

Abstract
A diagnosis of hereditary angioedema is usually made with recurrent episodes of swelling of the subcutaneous tissue with a family history. We herein report a case in which recurrent acute abdomen was the main manifestation of hereditary angioedema. A 45-year-old womon presented with a 10-year history of recurrent severe abdominal pain. Abdominal computed tomography revealed remarkable submucosal edema of the ileum. A blood examination revealed grossly reduced complement C4 and CH50 with deficiency of C1-inhibitor. Genetic testing revealed a heterozygous nonsense mutation of the SERPING1 gene, and a diagnosis of hereditary angioedema was made. Hereditary angioedema should be listed as a differential diagnosis of recurrent acute abdomen.
Keywords: hereditary angioedema, submucosal edema, C1-inhibitor, hypocomplementemia, acute abdomen
Introduction
Hereditary angioedema (HAE) is a rare genetic disorder characterized by recurrent episodes of angioedema. The prevalence is estimated at 1 in 50,000 approximately, with no marked differences by ethnic group or gender (1). Without effective therapies, the mortality rate is reported to be as high as 30% due to asphyxiation from laryngeal swelling (2).
HAE should be considered in patients who report recurrent episodes of subcutaneous or submucosal angioedema. Angioedema attacks mostly affect three anatomical locations: the skin, the upper airway and the gastrointestinal tract. Angioedema can be relatively easily noted if attacks involve the body surface but can be overlooked if the main attacks involve only the gastrointestinal tract.
We herein report a 45-year-old woman who presented with a 10-year history of recurrent severe abdominal pain.
Case Report
A 45-year-old woman presented with a 10-year history of recurrent severe abdominal pain. She developed hand swelling at nine years of age that resolved shortly. Thereafter, she came to develop similar hand or leg swelling occasionally.
She visited a core hospital in her prefecture, but no specific diagnosis was made. Her mother had died of asphyxiation from the larynx following upper endoscopy when the patient had been a child. In junior and senior high school, the frequency of swelling of the extremities had been extremely low, so patient had not cared about it. After her first childbirth, however, she started to develop colicky abdominal pain instead of hand or leg swelling. The abdominal pain occasionally became so severe that she had to request an ambulance. She had been admitted to hospitals on several occasions and had even visited a university hospital, but the cause of her abdominal pain had not been clarified. She underwent panhysterectomy at 42 years of age, since endometriosis was suspected as the cause of acute abdomen; however, she continued to experience abdominal pain afterwards. Once she began to take estrogen for menopausal troubles, the abdominal pain became more frequent than ever. She developed lower abdominal pain with constipation one week prior to admission that worsened day by day, until she ultimately had to be transported to our hospital by an ambulance.
On a physical examination, she appeared obviously ill, and her abdominal pain was 10/10 on the pain scale. Her blood pressure was 133/81 mmHg, pulse 79/min, respiratory rate of 14/min with an O2 saturation of 95% on ambient air and temperature of 36.9℃. Her abdomen was slightly distended with diffuse tenderness. There was no evidence of edema under the skin.
Blood examinations revealed grossly reduced complement C4 (3 mg/dL) and CH50 (6 U/mL) without leukocytosis or elevated inflammatory markers. Antinuclear antibodies and autoantibodies of connective tissue diseases were all negative. Computed tomography (CT) of the abdomen was performed, revealing localized intestinal submucosal edema with perihepatic ascites (Fig. 1). Due to concerns of HAE, an additional blood examination was performed, revealing C1-inhibitor deficiency (Table). Genetic testing revealed a heterozygous nonsense mutation in the SERPING1 gene (NCBI reference sequence: NM_000062.2:c.265C>T). This mutation changed the codon CAA (glutamine) to TAA (stop) at protein 89 of the plasma protein C1 inhibitor precursor (NCBI reference sequence: NP_000053.2:p.Gln89X) (Fig. 2). Accordingly, she was diagnosed with type 1 HAE. She stopped taking estrogen, and her recurrent abdominal was controlled with tranexamic acid and on-demand therapy of plasma-derived C1-inhibitor concentrate.
Figure 1.
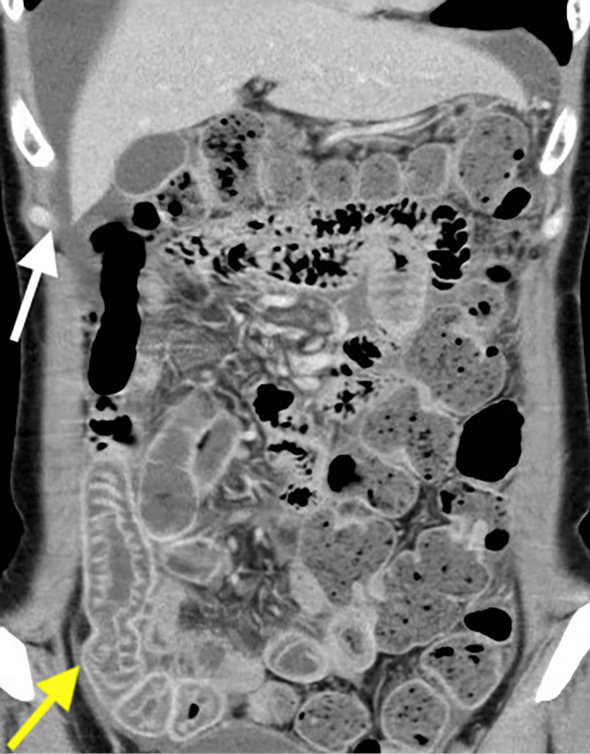
Abdominal CT findings on admission. The yellow arrow indicates submucous edema of the ileum. The white arrow indicates ascites around the liver.
