

Abstract
The global physiological function of specifically expressed genes of mitoxantrone (MTX)‐resistant prostate cancer (PCa) is unclear. In this study, gene expression pattern from microarray data was investigated for identifying differentially expressed genes (DEGs) in MTX‐resistant PCa xenografts. Human PCa cell lines DU145 and PC3 were cultured in vitro and xenografted into severe combined immunodeficiency (SCID) mice, treated with MTX intragastrically, three times a week until all mice relapsed. Gene expression profiles of the xenografts from castrated mice were performed with Affymetrix human whole genomic oligonucleotide microarray. The Cytoscape software was used to investigate the relationship between proteins and the signalling transduction network. A total of 355 overlapping genes were differentially expressed in MTX‐resistant DU145R and PC3R xenografts. Of these, 16 genes were selected to be validated by quantitative real‐time PCR (qRT‐PCR) in these xenografts, and further tested in a set of formalin‐fixed, paraffin‐embedded and optimal cutting temperature (OCT) clinical tumour samples. Functional and pathway enrichment analyses revealed that these DEGs were closely related to cellular activity, androgen synthesis, DNA damage and repair, also involved in the ERK/MAPK, PI3K/serine‐threonine protein kinase, also known as protein kinase B, PKB (AKT) and apoptosis signalling pathways. This exploratory analysis provides information about potential candidate genes and may bring new insights into the molecular cascade involvement in MTX‐resistant PCa.
Keywords: Castration‐resistant prostate cancer, differentially expressed genes, drug‐resistance, microarray, mitoxantrone
1. INTRODUCTION
Prostate cancer (PCa) is the most common cancer in men in western countries.1 The majority of patients with advanced PCa have disease that is initially sensitive to androgen deprivation therapy (ADT), which successfully reduces tumour burden, improves symptoms and can delay disease progression for several years,2, 3 although, responses are generally not durable and disease progression is inevitable. Mitoxantrone (MTX), a synthetic anthracenedione, has been routinely used for the treatment PCa for its palliative benefit which enhances clinical remission of the PCa patients. However, despite their initial response and survival benefits, the majority of patients eventually develop resistance to these therapies.4
The antineoplastic activity of MTX is believed to be related to its ability to bind DNA and inhibit DNA topoisomerase II, an essential enzyme in DNA synthesis and meiotic division which is highly expressed in cancer cells.5 Damage to DNA is a notable inducer of both transient and permanent alterations in cellular phenotypes. The accumulation of DNA lesions leads to genomic instability through chromosomes breaks, amplification of oncogenes and inactivation of tumour suppression genes, driving to the acquisition of a malignant cancer phenotype.6 However, cancer cells can overcome DNA damage by induction of a DNA damage secretory program such as proliferation, invasion, metastasis, especially treatment resistance can develop through a variety of signal pathways, including base excision repair, nucleotide excision repair, mismatch repair, direct repair and recombinational repair.7, 8 Comparative genomic hybridization can help to identify relevant genes involved in tumour chemotherapy‐resistance and to predict response and cancer prognosis.
In this study, the diversity and magnitude of transcriptional responses to genotoxic damage induced by MTX were assessed in two castration‐resistant prostate cancer (CRPC) xenografts types and their controls using gene expression profiling. Identification of up‐ and down‐regulated gene expression levels in MTX‐resistant CRPC could facilitate improved screening and understanding the underlying mechanisms of MTX resistance, paving the way for the development of targeted interventions that can circumvent such resistance to treatment.
2. MATERIALS AND METHODS
2.1. Cell viability assay prepared for SCID mice inoculation
Prostate cancer cell lines DU145 and PC3 were provided kindly by Fred Hutchinson Cancer Research Center. The cells were seeded in 96‐well plates in quintuplicate with Dulbecco's Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, USA) basal medium plus 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin, at 37°C in a humidified atmosphere with 5% CO2. After 24 hours of culture, cells were treated with MTX (0.01, 0.1, 1 or 10 mg/mL) for 24, 48 and 72 hours respectively. Cell medium was removed and 100 mL/well of MTT solution (0.5 mg/mL in PBS) were added and incubated for 3 hours (at 37°C, protected from light). After the end of incubation, the supernatants were removed carefully, 150 mL of dimethyl sulfoxide was added to each well. The cells were then shook for 10 minutes in the dark. Absorbance was measured at 450 nm in a Microplate Reader (Bio‐Rad 680). Analysis of the obtained results was done using GraphPad Prism 4 computer program to evaluate cell proliferation rate and cytostatic rate. Untreated cells were used as controls. For in vivo experiment, DU145 and PC3 cells were cultured in DMEM supplemented with 10% charcoal‐stripped FBS (Hyclone) and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. Confluent cells were harvested with trypsin‐ethylenediaminetetraacetic acid (EDTA) (0.05% trypsin and 0.53 mmol/L tetrasodium EDTA), centrifuged 5 minutes at 225 g and resuspended in the medium at 1 × 107/mL single cells. Aliquots of 0.1 mL were used for subcutaneous injection into CB‐17 severe combined immunodeficiency (SCID) mice (purchased from Guangzhou Provincial Medical Experimental Center).
2.2. Tumour inoculation and treatment
The animal study was carried out in a specific pathogen‐free room and was approved by the Medical Ethics Committee of the Zhengzhou University in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 80‐23, revised 1996). Four to six weeks old CB‐17 male SCID mice were used in the experiment. Cells (1 × 106 cells) were injected subcutaneously into both flanks resulting in two tumours per mouse to test the MTX sensitivity. Once tumours became palpable, the mice were randomly divided into four treatment groups (six mice per group). In the first three groups, MTX was administered three times a week at 0.35 mg/kg, 1 mg/kg and 3.5 mg/kg respectively. The fourth group was treated with physiological saline (control) at the same time‐points. In another set of experiment, animals with palpable tumours were also assigned into four groups: MTX (3.5 mg/kg), castration, MTX (3.5 mg/kg) in combination with castration and control. Surgical castration was performed after tumours have developed. MTX and saline were administered intragastriclly in a 100 µL volume three times a week in all experiments. The diameter of subcutaneously growing tumours was measured with a calliper twice a week until the animals were killed after 6 weeks of treatment. Tumour weight was calculated by the formula: Tumour weight (mg) = (length×width2)/2.
2.3. RNA extraction, Labelling, hybridization and scanning of microarray
Total tumour RNA was extracted using Trizol reagent (Takara, Dalian, China) and concentrations were determined by a spectrophotometer (NanoDrop, Nyxor Biotech). All the processes were carried out according to the manufacturers’ instructions. Enrichment of total RNA from samples was carried out using the RNeasy Micro kit (Qiagen, Germantown, MD, USA), and samples’ quality and quantity were assessed on a spectrophotometer. Hybridization was performed in Affymetrix Human Genome U133Plus2.0 Chambers. Washes and scanning of the arrays were carried out according to manufacturer's instructions. Images were autogridded and the chemiluminescent signals were quantified, corrected for background and spot and spatially normalized. Differentially expressed genes (DEGs) were identified through filtering the dataset using P‐value <0.01 and a signal‐to‐noise ratio>2 for use in anova statistical analysis.
2.4. Data preprocessing
The analysis was carried out using r package oligo (version 1.38.0, http://bioconductor.org/packages/release/bioc/html/oligo.html) to process the Affymetrix data files data by performing background correction, data normalization and expression calculation. Probes were annotated by annotation files, and without corresponding gene symbols the probes were removed. The DEGs between control and MTX‐treatment samples were screened by non‐paired t test in limma package.9 Genes within the threshold value |logFC (fold‐change)| >1 and P‐value <0.05 were identified as DEGs.
