

SUMMARY
The dopamine (DA) D2 receptor (D2R) is an important target for the treatment of neuropsychiatric disorders such as schizophrenia and Parkinson’s disease. However, the development of improved therapeutic strategies has been hampered by our incomplete understanding of this receptor’s downstream signaling processes in vivo and how these relate to the desired and undesired effects of drugs. D2R is a G protein-coupled receptor (GPCR) that activates G proteindependent as well as non-canonical arrestin-dependent signaling pathways. Whether these effector pathways act alone or in concert to facilitate specific D2R-dependent behaviors is unclear. Here, we report on the development of a D2R mutant that recruits arrestin but is devoid of G protein activity. When expressed virally in “indirect pathway” medium spiny neurons (iMSNs) in the ventral striatum of D2R knockout mice, this mutant restored basal locomotor activity and cocaine-induced locomotor activity in a manner indistinguishable from wildtype D2R, indicating that arrestin recruitment can drive locomotion in the absence of D2R-mediated G protein signaling. In contrast, incentive motivation was enhanced only by wildtype D2R, signifying a dissociation in the mechanisms that underlie distinct D2R-dependent behaviors, and opening the door to more targeted therapeutics.
Introduction
The neuromodulator dopamine (DA) plays major roles in motor function, motivation, rewardlearning, and cognition1–3, and its dysregulation has been implicated in psychiatric and neurological disorders including schizophrenia4, Parkinson’s disease5, addiction6, ADHD7, obsessive compulsive disorder8, and Tourette’s syndrome9. DA confers its actions through five seven-transmembrane, G protein-coupled receptors (GPCRs) (DARs; D1-D5R) that are distributed heterogeneously throughout the central nervous system. D2R is notable for its clinical importance as a therapeutic target. D2R is blocked by all current antipsychotic medications10, 11, is activated by anti-Parkinsonian medications12, and has been implicated as a target for the treatment of addiction13. However, medications that target this receptor have numerous on- and off-target effects that impair quality of life and medication compliance. Furthermore, the precise D2R localization and downstream intracellular signal transduction mechanisms that must be targeted to bestow clinical efficacy remain unknown.
Elucidating the roles of D2R in living animals is challenging in part because it is expressed heterogeneously across diverse cell types and brain regions14. For example, D2R is highly enriched in the striatum15, where it is expressed in functionally distinct neurons including the “indirect pathway” medium spiny projection neurons (iMSNs) as well as cholinergic interneurons (ChIs)14, 16. D2Rs also act as auto- and heteroreceptors at the terminals of dopaminergic midbrain17 and glutamatergic cortical neurons18, respectively, that innervate the striatum. Postand pre-synaptic striatal D2Rs are thought to play distinct roles, with the former regulating somato/dendritic excitability19 and the latter regulating transmitter synthesis and release20.
D2R can engage a multitude of signaling pathways within the same cell. D2R couples to Gi/o/z proteins15, which inhibit adenylyl cyclase to decrease cAMP levels, and also regulate signaling proteins such as DARPP-3221, ion channels22–26, and transcription factors27. D2R also recruits arrestins (arrestin-2 and −3)28, 29, which terminate G protein signaling, facilitate receptor internalization, and engage non-canonical, G protein-independent signaling pathways30. Whether G protein- and arrestin-dependent pathways act alone or in concert to facilitate specific D2R-dependent processes is not well understood.
Traditional pharmacological agents, i.e., agonists and antagonists of D2R, have two limitations. First, they target receptors in a cell type-independent manner and therefore it is difficult to identify their site of action, especially within the striatum. Second, they are largely pathway-independent so it is unclear which signaling pathway is responsible for an observed behavioral effect. Recently, novel ligands have been developed that are functionally selective, or biased, in their ability to activate either G proteins or arrestins downstream of D2R31–34. Though promising, these ligands are often partial agonists compared to DA and only have limited bias. Moreover, biased ligands cannot overcome the cell type specificity limitation, and like classical ligands, have off-target effects at other DARs and other targets. While the genetic knockout of specific signaling proteins can partially address these limitations35, 36, this approach is not specific to D2R signaling and is complicated by potential compensation during development.
Limitations associated with existing pathway-selective approaches are highlighted by studies investigating the role of arrestin signaling on D2R-dependent locomotion. Biased ligands that preferentially engage arrestins downstream of D2R were shown to inhibit DA-dependent, psychostimulant-induced locomotion in mice, suggesting that arrestin signaling inhibits locomotion31. However, the global knockout of arrestin-3 attenuated psychostimulant-induced locomotion, suggesting that arrestins are necessary for locomotion35. This discrepancy is likely due to the partial agonism of the biased compounds as well as the potential multitude of on- and off-target effects associated with these approaches.
We recently showed that upregulating D2R in iMSNs of the nucleus accumbens (NAc-iMSNs) of adult mice enhances both locomotion and motivation in mice37, 38. These overexpressed receptors are activated by endogenous DA, thereby obviating the need for exogenous ligands and maintaining the spatio-temporal aspects of signaling. Here, we further refined this approach to study, with cell type specificity, the precise role of arrestin recruitment to D2R and its presumed downstream signaling effects. We developed an arrestin-specific biased mutant of D2R by disrupting the receptor’s ability to activate G proteins. Virus-mediated expression of this mutant in NAc-iMSNs within a D2R-knockout background was sufficient to reverse blunted basal locomotor activity in these animals as well as to restore the locomotor response to cocaine to levels identical to that achieved by wildtype D2R. Remarkably, in contrast to wildtype, the expression of this mutant in NAc-iMSNs enhanced locomotion but not motivation, indicating that these behaviors are differentially regulated by D2R-mediated arrestin recruitment and that enhancing motivation requires G protein activation.
Materials and Methods
Modeling
The molecular representation of D2R co-crystalized with the antagonist risperidone (pdb: 6CM4)39 was prepared using Chimera, which was developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Molecular biology and heterologous expression
The short or long isoforms of human D2R were cloned into the expression vector pcDNA3.1 with a signal peptide to enhance receptor expression, a FLAG-tag at the receptor N-terminus, and with or without Renilla luciferase 8 (Rluc8) at the receptor C-terminus40. Point mutations in D2R were introduced using the QuikChange site-directed mutagenesis method (Stratagene, La Jolla, CA) per the manufacturer’s instructions. For the bioluminescence resonance energy transfer (BRET)- based assays used in this study, HEK293T cells were seeded onto 10 cm plates and transfected with a 1:1 ratio of DNA:polyethylenimine (linear, MW 25,000; PolySciences, Inc.). For the BRETbased D2R-mediated Gi1 recruitment assay, we transfected plasmids encoding D2R-Rluc8 (0.2 μg), human Gαi1 with mVenus inserted at position 91 (Gαi1–91-mVenus; 5 μg) and human Gβ1 and human Gγ2 (5 μg each). For the BRET-based D2R-mediated Gγ-mVenus recruitment assay, HEK293T cells were seeded onto 6-well plates and transfected with a 1:6 ratio of DNA:polyethylenimine with plasmids encoding D2R-Rluc8 (50 ng), the pertussis toxin insensitive variants (cDNA resource center, Bloomsburg University, PA, 50–125 ng) of human Gαi1 C351I, Gαi2 C352I, Gαi3 C351I, GαoA C351I, or WT human Gαz, human Gβ1 (125–250ng) and human Gγ2- mVenus (125–250 ng). For the D2R-mediated arrestin recruitment assay, we transfected D2RRluc8 (0.2 μg), bovine GRK2 (5 μg) and bovine arrestin-2 or human arrestin-3 fused to mVenus at their N-termini (8 μg)41, 42. For the D2R-AP2 interaction assay, we transfected D2R-Rluc8 (0.2 μg), arrestin-3 (5 μg), GRK2 (5 μg) and β2-adaptin subunit of AP-2 fused to YFP43 (10 μg). For the BRET-based D2R-mediated cAMP inhibition assay, we transfected plasmids encoding D2R (0.2 μg) and CAMYEL44 (6 μg; ATCC). For cAMP inhibition experiments in which Gi1 was overexpressed, HEK293T cells were seeded onto 6-well plates and transfected with CAMYEL (1 μg), D2R-WT or its mutants (0.06 μg), Gαi1 (0.165 μg), Gβ1 (0.5 μg) and Gγ2 (0.5 μg), For measuring receptor surface expression as well as internalization, D2R-WT and its mutants were cloned into pcDNA5/FRT/TO with a signal peptide and FLAG-tag at the receptor N-terminus. These plasmids were stably transfected into Flp-in T-Rex HEK293 cells using the Flp-in system (Thermo Fisher). Stable cell lines were seeded onto 10 cm plates 48 hours prior to the assay. Receptor expression was induced with 1 μg/mL tetracycline 24 hours prior to the assay. For the internalization assay, cells were also transiently transfected with pcDNA3.1 plasmids encoding arrestin-3 and GRK2 using a 1:1 ratio of DNA:polyethylenimine 24 hours prior to the assay.
HEK293T cells used in this study tested negative for mycoplasma contamination.
Bioluminescence resonance energy transfer (BRET) assays
All BRET studies were performed in HEK293T cells that were maintained in DMEM (Invitrogen) with 10% fetal bovine serum at 37 °C under 5% CO2. Cells were transiently transfected with the plasmids described above. Cells were prepared and assayed as described previously in detail42. Briefly, cells were washed, harvested and resuspended in DPBS containing 5 mM glucose at room temperature. Cells (~40 μg of protein per well according to a BCA protein assay kit, ThermoScientific) were distributed into a 96-well microplate (Wallac, PerkinElmer Life and Analytical Sciences). After incubation with coelenterazine H (5 μM) (Dalton Pharma Services) for 8 minutes, different ligands were injected and incubated for 2 to 10 minutes. For the CAMYEL assay, 30 μΜ of forskolin was added 10 minutes prior to the injection of DA. For the Gγ-mVenus recruitment assay, cells were harvested from 6-well plates 24 hours after transfection and plated into poly-D-lysine coated (Sigma-Aldrich) white-bottom 96-well optiplates (Wallac, PerkinElmer Life and Analytical Sciences) at a density of 50,000 cells per well. Cells were treated with 100 ng/mL pertussis toxin (Sigma-Aldrich) 16 hours prior to assay measurement. Twenty four hours after cells were transferred to plates, media was aspirated, cells washed once with DPBS and 80 μL DPBS containing 5 mM glucose was added to each well. Using a Pherastar FS plate reader (BMG Labtech), BRET1 signal was determined by quantifying and calculating the ratio of the light emitted by mVenus (510–540 nm) over that emitted by Rluc8 (485 nm).
