunction. More severe kidney disease results in circulating uremia-associated proinflammatory cytokines and impaired endothelial function and proliferation, an increase in vascular endothelial ROS generation, and vascular smooth muscle dysfunction. An alternative to this bidirectional approach is the hypothesis that both HFpEF and CKD may be concomitantly occur from the same underlying mechanism of endothelial dysfunction and inflammation (31).
Pulmonary Vascular Remodeling in PH-HFpEF compared to PVOD
A recent study performed by Fayyaz, et al., evaluated autopsy cases of HFpEF patients from the Mayo Clinic Tissue Registry from 1987-2015. Interestingly, the investigators noted significant pulmonary venous remodeling that appeared similar to that observed in the much rarer condition, pulmonary veno-occlusive disease (PVOD). Venous arterialization was more pronounced in patients with PH-HFpEF than patients with PVOD. Increased arterial and venous intimal and medial thickness was also observed which correlated with higher TPG and DPG by right heart catheterization (16).
Is Cpc-PH simply the end stage sequela of Ipc-PH?
The risk factors for the development of Cpc-PH and Ipc-PH considerably overlap and the echocardiographic changes are often indistinguishable between the two phenotypes. However, the average age of patients with Cpc-PH is 10-years younger than patients with Ipc-PH. This suggests that the chronic pulmonary vascular congestion of Ipc-PH is not sufficient to cause Cpc-PH (38). Furthermore, RV dysfunction was once thought to be an end stage manifestation of PH-HFpEF which occurred due to the late development of pre-capillary remodeling. However, more recent studies have demonstrated that there is no difference in the degree of pulmonary vascular remodeling when comparing patients with heart failure with and without RV dysfunction. Collectively, this questions the previously accepted paradigm that patients transition from an Ipc-PH phenotype to a Cpc-PH phenotype secondary to chronic passive pulmonary venous congestion alone and suggests that Cpc-PH may be an independent subphenotype of PH-HFpEF with a distinct pathobiology (16). Yet, there remains equipoise as to whether Cpc-PH and Ipc-PH exist on a spectrum of disease or if these two are distinct entities due to the cross-sectional nature of most studies performed to date. Assad and colleagues have provided longitudinal insight into the evolution of PH utilizing a cohort of 70 patients initially diagnosed with borderline PH and followed over time. Of patients with borderline PH, 61% went on to develop PH with an average time to disease development of 35 weeks. 79% of the patients who developed pulmonary hypertension developed post-capillary disease. The development of pre-capillary disease in 21%of patients within the brief follow up period suggests that chronic decompensated volume status and post capillary congestion alone are not entirely responsible for the rising PVR and TPG and lends further credence to Cpc-PH and Ipc-PH as two distinct disease states (52).
Is Cpc-PH more similar to PAH than to Ipc-PH?
Assad et al. probed the Vanderbilt electronic medical record, linked to a prospective DNA biorepository for all patients undergoing right heart catheterization over a 17-year period. They found that patients with Cpc-PH are frequently younger and female, more akin to patients with PAH than to patients with Ipc-PH. However, patients with Ipc-PH and Cpc-PH share the same co-morbidities including diabetes, obesity, atrial fibrillation, hypertension, heart failure, anemia, and coronary artery disease. Echocardiographically, patients with Cpc-PH and Ipc-PH demonstrated similar LV remodeling, LA enlargement, and LV ejection fraction that was distinct from patients with pulmonary arterial hypertension. When comparing right heart catheterization hemodynamics, patients with Cpc-PH had elevated RA pressure, PVR, PA compliance that more closely resembled patients with pulmonary arterial hypertension than Ipc-PH(38). Most notably, a gene ontology analysis found exonic single nucleotide polymorphisms (SNP) in genes which encode for actin binding, extracellular matrix, basement membrane, MHCII proteins, and other cytoskeletal structural and immune function proteins to a similar degree in lung tissue of patients with pulmonary arterial hypertension and Cpc-PH, but not Ipc-PH, suggesting a common etiology of pulmonary vascular disease (38,53).
Caravita and colleagues sought to evaluate whether patients with Cpc-PH have an exercise profile (specifically the ventilatory response to exercise and the exercise oscillatory breathing) which is closer to patients with PAH or patients with Ipc-PH. Seventy patients were included in this study. Exercise induced hyperventilation was most apparent in patients with PAH followed by Cpc-PH. Exercise oscillatory breathing, typically found in patients with PH secondary to left heart disease but rarely in patients with pulmonary arterial hypertension, also discriminated between patients with Ipc-PH and Cpc-PH. Patients with Cpc-PH had less exercise oscillatory breathing, similar to patients with PAH. These studies suggest that the exercise profile for patients with Cpc-PH is distinct from patients with Ipc-PH and further supports this as a distinct phenotype with underlying similarities to pulmonary arterial hypertension (54).