2.5. Functional and pathway enrichment analyses
DAVID (Database for Annotation, Visualization and Integrated Discovery http://david.abcc.ncifcrf.gov/, version 6.8)10 was used for Gene Ontology (GO) enrichment analysis. The overlapping DEG in MTX‐treatment xenografts DU145R and PC3R were screened out for functional enrichment. Gene Ontology enrichment analysis was used to predict the enrichment degree and the potential functions of the DEGs in biological processes (BP), cellular components (CC) and molecular functions. In addition, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was used for systematic analysis of gene functions indicating a statistically significant difference.
2.6. Pathway enrichment and network construction
Two hundred genes with significant differences were intercepted from differential gene expression data and statistically analyzed by the GeneSpring GX software package. The target gene expression data were analysed by Cytoscape and the signal pathway is derived from tumour‐related candidate genes in microarray data. In order to find out the DEGs closely related with signal pathway, additional filtering (minimum 3‐fold change) was applied to extract the most significant of these genes which were further analysed using Cytoscape software. Those genes with known gene symbols and their corresponding expression values were uploaded into the software. Networks of these genes were algorithmically generated based on their connectivity.
2.7. Western blot analysis
Preparation of total cell lysate and the procedures for Western blot analyses were performed. Protein samples were separated on 10% polyacrylamide resolving gels with the buffer system and transferred onto nitrocellulose membranes for 2 hours at 250 mA. Protein binding sites on the nitrocellulose were blocked for 1 hour at 25°C in 5% (w/v) Marvel/PBS/3% (v/v) Tween‐20 (PBST), then incubated overnight at 4°C with PARP1, ILB1, CDH1 and PLAUR monoclonal antibodies (1:1000 dilution; Invitrogen, California, USA). The membranes were washed 3 × 10 minutes in tris buffered saline tween and probed with horseradish peroxidise‐conjugated secondary antibodies (Amersham Life Sciences, Buckinghamshire, UK) for 1 hour at 25°C. Following 3 × 10 minutes washes in PBST, bands were detected using enhanced chemiluminescence (ECL+reagents, Amersham). Densitometric quantification of band intensities was performed with Kodak one‐dimensional image analysis software.
2.8. Gene validation by qRT‐PCR
Quantitative real‐time PCR was performed with QPK‐201 SYBR Green master mix (Toyobo, Osaka, Japan) and the ABI 7300 system from Applied Biosystems. The primers used in the study were obtained from Invitrogen (Beijing, China). Thermocycling parameters included a RT step at 50°C for 20 minutes, followed by a DNA polymerase activation step at 95°C for 2 minutes and 50 PCR cycles (95°C for 20 seconds, 60°C for 30 seconds). All reactions were conducted in triplicate. The fold‐change in differential expression for each gene was calculated using the comparative CT method.
2.9. Gene expression in tumour samples from patients
Tumour samples were collected from patients with metastatic PCa. We selected patients who were confirmed diagnostic of adenocarcinoma, evidence of progression despite castrate levels of testosterone, being eligible for systemic chemotherapy based on MTX. We were able to collect seven tumour samples embedded in 2 mL of optimal cutting temperature (OCT) medium, stored at −80°C until processing, and six samples formalin‐fixed, paraffin‐embedded (FFPE). These patients have been received transurethral prostatic resection before the start of MTX treatment.
Tissues from FFPE and OCT embedded were sectioned at 15 and 25 μm thicknesses, respectively, before the RNA extraction. Total RNA from FFPE samples was obtained using the GenElute™ FFPE RNA Purification Kit (Sigma). The RNAs from OCT samples were extracted using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. Quality and quantity of the total RNAs were measured by NanoDrop‐2000 Spectrophotometer (Nanodrop Technologies).
2.10. Statistical analysis
Results are presented as the mean ± SE of the mean. To determine whether differences between groups were statistically significant, Wilcoxon rank‐sum test of variance was performed, and P < 0.05 was considered to indicate a statistically significant difference. spss 12.0 software was used for statistical analyses.
3. RESULTS
3.1. MTX sensitivity testing
DU145 and PC3 cells were cultured with increasing concentrations of MTX, at different time‐point, IC50 was determined by MTT assay. Results demonstrated that MTX decreased cell proliferation in a time and dose‐dependent manner. As shown in Figure 1A,B, the highest cytotoxicity of MTX was at 72 hours, and IC50 is 0.1 mg/mL for the PCa cells. To investigate MTX sensitivity in vivo, 106 cells of the PCa cells were inoculated into the flank region of 5‐week‐old male CB‐17 SCID mice to generate subcutaneous tumours. In general, palpable tumour formation started at 4‐5 weeks after subcutaneous implantation. Once tumours grew to nearly 200 mm3, mice were treated intragastrically with different concentrations of MTX. All animal procedures were performed according to local guidelines on animal care and with appropriate institutional certification. Tumour volume and weight were measured twice weekly and calculated by the formula as previously described. As shown in Figure 2A,B; tumour volumes differed significantly between control and MTX‐treated mice (P < 0.01), while no significant effect was noted with less than 1 mg/kg of the drug administration. MTX decreased tumour growth in a dose‐dependent manner. Volume reduction for DU145 (45.8%) and PC3 (43.2%) xenografts was observed with 3.5 mg/kg MTX. However, all mice eventually relapsed and tumours became resistant to MTX.
Figure 1.
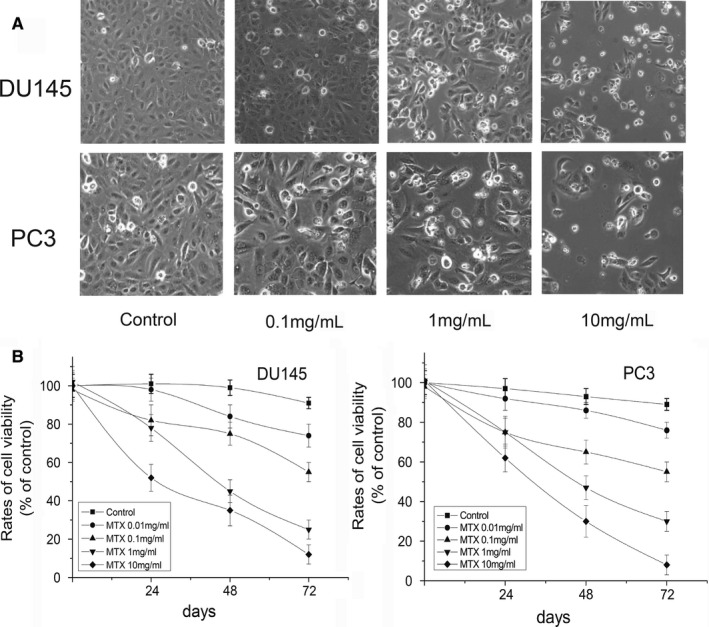
Cell morphology and viability of prostate cancer cells treated with different concentrations of mitoxantrone (MTX). A, DU145 cells: Controls (untreated cells), 0.1 mg/ml MTX, 1 mg/ml MTX, 10 mg/ml MTX; PC3 cells: Controls (untreated cells), 0.1 mg/ml MTX, 1 mg/ml MTX, 10 mg/ml MTX. B, Viability of DU145 and PC3 cells was determined by MTT assay. Error bars = SEM (n = 6)
Figure 2.