Table.
Blood Examination Findings.
| Complete blood count | Biochemistry | |||||
| RBC | 394×104 | /μL | TP | 6.4 | g/dL | |
| Hb | 11.8 | g/dL | Alb | 3.7 | g/dL | |
| Ht | 35.3 | % | AST | 27 | U/L | |
| WBC | 4,100 | /μL | ALT | 19 | U/L | |
| Neutro | 47 | % | LDH | 169 | U/L | |
| Eosino | 3 | % | γ-GTP | 11 | U/L | |
| Mono | 6 | % | T-bil | 0.24 | mg/dL | |
| Lympho | 44 | % | BUN | 12.9 | mg/dL | |
| Plt | 23.7×104 | /μL | Cr | 0.5 | mg/dL | |
| ESR | 17 | mm/h | Na | 141 | mEq/L | |
| Immunology | K | 4.2 | mEq/L | |||
| ANA | <40 | × | Cl | 104 | mEq/L | |
| Anti-DNA Ab | <1.7 | IU/mL | CRP | 0.26 | mg/dL | |
| Anti-SS-A Ab | <0.5 | index | Thyroid function | |||
| C3 | 108 | mg/dL | TSH | 0.631 | μIU/mL | |
| C4 | 3 | mg/dL | fT4 | 1.08 | ng/mL | |
| CH50 | 6 | U/mL | Infection | |||
| C1-INH | <25 | % | T-spot | (-) | ||
RBC: red blood cell, Hb: hemoglobin, Ht: hematocrit, WBC: white blood cell, Neutro: neutrophil, Eosino: eosinophil, Mono: monocyte, Lympho: lymphocyte, Plt: platelet, ESR: erythrocyte sedimentation rate, ANA: anti-nuclear antibody, CH50: 50% hemolytic unit of complement, C1-INH: C1-inhibitor, TP: total protein, Alb: albumin, AST: aspartate aminotransferase, ALT: alanine transaminase, LDH: lactate dehydrogenase, γ-GTP: γ-glutamyl transpeptidase, T-bil: total bilirubin, BUN: blood urea nitrogen, Cr: creatinine, CRP: C-reactive protein, TSH: thyroid-stimulating hormone
Figure 2.

Complementary DNA sequence of the SERPING1 gene. The red arrow indicates a cytosine (C) to thymine (T) substitution at nucleotide 265 of the coding sequence of the SERPING1 gene (NCBI reference sequence: NM_000062.2: c.265C>T), which changed glutamine (Gln) to a stop codon (X) (NCBI reference sequence: NP_53.2: p.Gln89X).
Discussion
The present case highlighted two critical clinical issues. HAE can be overlooked if the main attacks involve only the gastrointestinal tract, and overlooking HAE can increase the risk of mortality in the patient as well as in any blood relatives.
HAE is a rare genetic disorder caused by deficiency in a functional C1-inhibitor. Patients with HAE experience their first attack in childhood. The frequency of attacks increases around puberty, and a diagnosis is usually made in adolescence or postadolescence. The skin, upper respiratory tract and gastrointestinal tract are involved in 98% of attacks, which usually affect one organ at a time (3). Although attacks involving abdominal pain are a common manifestation of HAE, they are rarely the main manifestation (4). In such cases, the diagnosis of HAE can be challenging. Patients with HAE usually have normal routine laboratory values. CT of the abdomen can be useful for detecting gastrointestinal angioedema, such as in our case, and ultrasound may also be useful in its diagnosis (5), but imaging studies themselves are not specific enough for the diagnosis. If clinical histories or imaging studies are suggestive of HAE, complement screening should be performed. In patients with HAE, screening reveals normal C3, low C4, and low CH50 levels. However, confirmation of HAE requires a decreased C1-INH function. HAE is mainly categorized into two subtypes: Type 1 HAE, due to C1-INH deficiency (85% of cases) and Type II HAE, due to C1-INH dysfunction (15% of cases). The diagnosis of HAE does not always require genetic testing. However, genetic testing can be useful for confirming the diagnosis, especially for solitary cases, which account for up to 25% of HAE cases, and for understanding the pathogenesis. To date, more than 250 different mutations have been reported. Although a C1-INH gene mutation database (the HAEdb) (http://hae.enzim.hu/) has been established, the C1-INH gene mutation in our case is not listed (NCBI reference sequence: NM_000062.2:c.265C>T) (6). A database of the mutations in the Japanese population will be necessary for better understanding HAE.