Receptor surface expression and internalization
To measure surface expression of D2R-WT or its mutants, live cells were washed, harvested and resuspended in DPBS. For the internalization assay, live cells were preincubated with or without 10 μM DA for one hour at 37 °C under 5% CO2, and then washed, harvested and resuspended in DPBS. Cells were then incubated with mouse anti-FLAG M2 antibody (1:400 dilution; Sigma) for 30 minutes on ice, washed and resuspended in DPBS. Cells were then incubated with goat antimouse lgG(H+L)-Alexa 647 antibody (Thermo Fisher) for 30 minutes on ice, washed and resuspended in DPBS. Receptor expression was assessed using a BD Accuri C6 cytometer.
Animal
For D2R overexpression in wild-type mice we used adult hemizygous male and female D2-Cre (ER44; GENSAT, backcrossed for over 5 generations onto C57BL/6J background). For reexpression of D2Rs in indirect pathway of D2R KO mice, A2A-Cre(KG126;GENSAT) were crossed with Drd2−/−mice (B.6129S2-Drd2tm1low strain backcrossed for 20 generations onto C57BL/6J background45). Drd2−/+A2A-Cre (homozygous for A2A-Cre transgene) mice were intercrossed to obtain Drd2−/−A2A-Cre (hemizygous for A2A-Cre transgene). Mice were housed 3–5 per cage for most experiments on a 12 hour light/dark cycle. All experiments were conducted in the light cycle. All experimental procedures were conducted following NIH guidelines and were approved by Institutional Animal Care and Use Committees of Columbia University, New York State Psychiatric Institute, and the VA Portland Health Care System.
D2-Cre+/− or Drd2−/−A2A-Cre+/− mice (≥ 8 weeks old) were bilaterally injected with a Credependent double-inverted open reading frame (DIO) adeno-associated viruses (AAVs) encoding the long isoform of D2R-WT36, 37 or the arrrestin-biased mutant (D2R-ARB) followed by IRESmVenus, or a DIO AAV encoding EGFP (UNC Vector Core, Chapel Hill, NC) into the nucleus accumbens (NAc) using stereotactic Bregma-based coordinates: AP, +1.70 mm; ML, ±1.20 mm; DV, −4.1 mm (from dura) for behavioral experiments. For the ex vivo GTPγS incorporation assay, mice were injected bilaterally at two different coordinates in the dorsal striatum instead of the NAc to increase striatal tissue yield (AP, +1.3 mm; ML, +/−1.4 mm; DV, −3.3 mm, and AP, +0.9; ML, +/− 2.0; DV, −3.4 mm from dura). Groups of mice used for experiments were first assigned their AAVgenotype in a counterbalanced fashion that accounted for sex, age and home cage origin.
Histology
Mice were transcardially perfused with ice-cold 4% paraformaldehyde (Sigma, St. Louis, MO) in PBS under deep anesthesia. Brains were harvested, post-fixed overnight and washed in PBS. Free-floating 30-μm coronal sections were obtained using a Leica VT2000 vibratome (Richmond, VA). After incubation in blocking solution (10% fetal bovine serum, 0.5% bovine serum albumin in 0.5% TBS-Triton X-100) for 1 hour at room temperature, sections were labeled overnight at 4°C with primary antibodies against GFP/mVenus (chicken; 1:1000; AB13970 Abcam, Cambridge, MA) and D2R (rabbit; 1:500; in-house). Sections were incubated with fluorescent secondary antibodies (donkey anti-chicken FITC, 1:400; 703–096-155; Jackson ImmunoResearch, West Grove, PA and donkey anti-rabbit Alexa 546; 1:400; A10040; Invitrogen, Carlsbad, CA)for 2 hours at room temperature. Sections were then mounted on slides and coverslipped with Vectashield containing DAPI (Vector, Burlingame, CA). Digital images were acquired using a Nikon epifluorescence microscope, and processed with NIH Image J software. For D2R fluorescence intensity analysis, NIH Image J software was used to draw an ROI outlining the regions of the NAc with virus-induced D2R expression in each section, and mean intensity values were calculated. These values were normalized to background fluorescence in an adjacent unlabeled region (medial septum) for Drd2−/−A2A-Cre or to endogenous D2R immunofluorescence in striatum for D2-Cre mice. Values were obtained from individual hemispheres and used as individual data points for analysis.
Ex vivo GTPγS incorporation assay
Four weeks after surgery, brains were harvested into ice-cold PBS promptly following decapitation. Striatal tissue was dissected from each hemisphere on ice and then snap frozen in liquid nitrogen and stored at −80°C and shipped overnight on dry ice to Jupiter, FL where experimenters were blinded as to treatment groups. G protein-coupling in mouse brain was measured using a previously published protocol46,47. Membranes were prepared following glasson-glass dounce homogenization in homogenization buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCI, 1 mM EDTA, 1 mM DTT). The homogenate was passed through a 26-gauge needle, centrifuged twice at 20,000 × g for 30 min at 4 °C, and resuspended in ice-cold assay buffer (50 mM Tris-HCI pH 7.4, 100 mM NaCI, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT and 2 μΜ GDP for CB1 activation and 100 μM GDP for D2 activation). In a 96-well plate format, 2.5 μg of membrane protein was incubated in assay buffer containing ~0.1 nM [35S]GTPγS and increasing concentrations of test compound in a total volume of 200 μL for 1 h at room temperature. Test compounds (quinpirole or CP-55940) added to the assays at final DMSO concentration of 1%. Reactions were terminated by filtering through GF/B filters using a plate harvester (Brandel Inc., Gaithersburg, MD) and rinsing with cold dH2O. Filters were dried overnight, and radioactivity was determined with a microplate scintillation counter. The average of n=3 individual mouse striatum preparations were performed in duplicate presented as mean ± S. E. M.
Open field locomotor activity
Basal locomotion was assessed in open field boxes equipped with infrared photobeams to measure locomotor activity (Med Associates, St. Albans, VT) over 90 minute sessions. Data, expressed as distance traveled, were acquired using Kinder Scientific Motor Monitor software (Poway, CA). Cocaine-stimulated activity was measured in activity monitors (40 cm2; Accuscan Instruments, Inc., Columbus, OH), with 15 mg/kg of the drug injected i.p. after 90 minutes of acclimation to the chambers.
Operant apparatus
Eight operant chambers (model Env-307w; Med-Associates, St. Albans, VT) equipped with liquid dippers were used. Each chamber was located in a light- and sound-attenuating cabinet. The dimensions of the experimental chamber interior were 22 × 18 × 13 cm, with flooring consisting of metal rods placed 0.87 cm apart. A feeder trough was centered on one wall of the chamber. An infrared photocell detector was used to record head entries into the trough. Raising of the dipper inside the trough delivered a drop of evaporated milk. A retractable lever was mounted on the same wall as the feeder trough. A house light located on wall opposite to trough illuminated the chamber throughout all sessions.
Dipper and lever press training
Four weeks after AAV surgery, mice underwent operant training. Mice were weighed daily and food-restricted to 85–90% of baseline weight; water was available ad libitum. Mice were trained as previously described48. In the first training session, 20 dipper presentations were separated by a variable inter-trial interval (ITI) and ended after 20 rewards were earned or after 30 minutes, whichever occurred first. Mice reached criterion when head entries were made during 20 dipper presentations in one session. In the second training session, criterion was achieved when mice made head entries during 30 of 30 dipper presentations. Mice were trained to lever press on a continuous reinforcement (CRF) schedule. Levers were retracted after each reinforcer and were presented again after a variable ITI (average 30 seconds). The reward consisted of raising the dipper for 5 seconds, and the session ended when the mouse earned 60 reinforcements, or one hour elapsed, whichever occurred first. Sessions were repeated daily until mice achieved 60 reinforcements. Next, mice underwent fixed interval (FI) training wherein lever presses were reinforced until after a fixed interval (timed relative to the lever extension) had elapsed. Each reinforcement was followed by a variable inter-trial interval (average 30 seconds) during which the lever remained retracted, and then a new trial started, signaled by lever extension. Mice began with FI-4 s session and proceeded successively to longer interval sessions after earning ≥ 30 rewards in each session. The FI durations were 4, 8, 12, 16, and 24 seconds.
Progressive ratio (PR) task
Following FI schedules, mice were tested in a PR task, as previously described38, 49. Briefly, a reward was obtained after the mice made the required number of lever presses. The criterion was set at two lever presses for the first trial and the requirement doubled with each successive trial. The session ended after 2 hours or after 3 minutes had elapsed without a lever press. Breakpoint was defined as the last criterion successfully completed. Mean values from 4 daily PR sessions were analyzed. Investigators were blind to genotype of mice during the experiment and during data analysis. Mice were matched for age and sex.
Statistics and data analysis
For dose-response curves, nonlinear regression analysis was performed using the sigmoidal dose-response function in GraphPad Prism. All values reported are mean ± standard error mean (S.E.M.). Statistical analyses were performed using Graphpad Prism.
Sample sizes for behavioral studies were determined by performing statistical power analyses based on effect sizes observed in preliminary data or on similar work in the literature. Histological verification of viral expression was used to exclude mice with misplaced injections from behavioral data analysis in a manner blinded to genotype. Statistical analyses were performed using Graphpad Prism. Data are expressed as mean ± S.E.M.. Paired and unpaired two-tailed Student’s t-tests were used to compare 2-group data, as appropriate. Multiple comparisons were evaluated by one-way or two-way ANOVA and Bonferroni’s post hoc test, when appropriate. Log-rank tests were used to analyze survival curves. All statistical tests met their respective assumptions. A pvalue of < 0.05 was considered statistically significant. Behavioral findings were successfully replicated with mice from different litters, ages, or sexes, and in several instances, across independent cohorts or related mouse strains.