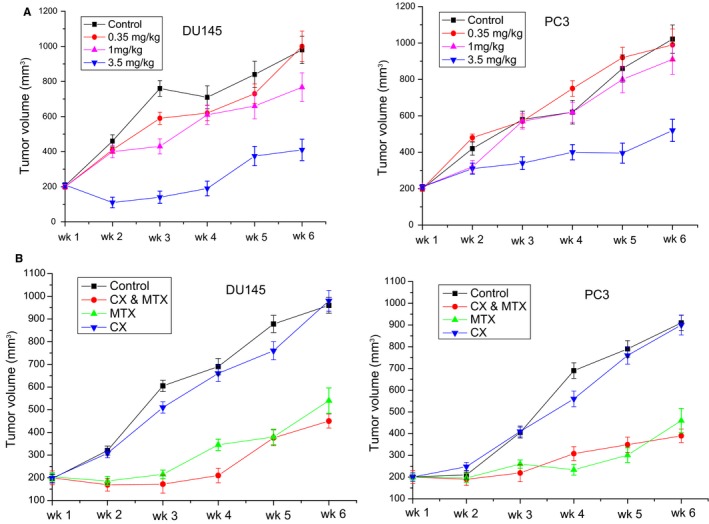
Effects of androgen deprivation and/or mitoxantrone (MTX) on prostate cancer (PCa) xenograft growth. A, Antitumour activities of different concentrations of MTX: Control, 0.35 mg/kg, 1 mg/kg and 3.5 mg/kg. B, Combined effects of androgen deprivation and MTX on PCa xenograft growth. (P < 0.015). (n = 5 mice per group)
To evaluate potential synergistic antitumour activities of androgen deprivation and MTX, SCID mice with palpable PCa xenografts were castrated and treated with 3.5 mg/kg MTX. We found that surgical castration did not significantly change the tumour size of either DU145 (P = 0.731) or PC3 (P = 0.794). However, as shown in Figure 2C,D; tumour volume was significantly decreased in the mice experienced with MTX (3.5 mg/kg) treatment (P < 0.01). On day 28, the mean tumor volume of the xenografts in control group grew from 186.65 ± 32.84 to 712.72 ± 41.26 mm3, whereas experienced castration and MTX (3.5 mg/kg) treatment, the mean tumor volumes of DU145 and PC3 were 231.23 ± 24.52 and 254.47 ± 25.71 mm3 respectively (P < 0.01). All mice eventually relapsed although consistently maintained their body weight during each study.
3.2. DEGs in MTX‐resistant xenografts
To identify the DEGs between MTX‐resistant xenografts and their controls, threshold |logFC| >1 and P‐value <0.05 were used in comparative analysis. A total of 1849, 2123 genes were extracted from the DU145R and PC3R respectively (Figure 3A,B). Among them, 986, 851 down‐regulated genes, and 863, 1272 up‐regulated genes were screened in the DU145R and PC3R xenografts respectively. We selected top 20 up‐regulated and down‐regulated genes according to the log ratio expression values (Tables 1 & 2), and among these, the ones in the MTX‐resistant groups whose expression levels were changed by more than 3‐fold compared with their control groups (P < 0.01). Upon comparison of the DEGs between both MTX‐resistant xenografts, 355 genes are overlapped, comprising 131 co‐downregulated genes and 224 co‐upregulated genes (Figure 3C,D). After that, the overlapping DEGs were clustered which can well differentiate the MTX‐treatment samples from the controls. The heatmap of the overlapping DEGs is shown in Figure 3E.
Figure 3.
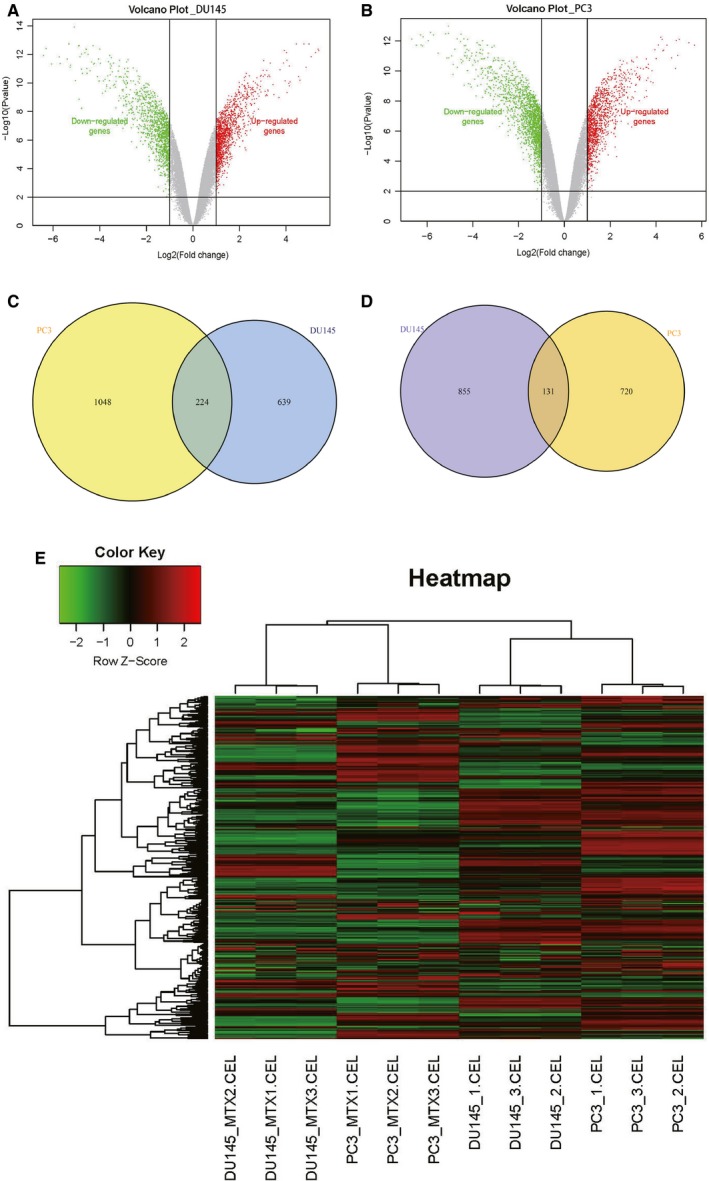
Volcano map and Venn diagram of differentially expressed genes (DEGs). A, Volcano plots of DEGs from DU145R. B, Volcano plots of DEGs from PC3R. Red spots represent up‐regulated genes, green spots represent down regulated genes, and grey dots represent non‐DEGs. C, VennPlot for the up‐regulated DEGs. D, VennPlot for the down‐regulated DEGs. E, Heatmap plot of the 355 overlapped genes between DU145R and PC3R dataset. Red represents higher expression and green represents lower expression. DEGs were selected by P < 0.05 and |log2 (fold‐change)| > 0.5. DEGs, differentially expressed genes
Table 1.