Further efforts were made by Opitz at al. to characterize demographics and treatment responses of patients with PH-HFpEF in comparison to patients with PAH. Patients were abstracted from the COMPERA registry of PH patients receiving targeted therapies. This did include patients with PH-HFpEF on PH-specific treatment (mPAP ≥ 25 mmHg, PAWP >15 mmHg, EF > 45%, echocardiographic evidence of diastolic dysfunction) or PAH (mPAP ≥ 25 mmHg, PAWP ≤ 15 mmHg). Pulmonary arterial hypertension was further divided into idiopathic and atypical pulmonary arterial hypertension, defined as ≥ 3 risk factors for cardiovascular disease, including arterial hypertension, coronary artery disease, diabetes, atrial fibrillation, and obesity. Patients with atypical pulmonary arterial hypertension and PH-HFpEF were older, more obese, had a longer six-minute walk test (6MWT) and had higher natriuretic peptide level. By right heart catheterization, the patients in the PH-HFpEF group were confirmed to have Cpc-PH with mean TPG 26 mm Hg and PVR of 7.0 WU. Hemodynamically, the RA pressure was significantly higher in the patients with PH-HFpEF when compared to both idiopathic and atypical pulmonary arterial hypertension. Phosphodiesterase 5 Inhibitors (PDE5i) were used as the first line therapy in the majority of patients with typical and atypical idiopathic pulmonary arterial hypertension (77% and 81%) and PH-HFpEF (94%). Endothelin Receptor Antagonists (ERAs) were used as the first line therapy in patients with typical idiopathic pulmonary arterial hypertension 31% of the time, with atypical idiopathic pulmonary arterial hypertension 22% of the time, and with PH-HFpEF 7% of the time. The preference for PDE5i therapy was continued throughout follow up in patients with PH-HFpEF, typical and atypical idiopathic pulmonary arterial hypertension. Combination therapy was initiated preferentially in patients with typical idiopathic pulmonary arterial hypertension and significantly less so in patients with atypical idiopathic pulmonary arterial hypertension and PH-HFpEF. All patients with PH had improvement of their World Health Organization (WHO) functional class, exercise capacity, 6-minute walk distance, and natriuretic peptide levels over a 12-month period. However, this effect was greatest in patients with idiopathic pulmonary arterial hypertension and least significant in patients with PH-HFpEF. There was no difference in five-year mortality after initiating therapy between the three groups. Based on a gradient treatment response and overlapping mortality curves this registry of PH treatment raises the question that PH-HFpEF, atypical pulmonary arterial hypertension, and idiopathic pulmonary arterial hypertension may reflect a continuum of the same disease whereby increasing comorbidities and increasing post-capillary disease gradually reduce the efficacy of typical pulmonary arterial hypertension therapies (5).
Therapeutics
Molecular Target #1: the NO-sGC-cGMP pathway
Nitric Oxide (NO) is an endogenously synthesized free radical which functions as an endothelium-derived relaxing factor. L-arginine is converted, via nitric oxide synthase (NOS), to NO. Endothelial NOS (eNOS) is primarily located in endothelial cells and eNOS derived NO is a potent vasodilator. The NO-sGC-cGMP pathway has been the focus of drug development for PH-HFpEF (Figure 4). Endothelial produced NO activates the enzyme soluble guanylate cyclase (sGC) and results in the formation of second messenger cyclic guanosine monophosphate (cGMP). cGMP increases the activity of protein kinase G (PKG) (55). In smooth muscle, PKG stimulates the opening of calcium-activated potassium channels, leading to cell hyperpolarization and relaxation. cGMP mediated- smooth muscle relaxation vasodilates the systemic and pulmonary circulation. The formed cGMP is rapidly degraded by the enzyme phosphodiesterase-5 to modulate NO dependent vasodilation. It has been proposed that in patients with HFpEF, cardiomyocyte and vascular NO bioavailability are reduced (56). Aberrancies in the NO-sGCcGMP pathway have been observed in myocardial homogenate of patients with HFpEF and may contribute to myocardial dysfunction, impaired myocardial relaxation, and myocardia stiffness (56).
Figure 4:
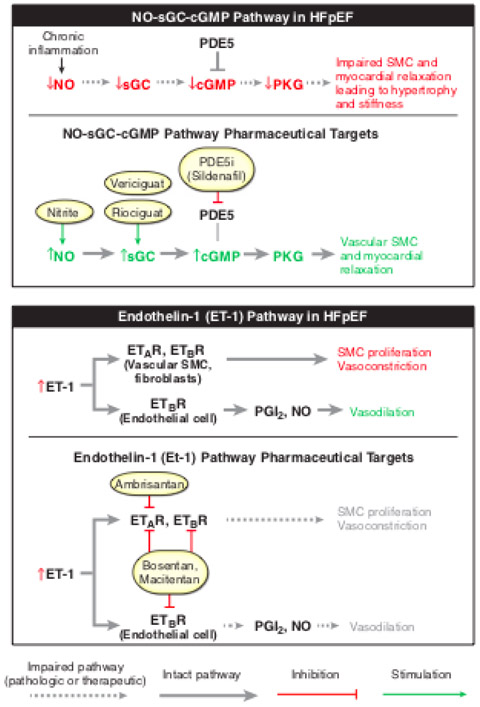
Schematic of the molecular pathways proposed to be aberrant in PH-HFpEF and the drugs which have targeted these pathways. Patients with HFpEF have abnormal nitric oxide (NO) bioavailability which results in an abnormal NO-sGC-CGMP pathway ultimately leading to impaired vascular smooth muscle cell (SMC) and myocardial relaxation which impairs myocardial relaxation and results in myocardial stiffness. Aberrancies in this pathway have been the molecular target of drugs including nitrite, vericiguat, riociguat, and sildenafil. The Endothelin-1 pathway has also been implicated in HFpEF. Endothelin-1, a potent vasoconstrictor, is increased in HFpEF. Via the interaction with the Endothelin receptor a (ETAR) and endothelin receptor B (ETBR) this results in a vascular smooth muscle cell proliferation and vasoconstriction which is more profound than the co-occurring ETBR mediated endothelial cell vasodilation. Vascular smooth muscle cell and fibroblast ETAR and ETBR are the targets of ambrisentan, bosentan, and macitentan.
A number of drugs enhance the NO-sGC-cGMP signaling pathway and are being evaluated as candidate therapies for PH-HFpEF (Figure 4). For example, Nitrite, particularly in the setting of hypoxia and academia, can be metabolized to Nitric Oxide (NO) in blood by reactions with hemoglobin and xanthine oxidoreductase, thus improving the NO deficiency that characterizes PH-HFpEF (56–58). Nitrite is being evaluated in a number of clinical trials in patients with both HFpEF and PH-HFpEF(59,60). PDE5 inhibitors, while FDA approved for patients with pulmonary arterial hypertension, have been evaluated with variable success and direct sGC stimulator and activator drugs are currently being evaluated as a therapy in PH-HFpEF (61).