Results
Development of an arrestin-biased D2R
Biased D2R mutants have been reported previously but their utility has been constrained by their incomplete bias towards either G proteins or arrestins41, 50 or diminished expression41, 51. The only reported arrestin-biased D2R mutant to date, D2R(A135R:M140D), enhanced locomotion when overexpressed in striatal D2R-expressing neurons of mice50, 52. However, this mutant was shown to be only partially biased toward arrestin recruitment50, which we confirmed using bioluminescence resonance energy transfer (BRET)-based G protein activation and arrestin recruitment assays in HEK293T cells. Under our assay conditions, maximal DA-induced G protein-dependent cAMP inhibition was impaired but still substantial (28 ± 2% of wildtype D2R (D2R-WT), n=3; Supplementary Figures 1A & C). The potency of DA was only slightly reduced compared to D2R-WT (~3-fold, Supplementary Figures 1A & D). Maximal DA-induced arrestin- 3 recruitment to D2R(A135R:M140D) was relatively less impaired, but still considerably lower than D2R-WT (55 ± 3% of D2R-WT, n=3; Supplementary Figures 1B & C). Thus, although D2R(A135R:M140D) is partially arrestin-biased, it is unclear whether arrestin or residual G protein signaling (or both) downstream of D2R is responsible for enhanced locomotion in vivo.
To develop a more arrestin-biased D2R mutant with levels of arrestin recruitment more comparable to D2R-WT, we took advantage of a previously reported arrestin-biased, muscarinic acetylcholine receptor (mAChR)-based Designer Receptor Exclusively Activated by Designer Drugs (DREADD)53. A single point mutation of an arginine (R3.50 according to the Ballesteros- Weinstein GPCR numbering system54) to leucine in this receptor’s “DRY motif” resulted in strong arrestin bias53. The DRY motif is critically involved in receptor activation55 and is conserved across Family A GPCRs, including mAChRs and DARs. Therefore, we mutated the DRY motif in D2R to DLY (D2R(R132L); Figures 1A & 1B), which greatly impaired DA-induced recruitment of the Gα subunit of Gi1 to the receptor (9 ± 5% of D2R-WT, n=5; Figure 1C)56. In contrast, R132L’s effect on DA-induced arrestin-3 recruitment was less severe, though it still significantly reduced maximal arrestin-3 recruitment (47 ± 8% of D2R-WT, n=6; two-tailed t-test, p<0.001) and potency (~16- fold lower compared to D2R-WT, n=6; two-tailed t-test, p<0.0001) (Figure 1D). Similar results were observed for arrestin-2 recruitment to D2R(R132L) (Supplementary Figure 2).
Figure 1. An extremely arrestin-biased D2R mutant receptor, D2R(L123W:R132L).
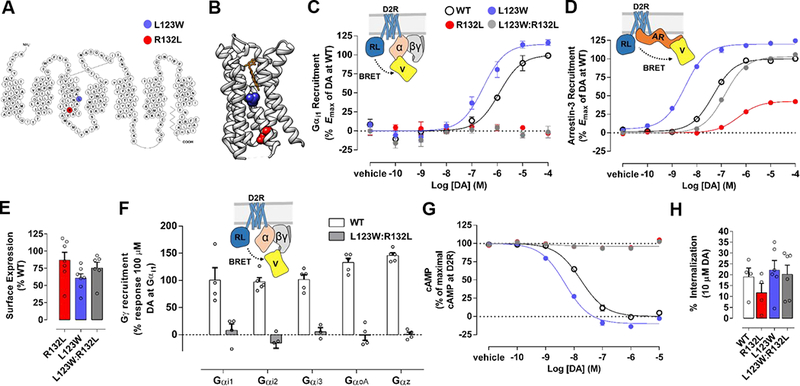
A) Snake-plot representation of D2R highlighting mutated residues R132 (red) to L and L123 (blue) to W in transmembrane segment 3 (TM3).
B) Ribbon representation of D2R co-crystallized with the D2-like receptor antagonist risperidone. Risperidone (orange) is shown bound to the DA-binding orthosteric binding site (OBS) of the receptor, which is formed by the TM bundle and lies above the mutated residues R132 (red) and L123 (blue).
C) Schematic representation of a bioluminescence resonance energy transfer (BRET)-based assay that measures agonist-induced recruitment of Gαi1 (α) fused to mVenus (V) in its α-helical domain to D2R fused to Renilla luciferase 8 (RL) at its C-terminus (inset). Gαi1-recruitment to D2R-WT, D2R(R132L), D2R(L123W), or D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of independent experiments (n= 6, 6, 3, and 3, respectively) performed in triplicate.
D) Schematic representation of a BRET-based assay that measures agonist-induced recruitment of arrestin-3 (AR) fused at its N-terminus to mVenus (V) to D2R-Rluc8 (inset). Arrestin-3- recruitment to D2R-WT, D2R(R132L), D2R(L123W), or D2R(L123W:R132L) in response to increasing concentrations of DA (refer to Figure 1C for figure legend). Dose-response curves are representative of independent experiments (n= 6, 6, 3, and 3, respectively) performed in triplicate.
E) Surface expression of D2R(R132L), D2R(L123W) and D2R(L123W:R132L) in HEK293T cells as a percentage of D2R-WT. Bars represent the average of six to seven independent experiments.
F) Gγ-mVenus recruitment to D2R-WT and D2R(L123W:R132L) with co-expression of pertussis toxin insensitive variants of human Gαi1–3 or GαoA or wildtype human Gαz in response to 100 μΜ DA. Cells were treated with 100 ng/mL pertussis toxin prior to measurement of response. Responses represent a change in BRET normalized to that of 100 μΜ DA at the D2R-WT with coexpression of Gαi1. Bars represent the average of four to five independent experiments.
G) Gi/o/z-mediated inhibition of forskolin (30 μM)-induced cyclic AMP accumulation (cAMPi) for D2R-WT, D2R(R132L), D2R(L123W), and D2R(L123W:R132L) in response to increasing concentrations of DA (refer to Figure 1C for figure legend). Dose-response curves are representative of three independent experiments performed in triplicate.
H) The percentage of D2R-WT, D2R(R132L), D2R(L123W), and D2R(L123W:R132L) that internalizes in response to DA (10 μM) after one hour. Bars represent the average of four to six independent experiments.
The short isoform of D2R (D2Rs) was used for all experiments with the exception of the Gγ- mVenus recruitment assay. Error bars represent S.E.M.
Even if D2R (R132L) preferentially recruits arrestin, its attenuated arrestin activity could still mask a role for arrestin-dependent processes in vivo. We hypothesized that combining R132L with mutations that increase receptor activity would enhance arrestin recruitment. The mutation of L3.41 to W (L123W in D2R) was shown previously to thermostabilize Family A GPCRs and facilitate their crystallization57, e.g., this mutation is present in the crystal structure of a close homolog of D2R, DA D3R58. Interestingly, DA was significantly more potent at D2R(L123W) (Figures 1A & 1B) compared to D2R-WT in assays measuring the recruitment of Gαi1 (~4-fold; two-tailed t-test; p<0.01; Figure 1C), arrestin-2 (~14-fold; two-tailed t-test, p<0.01; Supplementary Figure 2) and arrestin-3 (~14-fold; two-tailed t-test; p<0.0001; Figure 1D), suggesting that L123W enhances the affinity of the receptor for DA and/or the ability of the receptor to populate an active conformational state when bound to agonist.
As hypothesized, combining R132L and L123W created a mutant, D2R(L123W:R132L) (Figures 1A & B), that was capable of robustly recruiting arrestin-3 (100 ± 7% of D2R-WT, n=3; Figure 1D) and arrestin-2 (132 ± 9% of D2R-WT, n=3; Supplementary Figure 2). Consistent with its effect on D2R-WT, the L123W mutation combined with R132L enhanced the potency of DA by ~4-fold relative to that in R132L in both the arrestin-2 and −3 recruitment assays. When compared to D2R WT, D2R(L123W:R132L) was still ~2- to ~4-fold less sensitive to DA for the recruitment of arrestin-2 and −3, respectively. Remarkably, D2R(L123W:R132L) was devoid of detectable Gαi1 activity (n=3; Figure 1C). D2R(L123W:R132L) expressed at the plasma membrane of HEK293T cells at 75 ± 8% (n=6) of D2R-WT (Supplementary Figure S3 & Figure 1E). Furthermore, L123W:R132L conferred arrestin-bias to both the short (Figure 1) and long isoforms of D2R (Supplementary Figure 4).
Of note, D2R has been reported to couple more potently to Go than Gi isoforms59, 60, and it has been inferred based on knockout data that Go is a critical mediator of D2R activity in vivo61, 62. Thus, we carried out a BRET-based assay comparing the ability of each Gαi/o/z isoform to support DA-induced recruitment of the G protein subunit Gγ to D2R-WT or D2R(L123W:R132L). Consistent with our results for Gαi1 recruitment (Figure 1C), D2R(L123W:R132L) failed to recruit the Gγ subunit of Gi1, Gi2, Gi3, Goa, or Gz (Figure 1F).
We next tested whether G protein or arrestin recruitment to D2R-WT and its mutants correlated with the engagement of processes downstream of these signaling proteins. We first measured inhibition of cyclic AMP (cAMP) accumulation by endogenous Gi/o in HEK293T cells. Whereas DA robustly inhibited cAMP accumulation in cells expressing D2R(L123W) (107 ± 3% of D2R-WT, n=3), it had essentially no effect in those expressing D2R(R132L) and D2R(L123W:R132L) (1 ± 1% and 3 ± 3% of D2R-WT, respectively, n=3; Figure 1G). Similar effects were observed for D2R(L123W:R132L) even in cells overexpressing Gi1 (3 ± 5% of D2R-WT; n=4; Supplementary Figure 5). Overall, these results are consistent with the impaired G protein recruitment to D2R(R132L) and D2R(L123W:R132L) (Figure 1C).
We showed previously that arrestin-mediated internalization of D2R requires an interaction between the receptor and the β2-adaptin subunit of clathrin-associated adaptor protein-2 (AP- 2)41. In a BRET-based assay that we developed previously41, DA dose-dependently enhanced the interaction between AP-2 and D2R-WT (Supplementary Figure 6)41. There was no significant difference in the DA-induced interaction between AP-2 and D2R-WT or D2R(L123W:R132L) (twotailed t-test, p=0.98). Consistent with this observation, there was no significant difference in arrestin-dependent, DA-induced internalization of D2R-WT and D2R(L123W:R132L) (two-tailed t-test, p=0.49; Figure 1H). These results indicate that D2R(L123W:R132L) recruits arrestin that can then bind accessory proteins and facilitate processes downstream of receptor activation.