Top 10 up‐and down‐expressed genes in the xenograft of DU145R vs its control
| Primary accession | Gene symbol | Log2 ratio | Main function |
|---|---|---|---|
| NM_016192 | TMEFF2 | 5.784 | Function as both an oncogene and a tumour suppressor and may regulate prostate cancer cell invasion |
| NM_001618 | PARP1 | 5.742 | Involved in regulation of differentiation and proliferation and recovery of cell from DNA damage |
| NM_001005377 | PLAUR | 5.712 | Acts as a receptor for urokinase plasminogen activator |
| NM_001565 | CXCL10 | 5.532 | Chemotactic for monocytes and T‐lymphocytes |
| NM_012253 | TKTL1 | 5.208 | Catalyses the transfer of a two‐carbon ketol group from a ketose donor to an aldose acceptor |
| NM_000361 | THBD | 4.793 | Responsible for the conversion of protein C to the activated protein C |
| NM_007315 | STAT1 | 4.769 | Signal transducer and transcription activator |
| NM_001964 | EGR1 | 4.545 | Plays an important role in regulating the response to growth factors |
| NM_000442 | PECAM1 | 4.217 | Play a role in endothelial cell‐cell adhesion |
| NM_002585 | PBX1 | 3.736 | Acts as a transcriptional activator of PF4 in complex with MEIS1 |
| NM_198381 | ELF5 | −3.163 | Regulation of the later stages of terminal differentiation of keratinocytes and a number of epithelium‐specific genes |
| NM_020698 | TMCC3 | −3.703 | May be involved in the regulation of the proteolytic processing of the amyloid precursor protein |
| NM_001001924 | MTUS1 | −4.546 | Isoform 1 inhibits breast cancer cell proliferation, delays the progression and reduces tumour growth |
| NM_005242 | F2RL1 | −4.778 | A member of the G‐protein coupled receptor 1 family of proteins |
| NM_001624 | AIM1 | −4.875 | May function as suppressor of malignant melanoma. |
| NM_002276 | KRT19 | −5.091 | Responsible for the structural integrity of epithelial cells |
| NM_023938 | C1orf116 | −5.257 | Putative androgen‐specific receptor |
| NM_002354 | EPCAM | −5.386 | Functions as a homotypic calcium‐independent CAMs |
| NM_000165 | GJA1 | −5.695 | A gap junction protein involved in synchronized contraction of the heart and in embryonic development |
| NM_144777 | SCEL | −6.409 | May function in the assembly or regulation of proteins in the cornified envelope |
CAM, cell adhesion molecule.
Table 2.
Top 10 up‐and down‐expressed genes in the xenograft of PC3R vs its control
| Primary accession | Gene symbol | Log2 ratio | Main function |
|---|---|---|---|
| M_001005377 | PLAUR | 6.314 | Acts as a receptor for urokinase plasminogen activator |
| NM_006888 | CALM1 | 5.856 | Mediates the control of a large number of enzymes through calcium‐binding |
| NM_003133 | SRP9 | 5.484 | Plays a critical role in role in targeting secretory proteins |
| NM_016192 | TMEFF2 | 5.784 | Function as both an oncogene and a tumour suppressor and may regulate prostate cancer cell invasion |
| NM_004613 | TGM2 | 5.134 | Catalyses the cross‐linking of proteins and the conjugation of polyamines to proteins |
| NM_004061 | CDH12 | 4.678 | Cadherins are calcium‐dependent cell adhesion proteins |
| NM_001114753 | ENG | 4.482 | plays an important role in the regulation of angiogenesis |
| NM_001565 | CXCL10 | 4.369 | Chemotactic for monocytes and T‐lymphocytes |
| NM_003027 | SH3GL3 | 3.987 | Implicated in endocytosis |
| NM_001878 | CRABP2 | 3.856 | Transports retinoic acid to the nucleus |
| NM_005257 | GATA6 | −3.557 | Involved in gene regulation specifically in the gastric epithelium |
| NM_005118 | TNFSF15 | −4.078 | Mediates activation of NF‐kappa‐B |
| NM_005531 | IFI16 | −4.191 | Involved in transcriptional regulation |
| NM_024915 | GRHL2 | −4.682 | Transcription factor playing an important role in primary neurulation and in epithelial development |
| NM_004584 | RAD9A | −4.923 | inhibitor of zinc‐dependent metallocarboxypeptidases |
| NM_005797 | MPZL2 | −5.138 | Mediates homophilic cell‐cell adhesion |
| NM_004004 | GJB2 | −5.347 | Gap channels (gap junctions) are specialized cell‐cell contacts that provide direct intracellular communication |
| NM_144777 | SCEL | −5.495 | May function in the assembly or regulation of proteins in the cornified envelope |
| NM_001037330 | TRIM16 | −6.2 | Play a role in the regulation of keratinocyte differentiation |
| NM_017697 | ESRP1 | −6.676 | mRNA splicing factor that regulates the formation of epithelial cell‐specific isoforms |
3.3. Functional and pathway enrichment analysis of overlapping DEGs
Gene Ontology enrichment analysis revealed 355 overlapping genes that are involved in a number of BP including response to hypoxia, transforming growth factor β receptor signalling pathway, signal transduction and chemotaxis (Figure 4A). In terms of CC, DEGS were mostly enriched in the extracellular exosome, cell surface and lateral plasma membrane (Figure 4B). Molecular functions analysis indicated that the overlapping DEGs were mainly associated with protein binding, heparin binding and transcription factor binding (Figure 4C). Subsequential KEGG pathway enrichment analysis revealed that the common down‐regulated DEGs primarily enriched in the hippo signalling pathway, pathways in cancer, proteoglycans in cancer and cell adhesion molecules (CAMs) (Figure 4D). The top five enriched terms were presented in Table 3. These significantly enriched GO function and KEGG pathways could aid further understanding of the roles of these DEGs, involved in the development of MTX‐resistant CRPC.
Figure 4.
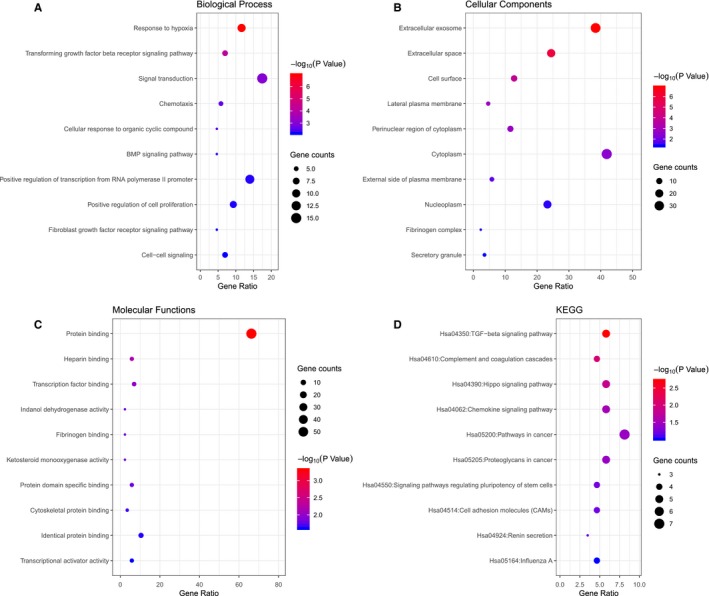
Gene Ontology‐enrichment analysis of overlapped differentially expressed genes. Here only show the top 10. A, Biological processes, B, Molecular functions, C, Cellular components, and D, Kyoto Encyclopedia of Genes and Genomes pathway analysis
Table 3.