Pre-clinical evaluation of drugs targeting the NO-sGC-cGMP pathway
Takimoto and colleagues exposed C57Bl/6 mice to transverse aorta constriction (TAC) surgery or sham surgery for 1-9 weeks. Animals were then administered the PDE5 inhibitor sildenafil or vehicle concurrently. TAC resulted in heart and cellular hypertrophy by 3 weeks which then yielded chamber dilation, myocardial fibrosis, and myocyte hypertrophy. Cardiac remodeling was inhibited in sildenafil treated transverse aortic constriction exposed animals. Sildenafil was also effective at completely reversing established hypertrophy and restoring function in animals exposed to transverse aortic constriction (62). Pre-clinical work performed by Mátyás et al. administered the PDE5 inhibitor Vardenafil for 7-weeks to Zucker diabetic fatty rats—an animal model of HFpEF. Vardenafil improved stiffening of the LV, LV dysfunction, and myocardial fibrotic remodeling. Lai, et al developed a two-hit model of PH-HFpEF where ZSF1 obese rats were administered a single dose of the VEGF receptor antagonist, Sugen. Animals developed an elevated pulmonary artery systolic pressure and left ventricular end diastolic pressure with a normal ejection fraction, modeling PH-HFpEF. These animals were treated with oral nitrite or oral metformin which improved cardiopulmonary hemodynamics and insulin sensitivity. Lai et al. proposed that activation of skeletal muscle SIRT3-AMPK as well as direct vascular NO formation mediates the therapeutic effects observed. Nitrite and metformin restored the levels of the mitochondrial protein deacetylase SIRT3 in skeletal muscle and, in doing so, treated components of the metabolic syndrome as well as PH-HFpEF. This suggests skeletal muscle SIRT3 may contribute to the pathology of pulmonary hypertension secondary to HFpEF and may represent a novel therapeutic target (63).
Clinical trials targeting Nitrite-NO-sGC-cGMP pathway
While several small, single center trials have reported positive results treating PH-HFpEF with PDE5i, a large multicenter study was negative, making it unlikely that PDE5i will ever be an approved therapy for PH-HFpEF. Guazzi et al. conducted a double-blind, randomized, placebo-controlled, 1-year study at a single center examining the effects of the PDE5 inhibitor sildenafil on PH-HFpEF. The 44 patients enrolled in this study had an average mPAP of 36 mmHg, mean PAWP of 21.9 mmHg, mean TPG of 14.5 mmHg, and PVR of 3.27 suggesting a preponderance of patients with the Cpc-PH phenotype secondary to HFpEF, although DPG was not reported. The study was designed to examine two primary endpoints, a reduction in pulmonary hemodynamics and RV performance. Treatment with sildenafil resulted in a significant reduction in pulmonary systolic, diastolic, and mean pressures at 6 months, and remained significant at 12 months of sildenafil administration. Additionally, sildenafil significantly lowered RA pressure at 6 and 12 months and improved RV function, as measured by tricuspid annular plane systolic excursion (TAPSE), and dimension (64). A potential limitation of this study was that most patients were male, in normal sinus rhythm, lacked some of the comorbidities seen in patient with PH-HFpEF (ie diabetes), and exhibited more profound RV systolic dysfunction and RV failure.
Despite these positive results, additional studies investigating the effects of PDE5 inhibitors in HFpEF and associated pulmonary hypertension have demonstrated less promise. Hoendermis et al. found no change in mPAP after 12 weeks of sildenafil administration in 52 patients. In contrast to the work done by Guazzi, this cohort included a mixed Ipc-PH and Cpc-PH phenotype with only 53% of patients with TPG >12 mmHg, 35% with PVR >3 WU, and 12% with DPG ≥7 mmHg. To assess whether the benefits of sildenafil were exclusive to patients with Cpc-PH, Hoendermis performed a subgroup analysis evaluating sildenafil in patients with PVR >3 WU. While the numbers were too small to be conclusive, sildenafil administration resulted in no hemodynamic improvements in the subgroup of patients with Cpc-PH. Work conducted by Redfield and colleagues as part of the RELAX trial sought to establish whether chronic sildenafil administration changed peak oxygen consumption at 24 weeks in patients with HFpEF. Inclusion criteria included prior heart failure hospitalization, or heart failure therapy, or IV diuretic therapy, or loop diuretic therapy for heart failure with left atrial enlargement, or elevated LV end diastolic pressure but did not include markers of PH secondary to HFpEF. Long term sildenafil failed to improve 6MWT, clinical status, or quality of life in this multicenter trial of 216 patients. Furthermore, LV mass or LV diastolic volume index and PA systolic pressure were unchanged with treatment as assessed by cardiac MRI and echocardiogram, respectively. Renal function, NT-proBNP, endothelin-1 and uric acid levels all increased in the treatment group compared to control. Similarly, there were more adverse vascular events and more patients in the treatment arm who withdrew from the study, died, or were too ill for cardiopulmonary testing (65). The SIOVAC trial was a multicentric placebo control trial of sildenafil in patients with PH-LHD secondary to valvular heart disease. Enrolled patients had persistent PH with mPAP >30 mmHg but had undergone surgical correction of valvular heart disease > 1 year prior to enrollment in the study. Patients were randomized to receive 40mg sildenafil three times a day for 6-months. Patients in the sildenafil group more frequent demonstrated a decline in the clinical composite score compared to the placebo group. The sildenafil arm also had an increase in major clinical events and hospitalizations due to heart failure at six months post randomization. The study included both patient with Cpc-PH (57%) and Ipc-PH. This study reinforces the risk associated with the use of sildenafil in patients with PH-LHD and supports the current clinical recommendations against the use of PDE-5 inhibitors in patients with PH-LHD (66). While the efficacy of PDE5i in PH-HFpEF, including specific hemodynamic phenotypes, remains unanswered by these studies it seems unlikely that such studies will be undertaken given the negative results seen to date.