It is conceivable that D2R(L123W:R132L) only appears biased towards arrestins because its basal (constitutive) activation of G proteins is extremely high, and thus the receptor is incapable of further activating these signaling proteins in response to DA. We used the D2R inverse agonist sulpiride63 to assess the constitutive G protein activity of D2R-WT, D2R(L123W:R132L) or D2R(E339A:T343R), a reported constitutively active mutant (CAM) of D2R64, in the cAMP assay. Sulpiride robustly reversed constitutive inhibition of cAMP accumulation by the CAM, and to a lesser degree that by D2R-WT (Supplementary Figure 7). However, it had no effect on cells expressing D2R(L123W:R132L) or mock transfected cells not expressing receptor (Supplementary Figure 7), indicating that this mutant does not constitutively activate G proteins. Overall, these results indicate that G protein activation by D2R(L123W:R132L) is disrupted whereas arrestin recruitment is intact.
Bias is maintained in the brain
To express the arrestin-biased D2R(L123W:R132L) mutant (henceforth called D2R-ARB) in vivo, we generated a Cre recombinase (Cre)-dependent AAV encoding a double-inverted open reading frame (DIO) containing this mutant followed by an internal ribosome entry site (IRES) and the YFP variant mVenus under the control of the synapsin promoter I (Synl; Figure 2A). We selectively expressed this receptor in iMSNs by injecting AAV in the NAc of adult Drd2−/−A2A-Cre mice (Figure 2B). We also injected AAVs containing EGFP or D2R-WT. Four weeks following injection of the AAVs, we observed EGFP or mVenus expression in the NAc (Figure 2C). The presence of D2R-WT or D2R-ARB was also verified by D2R immunofluorescence (Figure 2C). Comparable D2R dendritic-like immunoreactivity was observed in mice infected with D2R-WT and D2R-ARB AAVs (Figure 2C), consistent with their similar levels of expression in HEK293T cells (Figure 1E).
Figure 2. Arrestin bias downstream of D2R in iMSNs of the NAc.
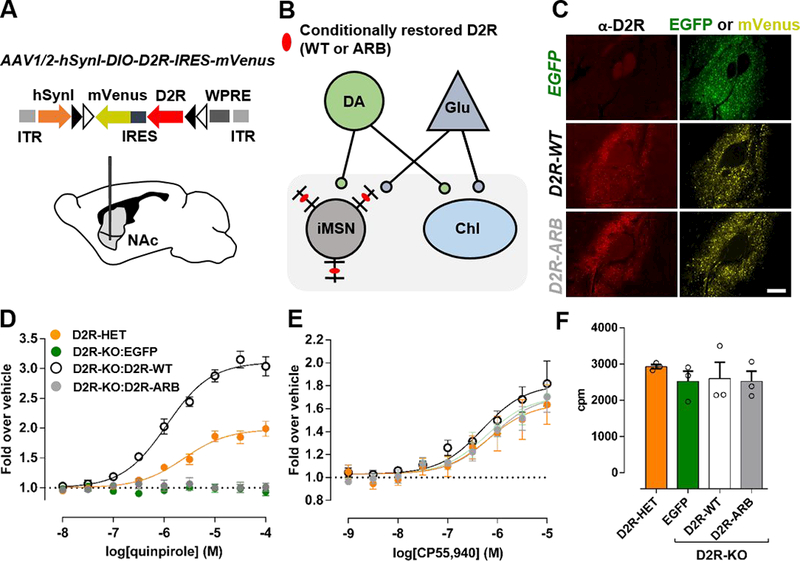
A) Schematic representation of AAV1/2-hSyn1-DIO-SF-D2R-IRES-mVenus. The expression cassette is oriented in the forward direction in the presence of Cre-recombinase, which recognizes flox sites (black and white arrows) (top). The AAV was injected into the NAc of Drd2−/−A2A-Cre mice (bottom).
B) Schematic of striatal neurons from mice lacking native D2Rs. D2R-WT or D2R-ARB (red ovals) was selectively expressed in NAc-iMSNs by injecting Cre-dependent AAVs into the ventral striatum of Drd2−/−A2A-Cre mice.
C) AAV-induced expression of EGFP, D2R-WT, or D2R-ARB in NAc four weeks after injection. Shown are D2R immunoreactivity (left) and EGFP (top right) or mVenus (middle, bottom right) fluorescence. Mean D2R immunofluorescence intensity analysis showed similar D2R levels in both D2R groups (D2R-WT: 1.70 ± 0.07 A.U., n=4 mice; D2R-ARB: 1.83 ± 0.07 A.U., n=5 mice; p = 0.21). Scale bar equals 100 μm.
(D) Quinpirole- or (E) CP55,940-induced 35S-GTPγS binding in dorsal striatal membranes prepared from Drd2+/-A2A-Cre (D2R-HET) or Drd2−/−mice. Drd2−/−A2A-Cre mice were injected with Cre-dependent AAVs encoding EGFP, D2R-WT, or D2R-ARB. Dose-response curves were fit globally from three independent experiments performed in duplicate. Dose-response curves were normalized to a vehicle control.
F) Baseline 35S-GTPγS incorporation did not differ between the samples in assays performed in parallel.
By expressing D2R-ARB in a D2R-KO background, we could assess its signaling properties independently of native D2R-mediated G protein signaling and thus determine whether the bias observed in heterologous cells is maintained ex vivo in the iMSNs. We measured G protein activity using a GTPγS incorporation assay in dorsal striatal homogenates46 (see Methods). Experiments were performed with the selective D2-like receptor agonist quinpirole to avoid activation of D1- like receptors and because quinpirole has similar efficacy as DA at both G protein activation (Supplementary Figure 8) and arrestin recruitment65. Not surprisingly, quinpirole induced robust G protein activation in heterozygotic D2R-KO mice (Drd2+/−A2A-Cre), but had no effect in homozygous D2R-KO (Drd2−/−A2A-Cre) mice infected with EGFP (Figure 2D). Whereas the selective restoration of D2R-WT in NAc-iMSNs rescued robust G protein activation in the D2RKO background, D2R-ARB had no effect (Figure 2D), consistent with its inability to activate G proteins in HEK293T cells (Figures 1C & F). To verify that AAV-mediated D2R overexpression did not affect the function of other Gi/O/z-coupled receptors, we activated cannabinoid receptors (CBRs) with the agonist CP55,94066. There was no significant difference in the ability of this ligand to activate G proteins downstream of CBRs in any of the groups, indicating that Gi/o/z activation was not generally disrupted (Figure 2E). Furthermore, baseline GTPγS was not significantly different between any of the groups (Figure 2F), suggesting that overexpression of EGFP, D2RWT, or D2R-ARB did not result in compensatory changes in basal G protein activity.
Selective restoration of D2R-mediated arrestin recruitment in accumbens-iMSNs reverses blunted locomotion in D2R-deficient mice
To address whether D2R-mediated G protein signaling is essential for D2R-mediated locomotion, we directed the expression of EGFP, D2R-WT or D2R-ARB to NAc-iMSNs of mice that lack native D2R (Drd2−/−A2A-Cre mice) using Cre-dependent AAVs, as described above (Figures 2A-C).
A number of studies have shown that ablation of the D2R gene in mice results in hypolocomotion as well as a blunted locomotor response to an acute administration of cocaine67, 68, which enhances DAR signaling by preventing the uptake of DA by its transporter (DAT)68. We tested whether D2R-WT or D2R-ARB is sufficient to reverse these phenotypes when expressed selectively in NAc-iMSNs of Drd2−/−A2A-Cre mice. Locomotor activity was assessed across three consecutive days for 90 minutes after intraperitoneal injections of either saline on days one and two or cocaine (15 mg/kg) on day three (Figure 3).
Figure 3. Selective expression of D2R-ARB in iMSNs of the NAc reverses blunted locomotor responses in D2R-knockout mice.
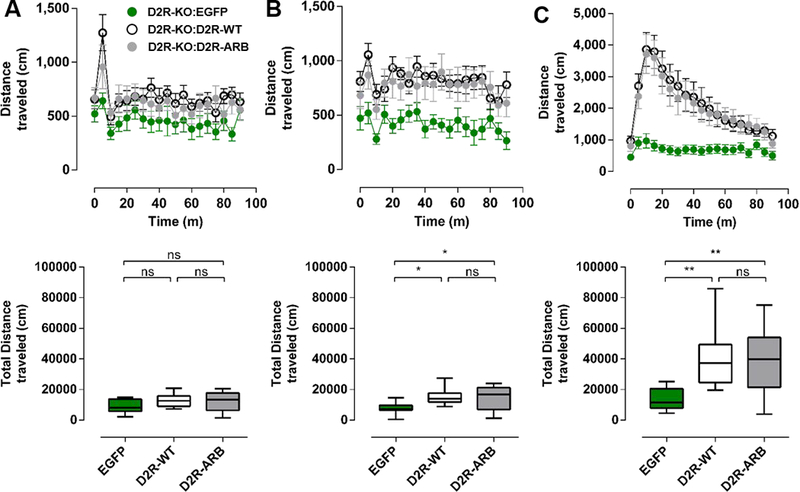
Time course of distance traveled (cm) by Drd2−/−A2A-Cre mice expressing EGFP, D2R-WT, or D2R-ARB 90 minutes following the injection of saline on day 1 (A, top), saline on day 2 (B, top), and cocaine (15 mg/kg) on day 3 (C, top). Total distance traveled by EGFP, D2R-WT, or D2RARB mice in an open field over 90 minutes after the injection of saline on day 1 (A, bottom), saline on day 2 (B, bottom), and cocaine on day 3 (C, bottom).
One-way ANOVA, Bonferroni. *p<0.05, **p<0.01, EGFP (n=11), D2R-WT (n=11), and D2R-ARB (n=12), respectively. Error bars represent S.E.M.
There was no significant difference in locomotor activity between EGFP, D2R-WT and D2RARB-virus-injected mice on day one (one-way ANOVA, F(2,31)=2.11, p=0.14, n=11–12 mice in each group; Figure 3A). However, on day two, locomotor activity was significantly higher in D2R-WT and D2R-ARB virus-injected mice compared to EGFP mice (one-way ANOVA, F(2,31)=5.23, p<0.05, Bonferroni, t=2.97 and 2.61, respectively, p<0.05, n=11–12 mice in each group; Figure 3B). Moreover, there was no significant difference in locomotor activity between D2R-WT and D2R-ARB virus-injected mice (one-way ANOVA, Bonferroni, t=0.43, p>0.05; Figure 3B).