GO function and KEGG pathway enrichment analysis of DEGs
| ID | Terms | Count | P value | Genes |
|---|---|---|---|---|
| Biological process | ||||
| GO:0001666 | Response to hypoxia | 10 | 1.12E‐07 | KCNMA1, EGR1, CCL2, EPAS1, SMAD4, THBS1, ENG, SRF, PLAU, MB |
| GO:0007179 | Transforming growth factor β receptor signalling pathway | 6 | 7.24E‐05 | CCL2, ID1, SMAD4, PARP1, GDF15, ENG |
| GO:0007165 | Signal transduction | 15 | 9.48E‐04 | SH3GL3, EPAS1, CRABP2, TANK, PLAUR, CXCL10, THBD, CD274, IL1B, INPP4B, … |
| GO:0006935 | Chemotaxis | 4 | 2.62E‐03 | ACKR3, PLAU, CXCL10, PLAUR |
| GO:0071407 | Cellular response to organic cyclic compound | 4 | 2.70E‐03 | CCL2, IL1B, STAT1, ARHGDIA |
| Cellular components | ||||
| GO:0070062 | Extracellular exosome | 33 | 1.31E‐07 | GALNT3, GNAI3, SLPI, PLAU, CDH1, CLDN11, EPCAM, TOR1A, IL1B, MB, … |
| GO:0005615 | Extracellular space | 21 | 1.23E‐06 | ADNP, CXCL10, FBLN1, THBD, CST6, HIST2H2BE, TACSTD2, SERPINB5, SLPI, IL1B, … |
| GO:0009986 | Cell surface | 9 | 1.41E‐04 | EPCAM, THBD, SLC1A3, ACKR3, AREG, THBS1, ALPP, FGFBP1, ENG |
| GO:0016328 | Lateral plasma membrane | 4 | 1.69E‐03 | EPCAM, TACSTD2, CDH1, GJB2 |
| GO:0048471 | Perinuclear region of cytoplasm | 9 | 1.72E‐03 | GALNT3,VAMP8, CDH1, MAP7, NDRG1, ACKR3, LAMC2, PRKACB, STAT1 |
| Molecular functions | ||||
| GO:0005515 | Protein binding | 57 | 4.87E‐04 | FOSL2, ATP6AP2, TNNC1, CRABP2, SDC2, CXCL10, EPCAM, SPRED2, SERPINA1, PRKACB, … |
| GO:0008201 | Heparin binding | 5 | 6.73E‐03 | CCL2, LAMC2, THBS1, FGFBP1, CXCL10 |
| GO:0008134 | Transcription factor binding | 6 | 1.07E‐02 | EPAS1, ID1, PBX1, ID3, PARP1, SRF |
| GO:0047718 | Indanol dehydrogenase activity | 2 | 1.40E‐02 | AKR1C3, AKR1C1 |
| GO:0070051 | Fibrinogen binding | 2 | 1.40E‐02 | FBLN1, THBS1 |
| KEGG pathyways | ||||
| hsa04350 | TGF‐β signalling pathway | 5 | 1.91E‐03 | ID2, ID1, SMAD4, ID3, THBS1 |
| hsa04610 | Complement and coagulation cascades | 4 | 9.42E‐03 | THBD, SERPINA1, PLAU, PLAUR |
| hsa04390 | Hippo signalling pathway | 5 | 1.51E‐02 | ID2, ID1, SMAD4, CDH1, AREG |
| hsa04062 | Chemokine signalling pathway | 5 | 2.99E‐02 | GNAI3, CCL2, PRKACB, STAT1, CXCL10 |
| hsa05200 | Pathways in cancer | 7 | 3.68E‐02 | GNAI3, EPAS1, SMAD4, CDH1, LAMC2, PRKACB, STAT1 |
DEG, differentially expressed genes; GO, Gene Ontology enrichment analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes.
3.4. Identification of candidate MTX‐resistant CRPC markers
To further clarify the core genes of DEGs identified in the microarray analysis, protein‐protein interaction (PPI) networks were generated using DU145R and PC3R significant proteins. To characterize the properties of the hub nodes based on analysis of the PPI network, maximal clique centrality were chosen to identify candidate MTX‐resistant CRPC markers. A node was identified as a hub protein if its degree is more than 2‐fold of the median degree of all nodes and highlighted with yellow colour (Figure 5A,B). For the hub significant proteins, it consists of 63 nodes and 285 edges in the DU145R network, 58 nodes and 255 edges in PC3R network. By calculating the value of the three features for each hub significant protein, the median values of “Degree,” “Betweenness” and “Closeness” for DU145R and PC3R were 9.55, 784.2, 0.019 and 10.74, 808.5, 0.018 respectively. Functional annotation and pathway analysis of the nodes in the above networks are displayed in Table 4. After performing edge percolated component and shortest path analysis, the most significant modules composed of 10 nodes were screened out from the PPI networks and the hub genes in the networks with a connectivity degree >16 were identified (Figure 5C,D). By comparing the hub genes between both MTX‐resistant xenografts, PARP1, IL8 and CDH1 are overlapped.
Figure 5.
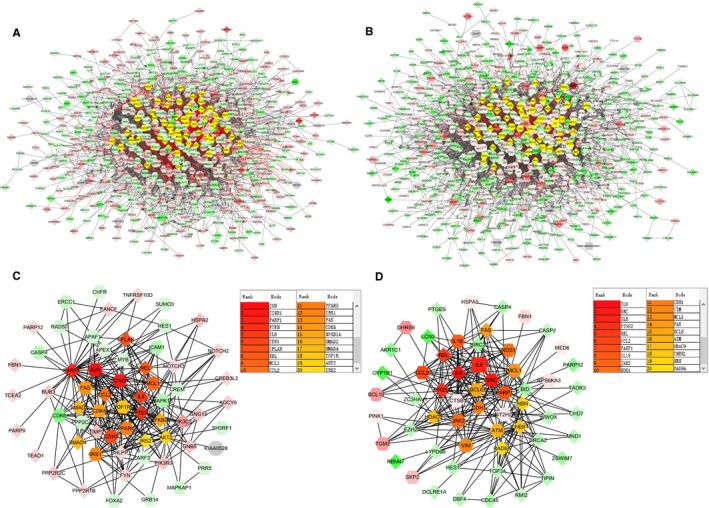
PPI networks constructed by the differentially expressed genes from mitoxantrone‐resistant xenografts. A, Network of significant proteins from DU145R. B, Network of significant proteins from PC3R. C, Network of hub significant proteins extracted from (A). D, Network of hub significant proteins extracted from (B). Red and green intensities indicate degree of up‐regulation and down‐regulation respectively. Genes in uncoloured notes were not identified as differentially expressed and were integrated into the networks indicating a relevance to this network
Table 4.