Borlaug and colleagues conducted a double-blind, randomized, placebo-controlled trial to study the acute cardiopulmonary hemodynamic effects of inorganic nitrite infusion. All patients enrolled had HFpEF, as defined by ejection fraction on echocardiogram and right heart catheterization. By PVR, patients enrolled in this trial primarily manifest the Ipc-PH phenotype with average baseline PVR of 2.3 mm Hg. Nitrite infusion reduced RA pressure, PAWP, and PA pressure at rest and with exercise. The reduction in the exercise mPAP was strictly dependent on the reduction of the PAWP which was more than 2-fold greater with exercise than at rest. Nitrite administration increased VO2 and cardiac output at peak exercise, secondary to an increase in stroke volume and left ventricular stroke work (60). A follow up study conducted by Borlaug demonstrated the hemodynamic benefits of inhaled sodium nitrite for PH-HFpEF. Participants were randomized to either placebo or inhaled nitrite and underwent baseline RHC with exercise before and after treatment. Of note, at baseline the patients who received placebo had a higher average PVR 3.1±1.7 compared to the patients who received inhaled nitrite 2.0±1.2. The primary outcome of the study was PAWP. Patients with PH-HFpEF demonstrated a significant increase in PAWP with exercise (20±6 to 34±7 mm Hg). Patients treated with inhaled nitrite had significant improvement in post-inhaled nitrite exercise PAWP (25±5 versus 31±6 mm Hg)(67). However, in the multicenter INDIE-HFpEF study, Borlaug and colleagues were unable to demonstrate any improvement in the primary endpoint of peak oxygen consumption during cardiopulmonary exercise or the secondary endpoints including activity level, quality of life score, or NT-proBNP in patients treated with three times a day inhaled sodium nitrite for 4 weeks (68). Simon et al. further evaluated the safety and efficacy of two doses of inhaled nitrite (45 and 90 mg) in patients with PH-HFpEF at rest, defined by a PAWP >15 and TPG >12 with an LVEF ≥ 40% and WHO functional class > I, when compared to patients with Group 1and Group 3 PH. In this study, a single dose of inhaled nitrite was effective at reducing RA pressure, PAWP, RV systolic and diastolic pressure, and PA systolic, diastolic, and mean PA pressure, with substantial improvement in PA compliance. Most hemodynamic parameters were impacted in a dose dependent manner. The effects of inhaled nitrite were most pronounced on PAWP in patients with PH-HFpEF (reduction from 18 to 10 mm Hg) when compared to patients with Group 1 or Group 3 PH. Inhaled nitrite produced a more pronounced reduction in RA pressure and PAWP than inhaled nitric oxide (iNO) (59). These studies, collectively, suggest the potential of inorganic nitrite supplementation as a viable therapy for PH related to HFpEF specifically with promising data related to cardiopulmonary hemodynamics, although improvements in clinical endpoints have not been demonstrated to date. Furthermore, improved cardiopulmonary hemodynamics after acute organic nitrite supplementation supports the hypothesis that a reduction in NO bioavailability is, mechanistically, the underpinning of PH-HFpEF development.
The DILATE trial assessed the acute hemodynamic effects of a single dose of the riociguat, a sGC stimulator, in PH-HFpEF. Patients in the placebo group, 0.5mg, and 2mg Riociguat group had baseline TPG >12 mm Hg with an average TPG of 11.7 mm Hg in the 1mg Riociguat group. The primary outcome of change in mPAP from baseline at 6 hours after drug administration was not achieved. Similarly, there was no significant change in TPG or PVR. However, there was a significant increase in stroke volume and cardiac index. SVR, mean arterial pressure, and systolic blood pressure were all significantly reduced in the treatment arm (69). Another sGC stimulator, vericiguat, has completed a phase II trial in HFpEF, the SOCRATES-PRESERVED trial. Participants received one of four possible doses of vericiguat or placebo for 12 weeks. The primary outcomes were the change from baseline in N terminal pro B-type natriuretic peptide (NT-proBNP) or the left atrial volume. At 12 weeks, there was no significant change in the primary endpoints when comparing the placebo to the three pooled highest doses of vericiguat. A secondary analysis demonstrating an improvement in NT-proBNP at the highest dose of vericiguat (10mg) coupled with improvements in patient-reported outcomes at the two highest doses of vericiguat and echocardiographic mitral e’ velocity has led to the pivotal VICTORIA trial (70).
Collectively, the clinical trials that have focused on the Nitrite-NO-sGC-cGMP pathway have included patients with both Ipc-PH and Cpc-PH. Initial trials with Sildenafil were promising but the results have not been reproducible in subsequent trials focusing on either Ipc-PH or Cpc-PH. The most promising hemodynamic response to drug therapy to date was seen in work performed by Borlaug with intravenous and subsequently inhaled sodium nitrite. The hemodynamic improvement seen in these trials seems related to an improvement in PAWP, leading to improvements in PA compliance suggesting that nitrite may benefit these patients by relieving the post-capillary disease and reducing the pulsatile load on the RV. Unfortunately, the patients enrolled in the intravenous and inhaled nitrite trial have right heart catheterization hemodynamics consistent with Ipc-PH and the Cpc-PH phenotype is excluded from these clinical trials limiting the generalizability of the benefit of nitrite to patients with Cpc-PH.