Both D2R-WT and D2R-ARB mice exhibited a robust increase in locomotor activity in response to administration of cocaine that was significantly different from that for EGFP mice (one-way ANOVA, F(2,31)=7.85, p=0.002, Bonferroni, t=3.54 and 3.34, respectively, p<0.05, n=11–12 mice in each group; Figures 3C). Moreover, there was no difference in the total distance traveled between D2R-WT and D2R-ARB mice in response to cocaine (one-way ANOVA, Bonferroni, t=0.28; Figure 3C). These results indicate the restoration of D2R-mediated arrestin recruitment in NAc-iMSNs, in the apparent absence of D2R-mediated G protein signaling, is sufficient to reverse basal hypolocomotion in D2R-KO mice. Moreover, with respect to the role of D2R, arrestin recruitment in iMSNs is sufficient to mediate the full psycholocomotor actions of cocaine.
Upregulated D2R-mediated arrestin recruitment enhances locomotion but not motivation
We recently upregulated D2R-WT in D2R-expressing NAc-iMSNs of D2-Cre mice that express native D2R-WT and observed a robust increase in basal locomotion in an open field assay as well as increased motivation in a progressive ratio task measuring motivation38, 48. Our recent mechanistic analysis suggests that D2R in iMSNs promotes motivation mainly by weakening canonical iMSN output to the ventral pallidum (VP) through their actions at the iMSN terminals48. This effect, by analogy with presynaptic D2-autoreceptor function in dopamine neurons, is most likely to be G protein-mediated69–71. Therefore, we hypothesized that the D2R-mediated enhancement of motivation would be dependent on G protein signaling at iMSN terminals, and thus compared the ability of the D2R-ARB to regulate these D2R-mediated behaviors with that of D2R-WT and the EGFP control when the receptors were upregulated in D2R wild-type mice.
We selectively overexpressed D2R-ARB as well as EGFP or D2R-WT in NAc-iMSNs of D2- Cre mice using the AAVs described above (Figure 4A). Four weeks after injection of the AAVs, we observed EGFP or mVenus expression in the NAc, as well as comparable levels of overexpressed D2R-WT or D2R-ARB (Figure 4B).
Figure 4. Overexpression of an arrestin-biased D2R in the NAc of mice enhances locomotion.
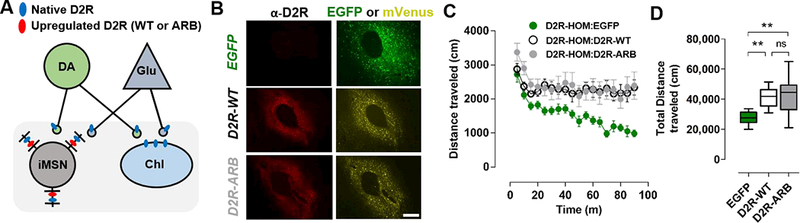
A) Schematic depicting neurons in the striatum that express native D2R (blue ovals). These include iMSNs, cholinergic interneurons (ChIs) as well as dopaminergic (DA) and glutamatergic (GIu) neurons, which express D2Rs at their terminals. D2R-WT or D2R-ARB (red ovals) were upregulated selectively in postsynaptic D2R-expressing iMSNs of D2-Cre mice.
B) AAV-induced expression of EGFP, D2R-WT, or D2-ARB in NAc four weeks following injection. Shown are D2R immunoreactivity (left), EGFP (top right), or mVenus (middle, bottom right) fluorescence. Mean D2R immunofluorescence intensity analysis showed similar D2R upregulation in both D2R groups (D2R-WT: 5.70 ± 0.30 A.U., n=3 mice; D2R-ARB: 5.95 ± 0.77 A.U., n=3 mice; p = 0.77). Scale bar equals 100 μm.
C, D) Distance traveled (cm) over 90 minutes in an open field by D2-Cre mice expressing EGFP, D2R-WT, or D2R-ARB.
One-way ANOVA, Bonferroni. **p<0.01, EGFP (n=10), D2R-WT (n=9), and D2R-ARB (n=8), respectively. Error bars represent S.E.M.
In agreement with our previous work38, basal locomotion was significantly higher in mice overexpressing D2R-WT than in those expressing EGFP (one-way ANOVA, F(2,24) = 9.06, p<0.05, Bonferroni, t=3.47, p<0.05, n=8–10 mice in each group; Figure 4C, 4D & Supplementary Figure 9A). Interestingly, overexpression of D2R-ARB significantly enhanced locomotion relative to the EGFP control (Bonferroni t=3.78, p<0.05) to the same level as D2R-WT overexpression (Figure 4D). Thus, the overexpression of a receptor capable of recruiting arrestins but not G proteins is sufficient to enhance basal locomotion, just as it can rescue basal locomotion in the D2R-KO background (Figure 3).
Using the same mice, we next investigated whether the D2R-ARB acts similarly to D2R-WT in the regulation of motivation. We showed previously that upregulation of D2R-WT in NAc-iMSNs increases motivation but does not alter consummatory behavior48, 49. Using a progressive ratio (PR) lever pressing task, we measured the willingness to expend effort to obtain a food reward by D2-Cre mice expressing EGFP, D2R-WT or D2R-ARB. Consistent with our previous findings, lever pressing (Figure 5A) and breakpoint (Figure 5B) were significantly higher in mice expressing D2R-WT than those expressing the EGFP control (one-way ANOVA, lever pressing: F(2,23)=11.66, p<0.05, Bonferroni, t =4.57, p<0.05; breakpoint: F(2,23)=10.39, p<0.05, Bonferroni, t=4.14, p<0.05). Remarkably, lever pressing and breakpoint were not significantly different between mice expressing EGFP and D2R-ARB (lever pressing: Bonferroni, t=0.63, p>0.05; breakpoint: Bonferroni, t=0.14, p>0.05, Figure 5A & 5B). A survival curve analysis of the percentage of mice that continued to respond as a function of time detected an overall group effect (Log-rank test; χ2=13.33, p<0.005), and pairwise log-rank tests comparing groups indicated that a greater number of D2R-ARB mice lasted significantly less time in the PR session than D2RWT mice but more time than EGFP mice (EGFP vs D2R-WT, χ2=12.54, p<0.0005; D2R-WT vs. D2R-ARB, χ2=7.407, p=0.0065; EGFP vs. D2R-ARB, χ2=2.222, p=0.1361; Figure 5C). Since a single press of the lever during a three-minute time period will extend the PR session, a very small increase in lever presses can extend session time without a significant increase in total lever presses or rewards received. Press rate, was unaltered by expression of either D2R-WT or D2RARB (one-way ANOVA, F(2, 23)=1.42, p=0.26; Figure 5D). Thus, these data indicate that overexpression of a D2R capable of selectively recruiting arrestin enhances locomotor activity, but is not sufficient to increase responding in a task measuring incentive motivation.
Figure 5. Overexpression of an arrestin-biased D2R in the NAc of mice does not enhance motivation.
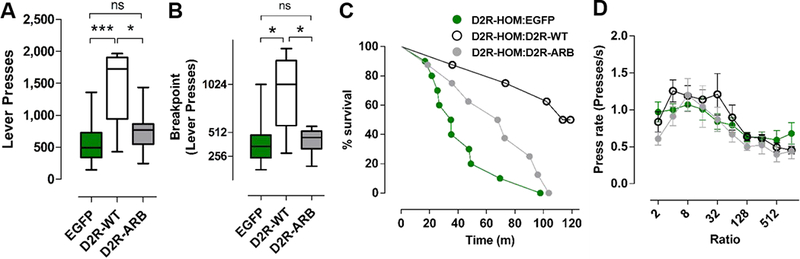
A) Average lever presses in a progressive ratio (PR)-task by D2-Cre mice expressing EGFP, D2R-WT, or D2R-ARB and (B) breakpoint were significantly increased in D2R-WT relative to D2R-ARB or EGFP. One-way ANOVA, Bonferroni. *p<0.05 and ***p<0.001, EGFP (n=10), D2RWT (n=9), and D2R-ARB (n=9), respectively.
C) Effect of overexpressing D2R-WT or D2R-ARB on PR session duration. Kaplan-Meier survival function showing the percentage of mice that continue to respond on the PR schedule as a function of session time. Average data of four PR sessions are shown. Log-rank test showed overall group effect (χ2=13.33, p<0.005), and pairwise log-rank tests comparing between groups indicated that D2R-ARB (n=8) and EGFP (n=10) mice lasted significantly less time in the PR session than D2R-WT (n = 8) mice (EGFP vs D2R-WT, χ2=12.54, p<0.0005; D2R-WT vs. D2RARB, χ2=7.407, p=0.0065; EGFP vs. D2R-ARB, χ2=2.222, p=0.1361).
D) Press rate, plotted here as a function of the ratio requirement, was not altered. One-way ANOVA, p=0.26, EGFP (n=10), D2R-WT (n=9), and D2R-ARB (n=9), respectively. Error bars represent S.E.M.
Discussion
Our understanding of the mechanisms by which D2R regulates diverse physiological processes has been limited by the nature of available genetic35, 50 and pharmacological tools31. While optogenetic and chemogenetic approaches are uncovering details regarding the circuitry underlying distinct behaviors72–75, very little is known about the roles in DA-mediated behaviors of distinct D2R-mediated signaling pathways in specific cell types. Using a novel arrestin-biased D2R mutant (L123W:R132L), termed D2R-ARB, we have shown in global D2R-KO mice that D2R-mediated arrestin recruitment in NAc-iMSNs enhances basal locomotion and supports cocaine-induced hyperlocomotion to the same extent as D2R-WT. These results indicate that arrestin recruitment to D2R in iMSNs can facilitate locomotor activity in the apparent absence of D2R-mediated G protein signaling. The D2R has also been shown to interact with a number of other proteins76, and it is conceivable that the mutations in the D2R-ARB also disrupt other as yet unidentified functions.