The pathways enriched for the MCC identified node in the PPI networks
| ID | Terms | Count | P value | Genes |
|---|---|---|---|---|
| DU145R | ||||
| GO:0007219 | Notch signalling pathway | 4 | 3.68E‐03 | HES1, NOTCH3, NOTCH2, CDK6 |
| GO:0006351 | Transcription, DNA‐templated | 12 | 3.49E‐05 | HES1, CCND1, MAPK13, CREM, JUN, PPARG, SMAD4, SMAD2, TCEA2, MYB, PARP1, APEX1 |
| GO:0032000 | Positive regulation of fatty acid β‐oxidation | 3 | 2.57E‐04 | IRS2, IRS1, AKT2 |
| GO:0008286 | Insulin receptor signalling pathway | 4 | 5.33E‐04 | IRS2, PIK3R3, IRS1, AKT2 |
| GO:0046328 | Regulation of JNK cascade | 3 | 6.11E‐04 | PHLPP1, IGF1R, SH3RF1 |
| GO:2001275 | Positive regulation of glucose import in response to insulin stimulus | 3 | 1.11E‐03 | PIK3R3, IRS1, AKT2 |
| GO:0042127 | Regulation of cell proliferation | 5 | 3.26E‐03 | FYN, TNFRSF10D, JUN, NFKBIA, FAS |
| GO:0045725 | Positive regulation of glycogen biosynthetic process | 3 | 1.11E‐03 | IRS2, IRS1, AKT2 |
| GO:0034097 | Response to cytokine | 3 | 4.50E‐03 | REL, JUN, TIMP2 |
| GO:0030513 | Positive regulation of BMP signalling pathway | 3 | 4.50E‐03 | HES1, SMAD4, SMAD2 |
| PC3R | ||||
| GO:0042127 | Regulation of cell proliferation | 9 | 4.71E‐08 | BID, PTGS2, EZH2, BRCA2, BCL6, JAK2, CHEK1, FAS, SRC |
| GO:0071260 | Cellular response to mechanical stimulus | 4 | 2.92E‐04 | BCL10, CHEK1, FAS, CASP2 |
| GO:0071347 | Cellular response to interleukin‐1 | 4 | 6.45E‐04 | IL6, CCL2, PTGS2, PTGES |
| GO:0050767 | Regulation of neurogenesis | 3 | 8.27E‐04 | NOS1, CHD7, BCL6 |
| GO:0006954 | Inflammatory response | 6 | 1.04E‐03 | CCL2, CASP4, PTGS2, REL, JAK2, FAS |
| GO:0000724 | Double‐strand break repair via HR | 4 | 1.14E‐03 | NBN, ZSWIM7, BRCA2, ATM |
| GO:0045087 | Innate immune response | 6 | 1.33E‐03 | BCL10, IL6, CASP4, REL, JAK2, SRC |
| GO:0097192 | Extrinsic apoptotic signalling pathway in absence of ligand | 3 | 3.94E‐03 | MCL1, FAS, CASP2 |
| GO:0070301 | Cellular response to hydrogen peroxide | 3 | 4.56E‐03 | IL6, CYP1B1, EZH2 |
| GO:0050727 | Regulation of inflammatory response | 3 | 8.72E‐03 | CASP4, BCL6, JAK2 |
MCC, maximal clique centrality.
3.5. Validation of gene expression data by Western blotting and qRT‐PCR
The expression patterns of four DEGs, PARP1, IL1B, CDH1 and PLAUR were evaluated by Western blot (Figure 6A) and quantitative real‐time PCR (qRT‐PCR) (Figure 6B). Results showed that up‐regulated ILB1 expression at the mRNA level, and enhanced positive expression of PARP1 and PLAUR in both DU145R and PC3R MTX‐resistant PCa xenografts. However, CDH1 was down‐expressed markedly in both MTX‐resistant tumour types as compared to their respective controls. In addition, a panel of 16 DEGs with the highest and lowest expression range in both androgen‐independent MTX‐resistant xenografts DU145R and PC3R vs their controls was selected and tested by qRT‐PCR (Figure 7A,B). Results showed that most of these genes exhibited a similar transcriptional profile to that of microarray data. The Pearson correction coefficient between the qRT‐PCR and microarray data for the 16 DEGs was 0.81. Hence, the microarray provided a reliable comparison of gene expression between androgen‐independent MTX‐resistant xenografts and untreated PCa.
Figure 6.
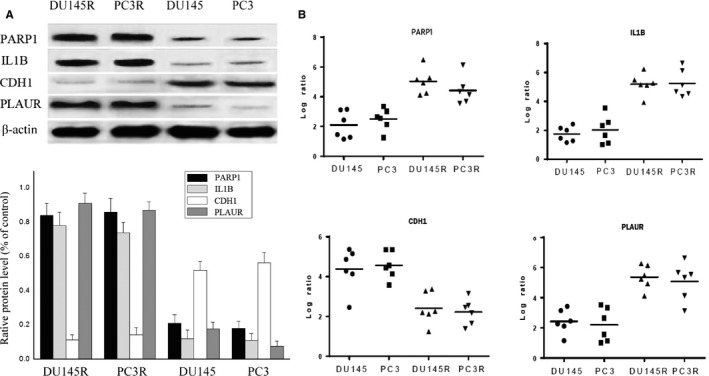
Expression profiles of PARP1, ILB1, CDH1 and PLAUR were evaluated by Western blot and qRT‐PCR. A, Western blot. B, Results expressed as western blotting band intensity. C, qRT‐PCR. Means ± SEM (n = 4)
Figure 7.
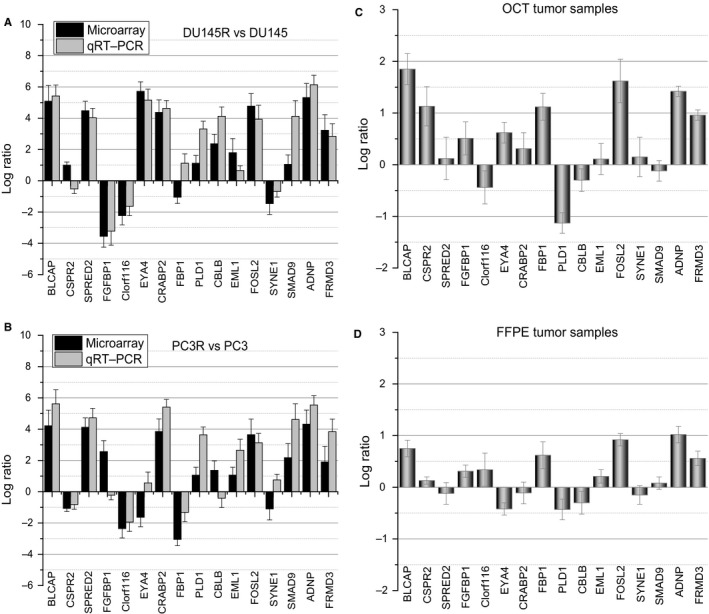
qRT‐PCR analysis of differentially expressed genes identified in the microarray. A, DU145R vs DU145, B, PC3R vs PC3, C, optimal cutting temperature (OCT) and D, formalin‐fixed, paraffin‐embedded (FFPE) tumour samples from patients with metastatic castration‐resistant prostate cancer. Expression data are represented by a log ratio calculated by comparing ΔCq from the xenograft with ΔCq from the controls. ΔCq was calculated as the difference between Cq of the targeted genes and Cq of the endogenous control gene ACTB
Moreover, the 16 DEGs were also analysed in MTX‐resistant (n = 4) vs MTX‐sensitive (n = 3) OCT tumour samples, and MTX‐resistant (n = 5) vs MTX‐sensitive (n = 4) FFPE tumour samples from patients with metastatic CRPC (Figure 7C,D). This analysis showed that BLCAP and FOSL2 are significantly up‐regulated in OCT samples. Of the 16 DEGs studied, 12 genes in the OCT tumours and 10 genes in the FFPE tumours were deregulated in the same way as the xenograft models. Interestingly, BLCAP, FOSL2, ADNP and FRMD3 were concurrently up‐expressed in the androgen‐independent, MTX‐resistant xenografts, as well as in both FFPE and OCT samples from MTX‐resistant CRPC patients.