Molecular Target #2: Endothelin Receptors
Endothelin-1 (ET-1) is a potent endogenous vasoconstrictor secreted by endothelial cells. In PAH, ET-1is upregulated. Via Endothelin Receptor A and B (ETAR and ETBR), expressed on vascular smooth muscle cells, ET-1 induces smooth muscle proliferation and functions as a potent vasoconstrictor. Bosentan and macitentan are endothelin receptor antagonists that target both ETAR and ETBR whereas ambrisentan selectively targets ETAR (Figure 4) (71,72). Higher levels of ET-1 have been reported in patients with HFpEF when compared to controls (2.61±0.81 versus 1.74±0.52 pg/mL; P<0.001) which demonstrate a positive correlation with pulmonary artery systolic pressure (71). Recent work by Meoli and colleagues specifically measured transpulmonary ratio of biomarkers including cAMP, cGMP, and ET-1 in patients with pulmonary arterial hypertension, without pulmonary hypertension, and patients with PH-LHD. PH-LHD was further subdivided into the Cpc-PH (PVR >3 and TPG >12 mm Hg) and Ipc-PH phenotype. The transpulmonary ratio of cAMPK and cGMPK is unchanged in all patient populations and subpopulations. The transpulmonary ratio of ET-1 was elevated only in the subset of patients with Cpc-PH and this was driven by an elevated pulmonary artery wedge ET-1 rather than a reduced PA ET-1 level. Pulmonary artery wedge ET-1 was correlated with elevated PAWP only in patients without pulmonary hypertension and the use of acute fluid challenge to increase PAWP failed to increase wedge ET-1. This suggests that neither PAWP elevation alone nor acute changes in PAWP are sufficient to drive the elevated wedge ET-1. In Cpc-PH only, pulmonary artery wedge ET-1 is strongly correlated with an elevated PVR (r=0.83). Transpulmonary ET-1 is not correlated with PVR. Cumulatively, this suggests that pulmonary artery wedge ET-1 and direct pulmonary vascular exposure to ET-1 may be involved in the development of Cpc-PH. Patients in the Cpc-PH and Ipc-PH group had comparable PAWP questioning the dogma that the pathobiology of Cpc-PH develops as a sequela of chronically elevated LV filling pressures. An identical PAWP between the Cpc-PH and Ipc-PH groups also refutes that pulmonary artery wedge ET-1 elevation is a secondary manifestation of elevated wedge pressure. These data hint to patient specific factors which increase pulmonary artery ET-1 production or reduce ET-1 clearance chronically coupled with pulmonary vascular hyperactivity to ET-1 (73).
Pre-clinical trials targeting Endothelin receptors
C57BL/6J male mice were administered chronic aldosterone for 4-weeks duration to induce HFpEF. Four groups were studied including 1) normal chow, 2) HFpEF with normal chow, 3) sham with chow containing macitentan, 4) HFpEF with chow containing macitentan. Total wall thickness, relative wall thickness, and posterior wall thickness were significantly degreased with macitentan administration, independent of body weight, blood pressure, and heart rate. Fulton index was unchanged between sham and macitentan treated animals. Macitentan treatment reduced cardiomyocyte size and decreased brain natriuretic peptide mRNA expression. Pretreatment with macitentan reduced aldosterone-induced cardiomyocyte hypertrophy. Mechanistically, this may be related to increased mef2a expression that is observed in adult rat ventricular myocytes which are aldosterone-stimulated. Macitentan pre-treatment eliminates this increased mef2a expression. In this mouse model, macitentan abrogated cardiac collagen deposition by normalizing levels of transforming growth factor-beta and collagen type-1 mRNA in the LV. Collectively, these pre-clinical data suggest that endothelin receptors may be an effective therapeutic target for PH associated with HFpEF (71).
Clinical Trials Targeting Endothelin Receptors
MELODY-1 was a small pilot study evaluating macitentan in patients with LHD and was the first randomized control trial to evaluate the use of an ERA in patients with Cpc-PH. A total of 63 patients were randomized to drug or placebo for 12 weeks. The primary outcomes were fluid retention or worsening of New York Heart Association (NYHA) functional class. Fluid retention and worsening functional class occurred more frequently in the macitentan treated arm. There was no change in hemodynamic parameters including PVR, mPAP, or PAWP at 12 weeks (74). The BADDHY-Trial evaluated the efficacy of 12 weeks of the ERA bosentan in patients with PH-HFpEF with the Cpc-PH phenotype as defined by an elevated TPG and PVR in both the treatment and placebo arm. 6MWT, echocardiogram, and laboratory data were collected at the start of the drug, at drug completion, and after an additional 12 weeks of monitoring without drug (week 24). There was no improvement of 6MWT or echocardiographic evaluation of pulmonary hypertension with drug and, in fact, patients treated with bosentan had worse clinical outcomes than those who received placebo (75). Despite data implicating elevated pulmonary artery wedge ET-1 to the development of Cpc-PH, neither the MELODY-1 or the BADDHY trial demonstrate any success in utilizing ERAs as a therapeutic drug class for patients with PH-HFpEF (73). The inconsistency between ET-1 elevations as a possible cause of Cpc-PH and the clinical failure of drugs that target the endothelin receptors remains to be answered further investigation and ongoing clinical trials.
Clinical Consensus
According to the 2009 ACCF/AHA and 2015 ESC/ERS Guidelines, there remains no clinical consensus on the treatment of PH-HFpEF (3,76). Given the failure of these therapies to reliably achieve primary clinical outcomes and the often adverse effects experienced by patients initiated on pulmonary vasodilators, it is currently not recommended to initiate pulmonary vasodilators in patients diagnosed with PH-HFpEF.
Future Trials
Clinical trials are currently ongoing examining treprostinil (United Therapeutics), a prostacyclin analogue which is FDA approved for use in PAH, for use in PH-HFpEF. SOUTHPAW is a double-blind placebo-controlled multicenter study to evaluate drug safety and efficacy with the primary outcome of 6MWT and secondary outcomes including changes in NT-proBNP, change in functional class, and time to clinical worsening (clinicaltrials.gov NCT03037580). SERENADE, a phase 2 clinical trial of macitentan (Actelion) is also ongoing in HFpEF exploring NT-proBNP as the primary outcome (clinicaltrials.gov NCT03153111). An oral formulation of nitrite is also being studied in PH-HFpEF (clinicaltrials.gov NCT03015402) and studies of therapies targeting metabolism in PH-HFpEF are being planned.
Conclusions
The morbidity, mortality, and healthcare costs secondary to PH related to HFpEF continue to climb at an alarming rate. Our understanding of the pathophysiology and therapy of this disease lags behind other, less common types of pulmonary hypertension. To date, there have been numerous failed clinical trials attempting to repurpose drugs currently used for the treatment of PAH for use in PH-HFpEF, potentially related to different endo-phenotypes within the HFpEF population. Additional preclinical and clinical trials are needed to better understand this disease process and identify additional therapeutic options for patients with PH-HFpEF.