The effects of D2R-ARB on locomotion are consistent with previous work showing that a global loss of arrestin-3 attenuates psychostimulant-induced locomotion35. These results, however, appear inconsistent with results showing that biased-D2R ligands, which selectively promote arrestin signaling, inhibit psychostimulant-induced locomotion31. This inconsistency may be due to the partial agonism of these first-generation biased ligands, which can confound the impact of bias by acting as partial antagonists to limit the ability of endogenously released DA to fully activate the arrestin pathway. Recent work with an engineered, partially arrestin-biased D2R showed that arrestin recruitment enhanced basal locomotion to only about half the level produced by D2R-WT52. This stands in contrast to our results and reflects the complexity of engineering biased receptors; indeed, consistent with the published behavioral results, we find that this receptor can only recruit arrestin to about 50% of the level of D2R-WT (Supplementary Figures 1B & C). In contrast, our arrestin-biased mutant, D2R-ARB, mediates full arrestin recruitment (Figure 1D), normal internalization (Figure 1G) and fully phenocopies the basal hyperlocomotor effects of D2R-WT (Figures 4C & D), even in a D2R-KO background (Figure 3). Furthermore, in the global D2R-KO background, cocaine enhanced locomotion to the same extent in mice expressing D2R-WT or D2R-ARB (Figures 3B & D), suggesting that acute D2R-mediated arrestin recruitment is sufficient to facilitate locomotion, whereas D2R-mediated G protein activation does not appear to be necessary. While we cannot rule out a role for some residual G protein activity in our D2R-ARB, any such activity is not detectable in the GTPγS assay (Figure 2D) or the G protein recruitment assays (Figure 1 and Supplementary Figure 4).
Our findings are consistent with an important role for arrestin-mediated signaling downstream of D2R. Most likely this signaling is mediated by arrestin scaffolding of Akt and PP2A, which decreases Akt activity, thereby reducing GSK3β phosphorylation and enhancing GSK3β activity35, 77. Indeed, an increasing body of work implicates GSK3β activation in DA-mediated locomotion77. Moreover, cocaine78- and amphetamine79-induced locomotion is inhibited by the specific GSK3β inhibitor SB 216763 as well as by heterologous knockout of GSK3β79. The target or targets of GSK3β phosphorylation that lead to enhanced locomotion are unknown, but some evidence points to GSK3β-mediated regulation of NMDA and AMPA receptors80, 81, which merits further study as a potential mechanism for how arrestin recruitment can affect locomotor activity.
In contrast to locomotion, incentive motivation was enhanced by the upregulation of D2R-WT but not by D2R-ARB. To our knowledge, this is the first study to dissociate between the mechanisms that underlie D2R-dependent locomotion and incentive motivation. Enhanced PR performance can result from enhanced arousal and an attendant enhancement in the activational component of motivation. In addition, enhanced PR performance can also be due to an enhancement in the directional component of motivation driven by the reward value. Our results suggest different mechanisms for each of these contributions. While the D2R-ARB did not produce a significant increase in lever presses or rewards, consistent with its inability to enhance incentive motivation, D2R-ARB did significantly extend survival in the PR protocol to a level intermediate between control and D2R-WT (Figure 5C). The survival analysis is a measure that is highly sensitive to the activational component of motivation, since a single press of the lever during a three-minute time period extends the PR session. Thus, behavioral activation, through a very small increase in lever presses, can extend session time without a significant increase in total lever presses or rewards received. This demonstrates that the PR protocol captures both the activational and goal-directed aspects of incentive motivation, which we have now disentangled with our pathway-selective receptor construct. Importantly, our results offer the possibility that therapeutic approaches that target distinct D2R-mediated signaling pathways in a cell typespecific manner may avoid adverse effects associated with conventional D2R-targeting medications by selectively engaging only a subset of D2R-mediated behavioral processes.
It is indeed remarkable that the activation of different signaling pathways in the same neurons can result in enhancement of distinct behaviors. Even though the receptors are expressed in the same population of iMSNs, we do not yet know whether a distinct subpopulation of neurons within the iMSN population mediates the different behaviors through different effector mechanisms, or whether activation of the different pathways in the same neurons can produce these divergent behaviors. Using slice electrophysiology paired with optogenetics, we have recently shown that D2R upregulation attenuates inhibitory transmission of iMSN output to the ventral pallidum (VP) 48. In vivo recordings confirmed the reduction in indirect pathway inhibitory transmission to the VP, and localized pharmacogenetic inhibition of iMSN terminals within the VP was sufficient to enhance motivation. These results suggest that D2R in iMSNs promotes incentive motivation mainly by weakening their canonical output to the VP through their actions at the iMSN terminals48.
In dopaminergic neurons, presynaptic D2-autoreceptors are thought to inhibit dopamine release from axon terminals via Gβγ-mediated inhibition of voltage-gated calcium channels and activation of Kv1.2 potassium channels69–71. Likewise, slice physiology studies have shown that pharmacological activation of D2R reduces inhibitory transmission at striatopallidal synapses82–85. These data suggest that D2R-mediated G protein activation is essential to inhibit iMSN terminals in the VP to enhance incentive motivation, in accordance with our findings that D2R-ARB does not enhance motivation. Analogous studies with a G protein-biased D2R mutant will be critical for providing insight into the roles of G protein signaling on incentive motivation and other D2Rdependent behaviors.
Of note, our studies have focused on the role of D2R-mediated signaling in the NAc. We previously have shown that inhibition of indirect pathway function produced by activating the Gi/arrestin coupled DREADD receptor hM4D enhances locomotion both in the dorsal and ventral striatum74. Thus, D2R upregulation in the dorsal striatum is also likely to enhance activity. It is conceivable, however, that the underlying signaling mechanisms may differ due to differences between the role of G protein-dependent and -independent signaling in iMSNs of the dorsal versus the ventral striatum41, 50, 51, and this will require further investigation. The expression of biased D2Rs in D2R in neuronal populations other than the NAc may provide further insight into the role of D2R, not only for the regulation of locomotion and motivation, but for other DAdependent behaviors as well. Such studies may ultimately shed light on the D2R populations and signaling pathways that must be targeted to maximize clinical efficacy and reduce adverse side effects.
Supplementary Material
A published arrestin-biased mutant, D2R(A135R:M140D), recruits arrestin-3 but also activates G proteins.
A) Inhibition of forskolin (30 μM)-induced cAMP accumulation (cAMPi). Dose-response curves are representative of three independent experiments performed in triplicate.
B)Arrestin-3 recruitment to D2R-WT and D2R(A135R:M140D). Dose-response curves are representative of three independent experiments performed in triplicate.
Summary of DA’s (C) Emax and (D) pEC50 in the cAMPi and arrestin assays.
The long isoform of D2R (D2Rl) was used for all experiments described in this figure. Error bars represent S.E.M.
A) Schematic representation of a BRET-based assay that measures agonist-induced recruitment of arrestin-2 fused to mVenus at its N-terminus (mVenus-arrestin-2) to D2R fused to Rluc8 at its C-terminus (D2R-Rluc8).
B) Arrestin-2-recruitment to D2R-WT, D2R(R132L), D2R(L123W), and D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of three independent experiments performed in triplicate.
C) Emax summary of DA-induced recruitment of arrestin-2 to D2R-WT and its mutants. Bars represent the average of three independent experiments.
D)Potency (pEC50) summary of DA-induced recruitment of arrestin-2 to D2R-WT and its mutants. Bars represent the average of three independent experiments
The short isoform of D2R (D2Rs) was used for all experiments. Error bars represent S.E.M.
D2R-WT and D2R(L123W:R132L) tagged with mVenus at their C-termini (yellow) co-expressed with cytosolic tdTomato in HEK293T cells. The short isoform of D2R (D2Rs) was used.
A) Gαi1-recruitment to the long isoforms of D2R-WT and D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of three independent experiments performed in triplicate.
B) Arrestin-3-recruitment to the long isoforms of D2R-WT and D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of four independent experiments performed in triplicate.
C) Emax summary of DA-induced recruitment of Gi1 or arrestin-3 to the long isoform of D2R(L123W:R132L) relative to the wildtype receptor. Bars represent the average of independent experiments for Gi1 (n=3) and arrestin-3 (n=4) recruitment.
D) Potency (pEC50) summary of DA-induced recruitment of arrestin-3 to the long isoforms of D2R-WT and D2R(L123W:R132L). Bars represent the average of four independent experiments. Error bars represent S.E.M.
Inhibition of forskolin (30 μM)-induced cAMP accumulation (cAMPi) in cells overexpressing Gi1. Dose-response curves are representative of three independent experiments performed in triplicate.
A) Schematic representation of a BRET-based assay that measures agonist-induced interaction between the β2-adaptin subunit of AP-2 fused to YFP at its C-terminus (β2-adaptin-YFP) and D2R fused to Rluc8 at its C-terminus (D2R-Rluc8).
B) Interaction between β2-adaptin-YFP and D2R-Rluc8, D2R(R132L), D2R(L123W), or D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of three independent experiments performed in triplicate.
C) Emax summary of DA-induced interaction between β2-adaptin-YFP and mutants of D2R relative to D2R-WT. Bars represent the average of three independent experiments.
D) Potency (pEC50) summary of DA-induced interaction between β2-adaptin-YFP and mutants of D2R relative to D2R-WT. Bars represent the average of three independent experiments. The short isoform of D2R (D2Rs) was used for all experiments. Error bars represent S.E.M.
A) cAMP-accumulation in HEK293T cells expressing D2R-WT, D2R(L123W:R132L) D2R(E339A:T343R) or no receptor (mock) in response to increasing concentrations of the D2R inverse agonist sulpiride. Dose-response curves are representative of two independent experiments performed in triplicate. cAMP-accumulation was measured using a BRET-based cAMP assay44.
B) Emax summary of sulpiride-induced inhibition of constitutive activity of D2R-WT and its mutants. Bars represent the average of two independent experiments.
The short isoform of D2R (D2Rs) was used for all experiments. Error bars represent S.E.M.
A) Inhibition of forskolin (30 μΜ)-induced cAMP accumulation (cAMPi) by dopamine (DA) and quinpirole (QP). Dose-response curves are representative of three independent experiments performed in triplicate.
Summary of DA’s (B) Emax and (C) pEC50 in the cAMPi assay.
The short isoform of D2R (D2Rs) was used for all experiments described in this figure. Error bars represent S.E.M.
A) Representative traces from one mouse per group showing the ambulation (beam crossing) during the last 10 minutes of the open field run. Grey circles indicate the final position of the mouse within the open field.
Center distance (B), periphery distance (C) and rearing in an open field over 90 minutes for EGFP, D2R-WT, or D2R-ARB overexpressing mice.