4. DISCUSSION
This study revealed global pathways and networks of DEGs involved in DU145R and PC3R MTX‐resistant PCa xenografts. Prostate cancer is a heterogeneous disease and many molecular methods have been used in the search to determine the mechanism behind its development, and find new therapeutic and prognostic targets.11 Microarray technology for gene expression profiling has proven to be successful in a variety of experimental settings having the potential to discover the diversified and dynamic molecules during tumour progression.12, 13 Despite enormous efforts made for differential expression detection and biomarker discovery, few methods have been investigated the gene expression level in tumour stage during MTX‐resistant progression. For this, our study attempt to identify putative molecules which may act as specific targets in cancer prognosis and therapy in future.
Our microarray study identified several hub genes and established their association with various genetic networks and biological pathways likely affecting MTX resistance in CRPC. Microarray analysed results revealed that PARP1, also known as a DNA nick sensor, overexpressed in DU145R and PC3R xenografts. Poly (ADP‐ribose) polymerase (PARP) is a family of 17 proteins involved in regulation of various cellular machineries, including necrosis, DNA repair, genomic stability, post‐translational modification of proteins and parthanatos.14, 15, 16 Recent analyses demonstrated that four of 17 human PARP (PARP1, PARP2, PARP4, PARP5) exhibit PARylating activity in vitro.17, 18 PARP activity is mainly due to PARP1 enzyme which detects DNA double‐strand breaks (DSBs) or DNA single‐strand breaks playing a role in protecting stalled replication forks from nuclease‐mediated degradation.19, 20 It was reported that PARP1 is also activated by some abnormal DNA structures or external signals through ERK pathway.21 Besides its canonical role in DNA repairing systems, PARP has demonstrated broader functions in controlling cell survival and death in modulating key components of angiogenesis in cancer cells.22 Inhibition of PARP1 is being exploited for the cancer treatment.23, 24 In fact, PARP inhibitors have showed an extremely promising anticancer treatment and are currently tested in phase I and II clinical trials in different solid tumours.25
Network analysis helps us obtain global and integrated molecular information about interactions among the significant DEGs. One important network was identified around the AKT2 genes. AKT2 is a putative oncogene encoding a protein belonging to the serine‐threonine protein kinase, also known as protein kinase B, PKB (AKT) subfamily of serine/threonine‐protein kinases, as well as a key node on the phosphatidylinositol 3‐kinase (PI3K/AKT) pathway which is recognized as a key pathway in carcinogenesis occurring commonly in diverse human cancer cells.26, 27 Previous studies have revealed that aberrant activation of PI3K/AKT pathway is also closely associated with the process of cancer metastasis.28 PI3K phosphorylates AKT and consequently facilitates tumourigenesis and cancer progression through its downstream targets.29 Report indicated that expressing active AKT can avoid apoptosis and checkpoint‐dependent cell cycle arrest due to suppression of homologous recombination (HR) and reliance on error‐prone NHEJ.30 Studies demonstrated that DNA damage activated AKT2 expression and subsequently conferred apoptotic resistance in ovarian cancer cells.31 At the same network, significantly decreased expression of CASP4, CASP2 and TNF‐receptor superfamily protein fas cell surface death receptor (FAS) was observed among the DEGs of MTX‐resistant xenografts.
In our study, Ataxia‐telangiectasia mutated kinase (ATM) was found to be highly up‐regulated in MTX‐resistant xenografts, and observed as a hub gene in the network. This gene is also highly expressed in non‐small cell lung cancer (NSCL) exposed to carbon ion irradiation.32 Ataxia‐telangiectasia mutated kinase, a member of PI3K/AKT family protein, its main function is to control the cell cycle progression following DNA damage, particularly DSBs.33 Reports indicated that MTX produce DNA cross‐links and DNA replication defects in tumour cells, once DNA damage occurs, ATM pathway for HR repair is activated.34 DNA repair is an essential prerequisite for the maintenance of genomic integrity and cellular viability.35 It is demonstrated that DNA‐damaging drugs (including MTX) can trigger an ATM‐dependent DNA damage response, leading to increased cytokine secretion and resistance to chemotherapy‐induced apoptosis.36 Report also revealed that ATM together with phosphotyrosine binding domain and a leucine zipper motif (APPL), a regulator of ATM phosphorylation, modulates DNA damage repair and consequently raises the survival of pancreatic carcinoma cells.37 In addition, ATM has been implicated in the control of RNA splicing that may play a role in genomic stability.38, 39
DNA‐damage response activates a secretory program that comprises a diverse spectrum of proteases, growth factors and, cytokines that have been shown to contribute to wound healing and altered immune responses.40, 41 Interleukin‐6 (IL‐6), at high concentrations, is found to protect cancer cells from therapy‐induced DNA damage, oxidative stress and apoptosis by facilitating the repair and induction of antioxidant response.42 In our study, IL6 is up‐regulated and screened as a hub gene in androgen‐independent, MTX‐resistant xenografts. Evidence support DNA damage and stress induce sustained IL‐6 and IL1B production in PCa through P2Y11 receptor‐p38 MAPK‐NF‐κB signalling pathway,6, 43 which may play an essential role in promoting cancer cells proliferation, survival, invasiveness and metastasis. Therefore, inhibition of IL‐6 or in combination with conventional anticancer therapies may be a potential therapeutic strategy for the treatment of MTX‐resistant PCa.
Sixteen DEGs were selected to be validated by qRT‐PCR according to their function and relative expression level in MTX‐resistant xenografts. These DEGs were further tested in MTX‐resistant CRPC patient samples (seven OCT and six FFPE samples). Results confirmed that BLCAP, EML1, FOSL2, ADNP and FRMD3 were deregulated in the same way among the xenografts, FFPE and OCT tumour samples. However, discordant results were observed in the expression of other genes such as PLD1, which was down‐expressed in MTX‐resistant tumour samples but was significantly overexpressed in MTX‐resistant xenografts. These conflicting results may be due to cellular heterogeneity between the xenografts from cell lines and the tumour sample from patients. Therefore, further clinical validation of these results is needed in a large cohort of patients.
In analysing, often individual genes were found in multiple categories of functions related to cancer development including cell signalling, cell death, cellular growth and proliferation. Besides, it reminds us there are certain limits in the analysis as there are many different gene interactions resulting from various cellular/experimental conditions. Nevertheless, this exploratory analysis may be still useful to bestow a theranostic perspective to the current trend of research in PCa or to develop targeted therapies to overcome MTX chemotherapy resistance.
DATA AVAILABILITY
The data of the study have been deposited into the Research Data Deposit (http://www.researchdata.org.cn), with the Approval Number as RDDB2018000423.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
AUTHORS CONTRIBUTIONS
All authors took part in writing, reviewing and editing the manuscript. S. Zhu, SQ Li and T. Huang designed experiments; RF Li, Y. Ma, C. Zhang, H. Wang, CJ Guo performed experiments; S. Zhu, Y. Ma, C. Zhang, T. Huang prepared figures; All authors reviewed the manuscript and approved it for publication.
ACKNOWLEDGEMENTS
This study was supported by grants from Medical Key Technologies R & D Program of Henan Province (201402004) and Natural Science and Technologies Program of Henan Province (172102410018).
Li S, Li R, Ma Y, Zhang C, Huang T, Zhu S. Transcriptome analysis of differentially expressed genes and pathways associated with mitoxantrone treatment prostate cancer. J Cell Mol Med. 2019;23:1987–2000. 10.1111/jcmm.14100
Contributor Information
Tao Huang, Email: huangtao_70@163.com.
Sha Zhu, Email: zsha@zzu.edu.cn.