Acknowledgments
Mark A. Simon: Grants: 1) Novartis; Personal Fees: 1) United Therapeutics, 2) Gilead, 3) St. Jude Medical, 4) Hovione Sciencia, 5) Actelion, 6) Bayer
Mark T. Gladwin: Grants: 1) Hemophilia Center of Western PA, 2) Bayer, 3) Institute for Transfusion Medicine; Patent: Use of nitrite salt in cardiovascular disease
Footnotes
COI:
Andrea R. Levine: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017. October;14(10):591–602. [DOI] [PubMed] [Google Scholar]
- 2.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011. March;32(6):670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016. January 1;37(1):67–119. [DOI] [PubMed] [Google Scholar]
- 4.Fang JC, DeMarco T, Givertz MM, Borlaug BA, Lewis GD, Rame JE, et al. World Health Organization Pulmonary Hypertension group 2: pulmonary hypertension due to left heart disease in the adult--a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2012. September;31(9):913–33. [DOI] [PubMed] [Google Scholar]
- 5.Opitz CF, Hoeper MM, Gibbs JSR, Kaemmerer H, Pepke-Zaba J, Coghlan JG, et al. Pre-Capillary, Combined, and Post-Capillary Pulmonary Hypertension: A Pathophysiological Continuum. J Am Coll Cardiol. 2016. July 26;68(4):368–78. [DOI] [PubMed] [Google Scholar]
- 6.Gerges M, Gerges C, Pistritto A-M, Lang MB, Trip P, Jakowitsch J, et al. Pulmonary hypertension in heart failure. epidemiology, right ventricular function, and survival. Am J Respir Crit Care Med. 2015. November 15;192(10):1234–46. [DOI] [PubMed] [Google Scholar]
- 7.Vanderpool RR, Saul M, Nouraie M, Gladwin MT, Simon MA. Association between hemodynamic markers of pulmonary hypertension and outcomes in heart failure with preserved ejection fraction. JAMA Cardiol. 2018. March 14;3(4):298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tampakakis E, Leary PJ, Selby VN, De Marco T, Cappola TP, Felker GM, et al. The diastolic pulmonary gradient does not predict survival in patients with pulmonary hypertension due to left heart disease. JACC Heart Fail. 2015. January;3(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerges C, Gerges M, Lang MB, Zhang Y, Jakowitsch J, Probst P, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in “out-of-proportion” pulmonary hypertension. Chest. 2013. March;143(3):758–66. [DOI] [PubMed] [Google Scholar]
- 10.Capomolla S, Febo O, Guazzotti G, Gnemmi M, Mortara A, Riccardi G, et al. Invasive and non-invasive determinants of pulmonary hypertension in patients with chronic heart failure. J Heart Lung Transplant. 2000. May;19(5):426–38. [DOI] [PubMed] [Google Scholar]
- 11.Diastolic Pulmonary Gradient In Pulmonary Hypertension - American College of Cardiology [Internet]. [cited 2018. January 13]. Available from: http://www.acc.org/latest-in-cardiology/articles/2017/02/09/14/39/diastolic-pulmonary-gradient-in-pulmonary-hypertension
- 12.Foundations of Anesthesia - 2nd Edition [Internet]. [cited 2018. February 21]. Available from: https://www.elsevier.com/books/foundations-of-anesthesia/hemmings/978-0-323-03707-5 [Google Scholar]
- 13.Chatterjee NA, Lewis GD. Characterization of pulmonary hypertension in heart failure using the diastolic pressure gradient. JACC: Heart Failure. 2015. January;3(1): 17–21. [DOI] [PubMed] [Google Scholar]
- 14.Ghio S, Crimi G, Temporelli PL, Traversi E, La Rovere MT, Cannito A, et al. Haemodynamic effects of an acute vasodilator challenge in heart failure patients with reduced ejection fraction and different forms of post-capillary pulmonary hypertension. Eur J Heart Fail. 2018. April;20(4):725–34. [DOI] [PubMed] [Google Scholar]
- 15.Adir Y, Guazzi M, Offer A, Temporelli PL, Cannito A, Ghio S. Pulmonary hemodynamics in heart failure patients with reduced or preserved ejection fraction and pulmonary hypertension: Similarities and disparities. Am Heart J. 2017. October;192:120–7. [DOI] [PubMed] [Google Scholar]
- 16.Fayyaz AU, Edwards WD, Maleszewski JJ, Konik EA, DuBrock HM, Borlaug BA, et al. Global Pulmonary Vascular Remodeling in Pulmonary Hypertension Associated with Heart Failure and Preserved or Reduced Ejection Fraction. Circulation. 2017. December 15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto N, Borlaug BA, Lewis GD, Hastings JL, Shafer KM, Bhella PS, et al. Hemodynamic responses to rapid saline loading: the impact of age, sex, and heart failure. Circulation. 2013. January 1;127(1):55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robbins IM, Hemnes AR, Pugh ME, Brittain EL, Zhao DX, Piana RN, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail. 2014. January;7(1):116–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borlaug BA. Invasive assessment of pulmonary hypertension: time for a more fluid approach? Circulation: Heart Failure. 2014. January 1;7(1):2–4. [DOI] [PubMed] [Google Scholar]
- 20.D’Alto M, Romeo E, Argiento P, Motoji Y, Correra A, Di Marco GM, et al. Clinical Relevance of Fluid Challenge in Patients Evaluated for Pulmonary Hypertension. Chest. 2017. January;151(1):119–26. [DOI] [PubMed] [Google Scholar]
- 21.Andersen MJ, Olson TP, Melenovsky V, Kane GC, Borlaug BA. Differential hemodynamic effects of exercise and volume expansion in people with and without heart failure. Circ Heart Fail. 2015. January;8(1):41–8. [DOI] [PubMed] [Google Scholar]
- 22.Berry NC, Manyoo A, Oldham WM, Stephens TE, Goldstein RH, Waxman AB, et al. Protocol for exercise hemodynamic assessment: performing an invasive cardiopulmonary exercise test in clinical practice. Pulm Circ. 2015. December;5(4):610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis GD, Bossone E, Naeije R, Grünig E, Saggar R, Lancellotti P, et al. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation. 2013. September 24;128(13):1470–9. [DOI] [PubMed] [Google Scholar]
- 24.Herve P, Lau EM, Sitbon O, Savale L, Montani D, Godinas L, et al. Criteria for diagnosis of exercise pulmonary hypertension. Eur Respir J. 2015. September;46(3):728–37. [DOI] [PubMed] [Google Scholar]