Acknowledgments
We thank Eric Teboul and Jeremy Sherman for technical assistance, and the Rodent Neurobehavioral Analysis Core at the New York Psychiatric Institute.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Author contributions
P.D., E.G, D.C.B., E.S., Y.Z., and J.R.L. performed the experiments. L.B., K.A.N., C.K. and J.A.J. supervised the project. P.D, E.G., C.K. and J.A.J. wrote the manuscript, with input from J.R.L., L.B. and K.A.N. This work was supported by NIH grants DA044696 to P.D., MH093672 to C.K., DA009158 to L.M.B., MH54137 and DA022413 to J. A. J., MH107648 to E.G, by Merit Review Award BX003279 to K.A.N. from the US Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research, and Development, and by the Lieber Center for Schizophrenia Research.
Competing financial interests
The authors declare no competing financial interests.
Supplementary information is available at MP’s website
References
- 1.Beninger RJ. The role of dopamine in locomotor activity and learning. Brain research 1983; 287(2): 173–196. [DOI] [PubMed] [Google Scholar]
- 2.Wise RA. Dopamine, learning and motivation. Nature reviews Neuroscience 2004; 5(6): 483–494. [DOI] [PubMed] [Google Scholar]
- 3.Brozoski TJ, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science 1979; 205(4409): 929–932. [DOI] [PubMed] [Google Scholar]
- 4.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophrenia bulletin 2009; 35(3): 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 2003; 39(6): 889–909. [DOI] [PubMed] [Google Scholar]
- 6.Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F. Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Archives of neurology 2007; 64(11): 1575–1579. [DOI] [PubMed] [Google Scholar]
- 7.DiMaio S, Grizenko N, Joober R. Dopamine genes and attention-deficit hyperactivity disorder: a review. J Psychiatry Neurosci 2003; 28(1): 27–38. [PMC free article] [PubMed] [Google Scholar]
- 8.Denys D, Zohar J, Westenberg HG. The role of dopamine in obsessive-compulsive disorder: preclinical and clinical evidence. The Journal of clinical psychiatry 2004; 65 Suppl 14: 11–17. [PubMed] [Google Scholar]
- 9.Buse J, Schoenefeld K, Munchau A, Roessner V. Neuromodulation in Tourette syndrome: dopamine and beyond. Neuroscience and biobehavioral reviews 2013; 37(6): 1069–1084. [DOI] [PubMed] [Google Scholar]
- 10.Seeman P Dopamine D2 receptors as treatment targets in schizophrenia. Clin Schizophr Relat Psychoses 2010; 4(1): 56–73. [PubMed] [Google Scholar]
- 11.Goodman LS, Gilman A, Brunton LL. Goodman & Gilman’s manual of pharmacology and therapeutics. McGraw-Hill Medical: New York, 2008, ix, 1219 p.pp. [Google Scholar]
- 12.Brooks DJ. Dopamine agonists: their role in the treatment of Parkinson’s disease. J Neurol Neurosurg Psychiatry 2000; 68(6): 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nature neuroscience 2010; 13(5): 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012; 76(1): 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological reviews 2011; 63(1): 182–217. [DOI] [PubMed] [Google Scholar]
- 16.Centonze D, Grande C, Usiello A, Gubellini P, Erbs E, Martin AB et al. Receptor subtypes involved in the presynaptic and postsynaptic actions of dopamine on striatal interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2003; 23(15): 6245–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ford CP. The role of D2-autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 2014; 282: 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Pickel VM. Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. The Journal of comparative neurology 2002; 442(4): 392–404. [DOI] [PubMed] [Google Scholar]
- 19.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends in neurosciences 2007; 30(5): 228–235. [DOI] [PubMed] [Google Scholar]
- 20.Burke DA, Rotstein HG, Alvarez VA. Striatal Local Circuitry: A New Framework for Lateral Inhibition. Neuron 2017; 96(2): 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP- 32/protein phosphatase-1 cascade. Neuron 1999; 23(3): 435–447. [DOI] [PubMed] [Google Scholar]
- 22.Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proceedings of the National Academy of Sciences of the United States of America 1992; 89(21): 10178–10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Surmeier DJ, Bargas J, Hemmings HC, Jr., Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 1995; 14(2): 385–397. [DOI] [PubMed] [Google Scholar]
- 24.Cepeda C, Chandler SH, Shumate LW, Levine MS. Persistent Na+ conductance in medium-sized neostriatal neurons: characterization using infrared videomicroscopy and whole cell patch-clamp recordings. J Neurophysiol 1995; 74(3): 1343–1348. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E. D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. The Journal of neuroscience : the official journal of the Society for Neuroscience 1997; 17(9): 3334–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiffmann SN, Lledo PM, Vincent JD. Dopamine D1 receptor modulates the voltagegated sodium current in rat striatal neurones through a protein kinase A. The Journal of physiology 1995; 483 ( Pt 1): 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersson M, Konradi C, Cenci MA. cAMP response element-binding protein is required for dopamine-dependent gene expression in the intact but not the dopamine-denervated striatum. The Journal of neuroscience : the official journal of the Society for Neuroscience 2001; 21(24): 9930–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL et al. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Molecular pharmacology 2014; 86(1): 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG. Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. The Journal of biological chemistry 2001; 276(40): 37409–37414. [DOI] [PubMed] [Google Scholar]
- 30.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol 2007; 69: 483–510. [DOI] [PubMed] [Google Scholar]
- 31.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M et al. Discovery of betaarrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proceedings of the National Academy of Sciences of the United States of America 2011; 108(45): 18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, McCorvy JD, Fischer MG, Butler KV, Shen Y, Roth BL et al. Discovery of G Protein-Biased D2 Dopamine Receptor Partial Agonists. Journal of medicinal chemistry 2016; 59(23): 10601–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Sassano MF, Zheng L, Setola V, Chen M, Bai X et al. Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D(2) receptor agonists. Journal of medicinal chemistry 2012; 55(16): 7141–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conroy JL, Free RB, Sibley DR. Identification of G protein-biased agonists that fail to recruit beta-arrestin or promote internalization of the D1 dopamine receptor. ACS chemical neuroscience 2015; 6(4): 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 2005; 122(2): 261–273. [DOI] [PubMed] [Google Scholar]
- 36.Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ et al. Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci U S A 2010; 107(33): 14845–14850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gallo EF, Meszaros J, Sherman JD, Chohan MO, Teboul E, Choi CS et al. Accumbens dopamine D2 receptors increase motivation by decreasing inhibitory transmission to the ventral pallidum. Nature communications 2018; 9(1): 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallo EF, Salling MC, Feng B, Moron JA, Harrison NL, Javitch JA et al. Upregulation of Dopamine D2 Receptors in the Nucleus Accumbens Indirect Pathway Increases Locomotion but does not Reduce Alcohol Consumption. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang S, Che T, Levit A, Shoichet BK, Wacker D, Roth BL. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018; 555(7695): 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo W, Shi L, Javitch JA. The fourth transmembrane segment forms the interface of the dopamine D2 receptor homodimer. The Journal of biological chemistry 2003; 278(7): 4385–4388. [DOI] [PubMed] [Google Scholar]
- 41.Clayton CC, Donthamsetti P, Lambert NA, Javitch JA, Neve KA. Mutation of three residues in the third intracellular loop of the dopamine D2 receptor creates an internalization-defective receptor. J Biol Chem 2014; 289(48): 33663–33675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donthamsetti P, Quejada JR, Javitch JA, Gurevich VV, Lambert NA. Using Bioluminescence Resonance Energy Transfer (BRET) to Characterize Agonist-Induced Arrestin Recruitment to Modified and Unmodified G Protein-Coupled Receptors. Curr Protoc Pharmacol 2015; 70: 2 14 11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamdan FF, Rochdi MD, Breton B, Fessart D, Michaud DE, Charest PG et al. Unraveling G protein-coupled receptor endocytosis pathways using real-time monitoring of agonist-promoted interaction between beta-arrestins and AP-2. JBiolChem 2007; 282(40): 29089–29100. [DOI] [PubMed] [Google Scholar]
- 44.Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. The Journal of biological chemistry 2007; 282(14): 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neve KA, Ford CP, Buck DC, Grandy DK, Neve RL, Phillips TJ. Normalizing dopamine D2 receptor-mediated responses in D2 null mutant mice by virus-mediated receptor restoration: comparing D2L and D2S. Neuroscience 2013; 248: 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bohn LM, Zhou L, Ho JH. Approaches to Assess Functional Selectivity in GPCRs: Evaluating G Protein Signaling in an Endogenous Environment. Methods in molecular biology 2015; 1335: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016; 167(3): 750–762 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallo EF, Meszaros J, Sherman JD, Chohan MO, Teboul E, Choi CS, Moore H, Javitch JA, Kellendonk C . Dopamine D2 receptors on ventral striatal projection neurons increase motivation by decreasing inhibitory transmission to the ventral pallidum. Nature communications In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trifilieff P, Feng B, Urizar E, Winiger V, Ward RD, Taylor KM et al. Increasing dopamine D2 receptor expression in the adult nucleus accumbens enhances motivation. Molecular psychiatry 2013; 18(9): 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peterson SM, Pack TF, Wilkins AD, Urs NM, Urban DJ, Bass CE et al. Elucidation of Gprotein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proceedings of the National Academy of Sciences of the United States of America 2015; 112(22): 7097–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lan H, Liu Y, Bell MI, Gurevich VV, Neve KA. A dopamine D2 receptor mutant capable of G protein-mediated signaling but deficient in arrestin binding. Molecular pharmacology 2009; 75(1): 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rose SJ, Pack TF, Peterson SM, Payne K, Borrelli E, Caron MG. Engineered D2R Variants Reveal the Balanced and Biased Contributions of G-Protein and beta-Arrestin to Dopamine-Dependent Functions. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakajima K, Wess J. Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Molecular pharmacology 2012; 82(4): 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ballesteros A, Weinstein H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci 1995; 25: 366–428. [Google Scholar]