REFERENCES
- 1. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24(14):2137‐2150. [DOI] [PubMed] [Google Scholar]
- 2. Rydzewska L, Burdett S, Vale CL, et al. Adding abiraterone to androgen deprivation therapy in men with metastatic hormone‐sensitive prostate cancer: a systematic review and meta‐analysis. Eur J Cancer. 2017;84:88‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. 1941. J Urol. 2002;167(2 Pt 2): 948‐951. [PubMed] [Google Scholar]
- 4. Flaig TW, Potluri RC, Ng Y, et al. Treatment evolution for metastatic castration‐resistant prostate cancer with recent introduction of novel agents: retrospective analysis of real‐world data. Cancer Med. 2016;5(2):182‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee CH, Hsieh MY, Hsin LW, et al. Anthracenedione‐methionine conjugates are novel topoisomerase II‐targeting anticancer agents with favorable drug resistance profiles. Biochem Pharmacol. 2012;83(9):1208‐1216. [DOI] [PubMed] [Google Scholar]
- 6. Gomez‐Sarosi L, Sun Y, Coleman I, et al. DNA damage induces a secretory program in the quiescent TME that fosters adverse cancer phenotypes. Mol Cancer Res. 2017;15(7):842‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864‐870. [DOI] [PubMed] [Google Scholar]
- 8. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366‐374. [DOI] [PubMed] [Google Scholar]
- 9. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang DW, Sherman BT, Tan Q, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(suppl_2): W169‐W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kato T, Hashimoto Y, Wong RK, et al. Influence of lifestyle choices on risks of CYP1B1 polymorphisms for prostate cancer. J Cell Mol Med. 2018;22(10):4676‐4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abulwerdi FA, Schneekloth JS Jr. Microarray‐based technologies for the discovery of selective. RNA‐binding molecules. Methods. 2016;103:188‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hussain SA, Palmer DH, Syn WK, et al. Gene expression profiling in bladder cancer identifies potential therapeutic targets. Int J Oncol. 2017;50(4):1147‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18(10):610‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stark JM, Pierce AJ, Oh J, et al. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol. 2004;24(21):9305‐9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kraus WL. Transcriptional control by PARP‐1: chromatin modulation, enhancer‐binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20(3):294‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vyas S, Matic I, Uchima L, et al. Family‐wide analysis of poly(ADP‐ribose) polymerase activity. Nat Commun. 2014;5:4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Otto H, Reche PA, Bazan F, et al. In silico characterization of the family of PARP‐like poly(ADP‐ribosyl)transferases (pARTs). BMC Genom. 2005;6:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schlacher K, Christ N, Siaud N, et al. Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ray Chaudhuri A, Callen E, Ding X, et al. Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature. 2016;535(7612):382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohen‐Armon M. PARP‐1 activation in the ERK signaling pathway. Trends Pharmacol Sci. 2007;28(11):556‐560. [DOI] [PubMed] [Google Scholar]
- 22. Ha HC, Snyder SH. Poly(ADP‐ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96(24):13978‐13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferrari N, Granata I, Capaia M, et al. Adaptive phenotype drives resistance to androgen deprivation therapy in prostate cancer. Cell Commun Signal. 2017;15(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bitler BG, Watson ZL, Wheeler LJ, et al. PARP inhibitors: clinical utility and possibilities of overcoming resistance. Gynecol Oncol. 2017;147(3):695‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matulonis UA, Wulf GM, Barry WT, et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high‐grade serous ovarian and breast cancer. Ann Oncol. 2017;28(3):512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petrulea MS, Plantinga TS, Smit JW, et al. PI3K/Akt/mTOR: a promising therapeutic target for non‐medullary thyroid carcinoma. Cancer Treat Rev. 2015;41(8):707‐713. [DOI] [PubMed] [Google Scholar]
- 27. Limb JK, Song D, Jeon M, et al. 2‐(trimethylammonium)ethyl (R)‐3‐methoxy‐3‐oxo‐2‐stearamidopropyl phosphate promotes megakaryocytic differentiation of myeloid leukaemia cells and primary human CD34(+) haematopoietic stem cells. J Tissue Eng Regen Med. 2015;9(4):435‐446. [DOI] [PubMed] [Google Scholar]
- 28. Lu C, Shan Z, Hong J, et al. MicroRNA‐92a promotes epithelial‐mesenchymal transition through activation of PTEN/PI3K/AKT signaling pathway in non‐small cell lung cancer metastasis. Int J Oncol. 2017;51(1):235‐244. [DOI] [PubMed] [Google Scholar]
- 29. Zhou Y, Gu P, Li J, et al. Suppression of STIM1 inhibits the migration and invasion of human prostate cancer cells and is associated with PI3K/Akt signaling inactivation. Oncol Rep. 2017;38(5):2629‐2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Piscitello D, Varshney D, Lilla S, et al. AKT overactivation can suppress DNA repair via p70S6 kinase‐dependent downregulation of MRE11. Oncogene. 2018;37(4):427‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin X, Shen J, Dan P, et al. RNA‐binding protein LIN28B inhibits apoptosis through regulation of the AKT2/FOXO3A/BIM axis in ovarian cancer cells. Signal Transduct Target Ther. 2018;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang L, Liu Y, Sun C, et al. Inhibition of DNA‐PKcs enhances radiosensitivity and increases the levels of ATM and ATR in NSCLC cells exposed to carbon ion irradiation. Oncol Lett. 2015;10(5):2856‐2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282(5395):1893‐1897. [DOI] [PubMed] [Google Scholar]
- 34. Skladanowski A, Konopa J. Mitoxantrone and ametantrone induce interstrand cross‐links in DNA of tumour cells. Br J Cancer. 2000;82(7):1300‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shibata A. Regulation of repair pathway choice at two‐ended DNA double‐strand breaks. Mutat Res. 2017;803‐805:51‐55. [DOI] [PubMed] [Google Scholar]
- 36. Zhao H, Traganos F, Darzynkiewicz Z. Phosphorylation of p53 on Ser15 during cell cycle caused by Topo I and Topo II inhibitors in relation to ATM and Chk2 activation. Cell Cycle. 2008;7(19):3048‐3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hennig J, McShane MP, Cordes N, et al. APPL proteins modulate DNA repair and radiation survival of pancreatic carcinoma cells by regulating ATM. Cell Death Dis. 2014;5:e1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tresini M, Warmerdam DO, Kolovos P, et al. The core spliceosome as target and effector of non‐canonical ATM signalling. Nature. 2015;523(7558):53‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wickramasinghe VO, Venkitaraman AR. RNA processing and genome stability: cause and consequence. Mol Cell. 2016;61(4):496‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gilbert LA, Hemann MT. DNA damage‐mediated induction of a chemoresistant niche. Cell. 2010;143(3):355‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rodier F, Coppe JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence‐associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11(8):973‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kumari N, Dwarakanath BS, Das A, et al. Role of interleukin‐6 in cancer progression and therapeutic resistance. Tumour Biol. 2016;37(9):11553‐11572. [DOI] [PubMed] [Google Scholar]
- 43. Glossop JR, Cartmell SH. Effect of fluid flow‐induced shear stress on human mesenchymal stem cells: differential gene expression of IL1B and MAP3K8 in MAPK signaling. Gene Expr Patterns. 2009;9(5):381‐388. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data of the study have been deposited into the Research Data Deposit (http://www.researchdata.org.cn), with the Approval Number as RDDB2018000423.