- 25.Kovacs G, Herve P, Barbera JA, Chaouat A, Chemla D, Condliffe R, et al. An official European Respiratory Society statement: pulmonary haemodynamics during exercise. Eur Respir J. 2017. November 22;50(5). [DOI] [PubMed] [Google Scholar]
- 26.Lam CSP, Donal E, Kraigher-Krainer E, Vasan RS. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail. 2011. January;13(1):18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018. March 20;137(12):e67–492. [DOI] [PubMed] [Google Scholar]
- 28.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014. January 21;129(3):e28–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerber Y, Weston SA, Redfield MM, Chamberlain AM, Manemann SM, Jiang R, et al. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern Med. 2015. June;175(6):996–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho JE, Lyass A, Lee DS, Vasan RS, Kannel WB, Larson MG, et al. Predictors of new-onset heart failure: differences in preserved versus reduced ejection fraction. Circ Heart Fail. 2013. March;6(2):279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ter Maaten JM, Damman K, Verhaar MC, Paulus WJ, Duncker DJ, Cheng C, et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail. 2016. February 10;18(6):588–98. [DOI] [PubMed] [Google Scholar]
- 32.Thenappan T, Shah SJ, Gomberg-Maitland M, Collander B, Vallakati A, Shroff P, et al. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ Heart Fail. 2011. May;4(3):257–65. [DOI] [PubMed] [Google Scholar]
- 33.Robbins IM, Newman JH, Johnson RF, Hemnes AR, Fremont RD, Piana RN, et al. Association of the metabolic syndrome with pulmonary venous hypertension. Chest. 2009. July 1;136(1):31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorlitzer M, Ankersmit J, Fiegl N, Meinhart J, Lanzenberger M, Unal K, et al. Is the transpulmonary pressure gradient a predictor for mortality after orthotopic cardiac transplantation? Transpl Int. 2005. April;18(4):390–5. [DOI] [PubMed] [Google Scholar]
- 35.Corte TJ, Wort SJ, Gatzoulis MA, Macdonald P, Hansell DM, Wells AU. Pulmonary vascular resistance predicts early mortality in patients with diffuse fibrotic lung disease and suspected pulmonary hypertension. Thorax. 2009. October;64(10):883–8. [DOI] [PubMed] [Google Scholar]
- 36.Rivera-Lebron BN, Forfia PR, Kreider M, Lee JC, Holmes JH, Kawut SM. Echocardiographic and hemodynamic predictors of mortality in idiopathic pulmonary fibrosis. Chest. 2013. August;144(2):564–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tedford RJ, Beaty CA, Mathai SC, Kolb TM, Damico R, Hassoun PM, et al. Prognostic value of the pre-transplant diastolic pulmonary artery pressure-to-pulmonary capillary wedge pressure gradient in cardiac transplant recipients with pulmonary hypertension. J Heart Lung Transplant. 2014. March;33(3):289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Assad TR, Hemnes AR, Larkin EK, Glazer AM, Xu M, Wells QS, et al. Clinical and Biological Insights Into Combined Post- and Pre-Capillary Pulmonary Hypertension. J Am Coll Cardiol. 2016. December 13;68(23):2525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tampakakis E, Shah SJ, Borlaug BA, Leary PJ, Patel HH, Miller WL, et al. Pulmonary effective arterial elastance as a measure of right ventricular afterload and its prognostic value in pulmonary hypertension due to left heart disease. Circ Heart Fail. 2018. April;11(4):e004436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brittain EL, Assad TR, Hemnes AR, Newman JH. The Diastolic Pressure Gradient Does Not-and Should Not-Predict Outcomes. JACC Heart Fail. 2015. October;3(10):845. [DOI] [PubMed] [Google Scholar]
- 41.Al-Naamani N, Preston IR, Paulus JK, Hill NS, Roberts KE. Pulmonary arterial capacitance is an important predictor of mortality in heart failure with a preserved ejection fraction. JACC Heart Fail. 2015. June;3(6):467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagueh SF, Smiseth OA, Appleton CP, Byrd BF, Dokainish H, Edvardsen T, et al. Recommendations for the Evaluation of Left Ventricular Diastolic Function by Echocardiography: An Update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2016. April;29(4):277–314. [DOI] [PubMed] [Google Scholar]
- 43.Machado RF, Gladwin MT. Pulmonary hypertension in hemolytic disorders: pulmonary vascular disease: the global perspective. Chest. 2010. June;137(6 Suppl):30S–38S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dokainish H. Left ventricular diastolic function and dysfunction: Central role of echocardiography. Glob Cardiol Sci Pract. 2015. January 26;2015:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grant ADM, Negishi K, Negishi T, Collier P, Kapadia SR, Thomas JD, et al. Grading diastolic function by echocardiography: hemodynamic validation of existing guidelines. Cardiovasc Ultrasound. 2015. June 24;13:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Left Atrial Function by Cardiac MRI in Idiopathic Pulmonary Arterial Hypertension(IPAH) and Pulmonary Hypertension Due to Heart Failure with Preserved Ejection Fraction(PH-HFpEF) | Cl06. MONEY DON’T MATTER TONIGHT: PULMONARY HYPERTENSION ASSESSMENT, PROGNOSTICATION, AND TREATMENT [Internet] [cited 2018. February 21]. Available from: https://www.atsjournals.org/doi/abs/l0.1164/ajrccm-conference.2017.195.1_MeetingAbstracts.A6903 [Google Scholar]
- 47.Schelbert EB, Piehler KM, Zareba KM, Moon JC, Ugander M, Messroghli DR, et al. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc. 2015. December 18;4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raeisi-Giglou P, Lam L, Tamarappoo BK, Newman J, Dweik RA, Tonelli AR. Evaluation of left ventricular diastolic function profile in patients with pulmonary hypertension due to heart failure with preserved ejection fraction. Clin Cardiol. 2017. June;40(6):356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosenkranz S, Gibbs JSR, Wachter R, De Marco T, Vonk-Noordegraaf A, Vachiéry J-L. Left ventricular heart failure and pulmonary hypertension. Eur Heart J. 2016. March 21;37(12):942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vachiéry J-L, Adir Y, Barberà JA, Champion H, Coghlan JG, Cottin V, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013. December 24;62(25 Suppl):D100–8. [DOI] [PubMed] [Google Scholar]