- 55.Rovati GE, Capra V, Neubig RR. The highly conserved DRY motif of class A G proteincoupled receptors: beyond the ground state. Molecular pharmacology 2007; 71(4): 959–964. [DOI] [PubMed] [Google Scholar]
- 56.Gales C, Rebois RV, Hogue M, Trieu P, Breit A, Hebert TE et al. Real-time monitoring of receptor and G-protein interactions in living cells. Nature methods 2005; 2(3): 177–184. [DOI] [PubMed] [Google Scholar]
- 57.Roth CB, Hanson MA, Stevens RC. Stabilization of the human beta2-adrenergic receptor TM4-TM3-TM5 helix interface by mutagenesis of Glu122(3.41), a critical residue in GPCR structure. Journal of molecular biology 2008; 376(5): 1305–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010; 330(6007): 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gazi L, Nickolls SA, Strange PG. Functional coupling of the human dopamine D2 receptor with G alpha i1, G alpha i2, G alpha i3 and G alpha o G proteins: evidence for agonist regulation of G protein selectivity. British journal of pharmacology 2003; 138(5): 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lane JR, Powney B, Wise A, Rees S, Milligan G. G protein coupling and ligand selectivity of the D2L and D3 dopamine receptors. The Journal of pharmacology and experimental therapeutics 2008; 325(1): 319–330. [DOI] [PubMed] [Google Scholar]
- 61.Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L. Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proceedings of the National Academy of Sciences of the United States of America 2001; 98(6): 3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marcott PF, Gong S, Donthamsetti P, Grinnell SG, Nelson MN, Newman AH et al. Regional Heterogeneity of D2-Receptor Signaling in the Dorsal Striatum and Nucleus Accumbens. Neuron 2018; 98(3): 575–587 e574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberts DJ, Lin H, Strange PG. Investigation of the mechanism of agonist and inverse agonist action at D2 dopamine receptors. Biochemical pharmacology 2004; 67(9): 1657– 1665. [DOI] [PubMed] [Google Scholar]
- 64.Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 2009; 5(9): 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klewe IV, Nielsen SM, Tarpo L, Urizar E, Dipace C, Javitch JA et al. Recruitment of beta-arrestin2 to the dopamine D2 receptor: insights into anti-psychotic and antiparkinsonian drug receptor signaling. Neuropharmacology 2008; 54(8): 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Griffin G, Atkinson PJ, Showalter VM, Martin BR, Abood ME. Evaluation of cannabinoid receptor agonists and antagonists using the guanosine-5’-O-(3-[35S]thio)-triphosphate binding assay in rat cerebellar membranes. The Journal of pharmacology and experimental therapeutics 1998; 285(2): 553–560. [PubMed] [Google Scholar]
- 67.Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G et al. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. The Journal of neuroscience : the official journal of the Society for Neuroscience 1998; 18(9): 3470–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chausmer AL, Elmer GI, Rubinstein M, Low MJ, Grandy DK, Katz JL. Cocaine-induced locomotor activity and cocaine discrimination in dopamine D2 receptor mutant mice. Psychopharmacology 2002; 163(1): 54–61. [DOI] [PubMed] [Google Scholar]
- 69.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 1996; 380(6571): 258–262. [DOI] [PubMed] [Google Scholar]
- 70.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 1996; 380(6571): 255–258. [DOI] [PubMed] [Google Scholar]
- 71.Martel P, Leo D, Fulton S, Berard M, Trudeau LE. Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PLoS One 2011; 6(5): e20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 2010; 466(7306): 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cazorla M, de Carvalho FD, Chohan MO, Shegda M, Chuhma N, Rayport S et al. Dopamine D2 receptors regulate the anatomical and functional balance of basal ganglia circuitry. Neuron 2014; 81(1): 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carvalho Poyraz F, Holzner E, Bailey MR, Meszaros J, Kenney L, Kheirbek MA et al. Decreasing Striatopallidal Pathway Function Enhances Motivation by Energizing the Initiation of Goal-Directed Action. The Journal of neuroscience : the official journal of the Society for Neuroscience 2016; 36(22): 5988–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ferguson SM, Neumaier JF. Grateful DREADDs: engineered receptors reveal how neural circuits regulate behavior. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2012; 37(1): 296–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bergson C, Levenson R, Goldman-Rakic PS, Lidow MS. Dopamine receptor-interacting proteins: the Ca(2+) connection in dopamine signaling. Trends in pharmacological sciences 2003; 24(9): 486–492. [DOI] [PubMed] [Google Scholar]
- 77.Beaulieu JM, Tirotta E, Sotnikova TD, Masri B, Salahpour A, Gainetdinov RR et al. Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience 2007; 27(4): 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miller JS, Tallarida RJ, Unterwald EM. Cocaine-induced hyperactivity and sensitization are dependent on GSK3. Neuropharmacology 2009; 56(8): 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proceedings of the National Academy of Sciences of the United States of America 2004; 101(14): 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li YC, Xi D, Roman J, Huang YQ, Gao WJ. Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 2009; 29(49): 15551–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li YC, Gao WJ. GSK-3beta activity and hyperdopamine-dependent behaviors. Neuroscience and biobehavioral reviews 2011; 35(3): 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cooper AJ, Stanford IM. Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology 2001; 41(1): 62–71. [DOI] [PubMed] [Google Scholar]
- 83.Tecuapetla F, Koos T, Tepper JM, Kabbani N, Yeckel MF. Differential dopaminergic modulation of neostriatal synaptic connections of striatopallidal axon collaterals. The Journal of neuroscience : the official journal of the Society for Neuroscience 2009; 29(28): 8977–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Floran B, Floran L, Sierra A, Aceves J. D2 receptor-mediated inhibition of GABA release by endogenous dopamine in the rat globus pallidus. Neurosci Lett 1997; 237(1): 1–4. [DOI] [PubMed] [Google Scholar]
- 85.Kohnomi S, Koshikawa N, Kobayashi M. D(2)-like dopamine receptors differentially regulate unitary IPSCs depending on presynaptic GABAergic neuron subtypes in rat nucleus accumbens shell. Journal of neurophysiology 2012; 107(2): 692–703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A published arrestin-biased mutant, D2R(A135R:M140D), recruits arrestin-3 but also activates G proteins.
A) Inhibition of forskolin (30 μM)-induced cAMP accumulation (cAMPi). Dose-response curves are representative of three independent experiments performed in triplicate.
B)Arrestin-3 recruitment to D2R-WT and D2R(A135R:M140D). Dose-response curves are representative of three independent experiments performed in triplicate.
Summary of DA’s (C) Emax and (D) pEC50 in the cAMPi and arrestin assays.
The long isoform of D2R (D2Rl) was used for all experiments described in this figure. Error bars represent S.E.M.
A) Schematic representation of a BRET-based assay that measures agonist-induced recruitment of arrestin-2 fused to mVenus at its N-terminus (mVenus-arrestin-2) to D2R fused to Rluc8 at its C-terminus (D2R-Rluc8).
B) Arrestin-2-recruitment to D2R-WT, D2R(R132L), D2R(L123W), and D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of three independent experiments performed in triplicate.
C) Emax summary of DA-induced recruitment of arrestin-2 to D2R-WT and its mutants. Bars represent the average of three independent experiments.
D)Potency (pEC50) summary of DA-induced recruitment of arrestin-2 to D2R-WT and its mutants. Bars represent the average of three independent experiments
The short isoform of D2R (D2Rs) was used for all experiments. Error bars represent S.E.M.
D2R-WT and D2R(L123W:R132L) tagged with mVenus at their C-termini (yellow) co-expressed with cytosolic tdTomato in HEK293T cells. The short isoform of D2R (D2Rs) was used.
A) Gαi1-recruitment to the long isoforms of D2R-WT and D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of three independent experiments performed in triplicate.
B) Arrestin-3-recruitment to the long isoforms of D2R-WT and D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of four independent experiments performed in triplicate.
C) Emax summary of DA-induced recruitment of Gi1 or arrestin-3 to the long isoform of D2R(L123W:R132L) relative to the wildtype receptor. Bars represent the average of independent experiments for Gi1 (n=3) and arrestin-3 (n=4) recruitment.
D) Potency (pEC50) summary of DA-induced recruitment of arrestin-3 to the long isoforms of D2R-WT and D2R(L123W:R132L). Bars represent the average of four independent experiments. Error bars represent S.E.M.
Inhibition of forskolin (30 μM)-induced cAMP accumulation (cAMPi) in cells overexpressing Gi1. Dose-response curves are representative of three independent experiments performed in triplicate.
A) Schematic representation of a BRET-based assay that measures agonist-induced interaction between the β2-adaptin subunit of AP-2 fused to YFP at its C-terminus (β2-adaptin-YFP) and D2R fused to Rluc8 at its C-terminus (D2R-Rluc8).
B) Interaction between β2-adaptin-YFP and D2R-Rluc8, D2R(R132L), D2R(L123W), or D2R(L123W:R132L) in response to increasing concentrations of DA. Dose-response curves are representative of three independent experiments performed in triplicate.
C) Emax summary of DA-induced interaction between β2-adaptin-YFP and mutants of D2R relative to D2R-WT. Bars represent the average of three independent experiments.
D) Potency (pEC50) summary of DA-induced interaction between β2-adaptin-YFP and mutants of D2R relative to D2R-WT. Bars represent the average of three independent experiments. The short isoform of D2R (D2Rs) was used for all experiments. Error bars represent S.E.M.
A) cAMP-accumulation in HEK293T cells expressing D2R-WT, D2R(L123W:R132L) D2R(E339A:T343R) or no receptor (mock) in response to increasing concentrations of the D2R inverse agonist sulpiride. Dose-response curves are representative of two independent experiments performed in triplicate. cAMP-accumulation was measured using a BRET-based cAMP assay44.
B) Emax summary of sulpiride-induced inhibition of constitutive activity of D2R-WT and its mutants. Bars represent the average of two independent experiments.
The short isoform of D2R (D2Rs) was used for all experiments. Error bars represent S.E.M.
A) Inhibition of forskolin (30 μΜ)-induced cAMP accumulation (cAMPi) by dopamine (DA) and quinpirole (QP). Dose-response curves are representative of three independent experiments performed in triplicate.
Summary of DA’s (B) Emax and (C) pEC50 in the cAMPi assay.
The short isoform of D2R (D2Rs) was used for all experiments described in this figure. Error bars represent S.E.M.
A) Representative traces from one mouse per group showing the ambulation (beam crossing) during the last 10 minutes of the open field run. Grey circles indicate the final position of the mouse within the open field.
Center distance (B), periphery distance (C) and rearing in an open field over 90 minutes for EGFP, D2R-WT, or D2R-ARB overexpressing mice.