The differential diagnoses for HAE include systemic lupus erythematosus (SLE), inflammatory bowel diseases (IBD), familial Mediterranean fever (FMF), infections and mesenteric ischemia. Enteritis, caused by SLE or lupus enteritis, is one of the most important differential diagnoses. In lupus enteritis, CT of the abdomen typically indicates submucosal edema of the intestine, and laboratory tests show hypocomplementemia (7). Both features are common to abdominal attacks of HAE. In fact, our case was referred to the Department of Rheumatology with suspected lupus enteritis due to characteristic CT findings and hypocomplementemia. The difference between SLE and HAE is in the levels of autoantibodies and complement profiles. In SLE, nearly 100% of patients have antinuclear antibodies (ANA), and the C3 levels are frequently low in the active phase, whereas in HAE, ANA is usually negative, and the C3 levels are within the normal range, even during attacks. In IBD or several infections, such as Salmonella and Shigella, CT of the abdomen shows bowel-wall thickening, with inflammatory markers on occasion. However, in HAE, hypocomplementemia is common, and inflammatory markers are usually negative. Patients with FMF frequently have recurrent abdominal pain with a periodic fever, whereas patients with HAE are typically afebrile. Mesenteric ischemia due to ischemic colitis, arterial thrombus/embolus or a closed loop obstruction can cause the sudden onset of abdominal pain and bowel edema. In these cases, the inflammatory marker levels are usually elevated, and the serum lactate or lactate dehydrogenase (LDH) levels increase as tissue injury progresses, which is unusual for HAE. In the differential diagnoses described above, the C1-inhibitor level is always within the normal range. Thus, when a patient presents with abdominal pain but normal C3 levels with low C4 and CH50 levels, the C1-inhibitor levels should be examined, as this can distinguish HAE from other diseases.
In the present case, the cause of recurrent abdominal pain was initially considered to be endometriosis. Thus, the patient underwent panhysterectomy and took estrogen-containing medications as hormone-replacement therapy for menopause-associated problems. Retrospectively, these treatments are considered to have endangered the patient. HAE attacks are triggered by physical and mental stress. Physical stress includes trauma, substantial dental work, endoscopy and endotracheal intubation. In particular, medical procedures in the upper airway can lead to asphyxiation. The World Allergy Organization (WAO) guidelines recommend the administration of C1-INH concentrate 1 to 6 hours before surgery (8). Our patient underwent panhysterectomy with endotracheal intubation without the administration of prophylactic C1-INH, which was associated with a risk of asphyxiation. Some medications- especially estrogen-containing medications and angiotensin-converting enzyme (ACE) inhibitors-are known to trigger HAE attacks and exacerbate their frequency and severity. After panhysterectomy, our patient took estrogen-containing medications for menopausal troubles, which indeed exacerbated the severity and frequency of her HAE attacks.
Undiagnosed HAE also endangers the lives of the patients' blood relatives. Our patient's mother died from asphyxiation from the larynx following upper endoscopy before our patient was diagnosed with HAE. Recently, one of her sons reported having occasional abdominal pain. The prophylactic administration of C1-INH concentrate before dental procedures, endoscopy and endotracheal intubation is advised if C1-INH deficiency is diagnosed.
In conclusion, HAE can present as acute abdomen and should therefore be listed as a differential diagnosis of acute abdomen, even if skin angioedema is not recognized. If an HAE diagnosis is confirmed in a patient, the physician should advise any blood relatives to undergo complement screening and C1-INH testing. This may help prevent unforeseen deaths among patients and their family members.
This case report was approved by the Ethics Committee of Tokyo Bay Urayasu/Ichikawa Medical Center. The Ethics Committee of Tokyo Medical and Dental University approved this genetic analysis.
Written informed consent was obtained from the patient for the genetic analysis and publication of this case report and accompanying images.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 359: 1027-1036, 2008. [DOI] [PubMed] [Google Scholar]
- 2. Frank MM, Gelfand JA, Atkinson JP. Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med 84: 580-593, 1976. [DOI] [PubMed] [Google Scholar]
- 3. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 119: 267-274, 2006. [DOI] [PubMed] [Google Scholar]
- 4. Bork K, Staubach P, Eckardt AJ, Hardt J. Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol 101: 619-627, 2006. [DOI] [PubMed] [Google Scholar]
- 5. Sheffer AL, Craig JM, Willms-Kretschmer K, Austen KF, Rosen FS. Histopathological and ultrastructural observations on tissues from patients with hereditary angioneurotic edema. J Allergy 47: 292-297, 1971. [DOI] [PubMed] [Google Scholar]
- 6. Kalmár L, Hegedüs T, Farkas H, Nagy M, Tordai A. HAEdb: a novel interactive, locus-specific mutation database for the C1 inhibitor gene. Hum Mutat 25: 1-5, 2005. [DOI] [PubMed] [Google Scholar]
- 7. Huang D-F, Chen W-S. Images in clinical medicine. Lupus-associated intestinal vasculitis. N Engl J Med 361: e3, 2009. [DOI] [PubMed] [Google Scholar]
- 8. Craig T, Aygören-Pürsün E, Bork K, et al. WAO Guideline for the Management of Hereditary Angioedema. World Allergy Organ J 5: 182-199, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]