- 51.Guazzi M, Borlaug BA. Pulmonary hypertension due to left heart disease. Circulation. 2012. August 21;126(8):975–90. [DOI] [PubMed] [Google Scholar]
- 52.Assad TR, Maron BA, Robbins IM, Xu M, Huang S, Harrell FE, et al. Prognostic effect and longitudinal hemodynamic assessment of borderline pulmonary hypertension. JAMA Cardiol. 2017. December 1;2(12):1361–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leopold JA. Biological Phenotyping of Combined Post-Capillary and Pre-Capillary Pulmonary Hypertension: Focus on Pulmonary Vascular Remodeling. J Am Coll Cardiol. 2016. December 13;68(23):2537–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caravita S, Faini A, Deboeck G, Bondue A, Naeije R, Parati G, et al. Pulmonary hypertension and ventilation during exercise: Role of the pre-capillary component. J Heart Lung Transplant. 2017. July;36(7):754–62. [DOI] [PubMed] [Google Scholar]
- 55.Lundberg JO, Gladwin MT, Weitzberg E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat Rev Drug Discov. 2015. September;14(9):623–41. [DOI] [PubMed] [Google Scholar]
- 56.Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction:comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013. July 23;62(4):263–71. [DOI] [PubMed] [Google Scholar]
- 57.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003. December;9(12):1498–505. [DOI] [PubMed] [Google Scholar]
- 58.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008. February;7(2):156–67. [DOI] [PubMed] [Google Scholar]
- 59.Simon MA, Vanderpool RR, Nouraie M, Bachman TN, White PM, Sugahara M, et al. Acute hemodynamic effects of inhaled sodium nitrite in pulmonary hypertension associated with heart failure with preserved ejection fraction. JCI Insight. 2016. November 3;1(18):e89620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borlaug BA, Koepp KE, Melenovsky V. Sodium nitrite improves exercise hemodynamics and ventricular performance in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2015. October 13;66(15):1672–82. [DOI] [PubMed] [Google Scholar]
- 61.Cruz L, Ryan JJ. Nitric oxide signaling in heart failure with preserved ejection fraction. JACC: Basic to Translational Science. 2017. June;2(3):341–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005. February;11(2):214–22. [DOI] [PubMed] [Google Scholar]
- 63.Lai Y-C, Tabima DM, Dube JJ, Hughan KS, Vanderpool RR, Goncharov DA, et al. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated With Heart Failure With Preserved Ejection Fraction. Circulation. 2016. February 23;133(8):717–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011. July 12;124(2):164–74. [DOI] [PubMed] [Google Scholar]
- 65.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013. March 27;309(12):1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bermejo J, Yotti R, García-Orta R, Sánchez-Fernández PL, Castaño M, Segovia-Cubero J, et al. Sildenafil for improving outcomes in patients with corrected valvular heart disease and persistent pulmonary hypertension: a multicenter, double-blind, randomized clinical trial. Eur Heart J. 2018. April 14;39(15):1255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borlaug BA, Melenovsky V, Koepp KE. Inhaled sodium nitrite improves rest and exercise hemodynamics in heart failure with preserved ejection fraction. Circ Res. 2016. September 16;119(7):880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reddy YNV, Olson TP, Obokata M, Melenovsky V, Borlaug BA. Hemodynamic correlates and diagnostic role of cardiopulmonary exercise testing in heart failure with preserved ejection fraction. JACC Heart Fail. 2018. May 21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonderman D, Pretsch I, Steringer-Mascherbauer R, Jansa P, Rosenkranz S, Tufaro C, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. 2014. November;146(5):1274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pieske B, Maggioni AP, Lam CSP, Pieske-Kraigher E, Filippatos G, Butler J, et al. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction:results of the Soluble guanylate Cyclase stimulatoR in heArT failurE patients with PRESERVED EF (SOCRATES-PRESERVED) study. Eur Heart J. 2017. April 14;38(15):1119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Valero-Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F. Dual Endothelin-A/Endothelin-B Receptor Blockade and Cardiac Remodeling in Heart Failure With Preserved Ejection Fraction. Circ Heart Fail. 2016. November;9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humbert M, Ghofrani H-A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax. 2016. January;71(1):73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meoli DF, Su YR, Brittain EL, Robbins IM, Hemnes AR, Monahan K. The transpulmonary ratio of endothelin 1 is elevated in patients with preserved left ventricular ejection fraction and combined pre- and post-capillary pulmonary hypertension. Pulm Circ. 2018. March;8(1):2045893217745019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vachiery J-L, Delcroix M, Al-Hiti H, Efficace M, Hutyra M, Lack G, et al. Melody-1: a pilot study of macitentan in pulmonary hypertension due to left ventricular dysfunction. J Am Coll Cardiol. 2017. March;69(11):1880.28385324 [Google Scholar]
- 75.Koller B, Steringer-Mascherbauer R, Ebner CH, Weber T, Ammer M, Eichinger J, et al. Pilot Study of Endothelin Receptor Blockade in Heart Failure with Diastolic Dysfunction and Pulmonary Hypertension (BADDHY-Trial). Heart Lung Circ. 2017. May;26(5):433–41. [DOI] [PubMed] [Google Scholar]
- 76.McLaughlin W, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association developed in collaboration with the American College of Chest Physicians; American Thoracic Society, Inc.; and the Pulmonary Hypertension Association. J Am Coll Cardiol. 2009. April 28;53(17):1573–619 [DOI] [PubMed] [Google Scholar]